

Abstract
Multilocus variable-number tandem-repeat analysis (MLVA) is efficient for routine typing and for investigating the genetic structures of natural microbial populations. Two distinct pathovars of Xanthomonas oryzae can cause significant crop losses in tropical and temperate rice-growing countries. Bacterial leaf streak is caused by X. oryzae pv. oryzicola, and bacterial leaf blight is caused by X. oryzae pv. oryzae. For the latter, two genetic lineages have been described in the literature. We developed a universal MLVA typing tool both for the identification of the three X. oryzae genetic lineages and for epidemiological analyses. Sixteen candidate variable-number tandem-repeat (VNTR) loci were selected according to their presence and polymorphism in 10 draft or complete genome sequences of the three X. oryzae lineages and by VNTR sequencing of a subset of loci of interest in 20 strains per lineage. The MLVA-16 scheme was then applied to 338 strains of X. oryzae representing different pathovars and geographical locations. Linkage disequilibrium between MLVA loci was calculated by index association on different scales, and the 16 loci showed linear Mantel correlation with MLSA data on 56 X. oryzae strains, suggesting that they provide a good phylogenetic signal. Furthermore, analyses of sets of strains for different lineages indicated the possibility of using the scheme for deeper epidemiological investigation on small spatial scales.
INTRODUCTION
Molecular typing of pathogen populations is essential to gain insight into their genetic diversity and population dynamics in order to elaborate efficient strategies for disease control (1, 2). In agricultural systems, pests are ideally controlled by integrated approaches, including eradication or treatment of diseased organisms and planting of resistant varieties. However, the durability of resistance can be challenged if pathogen diversity is significant. Importantly, gene flow between pathogen populations can facilitate the breakdown of resistance in crop plants (3). Hence, efficient and precise molecular-typing tools for identifying strains and differentiating among related bacterial isolates are essential for microevolutionary reconstruction as a population genetics approach for integrated plant protection.
Rice, one of the major crops worldwide, is affected by two bacterial diseases that are caused by strains of Xanthomonas oryzae, bacterial leaf blight (BLB), caused by X. oryzae pv. oryzae, and bacterial leaf streak (BLS), caused by X. oryzae pv. oryzicola. Collectively, these two diseases cause significant yield losses in tropical and temperate rice-growing areas. X. oryzae pv. oryzae colonizes xylem vessels upon entry into the vascular system. X. oryzae pv. oryzicola infects the plant via natural openings and colonizes the mesophyll (4). Genomes of members of both pathovars have been sequenced; however, the determinants of tissue specificity are still largely unknown (5, 6). While X. oryzae pv. oryzicola has been shown to be seedborne and seed transmitted (7, 8), the evidence that X. oryzae pv. oryzae is seedborne is still controversial (7, 9).
Because both pathogens infect the same host species and cause symptoms that at later stages of infection may be difficult to distinguish, both BLB and BLS diseases are not easy to unambiguously diagnose in the field. In the laboratory, strains of the two pathovars are identified by inoculation methods on susceptible host plants (10). Upon leaf inoculation, X. oryzae pv. oryzicola colonizes the mesophyll, resulting in water-soaked lesions, while X. oryzae pv. oryzae colonizes and spreads in the vascular system, resulting in long lesions along the leaf blade (4, 10). Besides the observation of visual symptoms, and for a more reliable diagnosis, a genomics-based multiplex PCR has been developed to differentiate the two pathovars (11). Recently, these diagnostic loci were converted into a loop-mediated isothermal amplification (LAMP) assay (12). In addition to pathovar discrimination, other assays were developed that can differentiate X. oryzae strains from Africa and Asia (13). Currently, phenotypic, multiplex PCR, and LAMP methods are routinely used by several laboratories to identify, to characterize, and to detect X. oryzae strains.
BLB was first described in Fukuoka Prefecture, Japan, in 1884 (14, 15) and has been reported in a number of rice-growing countries from Iran to Japan and Philippines (16). The disease was also reported in African countries, including Mali, Senegal, Niger, Gabon, Nigeria, Cameroon, Burkina Faso, Mauritania, and the Gambia (17). Reports for northern Australia (18) and South America (19) exist but are rather sporadic and less important (16).
BLS was first described in Philippines in 1918 and is prevalent in Asia (16), as well as in Africa in Mali (20) and Burkina Faso (21), and was recently reported in Madagascar (22), Burundi (23), and Uganda (24).
So far, studies have focused on X. oryzae pv. oryzae, describing the genetic and pathotypic diversity of different populations. Most of these studies analyzed regional populations of X. oryzae pv. oryzae, mainly using pathogenicity assays and/or restriction fragment length polymorphism (RFLP) profiling (25–30). For instance, Mishra and coworkers (2013) analyzed more than 1,000 isolates of X. oryzae pv. oryzae based on their reactions to 10 resistance genes. They also differentiated a subset of strains by an RFLP analysis using IS1112 as a probe. However, the study included only strains from India (30).
Much less is known about X. oryzae pv. oryzicola diversity. In Philippines, a study evaluated the diversity of 123 X. oryzae pv. oryzicola strains by RFLP analysis, concluding that the pathovar is endemic due to the large diversity of strains (31). A repetitive-element palindromic (rep)-PCR analysis of 141 Chinese X. oryzae pv. oryzicola strains revealed significant genetic diversity among X. oryzae pv. oryzicola strains in southwest China (32). One of the most exhaustive phylogenetic studies of X. oryzae was performed by Gonzalez and coworkers, who analyzed a set of 26 Asian X. oryzae pv. oryzae strains, 21 African X. oryzae pv. oryzae strains, and 14 X. oryzae pv. oryzicola strains from different origins (20). Using a polyphasic approach, including RFLP, rep-PCR, and fluorescent amplified fragment length polymorphism (AFLP), three lineages were defined within the species as Asian X. oryzae pv. oryzae, African X. oryzae pv. oryzae, and X. oryzae pv. oryzicola (from Asia and Africa). More recently, multilocus sequence typing (MLST) targeting nine housekeeping genes of a few strains of X. oryzae confirmed the designation of the three lineages (33). Other studies combining multilocus sequence analysis (MLSA) with the analysis of the type III effector repertoires of 40 X. oryzae strains belonging to different pathovars and from different geographical origins clearly confirmed that both X. oryzae pv. oryzae and X. oryzae pv. oryzicola pathogens belong to closely related but distinct phylogenetic groups formerly defined as lineages (20, 34). Finally, MLST and RFLP analyses focusing on a large collection of X. oryzae pv. oryzicola strains revealed a high level of genetic diversity among African strains (35).
Molecular methods previously used to evaluate the genetic diversity of X. oryzae were either cumbersome, poorly reproducible, insufficiently discriminative, or difficult to interpret evolutionarily. Over the past several years, MLST based on a set of housekeeping genes has become popular to investigate microbial populations of bacterial pathogens (36). However, this technique does not have enough resolution for an in-depth study of X. oryzae populations or epidemic outbreaks. Now, with easy access to nearly complete genome sequences, single-nucleotide polymorphism (SNP) analyses of genomes receive more attention due to the unprecedented increase in resolution they provide and the consequent possibility of investigating epidemics more accurately (37, 38). Although the costs of genome sequencing have decreased tremendously in the last few years, such typing methods are unlikely to become widely adopted as a standard due to bioinformatic and infrastructural constraints. Most developing countries that are concerned with epidemiological surveillance of plant diseases are not yet equipped for such analyses.
First reported in eukaryotic species, DNA motifs that are repeated in multiple copies were identified in bacterial genomes (39). Variation between strains is reflected by a change in the size of the repeat array, called variable-number tandem repeats (VNTRs) or mini- or microsatellites. Among the different models of mutations of microsatellites, the stepwise mutation model (SMM) was widely adopted for microsatellites, even if large jumps in repeat numbers may occasionally occur (40, 41). The SMM postulates that the size of a VNTR locus evolves through the addition or deletion of one repeat unit per mutation event. Consequently, VNTR loci provide us with connectible data reflecting patterns of evolutionary descent that can be used for epidemiological tracing of bacterial strains. The flanking regions next to the repeats are generally well conserved, sometimes even among different species. Consequently, PCR primers could be designed allowing the analysis of VNTR polymorphisms at different levels, e.g., the species or subspecies level (42). Since 2001, shortly after the first genome sequence became available, VNTR studies of bacterial plant pathogens became more and more popular, as exemplified by Xylella fastidiosa (43). In 2009, the first VNTR typing scheme was developed for Xanthomonas species (44). Later, a 25-locus-based VNTR scheme (named MLVA-25) was developed for X. oryzae pv. oryzicola (45) and evaluated on a limited collection of X. oryzae pv. oryzicola strains. Preliminary in silico analyses indicated that a few loci would be useful to characterize the X. oryzae pv. oryzae strains, as well, but the discriminatory power would have been rather low for the two X. oryzae pv. oryzae lineages in comparison to the X. oryzae pv. oryzicola lineage. Hence, a highly discriminatory multilocus variable-number tandem-repeat (MLVA) scheme that could at the same time identify the different X. oryzae pathovars and distinguish strains within pathovars would be very useful.
Here, we report on a new multilocus VNTR analysis that allows the production of robust and reproducible genetic data to efficiently characterize X. oryzae strains and to study epidemics of BLB and BLS on rice. For this purpose, a collection of 338 strains of the X. oryzae pathovars oryzae and oryzicola, originating from 20 countries, was analyzed. Both global and small-scale MLVAs of the three X. oryzae genetic lineages are discussed in the context of the geographical origins of the strains, the sampling dates, and the host plants. With this work, we wish to promote the worldwide use of MLVA-16 (a 16-locus-based VNTR scheme) for monitoring of BLB and BLS on various temporal and geographical scales.
MATERIALS AND METHODS
Bacterial strains and DNA isolation.
X. oryzae pv. oryzae and X. oryzae pv. oryzicola strains were obtained from different sources (see Table S1 in the supplemental material). The strains were representative of the different lineages (127 X. oryzae pv. oryzae strains from Asia, 59 X. oryzae pv. oryzae strains from Africa, and 152 X. oryzae pv. oryzicola strains from Africa and Asia). The isolates were maintained in 15% glycerol at −80°C. For DNA extraction, the bacteria were streaked on PSA plates (10 g/liter peptone, 10 g/liter sucrose, 1 g/liter glutamic acid, 15 g/liter Bacto Agar) and incubated for 24 h at 28°C. Two loops of bacterial cultures were washed in 1 M NaCl and then processed using the Wizard genomic DNA purification kit (Promega, Charbonnières, France) following the manufacturer's instructions.
Exploitation of genomic resources for VNTR locus extraction.
A bioinformatic pipeline (http://www.biopred.net/VNTR/) for the prediction of VNTR loci was applied to 10 X. oryzae genome sequences with the following parameters: algorithm, TRF; region length, 30 to 1,000 bp; unit length, 5 to 12 bp; and at least 6 tandem repeats with a similarity of at least 80% among the repeats (45). Four complete publicly available genome sequences were screened: three X. oryzae pv. oryzae strains from Asia, KACC 10331 (GenBank accession number NC_006834), MAFF 311018 (accession no. NC_007705), and PXO99A (accession no. NC_010717), as well as one Asian X. oryzae pv. oryzicola strain, BLS256 (accession no. NC_017267). In addition, we included six draft genome sequences: Asian X. oryzae pv. oryzae (PXO86), African X. oryzae pv. oryzae (BAI3, MAI1, and NAI8; accession no. AYSX00000000), and X. oryzae pv. oryzicola (GXO1 and MAI10; accession no. AYSY00000000). The pipeline first predicts VNTR loci for each individual genome sequence. In a second step, the VNTR loci are grouped by homology based on the conservation of flanking sequences. In the last step, genomes for which a certain VNTR locus was not predicted in step 1 (e.g., because they do not match the search criteria, such as the minimal number of repeats) are searched for homologous loci, as revealed by the presence of conserved flanking sequences. Finally, amplicon sizes were predicted for the 10 sequenced genomes at the website (http://minisatellites.u-psud.fr/ASPSamp/base_ms/blast/blast_primers_multi.php) in order to estimate if the loci are polymorphic for a given genetic lineage.
Four sets of PCR primers were designed for quadruplex PCRs with an optimal size range of the amplicons and minimal overlap in size, as shown in Table 1. The primers were designed based on a sequence alignment of the 500-bp flanking regions next to the VNTR loci, and the designed primers were subsequently tested (http://www.thermoscientificbio.com/webtools/multipleprimer/) for the optimal annealing temperature and avoidance of dimer formation.
TABLE 1.
Multiplexing scheme for primer pairs in pools
| Pool | Locus | Discriminative for (polymorphic): | Diagnostic for (monomorphic)a: | Primer 1 |
Primer 2 |
||
|---|---|---|---|---|---|---|---|
| Labelb | Sequence | Label | Sequence | ||||
| 1 | Xo_G06 | X. oryzae pv. oryzae Asia, X. oryzae pv. oryzae Africa, and X. oryzae pv. oryzicola | VIC | GCAGACGGATGGGCGTTG | GCTCGCCGGCACTCTCCT | ||
| Xo_G07 | NED | CAGGGCGAACGCGATGAG | GCTCCATGGTGCCGGAGA | ||||
| Xo_G58 | PET | CAACGAGGTGCCCGGCAA | GGAGCGCGCACATCATCG | ||||
| Xo_G81 | GTGCCGTCTCCCGACGCT | 6-FAM | GCCATCGCTACGCTGCGG | ||||
| 2 | Xo_G2553 | X. oryzae pv. oryzae Asia and X. oryzae pv. oryzae-Africa | X. oryzae pv. oryzicola (372) | GCTGGCGGTGACCACCAC | 6-FAM | GTCCAGCAGGTGCTCGCG | |
| Xo_G88 | X. oryzae pv. oryzicola (312) | VIC | CTCACTGCGGCGGTGTTG | TAAGCTGCGCAAGGCGCG | |||
| Xo_G44 | PET | GCCGTGCTTCCGTCTGCA | CCGCATCCATCGCGACAG | ||||
| Xo_G62 | X. oryzae pv. oryzicola (141) | GCTTCGCCGACCACGTGA | NED | ACGACAAGCAGCGCCTGC | |||
| 3 | Xo_G55 | X. oryzae pv. oryzae Africa and X. oryzae pv. oryzicola | X. oryzae pv. oryzae-As (294) | VIC | ACCCGGCAACTCGCAACC | GGCACGAGCAAGCGGCAT | |
| Xo_G59 | X. oryzae pv. oryzae-As (108) | PET | TCATTGCGTCACCCATCGG | GGCGGGCGCTATCAACTGTCC | |||
| Xo_G60 | X. oryzae pv. oryzae-As (197, 185) | GTTGCCGCCGCCGGTAC | 6-FAM | CGGCGTCTTGCCAACCCT | |||
| Xo_G67 | X. oryzae pv. oryzae-As (322) | NED | GCTTGGCGGGTCACATCG | TGGATCGACGCCGGACTG | |||
| 4 | Xo_G09 | X. oryzae pv. oryzae Asia and X. oryzae pv. oryzicola | X. oryzae pv. oryzae-Af (133) | CCGCGATAGGCCGAGGTC | 6-FAM | TGGCCTATCTGGCAGCGC | |
| Xo_G15 | X. oryzae pv. oryzae-Af (137) | CGCAACGATGTGCTGGCG | VIC | CGCAATCGCTTCGAACGC | |||
| Xo_G80 | X. oryzae pv. oryzae-Af (346) | PET | ACGGATGGCGTTGGCCAG | CGGCATGATCCTGGGCG | |||
| Xo_G83 | X. oryzae pv. oryzae-Af (422) | NED | CACCCGGCCGGCAATATC | TGCACCACCACGTTGGCG | |||
Numbers in parentheses indicate predicted sizes of PCR amplicons (in base pairs) based on the analysis of 10 genome sequences.
The following fluorescent labels of PCR primers were used (excitation/emission wavelengths in parentheses): VIC (535/555 nm), NED (550/570 nm), PET (570/590 nm), and 6-FAM (495/515 nm).
MLVA genotyping.
Genotyping was performed based on the method described for Xanthomonas citri pv. citri (46). Basically, four VNTR loci were amplified in a quadruplex PCR with four primers 5′ labeled with 6-carboxyfluorescein (6-FAM), NED, PET, and VIC fluorescent dyes (Applied Biosystems). Conditions were optimized based on test runs (Table 1). One microliter of diluted amplicons (10- to 50-fold dilution, determined from test runs) was mixed with 0.3 μl of the GeneScan −500 LIZ internal size standard (Applied Biosystems) and 10.7 μl of Hi-Di formamide (Applied Biosystems). Capillary electrophoresis was performed on an ABI Prism-3130xl genetic analyzer.
Amplicon sizes were scored with GeneMapper 4.0 (Applied Biosystems) and then converted to a number of tandem repeats (TRs) per lineage as described in Data Set S1 in the supplemental material. As a standard, when a TR copy was truncated, the TR number was rounded to the next higher number.
Bioinformatic analyses.
Hunter-Gaston Discriminatory Index (HGDI) scores were calculated (via http://insilico.ehu.es/mini_tools/discriminatory_power/) in order to evaluate the discriminatory power for each locus (47). Allelic richness was calculated for different group combinations using the rarefaction method with HP-Rare v6.6.6 (48). The genetic differentiation between geographic and genetic groups was estimated as fixation indices (FST values) calculated by Arlequin v.3.5.1.3 (49). As recombination can impact the apparent phylogeny of the strains, we quantified the degree of linkage disequilibrium by estimating the multilocus indices of association, IA, and the rD (corrected index of association) using Multilocus v1.3b (50) with 1,000 randomized data sets. To investigate the correlation between VNTR data and previously published typing data, Mantel tests were performed on pairwise distance matrices with 999 permutations using GenAIEx 6.5 (51). Two data sets were used: MLSA data for 35 individuals (11 African X. oryzae pv. oryzae strains, 15 Asian X. oryzae pv. oryzae strains, and 9 X. oryzae pv. oryzicola strains) (34) and RFLP data (Southern blots hybridized with a portion of the avrXa7 gene encoding a type III effector of the transcription activator-like family) for 56 X. oryzae pv. oryzicola strains (35).
Phylogenetic relationships within each lineage were determined using different approaches. The patterns of evolutionary descent were inferred using minimum spanning trees (MST) (categorical values or Manhattan distances). Manhattan neighbor-joining (NJ) phylogenetic trees using Jacquard correlation were constructed using BioNumerics v7.2 (Applied Maths, Sint-Martens-Latem, Belgium). Clonal complexes were analyzed with eBURST v3 to find descent patterns between the allelic types (sequence types [STs]) and founder types (52). Groups of strains differing by less than three loci, i.e., consisting of identical types, single-locus variants (SLVs), and double-locus variants (DLVs), were defined as clonal complexes. Discriminant analysis of principal components (DAPC), a clustering method without a priori assumptions, was performed using R version 3.0.1 (2013-05-16) with the “adegenet” package (53).
MLVA database.
MLVA data were deposited in the public database MLVAbank hosted at Institut de Recherche pour le Développement (IRD) Montpellier (http://www.biopred.net/MLVA/), which allows sharing and comparing of information with other scientists in an interactive manner and provides access to a few phylogenetic-analysis tools (46).
RESULTS
In silico analysis.
Using 10 available genome sequences (four Asian X. oryzae pv. oryzae strains, three African X. oryzae pv. oryzae strains, and three X. oryzae pv. oryzicola strains), 92 VNTR loci with 5-bp to 12-bp repeats were identified. Surprisingly, only five VNTR loci were conserved and polymorphic among the three genetic lineages previously defined by Gonzalez and coworkers (20). Thirty-seven additional VNTR loci were conserved and polymorphic among two different genetic lineages: 5 loci among Asian X. oryzae pv. oryzae and African X. oryzae pv. oryzae strains, 6 loci among African X. oryzae pv. oryzae and X. oryzae pv. oryzicola strains, and 26 loci among Asian X. oryzae pv. oryzae and X. oryzae pv. oryzicola strains. All 50 of the other VNTR loci were specific to one lineage (either predicted to be monomorphic or absent in the other lineages). Promising VNTR loci were selected according to their presence and polymorphism in at least two lineages. PCR amplicons that were generated on a set of 20 strains (test panel) per genetic lineage were sequenced (see Table S2 in the supplemental material). The test panel included strains that represent the worldwide diversity of the pathovar (to assess the conservation of loci), as well as a few strains that are geographically closely related (to assess the discriminatory power). DNA sequencing of two predicted loci, Xo_G25 and Xo_G53, among 40 Asian and African X. oryzae pv. oryzae strains revealed that they were located next to each other in the genome (at position 1434114 of strain PXO99A) (see Fig. S1 in the supplemental material). Xo_G25 (repeat size, 5 bp) was found to be polymorphic for Asian X. oryzae pv. oryzae (2 to 15 TRs) and monomorphic for African X. oryzae pv. oryzae (2 TRs). In contrast, Xo_G53 (repeat size, 8 bp) was found to be polymorphic for African X. oryzae pv. oryzae (5 to 10 TRs) and monomorphic for Asian X. oryzae pv. oryzae (2 TRs). Thus, the two loci were combined into a single VNTR locus called Xo_G2553.
In order to implement a universal MLVA scheme, 16 loci were selected that maximized the resolution between and within the different lineages of X. oryzae (Fig. 1 and Table 1). Importantly, all 16 loci were predicted to be present in all three lineages and polymorphic in at least two lineages. To ensure that the loci are well distributed along the bacterial chromosome, the 16 VNTR loci were mapped on the complete genome sequences of Asian X. oryzae pv. oryzae strains KACC10331, MAFF311018, and PXO99A and X. oryzae pv. oryzicola strain BLS256 (see Fig. S1 in the supplemental material). The loci were distributed over the genomes with good synteny between strains. All loci from Asian X. oryzae pv. oryzae strains were syntenic to each other, except for Xo_2553, which mapped at a different position in PXO99A. In contrast, BLS256 showed limited synteny with Asian X. oryzae pv. oryzae, which is in agreement with previously published genome comparisons (5).
FIG 1.
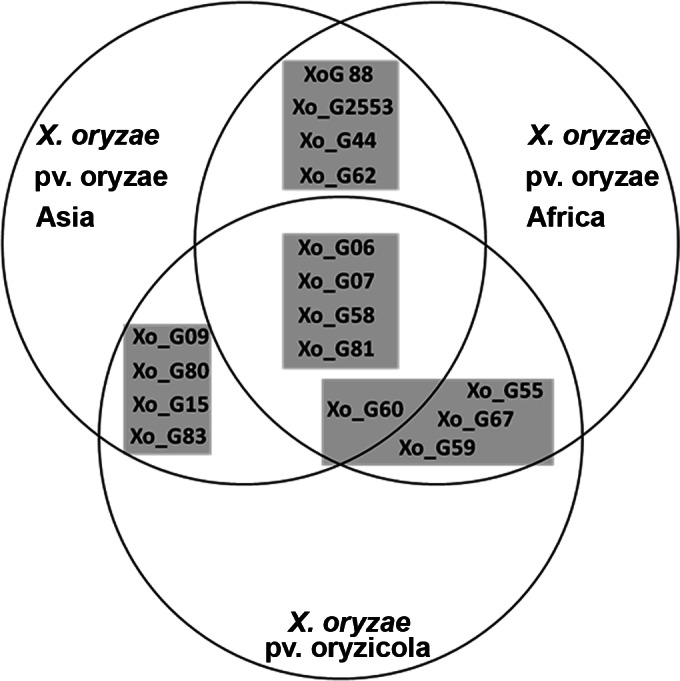
Venn diagram of VNTR loci that were included in the general typing scheme for X. oryzae. The positions of loci in the diagram correspond to their predicted polymorphism within each lineage. The four gray squares represent the four quadruplex-PCR pools.
Multiplexing in a 4-by-4 scheme, consisting of four quadruplex PCRs, was organized to include three highly discriminatory sets per lineage, while one set was predicted to include monomorphic loci that would serve as diagnostic markers for the respective lineages. For two loci, Xo_G07 and Xo_G44, large PCR amplicons were predicted due to the presence of insertion (IS) elements next to the VNTR locus for Asian X. oryzae pv. oryzae strains and X. oryzae pv. oryzicola strains, respectively, thus preventing their use for these lineages in further analyses.
MLVA-16 on a worldwide collection of Xanthomonas oryzae.
The multilocus VNTR analysis scheme based on the 16 loci (MLVA-16) was used to genotype 338 strains of X. oryzae. This scheme allowed us to distinguish 279 haplotypes. HGDI scores were calculated for all strains and ranged from 0.49 for Xo_G62 to 0.93 for Xo_G09 (Table 2). As expected from in silico analyses, one multiplexed set per lineage contained loci with low HGDI scores (Table 2). For example, Xo_G09, Xo_G15, and Xo_G80 were monomorphic for 59 African X. oryzae pv. oryzae strains. Likewise, Xo_G2553, Xo_G88, and Xo_G62 showed only low diversity for 152 X. oryzae pv. oryzicola strains. Only one locus, Xo_G59, which was predicted to be monomorphic based on the four available genome sequences, turned out to be polymorphic when 127 Asian X. oryzae pv. oryzae strains were analyzed, resulting in an HGDI score of 0.56. As suggested by the analysis of the four genome sequences, the locus Xo_G07 did not lead to PCR amplicons of less than 500 bp (which was the cutoff size for the electrophoretic analysis) for Asian X. oryzae pv. oryzae. It was impossible to design a primer between the IS element and the repeat array. Similarly, no signal was detected for almost all X. oryzae pv. oryzicola strains, except for seven African X. oryzae pv. oryzicola strains at the locus Xo_G44, because of amplicon sizes greater than 500 bp.
TABLE 2.
Genetic diversity of X. oryzae lineages estimated with the MLVA-16 scheme
| Locus |
X. oryzae pv. oryzae Africa (n = 59) |
X. oryzae pv. oryzae Asia (n = 127) |
X. oryzae pv. oryzicola Africa (n = 70) |
X. oryzae pv. oryzicola Asia (n = 82) |
Total (n = 338) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of alleles | Allelic rangea | HGDI score | No. of alleles | Allelic range | HGDI score | No. of alleles | Allelic range | HGDI score | No. of alleles | Allelic range | HGDI score | No. of Alleles | HGDI score | |
| Xo_G06 | 4 | 6–9 | 0.58 | 8 | 7–18 | 0.69 | 6 | 5–10 | 0.74 | 7 | 5–12 | 0.79 | 10 | 0.81 |
| Xo_G07 | 8 | 4–18 | 0.70 | −b | − | − | 4 | 5–8 | 0.55 | 8 | 5–13 | 0.65 | 13 | 0.83 |
| Xo_G58 | 6 | 2–7 | 0.64 | 8 | 3–13 | 0.77 | 10 | 6–16 | 0.83 | 6 | 6–11 | 0.32 | 14 | 0.81 |
| Xo_G81 | 4 | 7–15 | 0.60 | 6 | 6–25 | 0.28 | 13 | 11–26 | 0.85 | 12 | 3–20 | 0.81 | 20 | 0.84 |
| Xo_G2553 | 5 | 26–32 | 0.55 | 12 | 38–52 | 0.70 | 1 | 5 | 0 | 2 | 4–5 | 0.02 | 19 | 0.74 |
| Xo_G88 | 7 | 4–10 | 0.64 | 13 | 2–16 | 0.62 | 1 | 1 | 0 | 1 | 1 | 0 | 15 | 0.72 |
| Xo_G44 | 4 | 3–7 | 0.57 | 7 | 5–11 | 0.74 | 3 | 4–6 | 0.67 | − | − | − | 9 | 0.70 |
| Xo_G62 | 3 | 4–14 | 0.63 | 4 | 2–5 | 0.53 | 1 | 3 | 0 | 2 | 2–3 | 0.02 | 6 | 0.49 |
| Xo_G55 | 8 | 12–24 | 0.75 | 1 | 4 | 0 | 13 | 16–30 | 0.89 | 15 | 9–25 | 0.90 | 22 | 0.83 |
| Xo_G59 | 5 | 4–9 | 0.56 | 3 | 3–5 | 0.53 | 4 | 4–13 | 0.47 | 5 | 4–8 | 0.51 | 8 | 0.75 |
| Xo_G60 | 5 | 3–7 | 0.62 | 3 | 1–3 | 0.51 | 1 | 2 | 0 | 4 | 2–5 | 0.23 | 7 | 0.71 |
| Xo_G67 | 2 | 10–11 | 0.49 | 1 | 7 | 0 | 9 | 13–21 | 0.85 | 22 | 13–41 | 0.95 | 28 | 0.81 |
| Xo_G09 | 1 | 2 | 0 | 22 | 2–32 | 0.95 | 14 | 2–20 | 0.70 | 24 | 2–32 | 0.93 | 30 | 0.93 |
| Xo_G15 | 1 | 3 | 0 | 31 | 10–45 | 0.92 | 11 | 3–15 | 0.82 | 12 | 4–27 | 0.85 | 38 | 0.92 |
| Xo_G80 | 1 | 3 | 0 | 5 | 3–14 | 0.49 | 3 | 4–7 | 0.15 | 8 | 6–13 | 0.78 | 11 | 0.75 |
| Xo_G83 | 2 | 1–2 | 0.03 | 3 | 1–3 | 0.08 | 2 | 1–3 | 0.03 | 3 | 2–4 | 0.26 | 4 | 0.66 |
Alleles with the smallest number and with the largest number of repeats.
−, locus was not amplified due to the insertion of an IS element.
The allelic richness following a rarefaction procedure was calculated for three scenarios, considering two (X. oryzae pv. oryzae and X. oryzae pv. oryzicola), three (X. oryzae pv. oryzae from Africa, X. oryzae pv. oryzae from Asia, and X. oryzae pv. oryzicola, corresponding to the previously defined lineages), or four (X. oryzae pv. oryzae from Asia, X. oryzae pv. oryzicola from Asia, X. oryzae pv. oryzae from Africa, and X. oryzae pv. oryzicola from Africa) groups of strains (Table 3; see Table S3 in the supplemental material). In the last scenario, the allelic richness (A) was greater for X. oryzae pv. oryzae and X. oryzae pv. oryzicola strains from Asia, with A values of 9.35 and 8.66, respectively, than for X. oryzae pv. oryzae and X. oryzae pv. oryzicola strains from Africa, with A values of 4.19 and 7.52, respectively. Asian strains of X. oryzae pv. oryzae showed a relatively high private allelic richness (Ap) of 3.80 in comparison to African X. oryzae pv. oryzae (Ap = 1.04).
TABLE 3.
Average allelic richness and private allelic richness calculated by the rarefaction method on different possible population groupsa
| Genetic group | 4 groups (n = 59) |
3 groups (n = 59) |
2 groups (n = 152) |
|||
|---|---|---|---|---|---|---|
| A | Ap | A | Ap | A | Ap | |
| X. oryzae pv. oryzae Africa | 4.19 | 1.04 | 4.19 | 1.04 | 9.33 | 4.95 |
| X. oryzae pv. oryzae Asia | 9.35 | 3.80 | 9.35 | 3.80 | ||
| X. oryzae pv. oryzicola Africa | 7.52 | 1.53 | 9.12 | 4.76 | 9.12 | 4.76 |
| X. oryzae pv. oryzicola Asia | 8.66 | 1.56 | ||||
Four groups corresponding to the two pathovars and two continents, three groups corresponding to the three genetic lineages, and two groups corresponding to the two pathovars. n, number of strains retained for rarefaction calculation.
Monomorphic loci in each lineage tended to have a low number of repeats and to correspond to private alleles (see Table S3 in the supplemental material). A distinct combination of four alleles (Xo_G09, 2 TRs; Xo_G15, 3 TRs; Xo_G80, 3 TRs; and Xo_83, 2 TRs) was found to be unique to African X. oryzae pv. oryzae. X. oryzae pv. oryzicola was found to have two private monomorphic alleles (Xo_G25, 5 TRs, and Xo_G88, 1 TR). For Asian X. oryzae pv. oryzae, two alleles (Xo_G55, 4 TRs, and Xo_G67, 7 TRs) were lineage specific (see Table S3 in the supplemental material).
The association indices were highly significant within each lineage and at the country level, supporting strong linkage disequilibrium (Table 4). Additionally, the correlation of MLVA-16 and MLSA data sets (34) tested on a collection representative of the three lineages (n = 35) showed significant congruence (Mantel R = 0.86; P = 0.001). On the other hand, the congruence between MLVA-16 and RFLP avrXa7 data sets (35) was only slightly significant (Mantel R = −0.12; P = 0.056) on a collection of 56 African X. oryzae pv. oryzicola strains (see Fig. S2 in the supplemental material).
TABLE 4.
Linkage disequilibrium per lineage, with the number of individuals, IA, and rD
| Genetic group | na | IAb | rDb |
|---|---|---|---|
| X. oryzae pv. oryzicola Africa | 70 | 0.56 | 0.06 |
| Mali | 36 | 1.28 | 0.14 |
| Burkina Faso | 34 | 0.91 | 0.10 |
| X. oryzae pv. oryzicola Asia | 82 | 1.25 | 0.12 |
| China | 32 | 3.34 | 0.32 |
| Philippines | 48 | 1.10 | 0.10 |
| X. oryzae pv. oryzae Africa | 62 | 6.43 | 0.56 |
| Burkina Faso | 23 | 3.62 | 0.46 |
| Mali | 25 | 5.16 | 0.48 |
| X. oryzae pv. oryzae Asia | 127 | 1.323 | 0.10 |
| Philippines | 69 | 2.57 | 0.20 |
n, number of individuals.
P value < 0.001.
The three genetic lineages proposed by Gonzalez and coworkers were clearly differentiated (P < 0.001) (Table 5; see Fig. S3 in the supplemental material), and X. oryzae pv. oryzicola was less differentiated from African X. oryzae pv. oryzae (FST = 0.33466) than from Asian X. oryzae pv. oryzae (FST = 0.41889) (20).
TABLE 5.
Pairwise FST values between populations
| Genetic group |
FST |
|
|---|---|---|
| X. oryzae pv. oryzae Africa | X. oryzae pv. oryzae Asia | |
| X. oryzae pv. oryzae Africa | 0 | |
| X. oryzae pv. oryzae Asia | 0.41624a | 0 |
| X. oryzae pv. oryzicola | 0.33466a | 0.41889a |
P value < 0.001.
X. oryzae pv. oryzae from Africa.
As illustrated by the MST (Fig. 2) and the Manhattan NJ tree (see Fig. S4 in the supplemental material), strains from Niger and Burkina Faso were genetically relatively close to each other while strains from Cameroon and Mali were more distinct. Strains from two distant Burkina Faso regions, Bagre and Sourou, constituted a clonal group, and two Burkina Faso strains isolated from Bagre in 2004 clustered apart. Two strains originating from two different countries (BAI1 and NAI8), which were isolated in the same year (2004), shared the same haplotype. Strain BAI4, which was isolated from Oryza glaberrima and corresponds to race A2, was well separated from the other strains of the Burkina Faso-Niger complex (>4 locus differences and 1 to 5 repeat differences per locus), confirming previous results (20).
FIG 2.

Minimum spanning tree of African X. oryzae pv. oryzae strains using BioNumerics 7.1 and detailed view of the Malian clonal complex of strains from Niono and Sélingue (inset). The differences in repeat numbers per locus between two haplotypes is given between the circles. The circle sizes are proportional to the number of strains per haplotype. The red lines correspond to single-locus variants, green lines correspond to double-locus variants, black lines correspond to triple-locus variants, dashed lines correspond to quadruple-locus variants, and light-gray lines correspond to more than quadruple-locus variants.
DAPC identified three groups within the African X. oryzae pv. oryzae strains. DAPC-XooAf1 contained Malian strains and DAPC-XooAf2 contained strains from Cameroon and the Burkina Faso strain BAI4, while DAPC-XooAf1 comprised all the other strains from Burkina Faso and Niger. Using DAPC with four clusters, Malian strains could be further separated into two distinct clusters that correspond to X. oryzae pv. oryzae strains collected in Mali (i) between 1979 and 2009 (from Office du Niger and Kayes) and (ii) between 2010 and 2012 in Office du Niger. The second cluster formed a closely related genetic complex composed of 12 haplotypes (Fig. 2B; see Table S4 in the supplemental material). Eleven of the 12 haplotypes belonged to strains from Niono, while one haplotype belonged to two strains from Sélingue, a site that is located far from Niono (see Fig. S5 in the supplemental material).
X. oryzae pv. oryzicola.
Based on the categorical minimum spanning tree, almost all Asian and African X. oryzae pv. oryzicola strains were well separated into two groups (Fig. 3), except for two Chinese strains, HN-DA-1 and Xoc-China, which clustered with the African strains. Strain HN-DA-1 shared the same allele at 10 loci with the African X. oryzae pv. oryzicola strain BAI11, and Xoc-China had eight alleles in common with HN-DA-1. No haplotype was shared between strains from different countries.
FIG 3.

Minimum spanning tree of X. oryzae pv. oryzicola strains using BioNumerics 7.1. For details, see the legend to Fig. 2.
No clear clustering was revealed by the DAPC analysis following the k-means analysis, suggesting that there is no evident population structure (53). However, assuming two clusters (k-mean = 2), as supported by the MST and NJ tree, DAPC analysis grouped all African strains into one cluster (DAPC-Xoc1) and most Asian strains into the other cluster (DAPC-Xoc2). Only a few strains were excluded from this grouping: strains Xoc-China, GXO1, and HN-DA-1 from China and BLS103, BLS419, and BLS489 from Philippines clustered with the African strains in DAPC-Xoc1 (see Table S4 in the supplemental material). Chinese strains sampled in 2011 displayed two different haplotype groups, with only SLVs and DLVs that were amenable to eBURST analysis (see Fig. S5 in the supplemental material). The first haplotypic group comprised 10 strains with eight haplotypes, and the second haplotypic group comprised 8 strains with seven haplotypes. On the other hand, Philippine X. oryzae pv. oryzicola strains that were sampled over 30 years from various sites showed very few SLVs or DLVs and thus no visible structuring into clonal complexes.
The minimum spanning tree of African X. oryzae pv. oryzicola strains did not reveal close links among strains from Burkina Faso and/or Mali. Fifteen small clonal complexes, consisting of two to four STs, were found. Another, larger complex from Mali with eight STs, originating from two close sites, Kogoni and Niono, corresponded mainly to DLVs (Fig. 3). Importantly, isolates originating from the same field (MAI17 to MAI25 from Kogoni, MAI26 to MAI35 from Macina, and MAI40 to MAI45 from Sélingue) could be differentiated into up to three clonal complexes consisting of SLVs and DLVs.
X. oryzae pv. oryzae from Asia.
Asian X. oryzae pv. oryzae strains in our collection were found to be very diverse. No haplotypes belonging to different countries were connected as SLVs or DLVs in the minimum spanning tree (Fig. 4). We also studied two strains from South America, FXO 27 (LMG 9585) from Bolivia and CIAT1185 from Colombia. Both strains appeared to be related to Philippine strains, although they were distinct at five and three loci, respectively. A clonal complex of Philippine strains with SLVs and DLVs grouped 18 haplotypes corresponding to 24 strains that were sampled between 1994 and 2003 in the Laguna Bay region (Calauan and Mabitac) within a radial distance of about 20 km (Fig. 4B; see Fig. S5 in the supplemental material). The other 12 strains sampled in the same region over the same period and a strain from Los Baños, a neighboring Calauan village, that was sampled in 1979, were not related to this clonal complex. These findings could be explained by the fact that BLB is endemic, i.e., established for a long time in Philippines (54).
FIG 4.

Minimum spanning tree of Asian X. oryzae pv. oryzae strains using BioNumerics 7.1 and detailed view of a Philippine clonal complex of strains from Calauan and Mabitac (inset). The differences in repeat numbers per locus between two haplotypes are given between the circles. For details, see the legend to Fig. 2.
The DAPC analysis using a k-mean of 2 clustered a subset of Philippine strains together with Chinese, Malaysian, Indonesian, Thai, Burmese, and most Korean and Nepalese strains, as well as with the Colombian strain CIAT1185 (DAPC-XooAs1). The other cluster, DAPC-XooAs2, contained another subset of Philippine strains (mainly strains sampled before the 1990s), along with eight Indian strains, two Bangladeshi strains, two Nepalese strains, and one Korean strain, as well as the Bolivian strain FXO 27 (see Table S4 in the supplemental material).
DISCUSSION
We developed a new MLVA scheme that identifies the different lineages of X. oryzae, i.e., pathovars and those of continental origin, and shows high discriminatory power on small scales in space and time.
On a global scale, the MLVA-16 scheme confirmed the lineage differentiation that was largely described in previous typing and phylogenetic studies (20, 33, 34, 55). However, on a smaller scale, the MLVA-16 scheme was shown to be more discriminatory than other previously used molecular-typing tools. The MLVA-16 scheme could discriminate strains from the same region and even strains originating from the same field. Interestingly, epidemiologically related strains kept a signature of their relationship by descent on this small spatiotemporal scale.
Recombination does not strongly bias the phylogenetic signal.
Even though the association indices are quite low for some lineages, linkage disequilibrium was highly significant. Moreover, distance matrices obtained from MLVA-16 and from a different genotyping technique (MLSA) were significantly correlated. These lines of evidence support low levels of recombination, as has also been suggested for other Xanthomonas species (56). However, one needs to be cautious, because these observations could result from sampling biases. For instance, geographical isolation might have occurred in our collections, consequently promoting high linkage disequilibrium (57). To test this hypothesis, the locus association should be tested on a strongly sampled population, i.e., more strains isolated on a small spatiotemporal scale. Nevertheless, our results support the usefulness of the 16 VNTR markers to investigate demographic or epidemiological patterns.
As a special case, a distance matrix obtained from a tal gene-based RFLP data set was not correlated with an MLVA-16-based distance matrix when applied to a set of Malian and Burkina Faso X. oryzae pv. oryzicola strains. tal genes, which consist of tandem repeats with a large repeat unit (58), contribute to the colonization of host plants in a compatible interaction or trigger specific defenses in an incompatible interaction. Hence, tal gene-based markers are expected to be under selection. Moreover, the RFLP band sizes of tal genes do not always reflect their functional relatedness, i.e., identical tal RFLP bands can belong to functionally unrelated tal genes while different tal RFLP bands can correspond to functionally analogous tal genes. Together, these considerations may explain why a matrix generated by this marker is not correlated with a matrix obtained from neutral markers, such as most VNTRs.
MLVA-16 as a tool to identify lineages.
The MLVA-16 scheme produced some private and monomorphic alleles for each genetic lineage. Consequently, the different lineages of X. oryzae can be distinguished. In addition, other lineage-polymorphic loci also contribute to this differentiation. Hence, MLVA-16 directly identifies the pathogen responsible for a disease, i.e., BLB or BLS. Since BLB and BLS symptoms can be confused in the field at very early and late stages of the diseases, and also in cases of double infection, this information is important for efficient disease management. In the context of prior knowledge about prevalent BLB and BLS pathogens in a certain geographic area, the ability to distinguish between the two pathovars helps in deployment of disease-resistant rice cultivars and/or application of specific antibacterial agents. Moreover, the ability to distinguish between BLS and BLB might be helpful for quarantine purposes, especially with the increase in rice seed trade between countries.
FST indices of diversity suggest that African X. oryzae pv. oryzae strains are closer to X. oryzae pv. oryzicola than to Asian X. oryzae pv. oryzae, as shown previously by Gonzalez and coworkers using RFLP, AFLP, and rep-PCR. However, these results are challenged by an MLSA of the three housekeeping genes gyrB, rpoD, and glnA at a lower resolution, where the two X. oryzae pv. oryzae lineages group together and are more distant from X. oryzae pv. oryzicola (34). Interestingly, another MLSA using a different set of housekeeping genes, fusA, gyrB, and gapA, rather supported our scenario, i.e., that African X. oryzae pv. oryzae strains are closer to X. oryzae pv. oryzicola than to Asian X. oryzae pv. oryzae (33). Apparently, the choice of markers (with only a few polymorphisms) used for such analysis plays an important role in the discrimination of lineages. To resolve this problem, a genome-wide SNP analysis or an MLSA of the core genome could be performed (55). In conclusion, MLVA-16 differentiates lineages well on a global scale.
MLVA-16 has limited value for large-scale epidemiology.
First reported in Asia, X. oryzae pv. oryzae and X. oryzae pv. oryzicola were described decades later in Africa (4, 20). It has been assumed that BLB and BLS have been introduced accidentally from Asia to Africa. The fact that both African X. oryzae pv. oryzae and X. oryzae pv. oryzicola showed less allelic richness than Asian X. oryzae pv. oryzae and X. oryzae pv. oryzicola supports an Asian ancestor for African strains. However, even though this study was conducted on a large strain collection, additional and more extensive sampling will be necessary to confirm an Asian origin of the African populations of X. oryzae.
On a global scale, the MLVA-16 scheme revealed hardly any shared or related haplotypes (SLVs and DLVs) among strains from different countries. Similar findings were obtained with a microsatellite-based MLVA-14 of X. citri (46). Generally, no link can be drawn from a vast number of strains on a really large geographic scale or over a long time based on microsatellites that evolve rapidly. Therefore, the development of another MLVA scheme based on TRs with a slower molecular clock, i.e., minisatellites, where repeat units are longer, would be more appropriate for large-scale epidemiology. Indeed, as shown by N′Guessan and coworkers for Ralstonia solanacearum, the number of alleles in a large collection of strains decreases with an increase in the repeat unit size (59). Similarly, Pruvost and coworkers demonstrated the superiority of a minisatellite-based scheme over a microsatellite-based scheme for large-scale epidemiology of X. citri (46). Preliminary analyses have shown that no useful VNTR loci with repeat unit sizes above 12 bp are shared among the three X. oryzae lineages, thus preventing the development of a universal scheme. In the future, lineage-specific minisatellite schemes will be developed for global epidemiological monitoring.
Despite these concerns, MLVA-16 provided some important information on a larger geographical scale. For instance, we identified two Chinese X. oryzae pv. oryzicola strains (Xoc-China and HN-DA-1) that differed greatly from the other Asian X. oryzae pv. oryzicola strains (only 7 common alleles with the closest Asian X. oryzae pv. oryzicola strain). Our results suggest that these strains are rather related to African X. oryzae pv. oryzicola strains (10 common alleles with the closest African X. oryzae pv. oryzicola strain). This finding coincides with the recent increase of the seedborne BLS disease in Africa and with tighter commercial links between Africa and China. Moreover, the two strains from South America, which are related to two Philippine haplotypes, support a scenario with distinct events of introduction in South America, perhaps from Philippines. Finally, two African X. oryzae pv. oryzae strains from different countries, BAI3 from Burkina Faso and NAI8 from Niger, shared the same haplotype. This finding may reflect the exchange of material between western African countries.
In conclusion, even if the MLVA-16 scheme is not ideal for large-scale epidemiology with slow migration, it is still a valuable tool for more modern situations where migration is very fast due to human transport. To demonstrate the power of this tool, we would need a larger set of strains representative of the total diversity. Currently, access to strains or even genomic DNA of X. oryzae is limited due to quarantine restrictions or regulatory issues concerning biodiversity in the different countries. The MLVAbank database, which makes our data accessible for analyses and comparisons, will help to coordinate international efforts to understand the epidemiology of X. oryzae.
MLVA-16 as a new tool for local epidemiological monitoring.
Our results show that the MLVA-16 scheme is very useful for local epidemiological surveillance on a regional or country-wide scale for all three X. oryzae lineages. First, African X. oryzae pv. oryzae strains sampled from Malian regions (Niono and Sélingue) in the same period (2010 and 2012) showed relatively close haplotypes. Their allelic profiles reveal an expansion that has occurred clonally from an unidentified founder. However, the Malian collection showed a dynamic population structure, since Malian strains sampled in 2010 and 2012 are distant from Malian strains sampled up to 2009. However, a phylogenetic signature for relatedness remained while epidemiological information was lost, because strains of both complexes differ by at least nine alleles. Second, on a slightly larger scale, a set of X. oryzae pv. oryzicola strains sampled in southern China in 2011 shared identical or similar haplotypes, as shown by the minimum spanning tree and eBURST analysis (Fig. 4; see Fig. S5 in the supplemental material). Third, 24 Philippine X. oryzae pv. oryzae strains sampled over 9 years in Laguna Bay are linked in a clonal complex. It would be interesting to clarify how such a limited number of haplotypes was maintained for many years in this region. Moreover, since several very different haplotypes of Philippine X. oryzae pv. oryzae were found to coexist within this region, further investigation by extensive sampling would be of high interest.
Our results show that the MLVA-16 scheme holds great potential for small-scale reconstruction of population dynamics. Such an improved understanding of microevolution will be key for short- and medium-term management of control strategies for X. oryzae, particularly by deciphering the pathways of dispersal both by natural transmission and by human-mediated transmission (including seed transmission).
Conclusions.
The MLVA-16 scheme will be useful for high discrimination among X. oryzae strains and to identify the lineages and pathovars of these pathogens. MLVA-16 will further allow us to analyze new outbreaks and epidemics of both pathovars of X. oryzae. Additional samplings at the population level will be a further step to evaluate the efficiency of MLVA-16 by describing the patterns of descent of the strains and the population structures. Specifically, recently isolated X. oryzae pv. oryzicola strains from eastern and central Africa could be characterized (22–24).
Directions for use.
In cases where the pathovar and geographical origin are known, e.g., upon multiplex PCR (13), one could omit one of the four primer pools that correspond to loci that are largely monomorphic within a lineage. Hence, one would use a subset of primer pools in a multiplex MLVA-12 scheme that is adapted to the pathovars and origins of the isolates. Asian X. oryzae pv. oryzae strains need to be analyzed with pools 1, 2, and 4 (MLVA-12a); African X. oryzae pv. oryzae strains need pools 1, 2, and 3 (MLVA-12b); and X. oryzae pv. oryzicola strains need pools 1, 3, and 4 (MLVA-12c). Results can be analyzed and compared to our and other's data using the public database at IRD Montpellier (http://www.biopred.net/MLVA/), which allows sharing of information with other scientists in an interactive manner and provides access to a few phylogenetic-analysis tools (46).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the French Agence Nationale de la Recherche (ANR-2010-BLAN-1723) and the French Direction Générale de l'Armement (DGA) (2010 34 0006). Lucie Poulin is grateful to DGA for her Ph.D. fellowship (2011 60 091). Shuai Zhao is grateful to the Bourses Doctorales en Alternance from the French Embassy in China, the program of postgraduate education and research of Guangxi (T31036), and the plans for joint training of doctoral students and academic visiting of postgraduates of Guangxi University (L300278) for support.
We thank Jan Leach (CSU) and Nollie Vera-Cruz (IRRI) for providing us with strains or DNA. We are grateful to Gilles Vergnaud, Christine Pourcel, and Christophe Tourterel, all from Université de Paris-Sud, for helpful suggestions on the MLVA scheme and for providing support for the MLVAbank website. We also thank Charlotte Tollenaere (UMR RPB) and Virginie Ravigné (UMR BGPI) for helpful discussions and Karine Boyer (UMR PVBMT) for helpful technical assistance.
Footnotes
M.E.R. met authorship criteria but was unreachable for final approval of the byline and article.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02768-14.
REFERENCES
- 1.Gilmour MW, Graham M, Reimer A, Van Domselaar G. 2013. Public health genomics and the new molecular epidemiology of bacterial pathogens. Public Health Genomics 16:25–30. doi: 10.1159/000342709. [DOI] [PubMed] [Google Scholar]
- 2.Cai HY, Caswell JL, Prescott JF. 2014. Nonculture molecular techniques for diagnosis of bacterial disease in animals: a diagnostic laboratory perspective. Vet Pathol 51:341–350. doi: 10.1177/0300985813511132. [DOI] [PubMed] [Google Scholar]
- 3.McDonald BA, Linde C. 2002. Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40:349–379. doi: 10.1146/annurev.phyto.40.120501.101443. [DOI] [PubMed] [Google Scholar]
- 4.Niño-Liu DO, Ronald PC, Bogdanove AJ. 2006. Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol Plant Pathol 7:303–324. doi: 10.1111/j.1364-3703.2006.00344.x. [DOI] [PubMed] [Google Scholar]
- 5.Bogdanove AJ, Koebnik R, Lu H, Furutani A, Angiuoli SV, Patil PB, Van Sluys MA, Ryan RP, Meyer DF, Han SW, Aparna G, Rajaram M, Delcher AL, Phillippy AM, Puiu D, Schatz MC, Shumway M, Sommer DD, Trapnell C, Benahmed F, Dimitrov G, Madupu R, Radune D, Sullivan S, Jha G, Ishihara H, Lee SW, Pandey A, Sharma V, Sriariyanun M, Szurek B, Vera-Cruz CM, Dorman KS, Ronald PC, Verdier V, Dow JM, Sonti RV, Tsuge S, Brendel VP, Rabinowicz PD, Leach JE, White FF, Salzberg SL. 2011. Two new complete genome sequences offer insight into host and tissue specificity of plant-pathogenic Xanthomonas spp. J Bacteriol 193:5450–5464. doi: 10.1128/JB.05262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verdier V, Triplett LR, Hummel AW, Corral R, Cernadas RA, Schmidt CL, Bogdanove A, Leach JE. 2012. Transcription activator-like (TAL) effectors targeting OsSWEET genes enhance virulence on diverse rice (Oryza sativa) varieties when expressed individually in a TAL effector-deficient strain of Xanthomonas oryzae. New Phytol 196:1197–1207. doi: 10.1111/j.1469-8137.2012.04367.x. [DOI] [PubMed] [Google Scholar]
- 7.Mew TW, Misra JK. 1994. A manual of rice seed health testing. International Rice Research Institute, Manila, Philippines. [Google Scholar]
- 8.Xie GL, Mew TW. 1998. A leaf inoculation method for detection of Xanthomonas oryzae pv. oryzicola from rice reed. Plant Dis 82:1007–1011. doi: 10.1094/PDIS.1998.82.9.1007. [DOI] [PubMed] [Google Scholar]
- 9.Sakthivel N, Mortensen CN, Mathur SB. 2001. Detection of Xanthomonas oryzae pv. oryzae in artificially inoculated and naturally infected rice seeds and plants by molecular techniques. Appl Microbiol Biotechnol 56:435–441. doi: 10.1007/s002530100641. [DOI] [PubMed] [Google Scholar]
- 10.Yang B, Bogdanove A. 2013. Inoculation and virulence assay for bacterial blight and bacterial leaf streak of rice. Methods Mol Biol 956:249–255. doi: 10.1007/978-1-62703-194-3_18. [DOI] [PubMed] [Google Scholar]
- 11.Lang JM, Hamilton JP, Diaz MG, Van Sluys MA, Burgos MR, Vera Cruz CM, Buell CR, Tisserat NA, Leach JE. 2010. Genomics-based diagnostic marker development for Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola. Plant Dis 94:311–319. doi: 10.1094/PDIS-94-3-0311. [DOI] [PubMed] [Google Scholar]
- 12.Lang JM, Langlois P, Nguyen MHR, Triplett LR, Purdie L, Holton TA, Djikeng A, Vera Cruz CM, Verdier V, Leach JE. 2014. Sensitive detection of Xanthomonas oryzae pathovars oryzae and oryzicola by loop-mediated isothermal amplification. Appl Environ Microbiol 80:4519–4530. doi: 10.1128/AEM.00274-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauleon R, Triplett L, Snelling J, Vazquez S, Corral R, Leach JE. 2013. A pipeline for automated diagnostic primer design based on genomic sequence alignment of target and non-target genomes. Phytopathology 103:S2.92. [Google Scholar]
- 14.Tagami Y, Mizukami T. 1962. Historical review of the researches on bacterial blight of rice caused by Xanthomonas oryzae (Uyede and Ishiyama) Dowson. Spec Rep Plant Dis Ins Pests Forecasting Serv 10:112. [Google Scholar]
- 15.Mew TW. 1989. An overview of the world bacterial blight situation. Bacterial blight of rice, p 7–12. In Bacterial blight of rice: proceedings of the International Workshop on Bacterial Blight of Rice, 14 to 18 March 1988. International Rice Research Institute, Manila, Philippines. [Google Scholar]
- 16.Ou SH. 1985. Rice diseases, 2nd ed. Commonwealth Mycological Institute, Kew, United Kingdom. [Google Scholar]
- 17.Verdier V, Vera Cruz C, Leach JE. 2012. Controlling rice bacterial blight in Africa: needs and prospects. J Biotechnol 159:320–328. doi: 10.1016/j.jbiotec.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 18.Aldrick S, Buddenhagen IW, Reddy APK. 1973. The occurrence of bacterial leaf blight in wild and cultivated rice in Northern Australia. Crop Pasture Sci 24:219–227. doi: 10.1071/AR9730219. [DOI] [Google Scholar]
- 19.Lozano JC. 1977. Identification of bacterial leaf blight in rice, caused by Xanthomonas oryzae, in America. Plant Dis Rep 61:644–648. [Google Scholar]
- 20.Gonzalez C, Szurek B, Manceau C, Mathieu T, Sere Y, Verdier V. 2007. Molecular and pathotypic characterization of new Xanthomonas oryzae strains from West Africa. Mol Plant Microbe Interact 20:534–546. doi: 10.1094/MPMI-20-5-0534. [DOI] [PubMed] [Google Scholar]
- 21.Wonni I, Ouedraogo L, Verdier V. 2011. First report of bacterial leaf streak caused by Xanthomonas oryzae pv. oryzicola on rice in Burkina Faso. Plant Dis 95:72. doi: 10.1094/PDIS-08-10-0566. [DOI] [PubMed] [Google Scholar]
- 22.Poulin L, Raveloson H, Sester M, Raboin LM, Silué D, Koebnik R, Szurek B. 2014. Confirmation of bacterial leaf streak caused by Xanthomonas oryzae pv. oryzicola on rice in Madagascar. Plant Dis 98:1423. doi: 10.1094/PDIS-02-14-0132-PDN. [DOI] [PubMed] [Google Scholar]
- 23.Afolabi O, Milan B, Amoussa R, Koebnik R, Poulin L, Szurek B, Habarugira G, Bigirimana J, Silue D. 2014. First report of Xanthomonas oryzae pv. oryzicola causing bacterial leaf streak of rice in Burundi. Plant Dis 98:1426. doi: 10.1094/PDIS-05-14-0504-PDN. [DOI] [PubMed] [Google Scholar]
- 24.Afolabi O, Milan B, Poulin L, Ongom J, Szurek B, Koebnik R, Silue D. 2014. First report of Xanthomonas oryzae pv. oryzicola causing bacterial leaf streak of rice in Uganda. Plant Dis 98:1579. doi: 10.1094/PDIS-07-14-0745-PDN. [DOI] [PubMed] [Google Scholar]
- 25.Leach JE, Rhoads ML, Vera Cruz CM, White FF, Mew TW, Leung H. 1992. Assessment of genetic diversity and population structure of Xanthomonas oryzae pv. oryzae with a repetitive DNA element. Appl Environ Microbiol 58:2188–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson RJ, Baraoidan MR, Vera Cruz CM, Yap IV, Leach JE, Mew TW, Leung H. 1994. Relationship between phylogeny and pathotype for the bacterial blight pathogen of rice. Appl Environ Microbiol 60:3275–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi SH, Vera Cruz CM, Leach JE. 1998. Distribution of Xanthomonas oryzae pv. oryzae DNA modification systems in Asia. Appl Environ Microbiol 64:1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noda T, Du PV, Dinh V, Dinh ELH, Kaku H. 1999. Pathogenicity of Xanthomonas oryzae pv. oryzae strains in Vietnam. Ann Phytopathol Soc Japan 65:293–296. doi: 10.3186/jjphytopath.65.293. [DOI] [Google Scholar]
- 29.Chen XL, Yu L, Gao LL, Jiang T, Li QY, Huang Q. 2012. Elevational variation in diversity of Xanthomonas oryzae pv. oryzae in South-West China. J Phytopathol 160:261–268. doi: 10.1111/j.1439-0434.2012.01892.x. [DOI] [Google Scholar]
- 30.Mishra D, Vishnupriya MR, Anil MG, Konda K, Raj Y, Sonti RV. 2013. Pathotype and genetic diversity amongst Indian isolates of Xanthomonas oryzae pv. oryzae. PLoS One 8:e81996. doi: 10.1371/journal.pone.0081996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raymundo AK, Briones AM Jr, Ardales EY, Perez MT, Fernandez LC, Leach JF, Mew TW, Ynalvez MA, McLaren CG, Nelson RJ. 1999. Analysis of DNA polymorphism and virulence in Philippine strains of Xanthomonas oryzae pv. oryzicola. Plant Dis 83:434–440. doi: 10.1094/PDIS.1999.83.5.434. [DOI] [PubMed] [Google Scholar]
- 32.Lin L, Ji G, Ma G, Wang Y, Zhang L. 2011. Genotypic diversity analysis of Xanthomonas oryzae pv. oryzicola in Southwest China by RepPCR. Jiangxi Nongye Daxue Xuebao 33:264–269. [Google Scholar]
- 33.Triplett LR, Hamilton JP, Buell CR, Tisserat NA, Verdier V, Zink F, Leach JE. 2011. Genomic analysis of Xanthomonas oryzae isolates from rice grown in the United States reveals substantial divergence from known X. oryzae pathovars. Appl Environ Microbiol 77:3930–3937. doi: 10.1128/AEM.00028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hajri A, Brin C, Zhao S, David P, Feng J, Koebnik R, Szurek B, Verdier V, Boureau T, Poussier S. 2012. Multilocus sequence analysis and type III effector repertoire mining provide new insights into the evolutionary history and virulence of Xanthomonas oryzae. Mol Plant Pathol 13:288–302. doi: 10.1111/j.1364-3703.2011.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wonni I, Cottyn B, Detemmerman L, Dao S, Ouedraogo L, Sarra S, Tekete C, Poussier S, Corral R, Triplett L, Koita O, Koebnik R, Leach J, Szurek B, Maes M, Verdier V. 2014. Analysis of Xanthomonas oryzae pv. oryzicola population in Mali and Burkina Faso reveals a high level of genetic and pathogenic diversity. Phytopathology 104:520–531. doi: 10.1094/PHYTO-07-13-0213-R. [DOI] [PubMed] [Google Scholar]
- 36.Maiden MC. 2006. Multilocus sequence typing of bacteria. Annu Rev Microbiol 60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- 37.Holmes A, McAllister G, McAdam PR, Hsien Choi S, Girvan K, Robb A. 2014. Genome-wide single nucleotide polymorphism-based assay for high-resolution epidemiological analysis of the methicillin-resistant Staphylococcus aureus hospital clone EMRSA-15. Clin Microbiol Infect 20:O124–O131. doi: 10.1111/1469-0691.12328. [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Losada M, Cabezas P, Castro-Nallar E, Crandall KA. 2013. Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect Genet Evol 16:38–53. doi: 10.1016/j.meegid.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Lindstedt BA. 2005. Multiple-locus variable number tandem repeats analysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis 26:2567–2582. doi: 10.1002/elps.200500096. [DOI] [PubMed] [Google Scholar]
- 40.Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 41.Vogler AJ, Keys C, Nemoto Y, Colman RE, Jay Z, Keim P. 2006. Effect of repeat copy number on variable-number tandem repeat mutations in Escherichia coli O157:H7. J Bacteriol 188:4253–4263. doi: 10.1128/JB.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Belkum A, Scherer S, van Alphen L, Verbrugh H. 1998. Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev 62:275–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coletta-Filho HD, Takita MA, de Souza AA, Aguilar-Vildoso CI, Machado MA. 2001. Differentiation of strains of Xylella fastidiosa by a variable number of tandem repeat analysis. Appl Environ Microbiol 67:4091–4095. doi: 10.1128/AEM.67.9.4091-4095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bui Thi Ngoc L, Verniere C, Vital K, Guerin F, Gagnevin L, Brisse S, Ah-You N, Pruvost O. 2009. Development of 14 minisatellite markers for the citrus canker bacterium, Xanthomonas citri pv. citri. Mol Ecol Resour 9:125–127. doi: 10.1111/j.1755-0998.2008.02242.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhao S, Poulin L, Rodriguez-R LM, Forero-Serna N, Liu SH, Wonni I, Szurek B, Verdier V, Leach JE, He YQ, Feng JX, Koebnik R. 2012. Development of a variable number of tandem repeats typing scheme for the bacterial rice pathogen Xanthomonas oryzae pv. oryzicola. Phytopathology 102:948–956. doi: 10.1094/PHYTO-04-12-0078-R. [DOI] [PubMed] [Google Scholar]
- 46.Pruvost O, Magne M, Boyer K, Leduc A, Tourterel C, Drevet C, Ravigné V, Gagnevin L, Guérin F, Chiroleu F, Koebnik R, Verdier V, Vernière C. 2014. A MLVA genotyping scheme for global surveillance of the citrus pathogen Xanthomonas citri pv. citri suggests a worldwide geographical expansion of a single genetic lineage. PLoS One 9:e98129. doi: 10.1371/journal.pone.0098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol 26:2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalinowski ST. 2005. hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. doi: 10.1111/j.1471-8286.2004.00845.x. [DOI] [Google Scholar]
- 49.Excoffier L, Lischer HEL. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 50.Agapow P-M, Burt A. 2001. Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1:101–102. doi: 10.1046/j.1471-8278.2000.00014.x. [DOI] [Google Scholar]
- 51.Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spratt BG, Hanage WP, Li B, Aanensen DM, Feil EJ. 2004. Displaying the relatedness among isolates of bacterial species—the eBURST approach. FEMS Microbiol Lett 241:129–134. doi: 10.1016/j.femsle.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 53.Jombart T, Devillard S, Balloux F. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94–108. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mew TW, Alvarez AM, Leach JE, Swings J. 1993. Focus on bacterial blight of rice. Plant Dis 77:5–12. doi: 10.1094/PD-77-0005. [DOI] [Google Scholar]
- 55.Rodriguez-R LM, Grajales A, Arrieta-Ortiz ML, Salazar C, Restrepo S, Bernal A. 2012. Genomes-based phylogeny of the genus Xanthomonas. BMC Microbiol 12:43. doi: 10.1186/1471-2180-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vernière C, Bui Thi Ngoc L, Jarne P, Ravigné V, Guérin F, Gagnevin L, Le Mai N, Chau NM, Pruvost O. 2014. Highly polymorphic markers reveal the establishment of an invasive lineage of the citrus bacterial pathogen Xanthomonas citri pv. citri in its area of origin. Environ Microbiol 16:2226–2237. doi: 10.1111/1462-2920.12369. [DOI] [PubMed] [Google Scholar]
- 57.Maynard-Smith J, Smith NH, O'Rourke M, Spratt BG. 1993. How clonal are bacteria? Proc Natl Acad Sci U S A 90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boch J, Bonas U. 2010. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol 48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- 59.N′Guessan CA, Brisse S, Le Roux-Nio AC, Poussier S, Koné D, Wicker E. 2013. Development of variable number of tandem repeats typing schemes for Ralstonia solanacearum, the agent of bacterial wilt, banana Moko disease and potato brown rot. J Microbiol Methods 92:366–374. doi: 10.1016/j.mimet.2013.01.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.