

Abstract
Bacterial ghosts are empty cell envelopes of Gram-negative bacteria that can be used as vehicles for antigen delivery. Ghosts are generated by releasing the bacterial cytoplasmic contents through a channel in the cell envelope that is created by the controlled production of the bacteriophage ϕX174 lysis protein E. While ghosts possess all the immunostimulatory surface properties of the original host strain, they do not pose any of the infectious threats associated with live vaccines. Recently, we have engineered the Escherichia coli autotransporter hemoglobin protease (Hbp) into a platform for the efficient surface display of heterologous proteins in Gram-negative bacteria, HbpD. Using the Mycobacterium tuberculosis vaccine target ESAT6 (early secreted antigenic target of 6 kDa), we have explored the application of HbpD to decorate E. coli and Salmonella ghosts with antigens. The use of different promoter systems enabled the concerted production of HbpD-ESAT6 and lysis protein E. Ghost formation was monitored by determining lysis efficiency based on CFU, the localization of a set of cellular markers, fluorescence microscopy, flow cytometry, and electron microscopy. Hbp-mediated surface display of ESAT6 was monitored using a combination of a protease accessibility assay, fluorescence microscopy, flow cytometry and (immuno-)electron microscopy. Here, we show that the concerted production of HbpD and lysis protein E in E. coli and Salmonella can be used to produce ghosts that efficiently display antigens on their surface. This system holds promise for the development of safe and cost-effective vaccines with optimal intrinsic adjuvant activity and exposure of heterologous antigens to the immune system.
INTRODUCTION
Bacterial ghosts (BGs) are empty, nonliving cell envelopes of Gram-negative bacteria that still possess all surface structures, including immune-stimulating elements like lipopolysaccharides, lipoproteins, and flagella, while not posing any infectious threat (1–3). They are generated by the controlled production of bacteriophage ϕX174 lysis protein E, which leads to the formation of tunnel structures spanning the entire cell envelope (1, 2, 4). How exactly these tunnel structures are formed is not clear (see, e.g., references 1 and 5 to 11). Due to osmotic pressure, the cytoplasmic content of the bacteria is released through these structures, while the cell envelope is mostly preserved (1, 2).
BGs have been used as vaccines as well as vehicles for the delivery of antigens, drugs, and DNA (1, 2). For surface display of antigens in bacterial ghosts, the ice nucleation protein (6) and various outer membrane proteins, such as outer membrane protein A, have been used as anchors (12, 13). Recently, it has been shown that antibody responses to heterologous antigens that are secreted by Salmonella cells or exposed at the surface of Salmonella-derived outer membrane vesicles (OMVs) are higher than antibody responses to antigens that are present intracellularly or in the lumen of the OMVs, respectively (e.g., see references 14 to 16). This suggests that secreted and surface-exposed antigens elicit superior immune responses.
The self-sufficient autotransporter (AT) pathway, ubiquitous in Gram-negative bacteria, combines a relatively simple protein secretion mechanism with a high transport capacity (17). ATs consist of a secreted passenger domain and a cognate β-domain that facilitates transfer of the passenger across the cell envelope. Recently, capitalizing on the crystal structure of its passenger domain, we have engineered the Escherichia coli AT hemoglobin protease (Hbp) into a platform for the secretion and surface display of heterologous proteins using a side domain replacement strategy (18, 19). The Mycobacterium tuberculosis antigen and vaccine target ESAT6, a low-molecular-mass secreted T-cell antigen, was used as a model protein to develop the system (20). By interrupting the cleavage site between passenger and β-domain, Hbp display (HbpD) variants that remain cell associated and facilitate efficient surface exposure of ESAT6 in E. coli were constructed (19, 21). HbpD carrying ESAT6 (HbpD-ESAT6) was efficiently displayed not only in E. coli but also in an attenuated Salmonella enterica serovar Typhimurium strain (19). This demonstrates the potential of the Hbp platform for live vaccine development. However, there are serious safety concerns linked to the use of recombinant live bacterial vaccines, relating to potential dissemination in the environment and reversion to a virulent phenotype (22). This has prevented large-scale development and use of live recombinant vaccines. These concerns raised the question whether the highly efficient Hbp surface display system can be combined with nonreplicating carriers, like OMVs and BGs, which retain the ability to stimulate the innate immune response while being safe to use.
In this study, we show that the concerted production of the Hbp display platform and lysis protein E in E. coli and Salmonella leads to the formation of BGs efficiently displaying antigens on their surface. Hence, the Hbp-based display platform can be used to exploit BGs as carriers for antigens.
MATERIALS AND METHODS
Strains, plasmids, and culture conditions.
Escherichia coli strains MC4100 (23), EC452 [MC4100 Δ(λattL-lom)::bla lacIqP207-gfp] (24) and MC4100Δasd (this study) and Salmonella Typhimurium strains SL3261 (25) and SL3261HbpD-ESAT6 (19) were grown aerobically in standard lysogeny broth (LB). In the case of the Δasd mutant, the medium was supplemented with diaminopimelic acid (DAP; 50 μg/ml). When E. coli cells harbored pHbpD(p15A)-ESAT6 (see “Plasmid construction” below), the culture medium was supplemented with chloramphenicol (30 μg/ml), and when they harbored pRL1 (RL signifying rhamnose lysis) (see “Plasmid construction” below), the culture medium was supplemented with kanamycin (Km; 50 μg/ml). For the production of ghosts using pRL1, glucose (0.2%) was added to plates used for transformation and to precultures to reduce any background expression of lysis gene E. For Salmonella cells harboring pTL1 (TL signifying tetracycline lysis) (see “Plasmid construction” below), the culture medium was supplemented with ampicillin (100 μg/ml). Cultures were grown in shaker flasks (100-ml flasks with a culture volume of 35 ml) at 30°C in an Innova 4330 (New Brunswick Scientific) shaker at 200 rpm. Growth and BG formation were monitored by measuring the optical density at 600 nm (OD600) with a Shimadzu UV-1601 spectrophotometer. Production of green fluorescent protein (GFP) in EC452 was induced at an OD600 of 0.25 with 2.5 μM IPTG (isopropyl-β-d-thiogalactopyranoside). Production of lysis protein E was, in the case of pRL1, induced by the addition of 6 mM l-rhamnose and in the case of pTL1 by the addition of 0.2 μg/ml anhydrotetracycline at an OD600 of 0.5. HbpD-ESAT6 production from pHbpD(p15A)-ESAT6 was induced in E. coli at an OD600 of 0.25 with 0.4 mM IPTG. Cells/BGs were harvested immediately before and 120 min after lysis induction. Lysis efficiency was determined by comparing the number of CFU immediately before and 120 min after lysis induction (see “Monitoring CFU” below). For further analyses, cells and BGs were harvested by centrifugation in an Eppendorf 5415R centrifuge at 15,000 rpm for 10 min at 4°C and washed once in phosphate-buffered saline (PBS).
Monitoring CFU.
To monitor CFU in a culture, the culture was serially diluted (10-fold steps: 100 μl of culture/diluted material in 900 μl of LB) and 100 μl was plated onto LB agar plates. In the case of Δasd strains, DAP was added to the plates (50 μg/ml). In case there were still any CFU, plates containing 50 to 200 colonies were used for CFU counting. Lysis efficiency was calculated as follows: [1 − (CFUbefore/CFUafter)] × 100.
Plasmid construction.
The gene encoding lysis protein E (see Table S1 in the supplemental material) from bacteriophage ϕX174 (obtained from GenBank [NC_001422.1] and synthesized by GeneArt) was, using EcoRI and BamHI, cloned into the rhamnose promoter-based expression vector pLarge (pUC origin, Kmr), which is derived from pRha67 (26), yielding pRL1, and into the tetracycline promoter (tetA)-based expression vector pASK_IBA3 (pUC origin, Ampr), yielding pTL1 (27). To be able to clone the gene encoding lysis protein E directly from pRL1 into pASK_IBA3, its multiple cloning site was modified by amplifying the entire plasmid and introducing EcoRI and BamHI restriction sites using the Roche Expand long template PCR system (for primer sequences, see Table S1 in the supplemental material) (28). The pHbpD(p15a)-ESAT6 vector (p15A ori, Cmr) was constructed by substituting the pMB1 origin of replication of pHbpD(Δd1)-ESAT6 (19) by the PCR-amplified p15A origin of replication of pBAD33 using the EcoRI and SalI restriction sites (for primer sequences, see Table S1 in the supplemental material).
Strain construction.
To delete the essential asd gene (see Table S1 in the supplemental material) in E. coli MC4100 and Salmonella SL3261HbpD-ESAT6, we used the lambda Red bacteriophage-mediated homologous recombination method developed by Datsenko and Wanner (29). Inserted Kmr resistance markers were removed using FLP-mediated recombination (29). The asd Kmr insertions and deletions were confirmed by PCR and sequencing. Sequences of the primers used are listed in Table S1 in the supplemental material.
SDS-PAGE and immunoblotting.
Whole-cell/ghost lysates (0.05 OD600 units based on cells) were analyzed by standard SDS-PAGE using polyacrylamide gels followed by either Coomassie blue staining or immunoblotting as described before (30). Ghost formation was analyzed by detecting cytoplasmic (SecB, GroEL, DnaK), periplasmic (SurA), membrane (Lep, TolC), and ESAT6 (early secreted antigenic target of 6 kDa; the model antigen/vaccine target incorporated in the Hbp-based surface display system) marker proteins in pellet and supernatant fractions of ghost preparations and control cells by immunoblotting. Part of the pellet fraction was treated with proteinase K (pK) (to monitor surface localization of HbpD-ESAT6), and the supernatant was subjected to trichloroacetic acid (TCA) precipitation (10%, 1 h on ice and a subsequent wash with ice-cold acetone) (19). For immunoblotting experiments, the ECL system (GE Healthcare) was used for detection according to the instructions of the manufacturer, as well as a Fuji LAS-1000 charge-coupled-device (CCD) camera.
BPL-based inactivation of viable cells in ghost preparations.
For β-propiolactone (BPL)-based inactivation of any viable cells in ghost preparations, BPL (0.01%) was added directly to 1 ml of culture 120 min after lysis induction, and the mixture was incubated at 40°C (31). After 30 min, BPL (0.01%) was added again to the culture, and the culture was incubated for another 30 min at 40°C (31). Subsequently, BPL-treated cells/ghosts were washed 3 times with 1 ml PBS. Viability based on CFU was monitored by spreading 100 μl of culture onto LB plates.
Use of Δasd for inactivation of viable cells in ghost preparations.
For Δasd background-based inactivation of any viable cells in ghost preparations, ghosts/cells were harvested as described above, washed three times in PBS, and then resuspended in LB not containing DAP. Cultures were incubated at 30°C for 12 h, and viability was monitored by spreading 100 μl of culture onto LB plates containing DAP.
Freeze-drying of ghost preparations.
Ghost preparations from MC4100, MC4100Δasd, and Salmonella were freeze-dried using a standard freeze dryer. Ghost pellets from 5 ml of culture were resuspended in H2O and subsequently snap-frozen in liquid nitrogen. Freeze-drying was done overnight, and viability was then monitored by resuspending the lyophilized ghosts in LB and incubating the cultures for 7 days at 30°C followed by CFU counting as described above.
Fluorescence microscopy.
For monitoring of the release of the cytoplasmic content during BG formation and the structure of the remaining cell envelope of BGs, E. coli EC452 was used (see “Strains, plasmids, and culture conditions” above). EC452 was cultured as described above; IPTG was added to the culture at an OD600 of 0.25 for induction of gfp expression (see “Strains, plasmids, and culture conditions” above) and E gene expression was subsequently induced at an OD600 of 0.5 by the addition of 6 mM l-rhamnose. EC452 cells/BGs were harvested immediately before lysis induction and 120 min after induction, treated as described above, and stained with the membrane dye FM4-64 as described before (32). Prior to imaging, cells were placed on a microscope glass slide coated with a premade agarose pad (1% [wt/vol] agarose) and were left to immobilize for ∼5 min.
Prior to immunofluorescence microscopy to monitor HbpD-ESAT6 localization, cells/BGs were fixed using cross-linking reagents. Cells/BGs corresponding to 1 OD600 unit were harvested by centrifugation and resuspended in 1 ml of PBS. Subsequently, 1 ml fixing solution (5.6% formaldehyde and 0.08% glutaraldehyde in PBS) was added, and cells/BGs were incubated for 15 min at room temperature. Cells/BGs were thereafter washed three times with PBS, resuspended in 100 μl PBS, and incubated for 1 h with a mouse monoclonal ESAT6 antibody (HYB 76-8; 1:500 dilution in PBS–1% bovine serum albumin [BSA]) (20). The samples were washed once with PBS and subsequently incubated with a secondary goat anti-mouse antiserum labeled with Oregon Green 488 for 1 h and washed 3 times with PBS. Six microliters of cell/BG suspension was placed on a microscope glass, coated with thin premade agarose pads (1% [wt/vol] agarose) containing 0.6 μg/ml ethidium bromide, incubated at room temperature for ∼5 min in order to allow the cells/BGs to immobilize, and then directly inspected under the microscope.
All fluorescence imaging was performed on a Zeiss LSM700 system (Carl Zeiss, Jena, Germany) using an edge-to-edge pinhole of <70 μm and a 63× oil immersion objective (numerical aperture [NA], 1.4).
GFP, FM4-64, and Oregon Green 488 were all excited by a 488-nm diode laser; the excitation power was always kept under 2% of total laser power (10 mW). It was possible to image the pair GFP/FM4-64 (see Fig. 2) simultaneously due to the large Stokes shift in the emission spectra of FM4-64. We used a short-pass filter (SP550) and long-pass filter (PL560) in order to separate the respective emission signal. Ethidium bromide was excited by a 555-nm diode laser. The excitation power was kept under 5% of total laser power (10 mW). All appropriate filter settings for the fluorescence detection were chosen using the ZEN2011 software. The images were then exported to and analyzed in ImageJ (NIH, USA).
FIG 2.

Visualization of E. coli ghosts using confocal fluorescence microscopy and electron microscopy. (A) E. coli EC452 cells and ghosts derived from EC452 were visualized using fluorescence microscopy. The left panel shows EC452 cells that produce GFP in the cytoplasm and harbor pRL1. The cells were incubated with the red fluorescent membrane dye FM4-64 and subsequently visualized using fluorescence microscopy by simultaneously monitoring the fluorescent signals from GFP and FM4-64. The right panel shows a ghost isolated 120 min after the addition of l-rhamnose to the cells shown in the left panel. Bars, 1 μm. (B) The fluorescence microscopy-based analysis of cells and ghosts shown in panel A was verified on a population level using flow cytometry. Both GFP fluorescence and granularity (inset) were monitored. Green line, cells; black line, ghosts; a.u., arbitary units. (C) Cells (left panel) and ghosts (right panel) used for images in panel A were fixed, stained, and visualized by EM. Bars, 2 μm.
It is of note that all pictures of cells/ghosts shown in Fig. 2A, 5A, and 6E are representative of the whole population.
FIG 5.
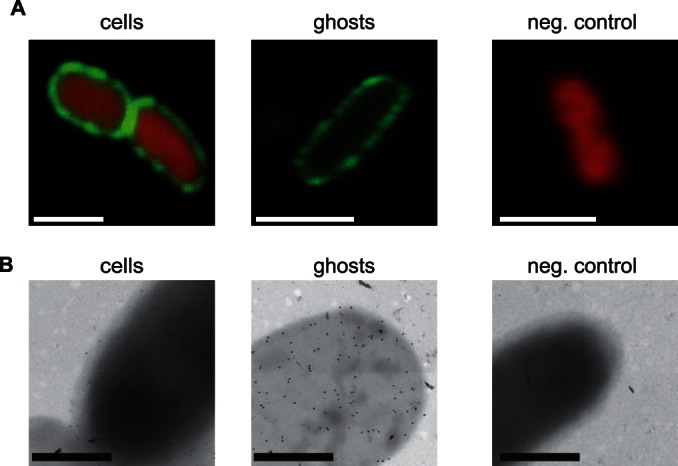
Visualization of E. coli bacterial ghosts displaying HbpD-ESAT6. (A) Confocal fluorescence microscopy images of cells and ghosts displaying HbpD-ESAT6. Left panel, E. coli MC4100 cells harboring pRL1 and pHbpD(p15A)-ESAT6 cultured in the presence of IPTG to induce expression of hbpd-esat6 were incubated with a mouse monoclonal antibody against ESAT6 and a green fluorescent (Oregon green 488) secondary antibody. As a cytoplasmic marker, the DNA binding red fluorescent dye ethidium bromide was used. Middle panel, E. coli MC4100 cells harboring pRL1 and pHbpD(p15A)-ESAT6 cultured in the presence of IPTG to induce expression of hbpd-esat6 and l-rhamnose to induce expression of lysis gene E were treated and visualized as the cells in the left panel. Right panel, as a control, plain MC4100 cells were subjected to the same treatment as cells in the left and middle panels and subsequently visualized as described above. These data were verified by flow cytometry (see Fig. S4 in the supplemental material). Bars, 1 μm. (B) EM images of cells and ghosts displaying Hbp-ESAT6. Left panel, E. coli MC4100 cells harboring pRL1 and pHbpD(p15A)-ESAT6 cultured in the presence of IPTG to induce expression of hbpd-esat6 and subsequently fixed and imaged by immunogold EM, using a mouse monoclonal antibody against ESAT6 and a gold-conjugated anti-mouse antibody (gold particle size, 10 nm). Middle panel, E. coli MC4100 cells harboring pRL1 and pHbpD(p15A)-ESAT6 cultured in the presence of IPTG to induce expression of hbpd-esat6 and l-rhamnose to induce expression of lysis gene E were treated and visualized as the cells in the left panel. Right panel, as a control, plain MC4100 cells were subjected to the same treatment as the cells in the left and middle panels and subsequently visualized by EM. Bars, 500 nm.
FIG 6.

Hbp-based display of ESAT6 in Salmonella ghosts. (A) In Salmonella strain SL3261HbpD-ESAT6, constitutive expression of hbpd-esat6 is governed by the lacUV5 promoter from the chromosome. Since Salmonella has no lac operon, there is no LacI available, which suggests that expression from the lacUV5 promoter is constitutive. Expression of the gene encoding lysis protein E from the high-copy-number plasmid pTL1 is induced upon the addition of anhydro-tetracycline. (B) Salmonella strain SL3261HbpD-ESAT6 harboring pTL1 was cultured in LB medium at 30°C. Expression of lysis gene E was induced with 0.2 μg/ml anhydrotetracycline during mid-log phase at an OD600 of 0.5 (time = 0 min). The optical density of the culture was monitored at the indicated time points during a period of time of 200 min after induction. The inset represents a culture of Salmonella strain SL3261HbpD-ESAT6 harboring pTL1 to which no anhydrotetracycline was added. n = 3, error bars indicate ±SD. (C) The presence of HbpD-ESAT6 in cells and ghosts and the media from which they were isolated was monitored by SDS-PAGE/immunoblotting using anti-ESAT6. (D) Top panel, surface exposure of HbpD-ESAT6 in cells and ghosts was monitored by means of proteinase K accessibility combined with SDS-PAGE/immunoblotting using anti-ESAT6. Bottom panel, cell envelope integrity during the procedure was demonstrated by showing the inaccessibility of the periplasmic chaperone SurA to proteinase K using anti-SurA. (E) Visualization of cells by confocal fluorescence microscopy. Left panel, Salmonella SL3261HbpD-ESAT6 cells harboring pTL1 were incubated with a monoclonal antibody against ESAT6 and a secondary anti-mouse antibody labeled with Oregon Green 488. As before, ethidium bromide was used as a cytoplasmic marker. Middle panel, Salmonella SL3261HbpD-ESAT6 cells harboring pTL1 cultured in the presence of anhydrotetracycline (for induction of the expression of lysis gene E) were treated and visualized as described for the cells in the left panel. Right panel, as a control, plain SL3261 Salmonella cells were subjected to the same treatment as the cells in the left and middle panels. Bars, 1 μm.
Flow cytometry.
Cell/BG size (forward scatter), granularity (side scatter), and fluorescence (FM4-64, cytoplasmic GFP, and Oregon Green 488 probe of the secondary antibody used for ESAT6 labeling) were analyzed by flow cytometry using a FACSCalibur instrument (BD Biosciences) essentially as described before (32). Cells/BGs were prepared as described under “Fluorescence microscopy.” The FlowJo software (Treestar) was used for raw data analysis/processing. Experiments were repeated at least 3 times, and in Fig. 1C, 2B, and 3, as well as in Fig. S4, S5, and S6 in the supplemental material, representative examples are shown.
FIG 1.

E. coli ghost formation by rhaBAD promoter-governed expression of lysis gene E. (A) Schematic representation of an E. coli cell harboring the rhaBAD promoter-based lysis gene E expression vector pRL1. Expression of lysis gene E is induced upon the addition of l-rhamnose. The production of lysis protein E leads to the formation of tunnel structures spanning the entire cell envelope. Through these tunnel structures, the cytoplasmic content of the bacteria is released due to osmotic pressure. (B) E. coli MC4100 cells harboring pRL1 were cultured in LB medium at 30°C. Expression of lysis gene E was induced with 6 mM l-rhamnose during mid-log phase (time = 0 min). The optical density (OD600) of the culture was monitored at the indicated time points during a period of time of 200 min after addition of l-rhamnose. The inset represents a culture of MC4100 cells harboring pRL1 to which no l-rhamnose was added. Error bars indicate ± standard deviations (SD); n = 3. (C) Using flow cytometry, cell and ghost sizes (forward scatter) and granularity (side scatter) were monitored. Straight lines, cells; dotted lines, ghosts. (D) Control cells, ghosts, and the culture medium were analyzed for the presence of a set of cytoplasmic (DnaK, GroEL, and SecB), periplasmic (SurA), and inner membrane protein (Lep) and outer membrane protein (TolC) markers by means of SDS-PAGE/immunoblotting as described in Materials and Methods. P, pellet; S, supernatant. For DNA localization as a marker for ghost formation, see Fig. S1 in the supplemental material.
FIG 3.

Morphology of BPL-treated and Δasd mutant-derived ghosts. (A) On the left, electron micrograph of an E. coli MC4100-derived ghost treated with BPL and freeze-dried. On the right, flow cytometry-based analysis of the size (forward scatter) of ghosts not treated and treated with BPL. Solid line, ghosts not treated with BPL; dotted line, ghosts treated with BPL and freeze-dried. (B) On the left, electron micrograph of an E. coli MC4100Δasd-derived ghost that upon isolation was further processed in the absence in DAP as described in Materials and Methods and freeze-dried. On the right, flow cytometry-based analysis of the size (forward scatter) of MC4100Δasd ghosts derived from cells cultured in the presence of DAP (solid line) and MC4100Δasd ghosts that upon isolation were further processed in the absence of DAP as described in Materials and Methods and finally freeze-dried (dotted line). Bars, 2 μm.
EM and immunoelectron microscopy.
For electron microscopy (EM) and immunoelectron microscopy, to mount cells/BGs on Formvar- and carbon-coated cupper grids (200 mesh, 125-μm pitch; Sigma-Aldrich), grids were floated on 20-μl drops of cell/BG suspensions (1 OD600 unit diluted in 500 μl PBS) for 10 min. The grids were then washed 3 times by letting them float on 20-μl drops of H2O. Liquid was removed between/after the washing steps using filter paper. Subsequently, for negative staining of the samples, the grids were floated on 20-μl drops of 0.5% uranyl acetate–H2O and washed again 3 times with H2O, and liquid was removed using filter paper. Finally, the grids were air dried. A Tecnai G2 Spirit Bio Twin (FEI Company) instrument, 120 kV (samples were imaged at 80 kV), with a Gatan US1000 2K camera was used to image cells/BGs. Images were exported to and analyzed in ImageJ (NIH, USA).
Cells/BGs displaying Hbp-ESAT6 were fixed in 1:1 PBS and 0.4 M PHEM buffer, consisting of 10 mM EGTA, 25 mM HEPES, 2 mM MgCl2, 60 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] containing 4% formaldehyde and 0.4% glutaraldehyde, for 2 h at room temperature. To mount fixed cells/BGs on Formvar- and carbon-coated gold grids (200 mesh, 125-μm pitch; Sigma-Aldrich), the grids were floated on 20-μl drops of fixed cells/BGs for 10 min and washed three times with PBS as described above. Subsequently, grids were incubated with a mouse monoclonal antibody against ESAT6 (HYB 76-8; 1:500 dilution in PBS–1% BSA) for 1 h followed by washing and 1 h of incubation with a 10-nm gold particle-conjugated secondary goat anti-mouse antibody (1:500 dilution in Tris-buffered saline [TBS]–1% BSA; Sigma-Aldrich). Finally, grids were washed, dried, and analyzed as described above.
It is of note that all the pictures of cells/ghosts shown in Fig. 2C, 3, 5B, as well as Fig. S3 in the supplemental material, are representative of the whole population.
RESULTS AND DISCUSSION
Efficient E. coli ghost formation by rhaBAD promoter governed expression of lysis gene E.
The recently developed Hbp-based surface display platform for Gram-negative bacteria makes use of the lacUV5-based promoter, which can be induced with IPTG if the Lac repressor LacI is present (19). To use this platform in combination with lysis protein E-mediated ghost formation, the expression of lysis gene E should be governed by a promoter system other than the lacUV5 promoter. Furthermore, this promoter system should be exceptionally tightly regulated since any background expression of the lethal lysis gene E would negatively affect the stability of the system. The tightly regulated, catabolite-repressed l-rhamnose-inducible promoter (rhaBAD) system has been previously used to express genes encoding toxic proteins (26). E. coli MC4100 cells harboring the pRhamnoseLysis1 (pRL1) vector, which contains lysis gene E under the control of the rhaBAD promoter, were cultured to mid-log phase, and subsequently 6 mM l-rhamnose was added to the culture to induce the expression of the gene encoding lysis protein E (Fig. 1A). To follow protein E-mediated lysis, the OD600 was monitored over a time period of 200 min after addition of l-rhamnose (Fig. 1B). The rapid decline in OD600 upon the addition of l-rhamnose strongly suggests that the rhaBAD promoter system governing the expression of lysis gene E can be used to convert E. coli cells into ghosts. Indeed, comparing CFU just before and 120 min after induction of the expression of lysis gene E showed a reduction in CFU of 99.82% ± 0.15% (n = 3).
Subsequently, ghosts and control cells (i.e., cells harvested just before the induction of the expression of lysis gene E) were studied using flow cytometry (Fig. 1C). Both the forward and side scatters of ghosts and control cells were monitored, which allowed comparing their size and internal complexity, respectively (32–34). The size (forward scatter) of a large fraction of the ghosts resembled the size of the control cells (Fig. 1C, left panel). However, it should be noted that the size of a smaller but considerable fraction of the ghosts was smaller than the size of the control cells, which is most likely due to the destabilizing forces exerted on the cell envelope during the release of the cytoplasmic content. The granularity (side scatter) of ghosts was significantly lower than the granularity of control cells (Fig. 1C, right panel), strongly suggesting that the ghosts were at least partially devoid of their cytoplasmic content.
To further characterize the efficiency of ghost formation by the rhaBAD promoter-driven expression of lysis gene E, as well as the integrity of the resulting ghosts, these were separated from their culture medium and from—if any—membrane fragments and/or vesicles by centrifugation and analyzed for the presence of a set of cytoplasmic, periplasmic, and inner/outer membrane protein markers and for the presence of DNA (Fig. 1D; see also Fig. S1 in the supplemental material). Cells not expressing lysis gene E were used as a control. The set of protein markers included three cytoplasmic proteins (the chaperones DnaK, GroEL, and SecB), one inner membrane protein (leader peptidase [Lep]), one periplasmic protein (the chaperone SurA), and one outer membrane protein (the channel protein TolC). The presence and localization of all six protein markers were monitored by immunoblotting (see Materials and Methods). The immunoblotting experiments showed that the cytoplasmic markers were released into the extracellular milieu (i.e., medium) upon expression of lysis gene E whereas the inner and outer membrane markers remained associated with the ghost fraction. Thus, it is unlikely that expression of lysis gene E from the rhaBAD promoter leads to disintegration of cells, which would have led to the presence of membrane fragments and/or vesicles in the supernatant fraction. Upon expression of lysis gene E, the periplasmic marker SurA partially localized to the soluble fraction, indicating that part of the periplasm was released during the process of ghost formation as has been observed before (9).
The presence/absence of DNA in isolated ghosts and control cells was monitored by gel electrophoresis and ethidium bromide staining (see Fig. S1 in the supplemental material). No DNA was detected in the ghosts, whereas DNA was detected in the medium fraction. In control cells, all three cytoplasmic markers (and also DNA) were found exclusively in the insoluble (i.e., pellet) fraction.
In conclusion, these data demonstrate that the expression of lysis gene E from the rhaBAD promoter in E. coli leads to the release of cytosolic content, indicating efficient formation of ghosts.
Characterization of E. coli ghosts by confocal fluorescence and electron microscopy.
To investigate the morphology of the ghosts, both confocal fluorescence microscopy and EM were used. For visualization of ghosts by confocal fluorescence microscopy, the E. coli strain EC452, which is an MC4100 variant that produces freely diffusing green fluorescent protein (GFP) in the cytoplasm, was used (24). The envelopes of ghosts derived from EC452 harboring pRL1 and control cells were stained with the red fluorescent membrane dye FM4-64 (35) (Fig. 2A). Upon the induction of expression of lysis gene E, the green signal emanating from the cytoplasmic GFP disappeared, while the red cell envelope remained visible (Fig. 2A). In many instances, parts of the envelope structure appeared to be disordered, most likely reflecting the formation of lysis tunnel structures that allow the release of cytoplasmic content. The same samples as those used for fluorescence microscopy were studied by flow cytometry to show that the fluorescent signal indeed disappeared from all cells in the population upon E-mediated lysis (Fig. 2B) (33). These observations were corroborated by EM imaging of ghosts and control cells (Fig. 2C). In the EM images, ghosts are distinguished from whole cells by a more transparent appearance due to decreased electron density, yet retaining, as also indicated by the flow cytometry data in Fig. 1C, a mostly intact envelope.
The uses of (i) β-propiolactone treatment and (ii) an asd deletion background both lead to complete loss of viability of ghost preparations.
As shown above, the rhaBAD promoter-governed expression of lysis gene E in E. coli leads to a reduction in viability that is close to 100%. Ghost preparations are routinely freeze-dried because this greatly facilitates storage and transport (1). Freeze-drying of ghost preparations leads to a complete loss of viability (reference 36 and our unpublished data). Notwithstanding this fact, a recent quality criterion for the bacterial ghost platform stipulates that any viable cells in ghost preparations must be inactivated before freeze-drying (1). Here, we have explored two different strategies to achieve this: (i) the use of β-propiolactone (BPL) and (ii) the use of an asd deletion background.
BPL cross-links ribonucleic acids and is widely used for sterilization of medical equipment and for the production of vaccines (37). The addition of BPL to MC4100 cells expressing lysis gene E indeed led to a complete loss of viability of ghost preparations prior to freeze-drying as monitored by CFU counting (data not shown). Using a combination of EM and flow cytometry, it was shown that BPL treatment does not affect the morphology of the ghosts (Fig. 3A).
As an alternative to chemical inactivation prior to freeze-drying, we explored the use of an asd deletion background. The essential asd gene encodes the enzyme aspartate semialdehyde dehydrogenase, which is involved in the synthesis of diaminopimelic acid (DAP) (38). DAP is required for the synthesis of the peptidoglycan in Gram-negative bacteria, and it is synthesized only by bacteria. We constructed an MC4100 asd deletion mutant strain (MC4100Δasd), which is, as expected, strictly dependent on the addition of DAP to the culture medium for growth. In the presence of DAP, this mutant strain's behavior was similar to that of the isogenic E. coli MC4100 with respect to growth and protein E-mediated lysis (see Fig. S2 in the supplemental material). To inactivate any viable cells before freeze-drying, the ghost preparations were washed and resuspended in culture medium not containing DAP and incubated for 12 h. The omission of DAP led to the complete inactivation of any viable cells as monitored by CFU counting (data not shown). Furthermore, using a combination of EM and flow cytometry, it was shown that removing DAP after ghost formation did not affect ghost morphology and size (Fig. 3B). On the other hand, EM analysis showed that exclusion of DAP from the MC4100Δasd culture medium prior to expression of lysis protein E led to the complete disintegration of cells (see Fig. S3 in the supplemental material).
Taken together, both the use of BPL and the use of an asd deletion strain background appeared to be effective strategies to achieve complete loss of viability of ghost preparations prior to freeze-drying without affecting the integrity of the ghosts.
Hbp-mediated surface display of antigen ESAT6 in E. coli ghosts.
Next, we investigated whether ghosts can be decorated with antigens using the Hbp platform. To this end, we cotransformed E. coli MC4100 cells harboring the rhaBAD promoter-based lysis gene E expression vector pRL1 with the compatible lacUV5 promoter-based vector pHbpD(p15A)-ESAT6, which allows the production of an HbpD derivative carrying ESAT6 at the position of passenger side domain 1 (HbpD-ESAT6) (Fig. 4A) (19).
FIG 4.

Hbp-mediated surface display of antigen ESAT6 in E. coli ghosts. (A) A schematic representation of how E. coli-derived ghosts displaying ESAT6 on their surface are generated. MC4100/pRL1 cells are transformed with the with pRL1-compatible plasmid pHbpD(p15A)-ESAT6, which harbors hbpd-esat6 under the control of the lacUV5 promoter. Expression of hbpd-esat6 is induced by the addition of IPTG. Expression of lysis gene E is induced by the addition of l-rhamnose and results in the formation of ghosts. (B) E. coli MC4100 cells harboring pRL1 and pHbpD(p15A)-ESAT6 were cultured in LB at 30°C. Expression of hbpd-esat6 was induced by the addition of 0.4 mM IPTG at an OD600 of 0.25. Expression of lysis gene E was induced with 6 mM l-rhamnose during mid-log phase, at an OD600 of 0.5 (time = 0 min). The optical density (OD600) of the culture was monitored at the indicated time points during a period of time of 200 min. The inset represents a culture of MC4100 cells harboring pRL1 and pHbpD(p15A)-ESAT6 to which no l-rhamnose was added. n = 3; error bars indicate ±SD. (C) The presence of HbpD-ESAT6 in cells and ghosts and the culture media was monitored by SDS-PAGE/immunoblotting using anti-ESAT6. P, pellet; S, supernatant. (D) The abundance of HbpD-ESAT6, marked by an asterisk (*), in cells and ghosts was monitored by SDS-PAGE followed by Coomassie blue staining. As controls, cells and ghosts without any HbpD-ESAT6 were used. (E) Top panel, surface exposure of HbpD-ESAT6 in cells and ghosts was monitored by means of proteinase K accessibility combined with SDS-PAGE/immunoblotting using a monoclonal antibody against ESAT6. Bottom panel, cell envelope integrity during the procedure was demonstrated by showing the inaccessibility of the periplasmic chaperone SurA to proteinase K using anti-SurA.
To monitor protein E-mediated lysis of MC4100 cells producing HbpD-ESAT6, expression of HbpD-ESAT6 was induced at an OD600 of 0.25 with 0.4 mM IPTG, and expression of lysis gene E was subsequently induced at an OD600 of 0.5 with 6 mM l-rhamnose. Upon the addition of l-rhamnose, culture turbidity was further monitored for over 200 min (Fig. 4B). The rapid decline in OD600 upon the addition of l-rhamnose indicates efficient formation of ghosts. Proper ghost formation was also supported by CFU counting showing a lysis efficiency of 99.47% ± 0.23% (n = 3) and by monitoring the location of HbpD-ESAT6 (Fig. 4C) and the same set of markers as those shown in Fig. 1D (data not shown). It should also be noted that lysis efficiency was maintained also when HbpD-ESAT6 was coproduced with lysis protein E in MC4100Δasd (data not shown). Furthermore, by SDS-PAGE combined with Coomassie blue staining it was shown that HbpD-ESAT6 is present at high levels in cells and ghosts (Fig. 4D) (19). Surface localization of HbpD-ESAT6 was monitored by treating control cells and ghosts with proteinase K (pK) followed by immunoblotting with a monoclonal antibody against ESAT6 (Fig. 4E, top panel). No HbpD-ESAT6 could be detected in ghosts and control cells after the pK treatment, indicating that HbpD-ESAT6 was efficiently displayed on the surface of ghosts and control cells. As a control to monitor the integrity of the cells/ghosts upon pK treatment, the levels of the periplasmic marker protein SurA were monitored. These experiments showed that SurA was protected against the proteolytic activity of pK, indicating that the pK treatment did not affect cell/ghost integrity (Fig. 4E, bottom panel). For the sake of clarity, SurA accumulation levels were here determined in isolated ghosts, i.e., after the partial release of SurA into the extracellular milieu during the process of ghost formation as seen in Fig. 1D.
Taken together, these data indicate that E. coli cells producing HbpD-ESAT6 can be used to generate ghosts without affecting ghost formation efficiency and ghost integrity.
Visualization of ghosts displaying HbpD-ESAT6 by fluorescence and electron microscopy.
To monitor the morphology of ghosts displaying HbpD-ESAT6 and the distribution of HbpD-ESAT6 on the surface, both fluorescence microscopy and immuno-EM were used.
For visualization by fluorescence microscopy, cells with HbpD-ESAT6 on their surface and ghosts derived from these cells were first incubated with a mouse monoclonal antibody against ESAT6 and subsequently a secondary anti-mouse antibody labeled with a green fluorescent probe (Oregon Green 488). The red fluorescent dye ethidium bromide, which enters the cytoplasm of cells and labels ribonucleic acids, was used to stain the cytoplasm. Cells displaying HbpD-ESAT6 feature a green halo representing the displayed ESAT6 antigen and a red cytoplasm representing the DNA/RNA in the cytoplasm (Fig. 5A, left panel [the right panel shows the control]). As expected, ghosts derived from these cells also show the green halo but lack the ethidium bromide signal, indicating that ghost formation is not affected by the high-level antigen display on their surface (Fig. 5A, middle panel [right panel, control]). The signal intensity and distribution of ESAT6 do not differ between whole cells and bacterial ghosts. The labeled cells and ghosts were also studied using flow cytometry (see Fig. S4 in the supplemental material), which showed that the ESAT6 levels in both whole cells and ghosts are homogeneous and HbpD-ESAT6 levels are similar. In addition, neither freeze-drying nor BPL treatment nor DAP depletion in combination with a Δasd background affected surface display of ESAT6 (see Fig. S4 in the supplemental material).
To monitor the distribution of HbpD-ESAT6 displayed on the surface of ghosts at a higher resolution, immuno-EM was used. A mouse monoclonal antibody against ESAT6 and subsequently a secondary anti-mouse antibody conjugated to a 10-nm gold particle were used to label ESAT6 (Fig. 5B). These data show that the HbpD-ESAT6 is evenly distributed over the cell envelope and that the distribution is maintained after E-mediated lysis.
Taken together, microscopic evaluation revealed that HbpD-ESAT6 production does not interfere with ghost formation and that ghost formation does not alter the efficiency and distribution of surface-displayed HbpD-ESAT6.
Hbp-based display of ESAT6 in Salmonella ghosts.
Recently, we have shown that the Hbp-based display platform can be used to efficiently display ESAT6 on the surface of the attenuated Salmonella Typhimurium strain SL3261 (19). Notably, in SL3261 HbpD-ESAT6 antigen surface display is based on the constitutive expression of hbpd-esat6 from the lacUV5 promoter from the chromosome (Fig. 6A). In Salmonella, expression from this promoter is constitutive since this bacterium does not have a lac operon and thus does not produce LacI repressor (39). To convert Salmonella cells producing HbpD-ESAT6 on the cell surface into bacterial ghosts, we first used the rhaBAD promoter system to express lysis gene E. However, in contrast to our observations in E. coli, OD600 measurements and CFU counting revealed that the rhaBAD promoter-based expression of gene E did not lead to the efficient formation of ghosts in Salmonella (results not shown). Therefore, we explored the use of the strong and tight tetracycline promoter (tetA) to express lysis gene E in Salmonella (Fig. 6A). SL3261HbpD-ESAT6 cells harboring vector pTetracyclinLysis (pTL1) were cultured to mid-log phase, and subsequently the tetracycline analog anhydrotetracycline was added to the medium to induce expression of lysis gene E (27). OD600 measurements and CFU counting indicated that expression of lysis gene E in this context resulted in efficient ghost formation (99.87% ± 0.10% [n = 3]) (Fig. 6B). Efficient formation of SL3261HbpD-ESAT6 ghosts was corroborated by flow cytometry (see Fig. S5 in the supplemental material) and by monitoring the presence of HbpD-ESAT6 (Fig. 6C) and the same set of cellular markers as those used for Fig. 1D, including DNA (data not shown), in ghosts, control cells, and culture medium.
Surface display of HbpD-ESAT6 on Salmonella cells and ghosts was monitored by proteinase K treatment and subsequent analysis by SDS-PAGE and immunoblotting using a monoclonal antibody against ESAT6 (Fig. 6D). On both cells and ghosts, HbpD-ESAT6 appeared fully accessible to pK and, hence, degraded, whereas no effect was observed on the levels of periplasmically localized SurA. This indicates that HbpD-ESAT6 was efficiently displayed on the surface of ghosts and control cells. Using a combination of fluorescence microscopy (Fig. 6E) and immuno-EM (data not shown), it was shown that HbpD-ESAT6 is distributed homogeneously on the surface of Salmonella cells and derived ghosts. This was verified on a population level by flow cytometry. In addition, neither freeze-drying, nor BPL treatment, nor an asd deletion background affected surface display of ESAT6 in SL3261HbpD-ESAT6-derived ghosts (Fig. 6 and results not shown).
Taken together, these results indicate that the Hbp-based surface display platform can be used to decorate the surface of Salmonella ghosts with antigens.
In summary, using M. tuberculosis ESAT6 as a model antigen, we have shown that the concerted production of an Hbp-display carrier and lysis protein E in both E. coli and Salmonella is a successful strategy for the formation of ghosts efficiently displaying antigens on their surface. Subsequent procedures to eliminate the last traces of live bacteria did not interfere with surface display of ESAT6. The novel combination of Hbp-mediated surface display of heterologous antigens and ghost formation constitutes a very safe, efficient, and accessible method for antigen delivery and vaccine development.
Supplementary Material
ACKNOWLEDGMENTS
The research leading to these results has received funding from the Swedish Research Council (VR-M), the Swedish Foundation for Strategic Research (SSF) through the Center for Biomembrane Research, and the European Union's Seventh Framework Programme (FP7/2007-2013) under Grant Agreement number 280873 ADITEC. In addition, W.S.P.J. was supported by a grant from the Dutch Technology Foundation STW.
We acknowledge A.-S. Höglund at the Imaging Facility of Stockholm University for support with EM microscopy.
W.S.P.J. and J.L. are involved in Abera Bioscience AB, which aims to exploit the Hbp platform for vaccine development. D.V. and J.-W.D.G. are involved in Xbrane Bioscience AB. Abera Bioscience AB and Xbrane Bioscience AB are both part of Serendipity Innovations.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02733-14.
REFERENCES
- 1.Langemann T, Koller VJ, Muhammad A, Kudela P, Mayr UB, Lubitz W. 2010. The Bacterial Ghost platform system: production and applications. Bioeng Bugs 1:326–336. doi: 10.4161/bbug.1.5.12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kudela P, Koller VJ, Lubitz W. 2010. Bacterial ghosts (BGs)–advanced antigen and drug delivery system. Vaccine 28:5760–5767. doi: 10.1016/j.vaccine.2010.06.087. [DOI] [PubMed] [Google Scholar]
- 3.Riedmann EM, Kyd JM, Cripps AW, Lubitz W. 2007. Bacterial ghosts as adjuvant particles. Expert Rev Vaccines 6:241–253. doi: 10.1586/14760584.6.2.241. [DOI] [PubMed] [Google Scholar]
- 4.Young R. 1992. Bacteriophage lysis: mechanism and regulation. Microbiol Rev 56:430–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasi U, Linke RP, Lubitz W. 1989. Evidence for membrane-bound oligomerization of bacteriophage phi X174 lysis protein-E. J Biol Chem 264:4552–4558. [PubMed] [Google Scholar]
- 6.Tanaka S, Clemons WM Jr. 2012. Minimal requirements for inhibition of MraY by lysis protein E from bacteriophage PhiX174. Mol Microbiol 85:975–985. doi: 10.1111/j.1365-2958.2012.08153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Y, Struck DK, Young R. 2009. Purification and functional characterization of phiX174 lysis protein E. Biochemistry 48:4999–5006. doi: 10.1021/bi900469g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernhardt TG, Roof WD, Young R. 2000. Genetic evidence that the bacteriophage phi X174 lysis protein inhibits cell wall synthesis. Proc Natl Acad Sci U S A 97:4297–4302. doi: 10.1073/pnas.97.8.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Witte A, Wanner G, Blasi U, Halfmann G, Szostak M, Lubitz W. 1990. Endogenous transmembrane tunnel formation mediated by phi X174 lysis protein E. J Bacteriol 172:4109–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witte A, Schrot G, Schon P, Lubitz W. 1997. Proline 21, a residue within the alpha-helical domain of phiX174 lysis protein E, is required for its function in Escherichia coli. Mol Microbiol 26:337–346. doi: 10.1046/j.1365-2958.1997.5781941.x. [DOI] [PubMed] [Google Scholar]
- 11.Schon P, Schrot G, Wanner G, Lubitz W, Witte A. 1995. Two-stage model for integration of the lysis protein E of phi X174 into the cell envelope of Escherichia coli. FEMS Microbiol Rev 17:207–212. [DOI] [PubMed] [Google Scholar]
- 12.Choi SH, Nam YK, Kim KH. 2010. Novel expression system for combined vaccine production in Edwardsiella tarda ghost and cadaver cells. Mol Biotechnol 46:127–133. doi: 10.1007/s12033-010-9277-2. [DOI] [PubMed] [Google Scholar]
- 13.Jechlinger W, Haller C, Resch S, Hofmann A, Szostak MP, Lubitz W. 2005. Comparative immunogenicity of the hepatitis B virus core 149 antigen displayed on the inner and outer membrane of bacterial ghosts. Vaccine 23:3609–3617. doi: 10.1016/j.vaccine.2004.11.078. [DOI] [PubMed] [Google Scholar]
- 14.Hess J, Gentschev I, Miko D, Welzel M, Ladel C, Goebel W, Kaufmann SH. 1996. Superior efficacy of secreted over somatic antigen display in recombinant Salmonella vaccine induced protection against listeriosis. Proc Natl Acad Sci U S A 93:1458–1463. doi: 10.1073/pnas.93.4.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang HY, Curtiss R III. 2003. Immune responses dependent on antigen location in recombinant attenuated Salmonella typhimurium vaccines following oral immunization. FEMS Immunol Med Microbiol 37:99–104. doi: 10.1016/S0928-8244(03)00063-4. [DOI] [PubMed] [Google Scholar]
- 16.Muralinath M, Kuehn MJ, Roland KL, Curtiss R III. 2011. Immunization with Salmonella enterica serovar Typhimurium-derived outer membrane vesicles delivering the pneumococcal protein PspA confers protection against challenge with Streptococcus pneumoniae. Infect Immun 79:887–894. doi: 10.1128/IAI.00950-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Ulsen P, Rahman SU, Jong WS, Daleke-Schermerhorn MH, Luirink J. 2014. Type V secretion: from biogenesis to biotechnology. Biochim Biophys Acta 1843:1592–1611. doi: 10.1016/j.bbamcr.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Jong WS, Sauri A, Luirink J. 2010. Extracellular production of recombinant proteins using bacterial autotransporters. Curr Opin Biotechnol 21:646–652. doi: 10.1016/j.copbio.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Jong WS, Soprova Z, de Punder K, ten Hagen-Jongman CM, Wagner S, Wickstrom D, de Gier JW, Andersen P, van der Wel NN, Luirink J. 2012. A structurally informed autotransporter platform for efficient heterologous protein secretion and display. Microb Cell Fact 11:85. doi: 10.1186/1475-2859-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorensen AL, Nagai S, Houen G, Andersen P, Andersen AB. 1995. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect Immun 63:1710–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jong WS, ten Hagen-Jongman CM, den Blaauwen T, Slotboom DJ, Tame JR, Wickstrom D, de Gier JW, Otto BR, Luirink J. 2007. Limited tolerance towards folded elements during secretion of the autotransporter Hbp. Mol Microbiol 63:1524–1536. doi: 10.1111/j.1365-2958.2007.05605.x. [DOI] [PubMed] [Google Scholar]
- 22.Detmer A, Glenting J. 2006. Live bacterial vaccines—a review and identification of potential hazards. Microb Cell Fact 5:23. doi: 10.1186/1475-2859-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 24.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol 181:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoiseth SK, Stocker BA. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- 26.Giacalone MJ, Gentile AM, Lovitt BT, Berkley NL, Gunderson CW, Surber MW. 2006. Toxic protein expression in Escherichia coli using a rhamnose-based tightly regulated and tunable promoter system. Biotechniques 40:355–364. doi: 10.2144/000112112. [DOI] [PubMed] [Google Scholar]
- 27.Skerra A. 1994. Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 151:131–135. doi: 10.1016/0378-1119(94)90643-2. [DOI] [PubMed] [Google Scholar]
- 28.Peubez I, Chaudet N, Mignon C, Hild G, Husson S, Courtois V, De Luca K, Speck D, Sodoyer R. 2010. Antibiotic-free selection in E. coli: new considerations for optimal design and improved production. Microb Cell Fact 9:65. doi: 10.1186/1475-2859-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Froderberg L, Rohl T, van Wijk KJ, de Gier JW. 2001. Complementation of bacterial SecE by a chloroplastic homologue. FEBS Lett 498:52–56. doi: 10.1016/S0014-5793(01)02494-2. [DOI] [PubMed] [Google Scholar]
- 31.Lubitz W. June 2011. US patent 7,968,323 B2 http://www.google.com/patents/US7968323.pdf.
- 32.Wagner S, Baars L, Ytterberg AJ, Klussmeier A, Wagner CS, Nord O, Nygren PA, van Wijk KJ, de Gier JW. 2007. Consequences of membrane protein overexpression in Escherichia coli. Mol Cell Proteomics 6:1527–1550. doi: 10.1074/mcp.M600431-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Haidinger W, Szostak MP, Beisker W, Lubitz W. 2001. Green fluorescent protein (GFP)-dependent separation of bacterial ghosts from intact cells by FACS. Cytometry 44:106–112. doi:. [DOI] [PubMed] [Google Scholar]
- 34.Haidinger W, Szostak MP, Jechlinger W, Lubitz W. 2003. Online monitoring of Escherichia coli ghost production. Appl Environ Microbiol 69:468–474. doi: 10.1128/AEM.69.1.468-474.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fishov I, Woldringh CL. 1999. Visualization of membrane domains in Escherichia coli. Mol Microbiol 32:1166–1172. doi: 10.1046/j.1365-2958.1999.01425.x. [DOI] [PubMed] [Google Scholar]
- 36.Mayr UB, Haller C, Haidinger W, Atrasheuskaya A, Bukin E, Lubitz W, Ignatyev G. 2005. Bacterial ghosts as an oral vaccine: a single dose of Escherichia coli O157:H7 bacterial ghosts protects mice against lethal challenge. Infect Immun 73:4810–4817. doi: 10.1128/IAI.73.8.4810-4817.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perrin P, Morgeaux S. 1995. Inactivation of DNA by beta-propiolactone. Biologicals 23:207–211. doi: 10.1006/biol.1995.0034. [DOI] [PubMed] [Google Scholar]
- 38.Galan JE, Nakayama K, Curtiss R III. 1990. Cloning and characterization of the asd gene of Salmonella typhimurium: use in stable maintenance of recombinant plasmids in Salmonella vaccine strains. Gene 94:29–35. doi: 10.1016/0378-1119(90)90464-3. [DOI] [PubMed] [Google Scholar]
- 39.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.