

Abstract
Africa is home to genetically diverse human populations. We compared the genetic structure of the Wolaita ethnic population from southern Ethiopia (WETH, n=120) with HapMap populations using genome-wide variants. We investigated allele frequencies of 443 clinically and pharmacogenomically relevant genetic variants in WETH compared to HapMap populations. We found that WETH were genetically most similar to the Kenya Maasai and least similar to the Japanese in HapMap. Variant alleles associated with increased risk of adverse reactions to drugs used for treating tuberculosis (rs1799929 and rs1495741 in NAT2), thromboembolism (rs7294, rs9923231 and rs9934438 in VKORC1), and HIV/AIDS and solid tumors (rs2242046 in SLC28A1) had significantly higher frequencies in WETH compared to African ancestry HapMap populations. Our results illustrate that clinically relevant pharmacogenomic loci display allele frequency differences among African populations. We conclude that drug dosage guidelines for important global health diseases should be validated in genetically diverse African populations.
Keywords: pharmacogenomics, global health, tuberculosis, HIV/AIDS, warfarin, Ethiopia
INTRODUCTION
Several variants in genes that encode for drug metabolizing and transport proteins have been demonstrated to affect inter-individual drug responses by regulating the absorption, distribution, metabolism, and excretion (ADME) of medications.1 Allele frequency differences for these variants are greater among African than European or Asian populations.2 Therefore, extrapolation of allele frequencies of these variants genotyped in one African population to another African population may be misleading.3 Given the wide genetic diversity and population structure of African populations,4 mapping the genetic structure of African populations that were not included in international genotyping and sequencing projects such as HapMap (http://hapmap.ncbi.nlm.nih.gov/) and the 1000 Genomes project (http://www.1000genomes.org/) will be valuable to understand the spectrum of genetic variation. Including diverse African populations in genetic mapping studies will also be useful to evaluate whether allele frequencies of loci associated with disease phenotypes are transferable across populations.
Moreover, documentation of the distribution of clinically and pharmacogenomically relevant variants across diverse African populations is critical to identify individuals and populations with relatively higher genetic risk for drug-induced toxicity and reduced drug response, and to make inference on the global health implications of those variants. For example, the HLA-B*57:01 allele that is strongly associated with hypersensitivity reactions to abacavir (a drug used to treat HIV/AIDS) shows wide frequency differences among African populations, ranging from 0% in Yoruba from Nigeria to 13.6% in Maasai from Kenya.5 Moreover, the CYP2B6*6 (rs3745274 T) allele known to be associated with high plasma concentration and adverse events to efavirenz (a drug used to treat HIV/AIDS) show significant allele frequency difference between East African populations (41.9% in Tanzanians vs. 31.4% in Ethiopians).6 The rs12979860 C allele associated with sustained hepatitis C virus clearance following treatment with the combination therapy of pegylated interferon-alpha and ribavirin is less common in populations of African ancestry compared to those of European or Asian ancestry,7 and varies in frequency among African populations (e.g. 23.5% in Biaka pygmies, 29.4% in Yoruba from Nigeria, and 59.6% in Luhya from Kenya) (http://hapmap.ncbi.nlm.nih.gov/).
Here we report genetic variations in the Wolaita ethnic population (WETH) from the Wolaita zone, Southern Ethiopia in comparison with African and non-African ancestry populations included in the International HapMap. The Wolaita are one of the indigenous population groups of Ethiopia that have inhabited the mid- and high-land areas of southern Ethiopia for several thousand years, and predominantly speak Wolaita, an Omotic branch of the Afroasiatic language family. Our aims were to determine the genetic structure of WETH as compared to global human population samples in HapMap, and to identify genetic variants of clinical and pharmacogenomic relevance that differentiate WETH from other HapMap populations.
MATERIALS AND METHODS
Data sets
We used genotype data from 120 randomly selected WETH individuals who were recruited to serve as controls in a genome-wide association study (GWAS) of podoconiosis (a type of lower limb lymphoedema resulting from long-term barefoot exposure to volcanic clay soil).8 Ethnically matched healthy controls were selected for all the cases, all of whom had self-identified Wolaita ethnicity. The analyzed samples did not have cryptic relatedness because significant excess sharing of marker alleles identical by descent from the same ancestral chromosome was ruled out because the PI_HAT values (a measure of the degree of genetic relationship between participants) were less than 0.05. Moreover, consanguinity is unexpected in the Wolaita population because marriage is practiced only among unrelated people. Genotyping was performed by deCODE Genetics using a chip (the Illumina HumanHap 610 Bead Chip) that contains more than 620 000 SNPs. Of the 551 840 autosomal SNPs in the raw genotype data, we excluded 39 249 SNPs that had a minor allele frequency (MAF) of <5%, 378 that were missing in more than 5% of individuals, and 321 that had a Hardy-Weinberg p-value <=0.001. The remaining 511 892 SNPs that passed quality filters were merged with the HapMap phase 3, release 2 database. The HapMap data contained 1 440 616 SNP genotypes representing the consensus dataset obtained after merging genotypes obtained from the Affymetrix Human SNP Array 6.0 and the Illumina Human1M-single beadchip in 1 184 individuals from 11 populations groups. A total of 464 642 SNPs were common to both datasets in 1 075 unrelated individuals (120 from WETH and 955 unrelated individuals included in HapMap 3). The ethno-geographical breakdown of the 955 HapMap individuals was as follows: African ancestry in Southwest USA (ASW, n=42), Utah residents with Northern and Western European ancestry from the CEPH collection (CEU, n=109), Han Chinese in Beijing, China (CHB, n=84), Chinese in Metropolitan Denver, Colorado (CHD, n=85), Gujarati Indians in Houston, Texas (GIH, n=88), Japanese in Tokyo, Japan (JPT, n=86), Luhya in Webuye, Kenya (LWK, n=83), Mexican ancestry in Los Angeles, California (MEX, n=50), Maasai in Kinyawa, Kenya (MKK, n=143), Tuscans in Italy (TSI, n=77), and Yoruba in Ibadan, Nigeria (YRI, n=108) (http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap3r2_B36/).9 For brevity, we use HapAFR when referring to the four HapMap populations of major recent sub-Saharan African ancestry (i.e., MKK, LWK, YRI, and ASW), and HapNonAFR when referring to the remaining seven HapMap populations.
Genetic structure
Multidimensional scaling (MDS) analysis
We obtained a list of SNPs that were not correlated with each other by performing linkage disequilibrium (LD) pruning of the 551 840 SNPs in the WETH dataset using an r2 threshold of 0.2, and a sliding window of 50 SNPs by skipping 5 SNPs between consecutive windows. A total of 123 182 SNPs remained after LD pruning, of which 112 480 SNPs were also in the HapMap data set and were used for MDS analysis as implemented in the program PLINK. To test whether middle-eastern populations that have shared genetic ancestry with Semetic-Cushitic Ethiopians also share similar level of ancestry with the Omotic WETH, we did a second MDS analysis by adding three middle-eastern populations from southwestern Asia (Bedouin from the Negev region of Israel, BED, n=45; Palestinian from the central region of Israel, PAL, n=44; and Druze from northern Israel, DRU, n=41) collected by the Human Genome Diversity Project (HGDP) as part of the Human Genome Diversity Cell Line Panel (http://hagsc.org/hgdp/files.html). We calculated squared correlation (r2) between adjacent SNPs and genome-wide mean r2in the program PLINK.
Fixation index (FST)
We randomly selected 10% of the 112 480 SNPs used in the MDS analysis (i.e., n=11 240 SNPs) and calculated pair-wise FST, a measure of the proportion of total variation in allele frequency that is due to population differences, between WETH and each HapMap population using GENEPOP, a population genetics software package that applies Weir and Cockerham’s multilocus FST estimates.10, 11
Admixture analysis
To estimate the extent of broad genetic clusters shared between WETH and HapMap populations, we conducted unsupervised model-based clustering analysis using the 11 240 SNPs used in FST analysis as implemented in the program STRUCTURE.12, 13 Analysis was performed using admixture model at burn-in period of 20 000 iterations followed by 10 000 Markov chain Monte Carlo replications. To determine the number of clusters that best fit our data, we evaluated the models using the estimated log likelihood of the data at each K, for K=2 to K=12. A graphical display of the resulting clusters was produced using the program DISTRUCT.14
Genome-wide and pharmacogenomic variant differentiations
We conducted allelic-based (1 d.f.) chi-squared tests on the 464 642 SNPs to test genome-wide allele frequency differences between WETH and each HapMap population. Next, we extracted known pharmacogenomically and clinically relevant SNPs from the Affymetrix drug metabolism enzymes and transporters (DMET) Plus platform (http://www.affymetrix.com),15 and the Pharmacogenomics Knowledgebase (PharmGKB, http://www.pharmgkb.org). We found that a total of 443 SNPs represented in our datasets were also included in these databases. We compared allele frequencies of WETH with each of the 11 HapMap populations using Pearson correlation, chi-squared, and FST tests across the 443 SNPs. P-values were considered to be statistically significant after the Bonferroni-correction was applied (P <=1 × 10−7 i.e., 0.05/464 642 for the genome-wide test, and P<= 1.13×10−4 i.e., 0.05/443 for the pharmacogenomic SNP test).
Ethical considerations
The study was approved by the institutional ethics review boards of the Medical Faculty of Addis Ababa University and the Armauer Hansen Research Institute–All Africa Leprosy, Tuberculosis and Rehabilitation Training Center as well as the ethics review committee of the Ethiopian Ministry of Science and Technology. Written informed consent was obtained from all participants prior to data collection. We have described, in detail, the tailored approach we followed for obtaining consent from the study participants elsewhere.16, 17
RESULTS
Genetic structure
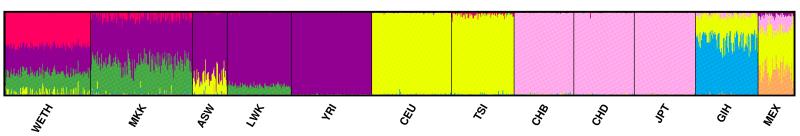
In the MDS analysis, the first dimension explained 7.95% of the total genetic variance, and separated African and non-Africans. The WETH lay at the farthest end of the African cluster along the first dimension, nearest to MKK and farthest from YRI. The second dimensions explained 3.72% of the genetic variance, and separated WETH from MKK (Figure 1). The WETH cluster was relatively farther from the middle-eastern population clusters (Supplementary Figure 1). FST analyses showed that WETH had the smallest differentiation with MKK (FST= 0.01) and the largest differentiation with JPT (FST = 0.12) (Figure 2). The genetic clusters produced in STRUCTURE for each K from K=2 to K=12 are shown in Figure 3 & Supplementary Figure 2. The best fit result was at K=7 (Supplementary Table 1). Three genetic clusters formed 96.8% of ancestry in WETH: the biggest genetic cluster in WETH (cluster 7) contributing 40.4% of ancestry was shared with MKK (5.7%) and TSI (1.9%); the second cluster (cluster 6, 32.2%) formed 99.6% of the variation in YRI; the third cluster (cluster 5, 24.2%) was common in East African HapMap populations (38.7% in MKK and 10.9% in LWK) (Supplementary Tables 2 and 3).
Figure 1. Plots of top three multidimensional scaling analysis of autosomal SNPs in WETH and 11 HapMap population groups.
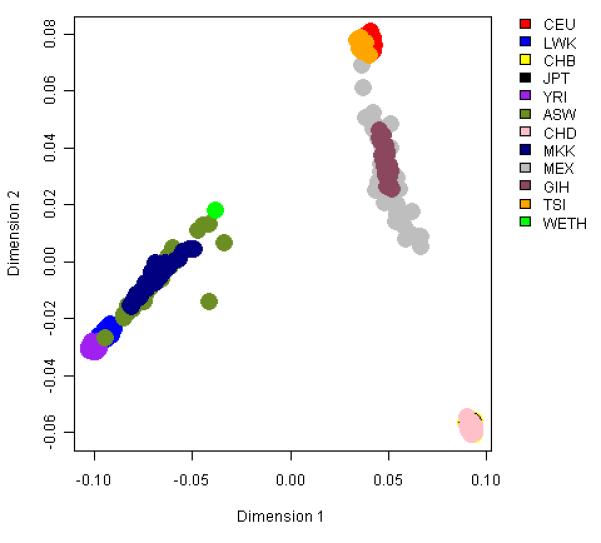
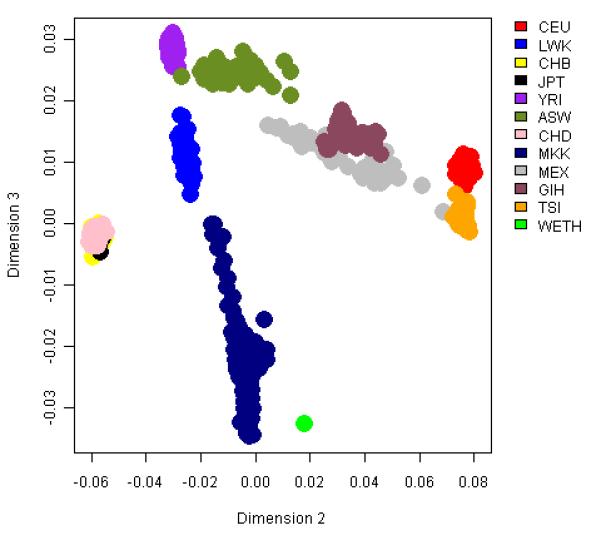
A Plot of dimensions 1 and 2 which captured 7.95% and 3.72% of the variation. B Plot of dimensions 2 and 3 which captured 3.72% and 0.71% of the variation.
Figure 2. Genetic differentiation between populations estimated by pair-wise FST distance.

Abbreviations: WETH, Wolaita from Wolaita zone, Ethiopia; ASW, African ancestry in Southwest USA; LWK, Luhya in Webuye, Kenya; MKK, Maasai in Kinyawa, Kenya; YRI, Yoruba in Ibadan, Nigeria; TSI, Tuscans in Italy; CEU, Utah residents with Northern and Western European ancestry from the CEPH collection; MEX, Mexican ancestry in Los Angeles, California; GIH, Gujarati Indians in Houston, Texas; CHB, Han Chinese in Beijing, China; CHD, Chinese in Metropolitan Denver, Colorado; JPT, Japanese in Tokyo, Japan.
Figure 3. Structure-defined population structure when the number of clusters (K)=7.
Genome-wide LD pattern
The adjacent SNP LD r2metrics of WETH were more strongly correlated with those of HapAFR (r2>0.80) than with those of HapNonAFR (r2<0.77). Moreover, the genome-wide mean r2of WETH was similar to that of HapAFR and lower than that of HapNonAFR (Supplementary Table 4). The number of SNPs in strong LD (r2≥0.8) with one or more SNPs was fewer in WETH compared with HapNonAFR, and slightly more in WETH compared with HapAFR. Within the HLA locus, in which a genome-wide significant association with podoconiosis was previously found, 8 LD was stronger and the r2 differences between populations were smaller (Figure 4; Supplementary Table 5).
Figure 4. Proportion of SNPs in strong linkage disequilibrium (r2>=0.8) with at least one other SNP in different locus window sizes.

A Genome-wide. B HLA locus.
Genome-wide and pharmacogenomic variant differentiations
Allele frequencies were significantly different between WETH and HapMap populations for several SNPs (Supplementary Figure 3), the fewest being with MKK (n=3 587 SNPs) and the most being with JPT (n=103 182 SNPs). Pair-wise comparisons of the five African-ancestry populations showed that each population had more SNPs with statistically significant allele frequency differences when compared to WETH than when compared to any other African population (Supplementary Table 6). Four SNPs (rs803904 and rs9697091 in ASTN2, rs10219883 in FRY, and rs10410147 in CTB-50E14.6) showed statistically significant allele frequency differences between WETH and all HapMap populations (Supplementary Table 7).
Pair-wise FST analysis on the 443 pharmacogenomic variants showed similar patterns and higher levels of genetic differentiation between WETH and HapMap populations compared to the genome-wide FST estimates (Supplementary Table 8 & Table 1). Plots of pharmacogenomic SNP allele frequency correlations between WETH and HapMap populations are shown in Supplementary Figure 4. WETH had stronger allele frequency correlation with MKK (r2= 0.75), low to moderate correlation with other HapMap Africa populations (r2= 0.47-0.54), and weaker correlation with HapNonAFR (r2<0.30). We found that SNPs rs2242046 and rs7294 had statistically significant allele frequency differences (p<0.05) and high differentiation (FST>0.05) between WETH and all four HapAFR. The SLC28A1 rs2242046 A allele was present at a frequency of 46% in WETH but absent in most African populations studied including HapMap YRI, San from Namibia, Biaka Pygmies from Central African Republic, Mbuti Pygmies from Democratic Republic of Congo, and Bantu speakers from Kenya (http://alfred.med.yale.edu/). The rs7294 A allele was less frequent in WETH (20%) than in HapAFR (45%-59%). The rs2242046 A allele (SLC28A1) is associated with increased dose-related toxicity of zidovudine and gemcitabine, pyrimidine analogues used to treating HIV/AIDS and several solid tumors respectively.18 The rs2242046 A allele (SLC28A1 1561 G>A) is a non-synonymous variant resulting in an amino acid change from aspartate to asparagine that has been shown to increase uptake of a pyrimidine nucleoside into cells,19 possibly explaining the mechanism of action of the variant. SNP rs7294 (VKORC1) is associated with variation in dose-related response to the oral anti-coagulant warfarin.20 We also found eight pharmacogenomically and clinically relevant SNPs which displayed significant allele frequency differences between WETH and three of the four HapAFR. Notably, the NAT2 rs1799929 T allele (481 C>T), associated with slow N-acetyltransferase (NAT2) catalytic activity,21 was more frequent in WETH (62%) than in three of the four HapMap Africa populations. We tested the strength of correlation (using an LD statistics, r2) between rs1799929 and the seven SNPs conventionally used to predict NAT2 acetylation phenotype and found in our genome-wide dataset. SNP rs1799929 displayed strong correlation (r2≥0.8) with rs1801280 (r2 = 0.98) and rs1208 (r2 = 0.81), and the haplotype frequencies were greatest for the NAT2*5B haplotype (rs1799929T- rs1801280C- rs1208G, frequency =58%). This haplotype (NAT2*5B) is associated with slow NAT2 acetylation phenotype, confirming that in WETH rs1799929 is on the haplotype background that codes for slow NAT2 acetylation status.
Table 1. Pharmacologically and clinically relevant variants in which WETH had statistically significant allele frequency differences with at least three of the four other HapMap African populations.
| WETH Frq | MKK | ASW | LWK | YRI | Predicted function | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||||||||
| SNP | Chr: pos | A1 | A2 | Frq | P | Frq | P | Frq | P | Frq | P | Gene | Drugs | Diseases | Effect | ||
| rs2242046* | 15:38298203 | A | G | 0.46 | 0.23 | 7.54×10−3 | 0.10 | 1.17×10−5 | 0.06 | 1.19×10−6 | 0.00 | 9.42×10−12 | SLC28A1 | Missense | Gemcitabine | NSCC | Dose-related toxicity |
| rs7294** | 16:31102321 | A | G | 0.20 | 0.52 | 1.93×10−3 | 0.53 | 9.40×10−4 | 0.45 | 0.02 | 0.59 | 1.30×10−5 | VKORC1 | 3′UTR | Warfarin, Acenocoumarol, Coumarin | Arteriosclerosis, Heart diseases, Myocardial infarction, Peripheral vascular diseases, Pulmonary embolism, Stroke, Thromboembolism | Dose-related toxicity |
| rs2108622 | 19:15990431 | T | C | 0.37 | 0.21 | ns | 0.09 | 2.01×10−3 | 0.11 | 1.28×10−2 | 0.05 | 2.31×10−5 | CYP4F2 | Missense | Warfarin, Acenocoumarol, Phenprocoumon, Fluindione | Arteriosclerosis, Heart diseases, Myocardial infarction, Peripheral vascular diseases, Pulmonary embolism, Stroke, Thromboembolism | Dose-related toxicity |
| rs3764006** | 12:21054369 | G | A | 0.33 | 0.48 | ns | 0.63 | 7.94×10−3 | 0.73 | 4.58×10−6 | 0.83 | 6.23×10−10 | SLCO1B3 | Synonymous | Rosuvastatin | Dyslipidemia | Clearance |
| rs20455 | 6:39325078 | A | G | 0.55 | 0.47 | ns | 0.24 | 4.12×10−3 | 0.22 | 1.20×10−3 | 0.10 | 8.25×10−9 | KIF6 | Missense | Pravastatin | Myocardial infarction | Efficacy |
| rs1799929* | 8:18257994 | T | C | 0.62 | 0.45 | ns | 0.26 | 9.73×10−5 | 0.33 | 1.48×10−2 | 0.20 | 1.56×10−6 | NAT2 | Synonymous | Isoniazid, Pyrazinamide, Rifampin, Sulfamethoxazole, Trimethoprim | Tuberculosis | Doserelated toxicity |
| rs533486* | 7:99440694 | T | C | 0.38 | 0.39 | ns | 0.11 | 6.96×10−3 | 0.09 | 1.05×10−3 | 0.04 | 3.02×10−6 | CYP3A43 | Intronic | Doceta×el | Neoplasms | Drug clearance |
| rs7483* | 1:110279701 | T | C | 0.36 | 0.10 | 9.64×10−3 | 0.10 | 9.64×10−3 | 0.15 | ns | 0.03 | 3.28×10−6 | GSTM3 | Missense | Cisplatin, Cyclophosphamide | Neoplasms | Efficacy and toxicity |
| rs1495741* | 8:18272881 | A | G | 0.87 | 0.82 | NS | 0.64 | 0.002 | 0.67 | 0.001 | 0.55 | 0.0005 | NAT2 | Downstream | Isoniazid, Sulfamethoxazole | Tuberculosis | Dose-related toxicity |
| rs437943 | 4:35372098 | T | C | 0.50 | 0.36 | ns | 0.22 | 2.66×10−2 | 0.13 | 1.42×10−5 | 0.17 | 5.85×10−4 | ARAP2 | Flanking 3′UTR | TNF-alpha inhibitors | Rheumatoid arthritis | Efficacy |
Abbreviations: Chr, Chromosome; Pos, Base-pair physical position (hg19); A1, Allele 1; A2, Allele 2; Frq, frequency of A1; WETH, Wolaita from Wolaita zone, Ethiopia; ASW, African ancestry in Southwest USA; LWK, Luhya in Webuye, Kenya; MKK, Maasai in Kinyawa, Kenya; YRI, Yoruba in Ibadan, Nigeria; P, Bonferroni corrected P-value (nominal P × 443); ns, not significant after Bonferroni correction; NSSC, non-small cell cancer.
Also significant difference with CHB, CHD and JPT.
Also significant difference with GIH.
In addition, the slow-acetylation associated A allele of rs1495741, a recently reported tag SNP for NAT2 acetylation status22, had high frequency in WETH (87.1%), which is greater than any HapMap populations and third-ranking following Amerindians (92%) and Bedouin (89%) in a comparison of 54 global populations (http://alfred.med.yale.edu/) (Table 1). Of the 120 individuals in WETH, 98.3% had at least one rs1495741 A allele (91 were AA homozygous, 27 were heterozygous and 2 were GG homozygous) (data not shown). A total of 17 pharmacogenomic SNPs showed statistically significant allele frequency differences between WETH and HapNonAFR but not between WETH and HapAFR (Table 2 & Supplementary Table 9).
Table 2. Pharmacologically and clinically relevant variants in which WETH had statistically significant allele frequency differences with all HapMap non-Africa populations but not with HapMap Africa populations.
| Allele A1 frequency |
Predicted function | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr: position | A1 | A2 | WETH | TSI | CEU | MEX | GIH | CHB | CHD | JPT | Gene | |
| rs6671692 | 1:171176879 | A | G | 0.17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | FMO2 | Synonymous |
| rs7536646 | 1:171174691 | A | G | 0.17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | FMO2 | Synonymous |
| rs3828193 | 2:101031561 | T | G | 0.10 | 0.39 | 0.58 | 0.47 | 0.37 | 0.60 | 0.71 | 0.65 | CHST10 | 5′UTR |
| rs7081 | 2:27422605 | C | T | 0.43 | 0.05 | 0.08 | 0.07 | 0.07 | 0 | 0 | 0 | SLC5A6 | 3′UTR |
| rs2295475 | 2:31589847 | A | G | 0.02 | 0.32 | 0.26 | 0.44 | 0.36 | 0.36 | 0.41 | 0.29 | XDH | Synonymous |
| rs9842091 | 3:38307510 | C | T | 0.44 | 0.11 | 0.04 | 0.05 | 0.04 | 0.04 | 0 | 0.02 | SLC22A13 | Synonymous |
| rs274548 | 5:131730807 | T | C | 0.63 | 0.18 | 0.21 | 0.16 | 0.17 | 0.10 | 0.10 | 0.03 | SLC22A5 | 3′UTR |
| rs3734254 | 6:35395010 | C | T | 0.62 | 0.24 | 0.22 | 0.09 | 0.21 | 0.27 | 0.32 | 0.23 | PPARD | 3′UTR |
| rs6457816 | 6:35362848 | C | T | 0.52 | 0.08 | 0.13 | 0.05 | 0.03 | 0.07 | 0.02 | 0.03 | PPARD | Intronic |
| rs7746988 | 6:35324741 | C | T | 0.37 | 0.01 | 0.03 | 0.06 | 0.01 | 0.04 | 0.01 | 0.03 | PPARD | Intronic |
| rs2242416 | 6:43273604 | G | A | 0.13 | 0.51 | 0.63 | 0.45 | 0.46 | 0.69 | 0.64 | 0.74 | CRIP3 | Missense |
| rs2230028 | 7:86894112 | C | T | 0.36 | 0.08 | 0.09 | 0.05 | 0.10 | 0.01 | 0.05 | 0.02 | ABCB4 | Synonymous |
| rs2515641 | 10:135351362 | T | C | 0.54 | 0.16 | 0.09 | 0.18 | 0.20 | 0.23 | 0.14 | 0.19 | CYP2E1 | Synonymous |
| rs1061040 | 14:23242828 | T | C | 0.42 | 0.09 | 0.11 | 0.08 | 0.11 | 0.10 | 0.08 | 0.11 | SLC7A7 | Synonymous |
| rs305968 | 19:41622189 | A | G | 0.72 | 0.26 | 0.34 | 0.42 | 0.34 | 0.29 | 0.22 | 0.30 | CYP2F1 | Synonymous |
| rs2070995 | 21:39086965 | T | C | 0.01 | 0.23 | 0.21 | 0.24 | 0.17 | 0.41 | 0.46 | 0.38 | KCNJ6 | Synonymous |
| rs1541290 | 21:43718483 | A | G | 0.17 | 0.55 | 0.50 | 0.61 | 0.49 | 0.60 | 0.56 | 0.62 | ABCG1 | 3downstream |
Abbreviations: Chr, Chromosome; Pos, Physical position (hg19); A1, Allele 1; A2,Allele 2; WETH, Wolaita from Wolaita zone, Ethiopia; TSI, Tuscans in Italy; CEU, Utah residents with Northern and Western European ancestry from the CEPH collection; MEX, Mexican ancestry in Los Angeles, California; GIH, Gujarati Indians in Houston, Texas; CHB, Han Chinese in Beijing, China; CHD, Chinese in Metropolitan Denver, Colorado; JPT, Japanese in Tokyo, Japan.
We compared the allele frequency distribution of the 443 pharmacogenomic SNPs between WETH and nine Ethiopian ethnic populations (Afar, Amhara, Anuak, Ari-blacksmith, Ari-cultivator, Ethiopian Somali, Gumuz, Oromo, and Tigre) using publicly available genotype data.23 We found three SNPs showing Bonferroni-corrected statistically significant allele frequency differences between WETH and Anuak, and one SNP showing significant allele frequency difference between WETH and Ari-blacksmith. The remaining 439 SNPs (99.1%) did not show significant allele frequency differences between WETH and all other ethnic groups. The small sample sizes of the individual ethnic groups (n=8-26) may have underpowered the analysis (Supplementary Table 10).
DISCUSSION
This large cohort of the Wolaita ethnic population extends the population genetic variation map of African populations by providing data on an Ethiopian population that is not represented in either the HapMap or the 1000 Genomes Projects. Specifically, we demonstrated the spectrum of genetic variation in the Wolaita population, one of the indigenous ethnic populations of Ethiopia, in the context of African and non-African HapMap populations, and explored the clinical and pharmacogenomic implications of these genetic variations. The MDS, FST, allele frequency, and LD analyses showed that WETH has more genetic similarity to African than non-African ancestry HapMap populations. Furthermore, compared with the Semetic-Cushitic Ethiopians,23 the Wolaita were found to be genetically more distant from the Southwest Asians. Compared to HapAFR, the WETH were genetically closest to the Kenyan Maasai and most distant to the Nigerian Yoruba. In addition, the Kenyan Maasai were genetically closer to the Ethiopian Wolaita than any other HapMap population including the Kenyan Luhya population, which is geographically closest to the Maasai. The closer genetic similarity of the Maasai to the Wolaita than the Luhya parallels broad linguistic stratification and demographic history of the populations – the Wolaita and the Maasai speak the Omotic and Nilo-Saharan branches, respectively, of the Afroasiatic language family, whereas the Luhya speak Bantu (http://linguistlist.org/). Together, these findings illustrate that a model that dictates inverse relationship between genetic similarity and geographic distance24 is inadequate to infer level of genetic differentiation among African populations, and illustrates that integration of demographic, linguistic and anthropological evidence is crucial for interpretation of the findings of population genetics studies in Africa.
Our finding of three major genetic clusters, each contributing >24% genetic ancestry, in the Wolaita shows that the Wolaita have a more diverse ancestral background than HapAFR, and concurs with the wide level of genetic admixture observed in East African populations.4 Our study participants were recruited from the Southern Ethiopia region that is known to be a mosaic of over 45 indigenous ethnic groups that have undergone inter-mixing over several thousands of years.25 Therefore, the diverse genetic background in the Wolaita may reflect this phenomenon consistent with suggestions that the genetic spectrum of the Ethiopian population has been shaped by a complex set of demographic, cultural and historical dynamics.23 To understand the sources of the major cluster that accounts for 40.4% of the ancestry in the Wolaita, but is far less common or absent in HapMap populations except in MKK (5.7%), it may be valuable to extend the populations studied to include other neighboring indigenous ethnic groups from southern Ethiopia. It should be noted that our goal in the STRUCTURE analysis was to estimate broad genetic clusters in the WETH in the context of HapMap populations, not to provide detailed or definitive genetic ancestral background of the WETH. Unbiased inference of genetic ancestry requires comprehensive sampling of neighboring and historically important distant populations, and should be validated in different clustering schemes using multiple data sets. In addition, the results of these analyses are highly variable depending on the population groups included and their sample size; therefore, the stability of the most appropriate value of K should be rigorously tested before ascribing ancestral origins.12
Several pharmacogenomically and clinically important SNPs that are associated with dose-limiting effects of therapies for HIV/AIDS, tuberculosis, thromboembolism and several forms of cancer were found to have statistically significant allele frequency differences between WETH and HapAFR. These observations demonstrate that the genetic diversity of African populations extends to genetic variants of clinical and functional importance. For example, rs2242046, a missense variant in the solute carrier family 28 gene (SLC28A1, also known as human concentrative nucleotide transporter 1 or hCNT1) was differentiated between WETH and HapAFR. The SLC28A1 gene is involved in the transportation of pyrimidine nucleosides and mediates the cellular uptake of antiviral and anticancer nucleoside analogs such as zidovudine which is used for treatment of HIV/AIDS and gemcitabine, which is the standard first-line agent for treatment of pancreatic cancer and solid tumors including bladder, breast and non-small cell lung cancer (NSCLC).26
A major dose limiting adverse effect of gemcitabine is hematological toxicity such as neutropenia and thrombocytopenia, which is often addressed by reducing the dose or increasing the intervals between gemcitabine administrations.18 The rs2242046 A allele is associated with increased hematological toxicity, and its frequency shows wide differences among populations: 3.4% in Asian Americans, 10% in African Americans, and 51.1% in European Americans.19, 27 Therefore, our finding of rs2242046 A allele’s high frequency in WETH and its absence in several African populations implies Wolaita NSCLC patients would be at greater risk of hematological toxicity on gemcitabine doses that are safe for other sub-Saharan African populations.
SNPs rs7294 and rs2108622 in VKORC1 and CYP4F2, two genes known to be principal genetic determinants of the therapeutic dose of warfarin showed significant allele frequency differences between WETH and HapAFR. Warfarin – the most widely prescribed oral anticoagulant for treating and preventing thromboembolic events – has a narrow therapeutic index and the dose of warfarin required to achieve a therapeutic response without causing severe bleeding shows large differences between individuals and populations.28 We found that the rs7294 A allele associated with a higher warfarin dose requirement 29 has significantly lower frequency in WETH compared to HapAFR and other HapMap populations except for South East Asians. Two other VKORC1 variants that are associated with the recommendation of a low warfarin dose (rs9923231 T and rs9934438 T) also have relatively higher frequency in WETH (26%) compared to HapAFR (2.2%-16.1%) and black South Africans (4%). Based on the low frequency of rs9934438 T in most South and West Africans, a previous study has indicated that this variant has limited pharmacogenomic application in Africa.30 In contrast, our study’s finding of higher allele frequency in Ethiopians shows its potential pharmacologic relevance in East African populations. Observation of a high frequency of rs9934438 T in the South African San (33%)31 as well as in the East African WETH concurs with previous studies suggesting the presence of ancestral affinity between Ethiopians and the Khoisan,32 and affirms the potential clinical and public health relevance of the variant in both populations. The finding also cautions that not all African populations can be co-classified as genetically warfarin-resistant based on shared continental origin, and that prescription of high doses of warfarin to some African populations may cause serious side effects. The variant frequency distributions of rs7294, rs9923231, and rs9934438 in our study population (WETH) is different from those found in a sample of Ethiopians of mixed ethnicity,33 further demonstrating the ethnic and genetic diversity of Ethiopians in warfarin sensitivity.
Classification of the WETH as genetically warfarin-sensitive or-resistant becomes less straightforward when we consider additional genetic markers of warfarin sensitivity/resistance. For example, we found that the CYP4F2 rs2108622 T allele, associated with higher required warfarin dose, has higher frequency in WETH than in HapMap and in HGDPs global populations except those from the Middle East and Oceania.31 A previous study has reported that a suggested marker of warfarin resistance in the VKORC1 gene (rs61742245 A or Asp36Tyr) has the highest frequency (15%) in Ethiopians.34, 35 Our findings of warfarin dose-associated allele frequency differences among African populations and among individuals within the Wolaita population indicate that when feasible, doses of warfarin should be personalized based on an individual’s genotypes at different loci, demographic, and clinical characteristics.
We also found that NAT2 gene alleles associated with slow acetylation status were significantly more frequent in WETH, suggesting the majority of Wolaita Ethiopians are at higher genetic risk for anti-tuberculosis drug-induced hepatotoxicity (ADIH).36, 37 For example, three quarters of WETH were homozygous for the rs1495741 AA genotype found to be associated with strong (OR=14) increased risk for ADIH in Taiwanese.38 ADIH is the most serious adverse reaction to the three first-line anti-tuberculosis drugs isoniazid, rifampicin, and pyrazinamide,39 and results in serious morbidity, mortality, treatment failure and interruption, relapse and drug resistance.36 Our finding of increased genetic susceptibility for ADIH in this Ethiopian population is consistent with previous reports of high incidence of ADIH (11.3%-30%)40-42 and anti-tuberculosis drug induced jaundice (8.9%)43 in Ethiopian patients. The World Health Organization ranked Ethiopia as the 7th high tuberculosis-burden country;44 therefore, pharmacogenomics-informed anti-tuberculosis drug dosages may be valuable in Ethiopia.
In all, our study has presented a genetic map of the Wolaita, an Omotic language speaking Southern Ethiopian ethnic population, in the context of other African and non-African populations. We found that genetic variants associated with dose-response in therapies for important infectious and non-infectious diseases (tuberculosis, HIV/AIDS, cardiovascular conditions, and cancer) were differentiated between the Wolaita and HapAFR, indicating that genetic tests for predicting drug dose requirements should be implemented in diverse African populations.
Supplementary Material
ACKNOWLEDGEMENTS
The research project was supported by the Wellcome Trust (grant #079791), and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, in the Center for Research on Genomics and Global Health (CRGGH). The CRGGH is also supported by the National Institute of Diabetes and Digestive and Kidney Diseases. We thank staff of the Mossy Foot Treatment and Prevention Association of Ethiopia for coordinating the fieldwork.
Footnotes
CONFLICT OF INTEREST: The authors declare no conflict of interest.
REFERENCES
- 1.Grossman I. ADME pharmacogenetics: current practices and future outlook. Expert Opin Drug Metab Toxicol. 2009;5(5):449–462. doi: 10.1517/17425250902902322. [DOI] [PubMed] [Google Scholar]
- 2.Ramos E, Doumatey A, Elkahloun AG, Shriner D, Huang H, Chen G, et al. Pharmacogenomics, ancestry and clinical decision making for global populations. Pharmacogenomics J. 2013 doi: 10.1038/tpj.2013.24. [DOI] [PubMed] [Google Scholar]
- 3.Matimba A, Del-Favero J, Van Broeckhoven C, Masimirembwa C. Novel variants of major drug-metabolising enzyme genes in diverse African populations and their predicted functional effects. Hum Genomics. 2009;3(2):169–190. doi: 10.1186/1479-7364-3-2-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rotimi CN, Jorde LB. Ancestry and disease in the age of genomic medicine. N Engl J Med. 2010;363(16):1551–1558. doi: 10.1056/NEJMra0911564. [DOI] [PubMed] [Google Scholar]
- 6.Ngaimisi E, Habtewold A, Minzi O, Makonnen E, Mugusi S, Amogne W, et al. Importance of ethnicity, CYP2B6 and ABCB1 genotype for efavirenz pharmacokinetics and treatment outcomes: a parallel-group prospective cohort study in two sub-Saharan Africa populations. PLoS One. 2013;8(7):e67946. doi: 10.1371/journal.pone.0067946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 8.Tekola Ayele F, Adeyemo A, Finan C, Hailu E, Sinnott P, Burlinson ND, et al. HLA class II locus and susceptibility to podoconiosis. N Engl J Med. 2012;366(13):1200–1208. doi: 10.1056/NEJMoa1108448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rousset F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 11.Weir BS, Cockerham CC. Estimating F-statistic for the analysis of population structure. Evolution. 1984;38(6):1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 12.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg NA. Distruct: a program for the graphical display of population structure. Molecular Ecology Notes. 2004;4:137–138. [Google Scholar]
- 15.Deeken J. The Affymetrix DMET platform and pharmacogenetics in drug development. Curr Opin Mol Ther. 2009;11(3):260–268. [PubMed] [Google Scholar]
- 16.Tekola F, Bull SJ, Farsides B, Newport MJ, Adeyemo A, Rotimi CN, et al. Tailoring consent to context: designing an appropriate consent process for a biomedical study in a low income setting. PLoS Negl Trop Dis. 2009;3(7):e482. doi: 10.1371/journal.pntd.0000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tekola F, Bull S, Farsides B, Newport MJ, Adeyemo A, Rotimi CN, et al. Impact of social stigma on the process of obtaining informed consent for genetic research on podoconiosis: a qualitative study. BMC Med Ethics. 2009;10:13. doi: 10.1186/1472-6939-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toschi L, Finocchiaro G, Bartolini S, Gioia V, Cappuzzo F. Role of gemcitabine in cancer therapy. Future Oncol. 2005;1(1):7–17. doi: 10.1517/14796694.1.1.7. [DOI] [PubMed] [Google Scholar]
- 19.Gray JH, Mangravite LM, Owen RP, Urban TJ, Chan W, Carlson EJ, et al. Functional and genetic diversity in the concentrative nucleoside transporter, CNT1, in human populations. Mol Pharmacol. 2004;65(3):512–519. doi: 10.1124/mol.65.3.512. [DOI] [PubMed] [Google Scholar]
- 20.D’Andrea G, D’Ambrosio RL, Di Perna P, Chetta M, Santacroce R, Brancaccio V, et al. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood. 2005;105(2):645–649. doi: 10.1182/blood-2004-06-2111. [DOI] [PubMed] [Google Scholar]
- 21.Ben Mahmoud L, Ghozzi H, Kamoun A, Hakim A, Hachicha H, Hammami S, et al. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatotoxicity in Tunisian patients with tuberculosis. Pathol Biol. 2012;60(5):324–330. doi: 10.1016/j.patbio.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Closas M, Hein DW, Silverman D, Malats N, Yeager M, Jacobs K, et al. A single nucleotide polymorphism tags variation in the arylamine N-acetyltransferase 2 phenotype in populations of European background. Pharmacogenet Genomics. 2011;21(4):231–236. doi: 10.1097/FPC.0b013e32833e1b54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagani L, Kivisild T, Tarekegn A, Ekong R, Plaster C, Gallego Romero I, et al. Ethiopian genetic diversity reveals linguistic stratification and complex influences on the Ethiopian gene pool. Am J Hum Genet. 2012;91(1):83–96. doi: 10.1016/j.ajhg.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bohonak AJ. IBD (Isolation by Distance): a program for analyses of isolation by distance. J Hered. 2002;93(2):153–154. doi: 10.1093/jhered/93.2.153. [DOI] [PubMed] [Google Scholar]
- 25.CSA & ORC Macro . The 2007 Population and Housing Census of Ethiopia. Addis Ababa; CSA: 2007. [Google Scholar]
- 26.Fukunaga AK, Marsh S, Murry DJ, Hurley TD, McLeod HL. Identification and analysis of single-nucleotide polymorphisms in the gemcitabine pharmacologic pathway. Pharmacogenomics J. 2004;4(5):307–314. doi: 10.1038/sj.tpj.6500259. [DOI] [PubMed] [Google Scholar]
- 27.Soo RA, Wang LZ, Ng SS, Chong PY, Yong WP, Lee SC, et al. Distribution of gemcitabine pathway genotypes in ethnic Asians and their association with outcome in non-small cell lung cancer patients. Lung Cancer. 2009;63(1):121–127. doi: 10.1016/j.lungcan.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5(3):e1000433. doi: 10.1371/journal.pgen.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin T, Miyata T. Warfarin dose and the pharmacogenomics of CYP2C9 and VKORC1 - rationale and perspectives. Thromb Res. 2007;120(1):1–10. doi: 10.1016/j.thromres.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Dandara C, Lombard Z, Du Plooy I, McLellan T, Norris SA, Ramsay M. Genetic variants in CYP (-1A2, -2C9, -2C19, -3A4 and -3A5), VKORC1 and ABCB1 genes in a black South African population: a window into diversity. Pharmacogenomics. 2011;12(12):1663–1670. doi: 10.2217/pgs.11.106. [DOI] [PubMed] [Google Scholar]
- 31.Ross KA, Bigham AW, Edwards M, Gozdzik A, Suarez-Kurtz G, Parra EJ. Worldwide allele frequency distribution of four polymorphisms associated with warfarin dose requirements. J Hum Genet. 2010;55(9):582–589. doi: 10.1038/jhg.2010.73. [DOI] [PubMed] [Google Scholar]
- 32.Semino O, Santachiara-Benerecetti AS, Falaschi F, Cavalli-Sforza LL, Underhill PA. Ethiopians and Khoisan share the deepest clades of the human Y-chromosome phylogeny. Am J Hum Genet. 2002;70(1):265–268. doi: 10.1086/338306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sominsky S, Korostishevsky M, Kurnik D, Aklillu E, Cohen Y, Ken-Dror G, et al. The VKORC1 Asp36Tyr variant and VKORC1 haplotype diversity in Ashkenazi and Ethiopian populations. J Appl Genet. 2014 doi: 10.1007/s13353-013-0189-2. [DOI] [PubMed] [Google Scholar]
- 34.Aklillu E, Leong C, Loebstein R, Halkin H, Gak E. VKORC1 Asp36Tyr warfarin resistance marker is common in Ethiopian individuals. Blood. 2008;111(7):3903–3904. doi: 10.1182/blood-2008-01-135863. [DOI] [PubMed] [Google Scholar]
- 35.Loebstein R, Dvoskin I, Halkin H, Vecsler M, Lubetsky A, Rechavi G, et al. A coding VKORC1 Asp36Tyr polymorphism predisposes to warfarin resistance. Blood. 2007;109(6):2477–2480. doi: 10.1182/blood-2006-08-038984. [DOI] [PubMed] [Google Scholar]
- 36.Tostmann A, Boeree MJ, Aarnoutse RE, de Lange WC, van der Ven AJ, Dekhuijzen R. Antituberculosis drug-induced hepatotoxicity: concise up-to-date review. J Gastroenterol Hepatol. 2008;23(2):192–202. doi: 10.1111/j.1440-1746.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- 37.Wang PY, Xie SY, Hao Q, Zhang C, Jiang BF. NAT2 polymorphisms and susceptibility to anti-tuberculosis drug-induced liver injury: a meta-analysis. Int J Tuberc Lung Dis. 2012;16(5):589–595. doi: 10.5588/ijtld.11.0377. [DOI] [PubMed] [Google Scholar]
- 38.Ho HT, Wang TH, Hsiong CH, Perng WC, Wang NC, Huang TY, et al. The NAT2 tag SNP rs1495741 correlates with the susceptibility of antituberculosis drug-induced hepatotoxicity. Pharmacogenet Genomics. 2013;23(4):200–207. doi: 10.1097/FPC.0b013e32835e95e1. [DOI] [PubMed] [Google Scholar]
- 39.Yew WW, Leung CC. Antituberculosis drugs and hepatotoxicity. Respirology. 2006;11(6):699–707. doi: 10.1111/j.1440-1843.2006.00941.x. [DOI] [PubMed] [Google Scholar]
- 40.Yimer G, Aderaye G, Amogne W, Makonnen E, Aklillu E, Lindquist L, et al. Anti-tuberculosis therapy-induced hepatotoxicity among Ethiopian HIV-positive and negative patients. PLoS One. 2008;3(3):e1809. doi: 10.1371/journal.pone.0001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassen Ali A, Belachew T, Yami A, Ayen WY. Anti-tuberculosis drug induced hepatotoxicity among TB/HIV co-infected patients at Jimma University Hospital, Ethiopia: nested case-control study. PLoS One. 2013;8(5):e64622. doi: 10.1371/journal.pone.0064622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yimer G, Ueda N, Habtewold A, Amogne W, Suda A, Riedel KD, et al. Pharmacogenetic & pharmacokinetic biomarker for efavirenz based ARV and rifampicin based anti-TB drug induced liver injury in TB-HIV infected patients. PLoS One. 2011;6(12):e27810. doi: 10.1371/journal.pone.0027810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mekonnen T, Abseno M, Meressa D, Fekde B. Prevalence and management outcomes of anti TB drugs induced hepatotoxicity, St. Peter TB Specialized Hospital. J Ethiopia Med Pract. 2002;4(1):32–38. [Google Scholar]
- 44.WHO . Global Tuberculosis Report 2012. WHO; Geneva: 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.