

Abstract
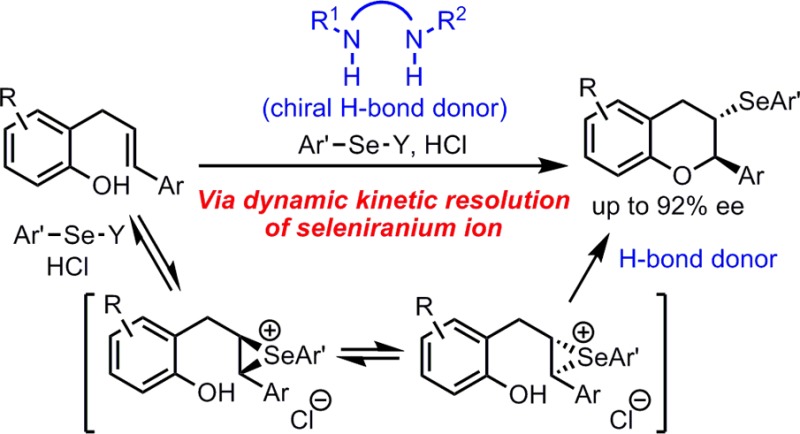
Highly enantioselective selenocyclization reactions are promoted by the combination of a new chiral squaramide catalyst, a mineral acid, and an achiral Lewis base. Mechanistic studies reveal that the enantioselectivity originates from the dynamic kinetic resolution of seleniranium ions through anion-binding catalysis.
Anion-binding catalysis by dual hydrogen-bond donors such as ureas and thioureas has been demonstrated as a powerful strategy for the development of highly enantioselective transformations involving cationic intermediates.1 The approach has been applied successfully to reactions involving an assortment of prochiral electrophiles, such as iminium ions, oxocarbenium ions, carbenium ions, and episulfonium ions.2 In these reactions, the dual hydrogen-bond donors have been shown to bind to the counterion of a prochiral electrophile and remain associated intimately enough with the cationic intermediate to induce high enantioselectivity in subsequent nucleophilic additions. We sought to explore the application of this principle to a stereochemically labile reactive substrate or intermediate, such that a racemic mixture of chiral cationic electrophiles might also be converted into highly enantioenriched products through anion-binding catalysis. Here, we demonstrate the application of this dynamic kinetic resolution strategy in the development of a highly enantioselective selenoetherification reaction.
Selenofunctionalization of alkenes proceeds via reaction of a π system with an electrophilic selenium source to generate a seleniranium ion, followed by nucleophilic ring opening.3 The resulting selenide product can be elaborated in a variety of synthetically useful transformations including oxidative elimination and radical substitution reactions. However, the development of catalytic, enantioselective variants of the selenofunctionalization reaction remains a challenge that is largely unmet.4,5 A principal difficulty lies in the configurational instability of seleniranium ions, as any enantioenrichment in seleniranium formation can be degraded prior to nucleophilic ring opening.5a,6 This problem was addressed by Denmark and co-workers through the design of an electrophilic selenium reagent that slows down the racemization pathway to a significant extent. By this strategy, a moderately enantioselective chiral Lewis base-catalyzed asymmetric selenoetherification reaction was devised that proceeds via ee-determining formation of the seleniranium ion (Scheme 1, Strategy A).5a The modest asymmetric induction is proposed to partly arise from incomplete suppression of the configurational scrambling of the seleniranium intermediate.
Scheme 1. Two Strategies for the Development of Enantioselective Selenofunctionalization Reactions.
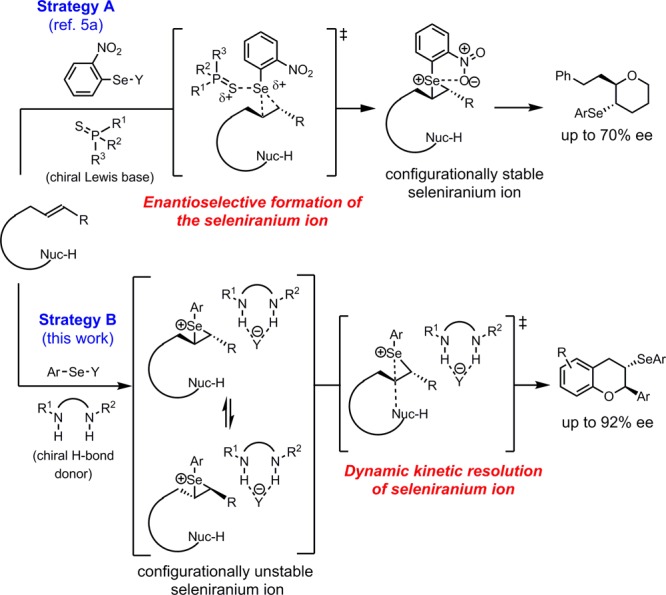
On the basis of these mechanistic insights, we sought to develop a highly enantioselective selenofunctionalization that takes advantage of rapid seleniranium racemization by means of the aforementioned dynamic kinetic resolution strategy. Through intimate association of an anion-bound chiral hydrogen-bond donor with the seleniranium ion, nucleophilic ring-opening of the two enantiomers of the electrophile could be accelerated to different extents, thereby allowing asymmetric induction to be achieved (Scheme 1, Strategy B).
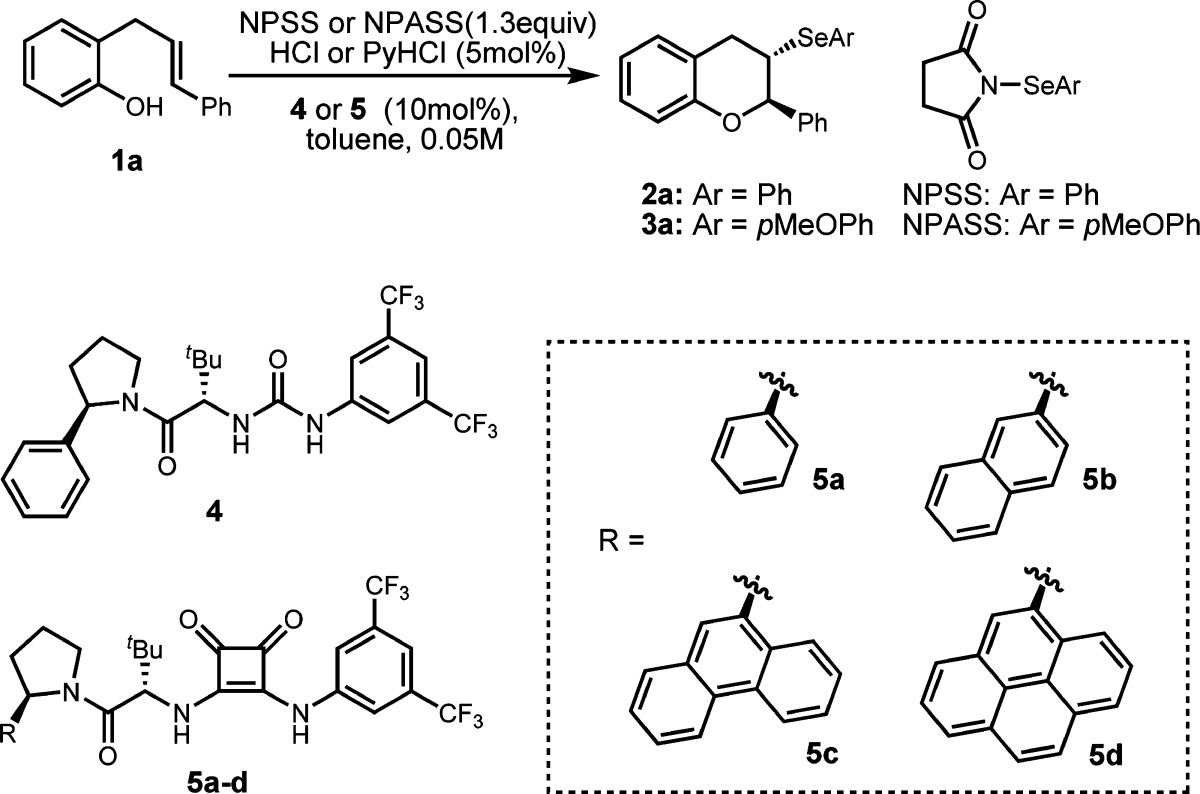
We chose o-allyl-substituted phenol 1a as the model substrate, which could undergo intramolecular selenoetherification to construct a chroman-type product with two contiguous stereogenic centers. A survey of chiral H-bond donor catalysts and reaction conditions led to the observation that urea 4 promoted the formation of 2a with 33% ee and near-quantitative yield in the presence of N-phenylselenyl succinimide (NPSS) as the selenium donor and hydrogen chloride as a co-catalyst (Table 1, entry 1). Acid is required to achieve synthetically useful yields, presumably by facilitating selenyl group transfer from NPSS to the olefin.7 However, under these conditions, significant product racemization was observed after prolonged reaction time. We speculate the strong acid promotes reversibility of the selenocyclization process by initial protonation of the ether product. Indeed, when a weaker acid, pyridinium chloride (PyHCl) was employed, the product was observed to be configurationally stable under the reaction conditions (entry 2).
Table 1. Reaction Optimizationa.
| entry | [Se] | acid | cat. | temp (°C) | time (h) | yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|---|
| 1 | NPSS | HCl | 4 | rt | 0.5 | 92 | 33(11d) |
| 2 | NPSS | PyHCl | 4 | rt | 2 | 85 | 36(36d) |
| 3 | NPSS | PyHCl | 5a | rt | 2 | 82 | 46 |
| 4 | NPSS | PyHCl | 5b | rt | 2 | 82 | 60 |
| 5 | NPSS | PyHCl | 5c | rt | 2 | 88 | 64 |
| 6 | NPSS | PyHCl | 5d | rt | 2 | 84 | 67 |
| 7 | NPSS | PyHCl | 5d | –30 | 48 | 67 | 72 |
| 8 | NPSS | PyHCl | 5d | –45 | 48 | <5 | nd |
| 9e | NPSS | PyHCl | 5d | –45 | 48 | 89 | 84 |
| 10e | NPASS | PyHCl | 5d | –45 | 48 | 85 | 87 |
| 11e | NPASS | PyHCl | 5d | –35 | 48 | 85 | 88 |
| 12e | NPASS | HCl | 5d | –35 | 48 | 87 | 88 |
Reactions conducted on 0.1 mmol scale.
Isolated yield of purified product.
Enantiomeric excess determined by HPLC analysis on commercial chiral columns.
ee after 12 h.
HMPA(S) (10 mol%) was added.
We evaluated whether stronger dual H-bond donors, such as thioureas and squaramides,8 might impart higher levels of stereoinduction in the selenocyclization reaction. While the thiourea analogue of catalyst 4 undergoes rapid decomposition under the reaction conditions,9 the corresponding squaramide 5a (Table 1, entry 3) indeed provides slightly improved enantioselectivity.10 We also hypothesized that expanding the aromatic substituent on the pyrrolidine unit might further increase the stereochemical communication in the transition structure by strengthening a cation−π interaction. Such a trend is precedented with the thiourea analogues of this class of catalysts in the context of polycyclizations and episulfonium ring-openings.2c,2f This strategy proved fruitful, as squaramides bearing larger aryl groups (5b–d) furnish further improvement of the enantioselectivity (entries 4–6).
Through evaluation of reaction temperature, we found that reactions conducted at −30 °C instead of room temperature provided an increase in product ee, albeit with substantially lower reactivity (Table 1, entry 7 vs entry 6). Further decrease in temperature led to almost complete shutdown of the reaction (entry 8). We hypothesized that the group transfer between NPSS and the substrate was inhibited at low temperatures due to the decreased solubility of the selenium reagent. This problem was circumvented by the introduction of a strong Lewis base in the reaction, which has been shown to activate NPSS in the presence of Brønsted acids.11 Tris(dimethylamino)phosphorus sulfide (HMPA(S)) promoted selenocyclization at −45 °C with full conversion after 48 h, and the product was generated in elevated enantiomeric excess (entry 9). Enantioselectivity is also responsive to substitution on the aryl group of the selenylating reagent, as the more electron-rich N-p-anisylselenyl succinimide (NPASS) allows the formation of 3a with improved ee (entry 11 vs entry 10). Reoptimization of the reaction temperature reveals that the optimal enantioselectivity can be achieved at −35 °C under otherwise identical conditions, with product 3a isolated in 88% ee (entry 11). Finally, at low temperature, the same result was observed when PyHCl was replaced by HCl as the acid catalyst, indicating that no significant acid-induced racemization of product occurred at this temperature during the course of the reaction (entry 12).
The scope of the selenocyclization reaction was evaluated under the optimized conditions. As shown in Table 2, substrates with different substituent patterns on the phenol motif (3b–g) provide cyclization products in high yield and enantioselectivity. Substitution ortho to the hydroxyl group (3h), however, results in significant reductions in enantioselectivity. The detrimental effect of an ortho substituent suggests a possible interaction between the hydroxyl group and catalyst that may play an important role in the mechanism of enantioinduction, as such an interaction may be sensitive to the steric environment around the hydroxyl group.2f Substrates functionalized at the meta or para position of the styrenyl ring can also undergo cyclization with high levels of reactivity and enantioselectivity (3i–l). However, generality with respect to styrenyl substitutents was limited, as lower enantioselectivities were observed with the o-methyl-substituted and p-anisyl derivatives (3m,n). In most cases, the enantiopurity of the selenocyclization products could be improved to >95% ee through a single recrystallization procedure with good recovery.
Table 2. Substrate Scopea.
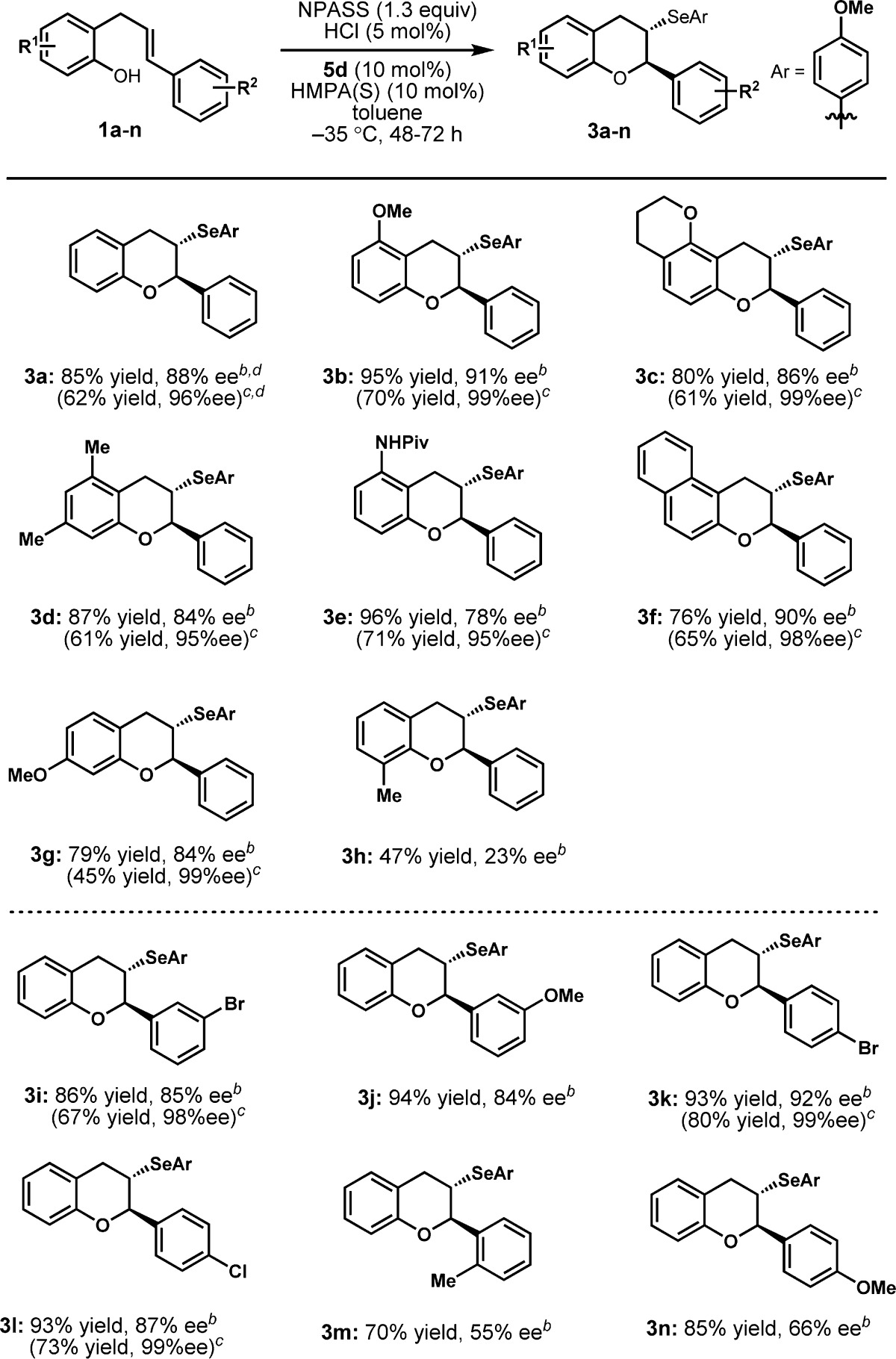
Reactions were conducted on 0.25 mmol scale.
Isolated yield and ee of purified product before recrystallization. The structure and absolute configuration of 3k were established by X-ray crystallography, and the stereochemistry of all other products was assigned by analogy.
Numbers in parentheses correspond to the isolated yield and ee after recrystallization. For details on the recrystallization procedure, see the Supporting Information.
Reaction was conducted on 1 mmol scale.
The products accessed by the squaramide-catalyzed selenocyclization can be elaborated through the variety of chemistries available to organoselenides (Scheme 2). Such operations could enable the synthesis of a diverse range of compounds of pharmaceutical interest, particularly since the selenocyclization products bear structural resemblance to the flavonoids, a family of natural products with a broad spectrum of biological activities.12 For example, we subjected the enantioenriched 3a to oxidative syn-elimination in the presence of H2O2 to generate flavene 6, which was subsequently subjected to osmium-catalyzed dihydroxylation to afford diol 7 with high diastereoselectivity (dr >20:1). In the presence of the radical initiator 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70), 3a also undergoes reaction with 8 under mild conditions to produce alkylated flavan 9 as 2:1 mixture of chromatographically separable diastereomers. The enantioenrichment of 3a is preserved in all the derived products.
Scheme 2. Derivatization of a Selenocyclization Product.

The high enantioselectivity we observed in the chiral H-bond donor-catalyzed selenoetherification can originate either from (i) face-selective selenyl group transfer to the olefin, or from (ii) dynamic kinetic resolution of the seleniranium ion in the ring-opening by the phenol. These two possibilities were evaluated by preparing the racemic seleniranium ion intermediate and subjecting it to the conditions of the enantioselective catalytic reaction. Racemic β-chloroselenide adduct rac-Int-k is formed quantitatively and instantaneously upon treating 1k with PhSeCl at −78 °C in the absence of catalyst, as determined by 1H NMR analysis.13 After addition of 20 mol% 5d and HMPA(S) to rac-Int-k, the reaction temperature was elevated to −45 °C to trigger the cyclization to form 2k (Scheme 3). The product ee generated through this stepwise procedure was similar to that of the reaction with PhSeCl added in the presence of catalysts 5d and HMPA(S).14 This result provides definitive evidence that rac-Int-k undergoes dynamic kinetic resolution in the presence of 5d, most likely via rapid equilibration of the enantiomeric seleniranium ions.
Scheme 3. DKR of Intermediate rac-Int-k with 5d.

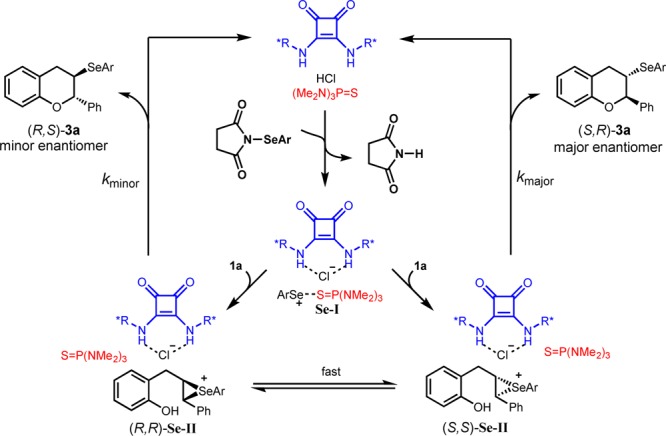
Based on our experimental findings, a possible catalytic cycle involving cooperative catalysis by a dual H-bond donor, a Brønsted acid, and a Lewis base may be advanced (Scheme 4). Cooperative Lewis base and Brønsted acid activation of the electrophilic selenium reagent results in formation of a reactive ion pair, Se-I, which may be associated with the squaramide catalyst.6a,13 This intermediate undergoes group transfer to olefin 1a to form enantiomeric seleniranium ions (R,R)-Se-II and (S,S)-Se-II, which are rapidly equilibrating. The subsequent cyclizations of (R,R)-Se-II and (S,S)-Se-II occurs at sufficiently different rates due to the association with chiral squaramide 5d to result in formation of highly enantioenriched product 3a.
Scheme 4. Proposed Catalytic Cycle.
In summary, a dual H-bond donor-catalyzed dynamic kinetic resolution protocol has been developed for highly enantioselective selenocyclization reactions. The scope of anion-binding catalysis has therefore been extended to stereochemically labile racemic electrophiles.
Acknowledgments
This work was supported by the NIGMS (GM-43214). S.L. is grateful for a predoctoral fellowship by Eli Lilly. We thank Dr. Robert Knowles for helpful discussions and Dr. Shao-Liang Zheng for X-ray crystallographic characterization of compound 3k.
Supporting Information Available
Complete experimental procedures and characterization data for products and all isolated intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Zhang Z. G.; Schreiner P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. [DOI] [PubMed] [Google Scholar]; b Robert J.; Phipps R. J.; Hamilton G. L.; Toste F. D. Nat. Chem. 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]; c Mahlau M.; List B. Angew. Chem., Int. Ed. 2013, 52, 518–533. [DOI] [PubMed] [Google Scholar]; d Brak K.; Jacobsen E. N. Angew. Chem., Int. Ed. 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Evans N. H.; Beer P. D. Angew. Chem., Int. Ed. 2014, 53, 11716–11754. [DOI] [PubMed] [Google Scholar]
- a Reisman S. E.; Doyle A. G.; Jacobsen E. N. J. Am. Chem. Soc. 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Klausen R. S.; Jacobsen E. N. Org. Lett. 2009, 11, 887–890. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Knowles R. R.; Lin S.; Jacobsen E. N. J. Am. Chem. Soc. 2010, 132, 5030–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Brown A. R.; Kuo W.-H.; Jacobsen E. N. J. Am. Chem. Soc. 2010, 132, 9286–9288. [DOI] [PMC free article] [PubMed] [Google Scholar]; e De C. K.; Mittal N.; Seidel D. J. Am. Chem. Soc. 2011, 133, 16802–16805. [DOI] [PubMed] [Google Scholar]; f Lin S.; Jacobsen E. N. Nat. Chem. 2012, 4, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Burns N. Z.; Written M. R.; Jacobsen E. N. J. Am. Chem. Soc. 2011, 133, 14578–14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For books and reviews on selenofunctionalization and related chemistry, see:; a Nicolaou K. C. and Petasis N. A.. Selenium in Natural Product Synthesis; CIS: Philadelphia, 1984. [Google Scholar]; b Liotta D., Ed. Organoselenium Chemistry; Wiley: New York, 1987. [Google Scholar]; c Back T. G., Ed. Organoselenium Chemistry—A Practical Approach; Oxford University Press: Oxford, 1999. [Google Scholar]; d Topics in Current Chemistry: Organoselenium Chemistry; Wirth T., Ed.; Springer: Heidelberg, 2000. [Google Scholar]; e Petragnani N.; Stefani H. A.; Valduga C. J. Tetrahedron 2001, 57, 1411–1448. [Google Scholar]; f Ranganathan S.; Muraleedharan K. M.; Vaish N. K.; Jayaraman N. Tetrahedron 2004, 60, 5273–5308. [Google Scholar]; g Wirth T., Ed.; Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, 2012. [Google Scholar]; h Santi C.; Tidei C.. Electrophilic Selenium/Tellurium Reagents: Reactivity and their Contribution to Green Chemistry. PATAI’s Chemistry of Functional Groups; Wiley Online Library, 2013; DOI 10.1002/9780470682531.pat0720. [DOI] [Google Scholar]
- For reviews on diastereoselective selenofunctionalization using chiral selenenylating reagents, see:; a Wirth T. Tetrahedron 1999, 55, 1–28. [Google Scholar]; b Wirth T. Angew. Chem., Int. Ed. 2000, 39, 3740–3749. [DOI] [PubMed] [Google Scholar]; c Tiecco M.; Testaferri L.; Marini F.; Bagnoli L.; Santi C.; Temperini A.; Sternativo S.; Tomassini C. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 729–740. [Google Scholar]; d Browne D. M.; Wirth T. Curr. Org. Chem. 2006, 10, 1893–1903. [Google Scholar]
- For recent reports on catalytic enantioselective selenofunctionalization reaction, see:; a Denmark S. E.; Kalyani D.; Collins W. R. J. Am. Chem. Soc. 2010, 132, 15752–15765. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guan H.; Wang H.; Huang D.; Shi Y. Tetrahedron 2012, 68, 2728–2735. [Google Scholar]; c Wei Q.; Wang Y.; Du Y. L.; Gong L. Beilstein J. Org. Chem. 2013, 9, 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Denmark S. E.; Collins W. R.; Cullen M. D. J. Am. Chem. Soc. 2009, 131, 3490–3492. [DOI] [PubMed] [Google Scholar]; b Denmark S. E.; Edwards M. G. J. Org. Chem. 2006, 71, 7293–7306. [DOI] [PubMed] [Google Scholar]
- a Nicolaou K. C.; Claremon D. A.; Barnette W. E.; Seitz S. P. J. Am. Chem. Soc. 1979, 101, 3704–3706. [Google Scholar]; b Nicolaou K. C.; Petasis N. A.; Claremon D. A. Tetrahedron 1985, 41, 4835–4841. [Google Scholar]
- For comparison of anion-binding strength between squaramides and ureas, see:; a Amendola V.; Bergamaschi G.; Boiocchi M.; Fabbrizzi L.; Milani M. Chem.—Eur. J. 2010, 4368–4380. [DOI] [PubMed] [Google Scholar]; b Amendola V.; Fabbrizzi L.; Mosca L.; Schmidtchen F.-P. Chem.—Eur. J. 2011, 5972–5981. [DOI] [PubMed] [Google Scholar]; c Lu T.; Wheeler S. E. Chem.—Eur. J. 2013, 15141–15147. [DOI] [PubMed] [Google Scholar]
- Decomposition is most likely attributable to oxidation of the thiourea by the high-valent selenium reagent.
- For examples of chiral squaramide-catalyzed enantioselective reactions involving neutral electrophiles, see:; a Malerich J. P.; Hagihara K.; Rawal V. H. J. Am. Chem. Soc. 2008, 130, 14416–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Konishi H.; Lam T. Y.; Malerich J. P.; Rawal V. H. Org. Lett. 2010, 12, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dai L.; Wang S.-X.; Chen F.-E. Adv. Synth. Catal. 2010, 352, 2137–2141. [Google Scholar]; d Zhu Y.; Malerich J. P.; Rawal V. H. Angew. Chem., Int. Ed. 2010, 49, 153–156. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yang K. S.; Nibbs A. E.; Türkmen Y. E.; Rawal V. H. J. Am. Chem. Soc. 2013, 135, 16050–16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmark S. E.; Collins W. R. Org. Lett. 2007, 9, 3801–3804. [DOI] [PubMed] [Google Scholar]
- For studies on the bioactivities of flavonoids, see:; a Flavonoids in health and disease, 2nd ed.; Rice-Evans C. A., Packer L., Eds.; Marcel Dekker, New York, 2003. [Google Scholar]; b Maloney D. J.; Deng J. Z.; Starck S. R.; Gao Z.; Hecht S. M. J. Am. Chem. Soc. 2005, 127, 4140–4141. [DOI] [PubMed] [Google Scholar]; c Yáñez J. A.; Remsberg C. M.; Takemoto J. K.; Vega-Villa K. R.; Andrews P. K.; Sayre C. L.; Martinez S. E.; Davies N. M. In Flavonoid pharmacokinetics: Methods of Analysis, Preclinical and Clinical Pharmacokinetics, Safety, and Toxicology; Davies N. M., Yáñez J. A., Eds.; Wiley: Chicester, 2013; Chapter 1, pp 1–69. [Google Scholar]
- See Supporting Information for the conditions and spectroscopic data of the NMR experiments.
- The small discrepancy between the ee’s under the two conditions is due to the formation of a trace amount of racemic product in reaction 2 prior to the addition of the chiral catalyst, as established by NMR analysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.