

Abstract
The p75 neurotrophin receptor (p75NTR ) is involved in degenerative mechanisms related to Alzheimer’s disease (AD). In addition, p75NTR levels are increased in AD and the receptor is expressed by neurons that are particularly vulnerable in the disease. Therefore, modulating p75NTR function may be a significant disease-modifying treatment approach. Prior studies indicated that the non-peptide, small molecule p75NTR ligands LM11A-31, and chemically unrelated LM11A-24, could block amyloid-β-induced deleterious signaling and neurodegeneration in vitro, and LM11A-31 was found to mitigate neuritic degeneration and behavioral deficits in a mouse model of AD. In this study, we determined whether these in vivo findings represent class effects of p75NTR ligands by examining LM11A-24 effects. In addition, the range of compound effects was further examined by evaluating tau pathology and neuroinflammation. Following oral administration, both ligands reached brain concentrations known to provide neuroprotection in vitro. Compound induction of p75NTR cleavage provided evidence for CNS target engagement. LM11A-31 and LM11A-24 reduced excessive phosphorylation of tau, and LM11A-31 also inhibited its aberrant folding. Both ligands decreased activation of microglia, while LM11A-31 attenuated reactive astrocytes. Along with decreased inflammatory responses, both ligands reduced cholinergic neurite degeneration. In addition to the amelioration of neuropathology in AD model mice, LM11A-31, but not LM11A-24, prevented impairments in water maze performance, while both ligands prevented deficits in fear conditioning. These findings support a role for p75NTR ligands in preventing fundamental tau-related pathologic mechanisms in AD, and further validate the development of these small molecules as a new class of therapeutic compounds.
Keywords: Alzheimer’s disease, LM11A-31, LM11A-24, p75 neurotrophin receptor
INTRODUCTION
Alzheimer’s disease (AD) is characterized by: widespread neurodegeneration throughout the association cortex, basal forebrain, hippocampus, and limbic system [1-4]; abnormal accumulation of amyloid-β (Aβ); excess phosphorylation, misfolding, and aggregation of tau protein [1, 5-7]; and increased microglial activation [8-11] and reactive astrogliosis [12-14]. While attempts to lower Aβ levels make up the majority of therapeutic developmental efforts, multiple phase three clinical trials with anti-amyloid therapies have thus far failed to achieve pre-determined clinical end-points [15-18]. It is likely that an effective therapy will require multiple approaches, including managing Aβ accumulation along with reducing neuronal vulnerability to Aβ and other causative factors [19, 20]. An important more recent strategy consists of inhibiting tau-mediated degenerative mechanisms [21, 22], and antibody-based approaches targeting pathological forms of tau are under development [23]. It is possible that modulating upstream mechanisms promoting the formation of pathological tau will be particularly effective; however, the search for small molecules capable of regulating such mechanisms remains at nascent stages. Engagement of strategic receptors upstream of AD-relevant signaling pathways may produce simultaneous effects on multiple degenerative mechanisms, including those modulating tau.
The p75 neurotrophin receptor (p75NTR ) is one of the few receptors directly connected to multiple nodes of signaling networks that undergo abnormal modulation in AD [24]. Its expression occurs in CNS cell populations that degenerate in AD, including cortical, hippocampal, basal forebrain, raphe, and locus coeruleus neurons [25-31]. Multiple studies indicate that p75NTR levels increase in human AD [25-27, 32, 33] and mouse AD [34, 35] models. Levels of pro-nerve growth factor (proNGF), a p75NTR ligand that promotes degenerative signaling through a number of mechanisms, including activation of JNK, are also increased [36-38]. Consistent with these findings, p75NTR has been found to play a key role in enabling Aβ-induced degeneration [39-42]. Removal of exon III in p75NTR , which deletes its neurotrophin binding domain, prevents basal forebrain cholinergic neurite degeneration in Thy1-hAβPPLond/Swe (AβPPL/S) mice [43]. Increased expression of p75NTR relative to TrkA is believed to contribute to degenerative signaling in AD-affected basal forebrain cholinergic neurons (BFCNs) [44, 45].
Our laboratories have developed small molecule ligands to regulate p75NTR signaling, and thereby AD-related degenerative mechanisms [46]. We demonstrated in previous in vitro studies that these small molecules compete with NGF and proNGF for binding to p75NTR but not to TrkA [47]. Although they are ostensibly related only by the property of containing a pharmacophore designed to mimic the NGF loop 1 p75NTR binding domain, the otherwise chemically diverse LM11A compounds have similar degeneration-preventing activities and potencies [47]. Further in vitro studies showed that two ligands, LM11A-31 and LM11A-24, prevented Aβ-induced degeneration of hippocampal, cortical, and basal forebrain neurons. The compounds also blocked Aβ-induced activation of GSK3β, cdk5, and c-Jun, and prevented Aβ-induced inhibition of AKT and CREB activation [48]. In in vivo studies employing AβPPL/S mice, LM11A-31 inhibited neurite degeneration and improved performance in novel object recognition and Y-maze testing after 2.5–3 months of once daily oral administration, without affecting Aβ levels [49]. Given the ability of p75NTR ligands to inhibit the Aβ induction of multiple tau kinases in vitro, we determined in the present study whether p75NTR small molecule ligands are capable of inhibiting excess tau phosphorylation and aberrant folding along with potentially related [29] excess microglial and astrocytic activation. We also determined whether the structurally distinct p75NTR ligand LM11A-24 would have one or more anti-degenerative activities, and hence support a potential ligand class effect as opposed to an idiosyncratic effect of a single compound. In this study, we first examined the brain distribution of p75NTR and target engagement of ligands, and then assessed the effects of lead compound LM11A-31 and additional class-member LM11A-24 on tau phosphorylation and misfolding, glial activation, neurite degeneration, and AD-relevant behavioral tests. These studies suggest that modulation of AD degenerative mechanisms represents a class effect of p75NTR ligands, and suggest a novel small molecule approach for preventing Aβ-induced excess tau phosphorylation, tau misfolding, and glial activation.
MATERIALS AND METHODS
AβPPL/S mice
Thy1-hAβPPLond/Swe (AβPPL/S) mice, transgenic line 41 mice overexpressing human amyloid-β protein precursor 751 containing the London (V717I) and Swedish (K670N/M671L) mutations under the control of the Thy1 promoter [43, 50-57], were maintained on a C57BL/6J background and housed under a 12-h light/dark schedule with ad libitum access to food and water. Male mice were treated beginning at 3.5–4.5 months of age. In AβPPL/S mice, plaque deposition is evident by 3–4 months of age in the frontal cortex and by 5–6 months in the hippocampus [55]. AβPPL/S and wild-type (wt) littermates were randomized to vehicle, LM11A-31, or LM11A-24 treatment groups such that there were no significant differences in age between treatments. LM11A-31 [2-amino-3-methyl-pentanoic acid (2-morpholin-4-yl-ethyl)-amide], an isoleucine derivative, and LM11A-24 (N-(3-dimethyl aminopropyl)-1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxopurine-7-acetamide), a caffeine derivative [47], were synthesized by Ricerca Biosciences at >97% purity as assessed by liquid chromatography/mass spectroscopy (structures shown in Supplementary Figure 1A). Additionally, all lots were tested for bioactivity (increased hippocampal neuron survival) in cell culture as characterized in previous work [47]. LM11A-31 (at 5, 25, 50, or 100 mg/kg) or LM11A-24 (at 50 or 100 mg/kg) was administered in sterile water by oral gavage (10 μl per gram of body weight) following a 4-h fast once each day, while vehicle-treated mice received an equivalent volume per weight of sterile water on the same schedule. Mice were treated for a total of 3 months. Behavioral testing was performed at 2.5 months after treatment onset when mice were 6–7 months of age. In all behavioral experiments, mice were handled by the experimenter for 5 days before testing, and habituated to the testing room 2-h prior to testing. At study completion, mice were injected intraperitoneally with 1.25% Avertin and perfused intracardially with 4% paraformaldehyde. A subset of brains from each treatment group generated from behavioral experiments was used for morphological and western blot analyses. All dosing, behavioral testing, and analyses were performed with blinding to genotype and test compound. All experiments were in accordance with protocols approved by the Institutional Animal Care and Use Committee of Stanford University, and were performed based on the NIH Guide for the Care and Use of Laboratory Animals.
Immunohistochemistry
Frozen coronal sections (40 μm) were taken through the entire brain using a Microm HM 450 sliding microtome (Thermo Scientific) and immunostained according to standard protocols. The following antibodies were used: mouse anti-AT8 [recognizes tau phosphorylated at serine 202 and threonine 205 (p-tauSer202/Thr205); Thermo Scientific], mouse anti-MC-1 (recognizes aberrantly folded tau [58]; a kind gift from Dr. Peter Davies, Albert Einstein School of Medicine, NY), rat anti-CD68 (AbD Serotec), mouse anti-glial fibrillary acidic protein (GFAP; Dako), and goat anti-choline acetyl transferase (ChAT; Millipore). Briefly, free floating sections were immunolabeled with antibodies (1:1500 for AT8, 1:250 for MC-1, 1:1000 for CD68, 1:1000 for GFAP, and 1:800 for ChAT) in conjunction with M.O.M. (for AT8 and MC-1), ABC Vector Elite, and 3,3′-diaminobenzidine or Vector VIP kits (Vector Laboratories) for visualization. For confocal imaging, sections at the level of bregma −1.70 to −1.94 were immunolabeled with goat anti-p75NTR extracellular domain (ECD; Neuromics) at 1:250, mouse anti-neuronal nuclei (NeuN; Millipore) at 1:500, CD68 at 1:500, or GFAP at 1:500 followed by Alexa Fluor secondary antibodies (Life Technologies). Images of sections co-stained with p75NTR and NeuN, p75NTR and CD68, and p75NTR and GFAP were captured with a Leica TCS SPE DM5500 laser scanning confocal microscope using 647 nm (for p75NTR ) and 488 nm (for NeuN, CD68, and GFAP) filters. Specificity of the p75NTR ECD antibody was evaluated with western blot analysis. Amyloid plaques were stained with 1% Thioflavin-S (Thio-S; Sigma-Aldrich).
Western blotting
NIH-3T3 cell pellets (cells stably transfected with empty or p75NTR vector), PC12 cell pellets, or hippocampal or cortical tissue were sonicated in RIPA buffer (20 nM Tris, pH 8.0, 137 mM NaCl, 1% NP-40, 10% glycerol, 1 mM PMSF, 500 μM ortho-vanadate, 10 μg/ml aprotinin, and 1 μg/ml leupeptin). Briefly, lysates were resolved by electrophoresis, transferred to a PVDF membrane, and probed with anti-p75NTR ECD (1:2500), rabbit anti-p75NTR intracellular domain (ICD, 1:2000; Promega), or mouse anti-actin (1:10,000; Sigma-Aldrich). Proteins were visualized using a chemiluminescence detection system (GE Healthcare Life Sciences). Immunoreactive bands were quantified using UN-SCAN-IT software (Lane Analysis Positive function; Silk Scientific). The amount of C-terminal fragment (CTF) or ICD was determined as a ratio to full-length (FL) p75NTR within each lane, and the amount of FL was determined as a ratio to actin (n = 6 mice per genotype-treatment group; values from immunoblots from 4 independent runs for each mouse sample were averaged to yield one value per mouse).
Quantitation of tau pathology in the cortex
For assessment of AT8 (p-tau)-labeled dystrophic neurites, three coronal sections per mouse were analyzed (n = 9–13 mice per genotype-treatment group). In each section, two non-overlapping, adjacent 10×fields (550 μm × 425 μm) of the cortex comprising the cingulate, motor, and primary somatosensory areas in each hemisphere between bregma +0.86 and −0.34 based on the mouse brain atlas of Franklin and Paxinos 3rd edition [59] were analyzed for a total of 12 fields per mouse. Clusters of dystrophic neurites were traced by manually outlining their perimeters using Neurolucida software (MBF Biosciences) and the area of clusters computed. Pixel intensity of AT8 staining from the same fields was measured by optical densitometry (OD) using UN-SCAN-IT (Segment Analysis function). Fields containing both dystrophic neurite clusters and non-cluster regions made up largely of cell bodies, subfields (175 μm × 175 μm, one subfield per field) containing only non-cluster/cell body regions within cortical layers II-IV, and subfields covering each dystrophic neurite cluster were each analyzed separately. For each mouse, the values from each field or subfield were averaged to yield one value for each measurement per mouse.
The same region of the cortex, and the same number and size of fields were analyzed to assess MC-1-labeled dystrophic neurite clusters. Clusters were traced and the area of clusters computed (n = 12 mice per genotype-treatment group), then subfields covering each dystrophic neurite cluster in a field were each analyzed for total pixel intensity (n = 12 mice per genotype-treatment group).
Quantitation of CD68 and GFAP immunoreactivity
For assessment of microglial and astrocytic activation, CD68-labeled microglia (appearing as either single cells or in clusters, with most signal associated with clusters of cells) and GFAP-labeled astrocytes were analyzed (two sections per mouse, CD68: n = 5– 6 and GFAP: n = 6–7 mice per genotype-treatment group). One 10× field containing the primary and secondary somatosensory areas of each hemisphere between bregma −1.58 and −1.82 was analyzed per section, for a total of 4 fields per mouse. Values for each field within a given mouse were averaged to yield one value per mouse. Percent area occupied by CD68 or GFAP staining was determined using ImagePro-Plus thresholding software (Media Cybernetics). For analysis of hippocampal CD68 burden, one 20× field containing the CA1 region of the hippocampus of each hemisphere in the same sections that were used for cortical quantitation was analyzed per section, for a total of two sections per mouse (n = 8 mice per genotype-treatment group).
Quantitation of BFCN neurites
ChAT-stained neurites of cholinergic neurons in the vertical limb of the diagonal band of Broca (VDB), which innervate hippocampal and cortical areas, were analyzed using Neurolucida software with a Leica DM5000B light microscope coupled with a Q Imaging camera according to methods adapted from previous work [43]. An automated, unbiased scanning procedure (Neurolucida ‘Meander Scan’) allowed areas of user-defined contour to be viewed without overlap. In every other field, all cell bodies and dendrites originating from them were manually traced, while focusing up and down the z-plane through the tissue with a 40× objective, which allowed an accurate discrimination of neurites emanating from neurons and assessment of volume. The ‘Branch Structure Analysis’ routine was used to compute the length and volume of neurites. One section per mouse (n = 6–8 mice per genotype-treatment group) containing the VDB at approximately bregma +1.10 was analyzed. Anatomical landmarks contained within each section delineating the VDB included the anterior part of the anterior commissure and the major island of Calleja, before (anterior to) the emergence of the horizontal diagonal brand (approximate bregma +0.98); typically, this criteria was met by only one section when every 8th section in a 1 to 16 series was processed.
Quantitation of cortical cholinergic terminals
Two sections per mouse (n = 8 mice per genotype-treatment group) were analyzed. One 40× field covering the majority of the cingulate cortex containing ChAT-stained cholinergic projection fibers in each hemisphere was analyzed per section between bregma +1.10 and +0.38, for a total of 4 fields per mouse. Percent area occupied by cholinergic terminals was determined using ImageProPlus [43].
Quantitation of cholinergic dystrophic neurites and amyloid plaques in the cortex and hippocampus
For assessment of dystrophic neurites, three sections per mouse (n = 8–16 mice per genotype-treatment group) were analyzed. Two non-overlapping, adjacent 10× fields of the cortex comprising the cingulate, motor, and primary somatosensory areas in each hemisphere between bregma +1.34 and +0.14 were analyzed per section, for a total of 12 fields per mouse. Clusters of dystrophic neurites were traced by manually out-lining the perimeters using Neurolucida and the area of clusters was computed using the Branched Structure Analysis routine. The area of dystrophic neurite clusters in the central part of the hippocampus comprising the stratum lacunosum-moleculare, molecular layer of the dentate gyrus, and stratum radiatum was also analyzed. Three to five sections were analyzed per mouse (n = 4 mice per genotype-treatment group). One to five 10× fields covering the entire hippocampal area of each hemisphere per section were analyzed. The same region of the hippocampus was analyzed to determine the percentage of area occupied by Thio-S-positive amyloid plaques using ImageProPlus (n = 7–8 mice per genotype-treatment group) [43].
Delayed-matching-to-place water maze
The delayed-matching-to-place water maze was originally designed to assess spatial working/episodic-like learning and memory in rats by Morris and colleagues [60, 61], and has been adapted for use in mice [50]. The apparatus consisted of a 200 cm diameter tank filled with opaque water at a temperature of 22.0 ± 1.5°C with a circular platform (10 cm in diameter) submerged 1–2 cm below the water surface and placed randomly in the pool with its position changing daily. Mice (n = 12–18 per genotype-treatment group) were given a series of four trials per day, 10 min apart, with a maximum of 120 seconds to find the submerged platform. If a mouse was unable to find the platform in that period of time, it was physically guided to it. After remaining on the platform for 10 seconds, mice were removed and placed in a dry cage. This process was repeated once daily for 6 days. Successful learning of the delayed-matching-to-place task was determined by the gradual decrease in escape latency in a trial-to-trial comparison averaged over day 2–6 (day 1 was considered as habituation to the experimental set-up, and therefore excluded from the analysis). Mice were given visible platform training to ensure the lack of gross sensorimotor and visual deficits. Swim paths and velocities were recorded with an Ethovision video tracking system.
Fear conditioning
Two context chambers varying in olfactory and visual cues, textures of the floor and room (Coulbourn Instruments), termed Context A and Context B, were used to assess associative learning. On day 1, mice (n = 15–24 per genotype-treatment group) were placed in Context A and after 3 min of baseline recording, received five tone-shock pairings. The tone (2 kHz, 70 dB, 20 seconds) was followed by an 18 seconds empty interval, and then a 2 seconds 0.5 mA shock [50]. On day 2, mice were placed in novel Context B for 3 min and subsequently given three tone presentations without shocks in order to test tone-cued responses. On day 3 of the experiment, mice were placed in the original Context A for 5 min without presentation of the tone or shock to test contextual memory. Freezing was an indication of learning and defined as the complete lack of motion for a minimum of 0.75 seconds, and recorded with an overhead FearConditioningVideo tracking system (Med-Associates).
Activity chamber
Assessment of general activity took place in a square arena (43.2 cm × 43.2 cm) located in a sound-attenuated chamber (66 cm × 55.9 cm × 55.9 cm; Med Associates). Each mouse (n = 15–24 per genotype-treatment group) was placed in the center of the arena and movement recorded for 10 min by an automated tracking system (Ethovision, Noldus Information Technology) with three planes of infrared detectors.
Hot plate test
To assess whether there is a genotype difference in nociception, each mouse (n = 9 per genotype) was placed on a metal plate covered by a transparent glass cylinder (25 cm in height × 12 cm in diameter) set at 55 ± 0.2°C. The latency to first hind paw lick or jump was recorded. A 30 seconds cut-off time minimized exposure of the mice to painful stimuli and distress.
Statistics
All data were presented as mean ± SEM. A majority of the data was analyzed using a one-way ANOVA followed by post-hoc Newman-Keuls testing. Analyses of AT8- and MC-1-labeled dystrophic neurite clusters used an ANOVA and for planned comparisons with explicit predictions as to direction of difference, a one-tailed t-test. The delayed-matching-to-place water maze test was evaluated using a two-way ANOVA (factors: genotype and treatment) with post-hoc Bonferroni testing. The fear conditioning test was evaluated using an ANOVA and for planned comparisons, a two-tailed t-test. Significance was set at p < 0.05. For all figures, *p or +p is <0.05, **p or ++p is <0.01, and ***p or +++p is <0.001.
RESULTS
p75NTR is expressed by cortical and hippocampal neurons in AβPPL/S mice
Immunohistological studies have shown that p75NTR levels are increased in human AD brains [26, 27]. Though most consistently found to be expressed by BFCNs in the adult human and rodent brain, p75NTR has in some cases, using various p75NTR antibodies, been reported to be expressed by cell types beyond BFCNs and relevant to AD, such as cortical and hippocampal neurons [25, 30, 31, 62]. It has also been reported to be expressed by glia such as microglia [63, 64] and astrocytes [65, 66]. We sought to examine the CNS distribution of p75NTR expression using an antibody evaluated for p75NTR specificity. Western blot analysis with antibody directed to full length (FL) p75NTR detected a single band consistent with FL size in p75NTR -NIH-3T3 and PC12 cells but not in control NIH-3T3 cells (Fig. 1A). Assessment of cortical and hippocampal tissue samples from wt and AβPPL/S mice similarly revealed a single band corresponding in size to FL p75NTR . Confocal co-labeling of p75NTR and NeuN revealed high levels of somal and neurite staining for p75NTR especially in pyramidal neurons of cortical layers III-V (Fig. 1B) and in the dentate gyrus and CA1-CA3 regions of the hippocampus (Fig. 1C). Interestingly, no apparent qualitative difference was observed in the number of p75NTR -immunoreactive neurons or intensity of staining between wt and AβPPL/S mice, and quantitative western blotting (Fig. 2B) did not detect a difference in p75NTR levels between wt and AβPPL/S mice at the 6.5–7.5 month age point assessed in the present study. p75NTR was not detected in CD68 co-labeled clusters of microglia (Fig. 1D) or CD68 co-labeled astrocytes (Fig. 1E).
Fig. 1.

p75NTR is expressed in cortical and hippocampal neurons of AβPPL/S mice. A) Western blotting with a p75NTR extracellular domain antibody detected a single band in 3T3 cells stably transfected with p75NTR , PC12 cells, and hippocampal (hipp) and cortical (cx) tissues from wild-type (wt) and AβPPL/S mice, corresponding to the full-length receptor. Representative 40× confocal images from AβPPL/S mouse sections showing p75NTR staining (red) in a primarily neurite and cell surface distribution within cells staining for the neuronal nuclear marker NeuN (green), including somatosensory cortex layer V neurons (B) and hippocampal CA1 (C) pyramidal neurons. No p75NTR signal was detected in microglia (CD68, green; D) or astrocytes (GFAP, green; E). Magnified outset, i, showing a cluster (arrow) of microglia with a few DAPI-labeled nuclei (blue) and an individual (arrowhead) CD68-positive microglia cell. Scale bar, 20 μm.
Fig. 2.
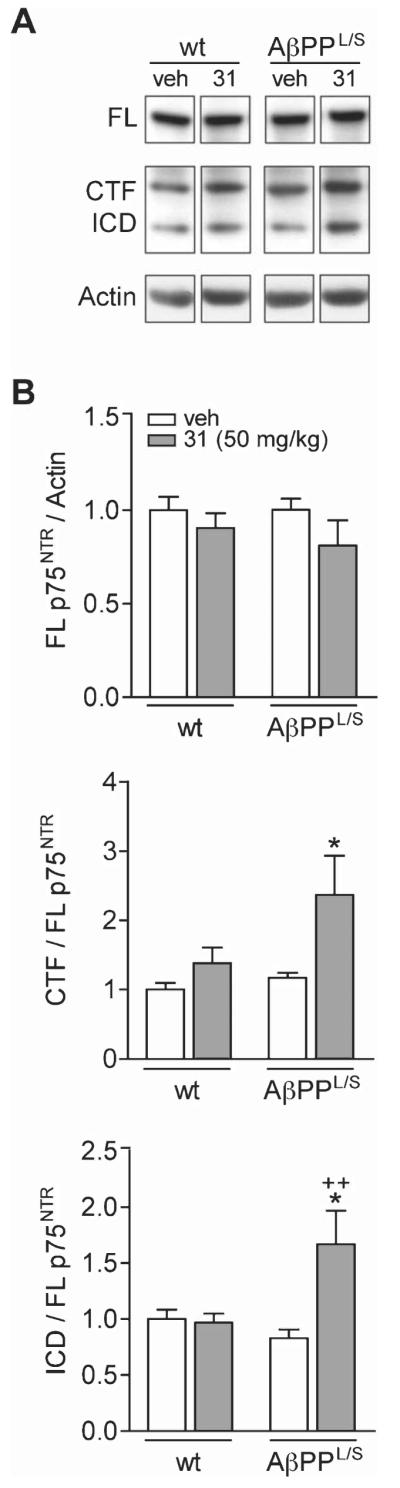
LM11A-31 modulates cleavage of p75NTR in the hippocampus of AβPPL/S mice. A) Western blotting using a p75NTR intracellular domain (ICD) antibody detected bands corresponding to full-length (FL) p75NTR , the C-terminal fragment (CTF) and ICD in hippocampal tissue from wild-type (wt) and AβPPL/S mice treated with vehicle (veh) or LM11A-31 (31) at 50 mg/kg. B) Data represent mean ± SEM densitometry values for each group (n = 6 mice) of immunoblots and expressed as a ratio of FL to actin (top graph), CTF to FL (middle graph), and ICD to FL (bottom graph), normalized to the wt-veh group. For CTF/FL, *p < 0.05 versus wt-veh, wt-31, and AβPPL/S-veh. For ICD/FL, *p < 0.05 versus wt-veh and wt-31, ++p < 0.01 versus AβPPL/S-veh.
p75NTR ligand specificity and target engagement in AβPPL/S mice
Prior studies demonstrated the specificity of LM11A-31 and LM11A-24 for p75NTR , with a lack of binding to Trks [47] and LM11A-31 was found to have no significant interactions in a broad screen of approximately 50 pharmacologically well-characterized receptors (Cerep ExpresSProfile) [48]. In the present study, a Cerep ExpresSProfile screen of LM11A-24 similarly showed no significant binding interactions (Supplementary Table 1). Previous in vitro studies demonstrated that the protective effects and signaling of LM11A-31 was dependent on the presence of p75NTR [47, 48].
Studies were conducted to examine the engagement in vivo of p75NTR by LM11A-31. Among the most proximate effects of ligand binding to p75NTR is ligand-induced receptor intramembrane proteolysis [67-69]. Cleavage in the ectodomain by γ-secretase results in a free extracellular domain and membrane bound C-terminal fragment (CTF). Cleavage of the CTF by -y-secretase releases the intracellular domain (ICD), which modulates neuronal survival/death and neurite outgrowth. Western analysis of hippocampal tissue from wt and AβPPL/S mice with anti-p75NTR -ICD revealed bands corresponding to full length (FL), CTF, and ICD (Fig. 2A) and quantitation revealed no difference in the levels of FL p75NTR across genotypes or treatments (Fig. 2B, top graph). In AβPPL/S mice treated with LM11A-31 compared with AβPPL/S mice treated with vehicle, or wt littermates given either treatment, there was significantly more CTF (~2-fold; Fig. 2B, middle graph) and ICD (1.5- to 2-fold; Fig. 2B, bottom graph), suggesting that the compound promotes proteolytic processing of p75NTR similar to that occurring due to neurotrophin ligand interactions, consistent with the engagement of the receptor in vivo.
Orally administered LM11A-24 crosses the blood-brain barrier
Prior studies had demonstrated that administration of LM11A-31 at a dose of 50 mg/kg leads to brain levels well above those achieving maximal effects in vitro [49]. In the present study, LM11A-24 achieved brain concentrations above those known to be neuroprotective against Aβ in vitro (EC50 = 20nM) [48] following oral administration at 50 mg/kg; however, it produced substantially lower brain exposure than LM11A-31 (Supplementary Figure 1B). Plasma measurements demonstrated that levels of LM11A-24 are greater over time than those of LM11A-31, indicating that the lower brain levels of LM11A-24 are likely due to decreased ability to cross the blood brain barrier. With these considerations, wt and AβPPL/S mice were treated with LM11A-31 at a dose of 50 mg/kg/day and LM11A-24 at doses of 50 or 100 mg/kg/day for 3 months starting at ages 3.5–4.5 months.
p75NTR ligand reduces tau hyperphosphorylation in AβPPL/S mice
Neurofibrillary tangles and dystrophic neurites contain abnormal filaments of hyperphosphorylated tau proteins [70-74]. In vitro studies with hippocampal neurons showed that LM11A-31 and LM11A-24 inhibit Aβ-induced phosphorylation of tau as measured by AT8 western blot analysis [48]. Given the potential therapeutic implications, it was determined whether p75NTR ligands mitigate excessive levels of p-tau present in vivo in AβPPL/S mice. Six-month-old AβPPL/S mice have substantially increased neuronal levels of p-tau in the cortex and hippocampus [54]. In the present study, as seen in Fig. 3A, animals at age 6.5–7.5 months had increased p-tau immunoreactivity, and specifically, p-tau-labeled dystrophic neurite clusters were observed in the cortex of vehicle-treated AβPPL/S mice but not in wt mice. Qualitative assessment suggested that treatment with LM11A-31 reduced p-tau staining in both neurite clusters and also non-cluster regions containing pre-dominately cell bodies (Fig. 3A). Quantitation of p-tau dystrophic neurite clusters in AβPPL/S mice revealed a decrease of 42% in the total dystrophic neurite cluster area with LM11A-31 treatment relative to vehicle while LM11A-24 had no significant effect (Fig. 3B).
Fig. 3.

LM11A-31 and LM11A-24 reduce excessive levels of tau phosphorylation in the cortex of AβPPL/S mice. Wild-type (wt) and AβPPL/S mice were treated with vehicle (veh), LM11A-31 at 50 mg/kg, or LM11A-24 at 50 or 100 mg/kg. A) Representative 10× images of AT8 (phosphorylated tau, p-tauSer202/Thr205)-immunolabeled dystrophic neurite clusters and non-cluster regions that predominately contain cell bodies in the motor cortex. Scale bar, 50 μm. Compared to wt mice, AβPPL/S mice exhibited prominent dystrophic neurite clusters (magnified outset, i) and a higher level of somal accumulation of p-tau (arrows, ii), which treatment with LM11A-31 appeared to reduce (clusters, iii; cell bodies, iv). B) Dystrophic neurite clusters were manually outlined using Neurolucida software. The total area of p-tau-labeled dystrophic neurite clusters in AβPPL/S mice was significantly reduced by LM11A-31 treatment. Data represent mean ± SEM from n = 9–13 mice per group. *p < 0.05 versus AβPPL/S-veh. C) Densitometry of p-tau immunoreactivity in dystrophic neurite clusters (left and right graphs) and non-cluster/cell body regions (left and middle graphs) showed significantly lower staining intensity in AβPPL/S mice treated with LM11A-31 versus vehicle; LM11A-24 only significantly reduced staining intensity at 100 mg/kg and only in non-cluster/cell body regions (middle graph) but not in neurite clusters (n = 9–14 mice per group). *p < 0.05, **p < 0.01, and ***p < 0.001 versus wt-veh; +p < 0.05, ++p < 0.01, and +++p < 0.001 versus AβPPL/S-veh.
In order to further quantitate the effects of LM11A-31 and LM11A-24 on tau phosphorylation, AT8 immunoreactivity was also measured in terms of pixel intensity. In regions of combined dystrophic neurite clusters and non-cluster/cell bodies, vehicle-treated AβPPL/S mice demonstrated a 604% increase in p-tau staining intensity relative to wt mice (Fig. 3C, left graph) and by 54% in non-cluster/cell body regions (Fig. 3C, middle graph). Treatment with LM11A-31 significantly reduced excessive p-tau in AβPPL/S mice by 59% for regions of combined dystrophic neurite clusters and non-cluster/cell bodies (Fig. 3C, left graph), by 43% for non-cluster/cell bodies (Fig. 3C, middle graph), and by 45% for dystrophic neurite clusters (Fig. 3C, right graph), while vehicle had no effect. LM11A-24 caused a 34% reduction in p-tau accumulation only in non-cluster/cell body regions at the higher 100 mg/kg dose (Fig. 3C, middle graph). Neither compound affected p-tau levels in non-cluster/cell body regions of wt mice indicating a lack of effect on normal, physiological tau phosphorylation.
p75NTR ligand reduces pathological tau folding in AβPPL/S mice
Studies suggest that the Ser202 and Thr205 (the epitope recognized by the antibody AT8) are among the residues through which excess phosphorylation contributes to pathological conformation of tau [75]. Therefore, the finding that p75NTR ligands reduce tau phosphorylation at the AT8 epitope predicted that the compounds might produce a reduction in pathological tau conformers as well. Immunostaining was performed with MC-1, an antibody that recognizes misfolded tau [58]. LM11A-31 appeared to reduce the area and staining intensity of MC-1-labeled dystrophic neurites in the cortex of AβPPL/S mice (Fig. 4A). Quantitation of MC-1 signal showed that LM11A-31 caused a 41% decrease in total stained cluster area (Fig. 4B). Furthermore, optical density measurements revealed that LM11A-31 reduced MC-1 signal by 37% in dystrophic neurite clusters of AβPPL/S mice (Fig. 4C). This reduction in the level of misfolded tau supports the idea that p75NTR ligands might decrease tau pathology, at least in part via mitigation of aberrant tau phosphorylation at the AT8 epitope.
Fig. 4.

LM11A-31 reduces MC-1-labeled pathological tau folding in the cortex of AβPPL/S mice. A) Representative 10× images of MC-1-immunolabeled dystrophic neurites in the motor cortex. Scale bar, 50 μm. AβPPL/S mice exhibited abundant MC-1 immunostaining consistent with misfolded tau within dystrophic neurite clusters, which is not present in wt mice (data not shown) and appeared to be reduced by LM11A-31 treatment. B) The total area of MC-1-labeled dystrophic neurite clusters in AβPPL/S mice was significantly decreased with LM11A-31 (n = 12 mice per group). *p < 0.05 versus AβPPL/S-veh. C) Densitometry of MC-1 immunoreactivity within clusters showed significantly lower staining intensity in AβPPL/S mice treated with LM11A-31 (n = 12 mice per group). *p < 0.05 versus AβPPL/S-veh.
p75NTR ligands decrease inflammatory changes in AβPPL/S mice
Neuropathologic and neuroradiologic studies in AD have demonstrated that inflammatory responses such as microglial activation and astrocyte reactivity are present early in the course of the disease [76]. Activated microglia and reactive astrocytes surrounding senile plaque cores have been consistently reported in AD brains [10, 77-79]. Moreover, there is evidence for microglial activation [80-82] and reactive astrocytes [83] in the proximity of tau deposits, and for the interplay of tau pathology and the inflammatory cascade [84, 85]. In order to better assess the biological significance of the ability of p75NTR ligands to reduce pathologic tau accumulation, it was of interest to determine whether they would diminish these inflammatory processes. Previous studies using 9- and 16-month old AβPPL/S mice reported cortical microglial activation (reflected by increased Iba-1 or CD68 staining) [55, 56] and hippocampal astrogliosis (reflected by GFAP upregulation) [51].
In the current study, in mice 6.5–7.5 months of age, numerous clusters of CD68-immunoreactive cells displaying the typical resting microglial morphology (small cell body with long branching processes) were seen distributed throughout the cortex and in particular the somatosensory region of all wt mice (Fig. 5A). In AβPPL/S mice, an increase in activated microglia morphology (with larger cell bodies and bushy, thick processes) was observed (Fig. 5A). Many, but not all CD68-positive clusters were associated with Thio-S-positive plaques. Both LM11A-31 and LM11A-24 appeared to reduce activation of these cells (Fig. 5A). Quantification of the area occupied by CD68-positive clusters of microglia revealed a 143% increase in CD68 staining in the cortex of AβPPL/S mice compared to their wt littermates (Fig. 5B). Both ligands significantly reduced CD68 staining relative to vehicle in AβPPL/S mice (33% for LM11A-31, 40% for LM11A-24 at 50 mg/kg, and 41% for LM11A-24 at 100 mg/kg) and had no significant effect in wt lit-termates (Fig. 5B). Similar to findings in the cortex, within the hippocampus, there was 89% more CD68 staining in vehicle-treated AβPPL/S mice relative to wt littermates, which LM11A-31 reduced by 46% (Fig. 5C).
Fig. 5.

LM11A-31 and LM11A-24 reduce activated microglia in the cortex and hippocampus of AβPPL/S mice. Wild-type (wt) and AβPPL/S mice were treated with vehicle (veh), LM11A-31 at 50 mg/kg, or LM11A-24 at 50 or 100 mg/kg. A) Representative 20× images of CD68-immunolabeled microglia in the primary and secondary somatosensory cortex. Scale bar, 50 μm. CD68 expression appeared to be increased along with activated microglia morphology (larger cell bodies and bushy, thick processes) in AβPPL/S mice, and this response was reduced with LM11A-31 and LM11A-24. B) Quantification of cortical CD68 immunoreactivity. CD68 staining was significantly decreased in AβPPL/S mice treated with LM11A-31 or LM11A-24. Data represent mean ± SEM from n = 5–6 mice per group. ***p < 0.001 versus wt-veh; +p < 0.05 and ++p < 0.01 versus AβPPL/S-veh. C) In AβPPL/S hippocampus, LM11A-31 significantly reduced CD68 staining (n = 8 mice per group). *p < 0.05 versus wt-veh; +p < 0.05 versus AβPPL/S-veh.
In wt mice, GFAP-immunoreactive cells displaying the typical stellate shape and multiple branched processes of astrocytes were distributed throughout the cortex, though they were more abundant in the somatosensory cortex (Fig. 6A). In contrast, vehicle-treated AβPPL/S mice had more cells displaying a reactive astrocytic morphology with thicker and more extended processes than wt littermates, and LM11A-31 appeared to reduce this reactive morphology (Fig. 6A). Quantification of the area occupied by GFAP-positive astrocytes in vehicle-treated mice revealed 906% more GFAP staining in AβPPL/S mice compared to wt (Fig. 6B). LM11A-31, but not LM11A-24, significantly decreased GFAP staining (by 68%) in AβPPL/S mice and had no significant effect in wt mice (Fig. 6B).
Fig. 6.

LM11A-31 reduces reactive astrocytes in the cortex of AβPPL/S mice. Wild-type (wt) and AβPPL/S mice were treated with vehicle (veh), LM11A-31 at 50 mg/kg, or LM11A-24 at 50 or 100 mg/kg. A) Representative 20× images of glial fibrillary acidic protein (GFAP)-immunolabeled astrocytes in the primary and secondary somatosensory cortex. Scale bar, 50 μm. GFAP expression appeared to be increased along with characteristic activated astrocytic morphology with cells displaying thicker and more extended processes in AβPPL/S mice and this response was reduced with LM11A-31. B) Quantification of GFAP immunoreactivity. GFAP staining was significantly decreased in AβPPL/S mice treated with LM11A-31 but not with LM11A-24. Data represent mean ± SEM from n = 6–7 mice per group. *p < 0.001 versus wt-veh; +p < 0.05 versus AβPPL/S-veh.
p75NTR ligands prevent degeneration of basal forebrain cholinergic neurites and cortical projection fibers in AβPPL/S mice
Cholinergic neurons of the basal forebrain projecting to cortical and hippocampal targets are critical for attention and cognition, and are particularly vulnerable in aging and AD [86, 87]. It is well established that p75NTR regulates the somal size and projection fiber volume of BFCNs [88, 89], and a recent study showed that LM11A-31 was able to prevent cholinergic neurite degeneration in AβPPL/S mice [49]. To determine whether LM11A-24 would also inhibit such degeneration, cholinergic neurite morphology was evaluated and compared in AβPPL/S animals treated with vehicle and each compound. The structure of neuritic trees from cholinergic neurons in the VDB, which contains a proportionally larger projection to the cortex and hippocampus than the medial septum [90-92] was analyzed. In AβPPL/S mice compared to vehicle-treated wt mice, cholinergic neurites labeled with ChAT appeared shorter and thinner (Fig. 7A). These degenerative changes were mitigated in AβPPL/S mice treated with LM11A-24 as well as LM11A-31 (Fig. 7A). Quantitative analysis revealed a 37% decrease in neurite length (Fig. 7B, right graph) and 64% volume loss (Fig. 7B, left graph) in vehicle-treated AβPPL/S mice relative to wt littermates, which were completely prevented by both LM11A-31 and LM11A-24. Neither ligand had significant effects on neurites of wt mice (Fig. 7B).
Fig. 7.

LM11A-31 and LM11A-24 reduce degeneration of cholinergic neurites in the basal forebrain and cortical projection target of AβPPL/S mice. Wild-type (wt) and AβPPL/S mice were treated with vehicle (veh), LM11A-31 at 50 mg/kg, or LM11A-24 at 50 or 100 mg/kg. A) Representative 10× images of choline acetyltransferase (ChAT)-immunolabeled neurites in the basal forebrain. Scale bar, 50 μm. Many neurites (arrows) of AβPPL/S-veh mice exhibited degenerative changes including interrupted segments and decreased apparent length compared to those in wt mice and these effects were markedly less detectable with LM11A-31 or −24 treatment. B) Neurites projecting from cholinergic neuron soma were traced in 3-dimensions. Relative to wt-veh mice, neurites of AβPPL/S-veh mice exhibited significant reductions in length (right graph) and volume (left graph), while these changes were absent in AβPPL/S mice treated with LM11A-31 or LM11A-24. Data represent mean ± SEM from n = 6–8 mice per group. ***p < 0.001 versus wt-veh; +++p < 0.001 versus AβPPL/S-veh. C) Representative 40× images of ChAT-immunolabeled fibers in the cingulate cortex. Scale bar, 20 μm. AβPPL/S-veh mice displayed decreased levels of continuous fibers compared to wt mice and this was not apparent in AβPPL/S mice treated with LM11A-31 or −24. D) Cholinergic fiber quantification. Relative to wt-veh mice, cholinergic fiber density was significantly decreased in AβPPL/S-veh mice, while this reduction was not present in AβPPL/S mice treated with LM11A-31 or LM11A-24. Data represent mean ± SEM from n = 8 mice per group. ***p < 0.001 versus wt-veh; +++p < 0.01 versus AβPPL/S-veh.
Previously, it was observed that 5.5–7.5-month old AβPPL/S mice exhibit a loss of cholinergic fibers in the cingulate cortex [43]. To determine whether the rescue of cholinergic neurite degeneration observed in the basal forebrain was also beneficial for cortical target innervation, the density of ChAT-positive fibers was assessed in LM11A compound-treated animals. Cholinergic fibers in the cingulate cortex of vehicle-treated AβPPL/S mice appeared diminished in density relative to wt mice, whereas no decrease was observed in mice treated with either LM11A-31 or LM11A-24 (Fig. 7C). Quantitation revealed a 72% decrease in cortical fiber density of vehicle-treated AβPPL/S mice relative to wt littermates, which was entirely reversed by either compound, with no effects on fibers in wt mice (Fig. 7D).
p75NTR ligands reduce cholinergic dystrophic neurites in AβPPL/S mice
Accumulation of Aβ in AD is associated with the appearance of dystrophic neurites [93], which likely impair neuronal and synaptic function. Dystrophic neurites are bulbous, tortuous processes that coalesce together [94], are part of a spectrum of degenerative changes that result from cytoskeletal derangement [95], manifest early in AD [96] and precede neuronal death [97]. Earlier studies found that AβPPL/S mice show prominent ChAT-labeled dystrophic neurites particularly in the vicinity of Thio-S-positive amyloid deposits in the cortex and 50 mg/kg/day of LM11A-31 decreased this dystrophy without affecting Aβ42 levels or plaque area [49]. To examine the effects of LM11A-24 on neuritic dystrophy, ChAT-positive dystrophic neurites and Thio-S accumulation were evaluated. Amyloid deposits and associated dystrophic neurites were prominent in AβPPL/S mice (Fig. 8A). In AβPPL/S mice treated with LM11A-31, dystrophic neurite area appeared diminished, while amyloid deposits were not affected (Fig. 8A). The lack of effect on amyloid plaques is consistent with our prior studies showing no effect on Aβ levels as determined by ELISA measurements [98]. In a dose-response study quantitating dystrophic neurite area in the cortex, 5 mg/kg of LM11A-31 had a modest effect that did not reach statistical significance (Fig. 8B). Doses of 25, 50, and 100 mg/kg showed highly significant and similar effect sizes, indicating that a maximal effect of an approximately 80% reduction in cholinergic dystrophy is reached by the dose range of 25–50 mg/kg (Fig. 8B). In studies of LM11A-24, 50 and 100 mg/kg doses led to a level of maximum efficacy similar to the maximum effect found with LM11A-31 (Fig. 8B) despite lower brain levels of LM11A-24.
Fig. 8.

LM11A-31 and LM11A-24 reduce cholinergic dystrophic neurites in the cortex and hippocampus of AβPPL/S mice. AβPPL/S mice were treated with vehicle (veh), LM11A-31 at 5, 25, 50, or 100 mg/kg, or LM11A-24 at 50 or 100 mg/kg. A) Representative 20× images of choline acetyltransferase (ChAT)-immunolabeled dystrophic neurites in the primary somatosensory cortex. Scale bar, 20 μm. AβPPL/S-veh mice exhibited prominent clusters of dystrophic neurites. Co-staining with Thioflavin-S (Thio-S) showed localization of most dystrophic neurite clusters with amyloid plaques. Treatment of AβPPL/S mice with LM11A-31 appeared to diminish cholinergic neurite dystrophy, while there was no apparent change in amyloid plaque size. B) Relative to AβPPL/S-veh mice, total cholinergic dystrophic neurite area was significantly reduced in the cortex with 25, 50 and 100 mg/kg of LM11A-31, as well as 50–100 mg/kg of LM11A-24. Data represent mean ± SEM from n = 8–16 mice per group. ***p < 0.001 versus AβPPL/S-veh. C) Within the hippocampus, there was also a significant reduction in the total area of dystrophic neurites in AβPPL/S mice treated with LM11A-31 or LM11A-24 (n = 4 mice per group). *p < 0.05 and **p < 0.01 versus AβPPL/S-veh. D) Quantitation of amyloid deposits in the hippocampus revealed no significant differences between AβPPL/S-veh and AβPPL/S-31 or AβPPL/S-24 mice (n = 7-8 mice per group).
Dystrophy of cholinergic neurites was also quantitatively determined in the stratum lacunosum-moleculare, molecular layer of the dentate gyrus, and stratum radiatum of the hippocampus, regions that contain projection fibers of hippocampal pyramidal neurons and granule cells, as well as septal cholinergic and entorhinal cortical efferent fibers. Similar to findings in the cortex, quantitation revealed that both p75NTR ligands significantly reduced the total dystrophic cluster area (Fig. 8C). As in the cortex, the percentage of hippocampal area stained by Thio-S was not affected by either LM11A-31 or LM11A-24 (Fig. 8D), consistent with our prior study using Aβ ELISA measurements showing a lack of effect of LM11A-31 on hippocampal Aβ42 levels [49]. These findings suggest that LM11A-24, like LM11A-31, is capable of preventing degeneration of cholinergic neurites in the basal forebrain and in cortical and hippocampal projection targets in an in vivo model of chronic Aβ-induced degeneration.
p75NTR ligands prevent behavioral deficits in AβPPL/S mice
Given p75NTR ligand amelioration of neurode-generation in AβPPL/S mice, we evaluated whether LM11A-31 and LM11A-24 would affect the behavioral manifestations seen in this AD model. AβPPL/S mice exhibit several behavioral deficits, some as early as 6 months of age, on a panel of learning and memory tests, including contextual fear conditioning, delayed-matching-to-place in a modified Barnes maze, Morris water maze, hole-board, social novelty, T-maze, Y-maze, and novel object recognition [50, 52-55, 57]. Previously, LM11A-31 was shown to decrease performance deficits in the Y-maze and novel object recognition tests in AβPPL/S mice [43].
The delayed-matching-to-place water maze test [60, 61] was used to assess memory and cognitive flexibility. Cognitive flexibility is the ability to deal with an increasingly demanding task as mice learn that the platform position is changed each day but remains the same during any given day. Learning was measured as changes in escape latency (time to find the platform) in successive trials. Figure 9A shows swim paths of mice on trials 1 and 4. Paths on trial 1 were circuitous for all mice when the platform position was unknown. Paths on trial 4 reflected the influence of both genotype and treatment. Wt mice given vehicle or LM11A-31 took short, relatively direct paths to the platform on trial 4. Vehicle-treated AβPPL/S mice performance was impaired as reflected in lengthier paths over larger areas of the pool. LM11A-31-treated AβPPL/S mice, however, performed well on trial 4 as demonstrated by paths that appeared comparable to wt mice. Figure 9B shows the quantitation of escape latencies of mice in treatment groups. Mice were given four trials per day. The escape latency in each trial was averaged over day 2–6 with day 1 considered as habituation. There was no significant difference on trial 1 latencies between groups, however, trial 4 showed 53% longer latencies of vehicle-treated AβPPL/S mice relative to wt (Fig. 9B). AβPPL/S mice treated with LM11A-31, however, had latencies indistinguishable from those of wt mice (Fig. 9B, top graph). In contrast, there was no significant difference between AβPPL/S mice treated with vehicle or LM11A-24 at either the 50 or 100 mg/kg dose (Fig. 9B, middle and bottom graphs). No genotype difference was detected in the swim velocities of mice in any treatment condition and no sensorimotor abnormalities in the water maze or visual deficits were observed (data not shown).
Fig. 9.

LM11A-31 prevents impairment in the delayed-matching-to-place water maze in AβPPL/S mice. Wild-type (wt) and AβPPL/S mice were treated with vehicle (veh), 50 mg/kg LM11A-31, or 50 or 100 mg/kg LM11A-24. A) Representative images of swim paths from trials 1 and 4 on day 3. B) Trial average comparison showed a significantly longer escape latency of vehicle-treated AβPPL/S relative to wt mice. Treatment with LM11A-31 (top graph) but not with LM11A-24 at 50 (middle graph) or 100 (bottom graph) mg/kg resulted in escape latencies of AβPPL/S mice indistinguishable from those of wt mice. Data represent mean ± SEM from n = 12–18 mice per group. **p < 0.01 versus wt-veh; ++p < 0.01 versus AβPPL/S-veh.
Following delayed-matching-to-place testing, mice were assessed for amygdala/hippocampus-dependent associative learning or contextual memory in fear conditioning [99, 100]. In fear conditioning [50], a mouse learns to associate a neutral conditioned stimulus (CS; a tone) with an aversive unconditioned stimulus (US; a foot-shock) and displays a conditioned response (CR; freezing). After repeated CS-US pairings, the mouse learns to fear both the tone and training context. The amygdala has been shown to play an important role in both the acquisition and expression of conditioned fear and the hippocampus is necessary for contextual and tone conditioning. Relative to baseline measurements, both genotypes acquired the task equally well, as demonstrated by 20% freezing on day 1 during acquisition testing (data not shown). There was no significant difference in freezing between genotypes and treatment conditions during the tone presentation in a novel context on day 2, thereby indicating intact amygdala function (Fig. 10A). However, AβPPL/S mice exhibited significantly decreased freezing behavior (by 43%) on day 3 in the original training context relative to vehicle-treated wt mice indicating a deficit in contextual memory retrieval (Fig. 10B). This learning impairment in AβPPL/S mice was rescued entirely by treatment with both LM11A-31 and LM11A-24 relative to vehicle as demonstrated by increased freezing performance, comparable to that of wt littermates (Fig. 10B). The activity levels (i.e., distance moved) of AβPPL/S mice were significantly higher (by 26%) than that of wt mice though neither ligand had significant effects on the activity of either genotype (Fig. 10C). Therefore, the degree of freezing was not confounded by drug-induced changes in overall amount of activity. Additionally, no significant difference in latency to respond to a hot surface was found between wt and AβPPL/S mice and with either LM11A-31 or LM11A-24 treatment (Fig. 10D), suggesting no disparity in responsiveness to aversive stimuli such as the foot-shock in all groups.
Fig. 10.

LM11A-31 and LM11A-24 prevent impairment in contextual fear conditioning in AβPPL/S mice. A) Graphs show performance of wild-type (wt) and AβPPL/S mice treated with vehicle (veh), LM11A-31 at 50 mg/kg, or LM11A-24 at 50 or 100 mg/kg on trace fear conditioning. All mice acquired the fear conditioning task without significant differences in freezing (data not shown). A) In tone memory testing, no freezing differences between genotypes or treatments were apparent. B) In context memory testing, AβPPL/S mice showed significant impairment (less freezing), compared to vehicle-treated wt mice, while treatment with LM11A-31 or LM11A-24 normalized (increased freezing) memory. ***p < 0.001 versus wt-veh; +p < 0.05 versus AβPPL/S-veh. C) AβPPL/S-veh mice traveled a significantly longer distance in the activity chamber than wt-veh mice, demonstrating that lower freezing was not a result of lower activity. Data represent mean ± SEM from n = 15–24 mice per group. **p < 0.01 versus wt-veh. D) Wt and AβPPL/S mice showed no difference in latency of response to the hot plate surface, suggesting that differences in pain perception did not contribute to differences in degrees of freezing (n = 9 per genotype).
DISCUSSION
The present studies demonstrated that two chemically dissimilar p75NTR ligands are capable of inhibiting excess tau phosphorylation and aberrant folding in a well characterized high-Aβ mouse model. Along with effects on tau, both compounds attenuated activation of microglia and astrocytes, reduced neurite degeneration, and prevented deficits in contextual fear conditioning. The ability of these two small molecules with distinct structures other than the sharing of an NGF loop 1 pharmacophore, indicates a compound class effect in the prevention of Aβ- and tau-related neurodegenerative mechanisms.
Key steps in novel target development include verification of target distribution and target engagement. Our findings here, using an antibody characterized for specificity, show that p75NTR is expressed by hippocampal pyramidal and cortical layer V neurons in an AD model. p75NTR has been reported to be expressed in glia particularly after CNS injury but also under normal conditions [65, 66, 101-104]. However, in this study it was not detected in either microglia or astrocytes in wt or AβPPL/S mice. This raises the possibility that the effect of LM11A-31 and/or LM11A-24 on inflammation is an indirect one, as suggested by Meeker and colleagues in in vitro studies [105]. Low levels of glial p75NTR expression, and a potential direct effect by p75NTR ligands, however, cannot be ruled out.
LM11A-31 was found to exhibit p75NTR target engagement in vivo. Unlike many growth factor receptors, p75NTR activation cannot be monitored by ligand-induced phosphorylation; therefore, we assessed an alternative proximate event consisting of ligand-induced proteolytic receptor cleavage yielding CTF and ICD fragments. The p75NTR ICD interacts with a number of intracellular proteins to modulate various pathways (including NF-κB, AKT, and JNK) associated with cell survival [69, 106]. Emerging evidence suggests that the ICD also regulates p75NTR -mediated neurite outgrowth [69]. In one recent study, a specific peptide sequence derived from the ICD was found to promote binding of NGF to TrkA, resulting in increased ERK1/2 and AKT signaling along with neurite outgrowth [107]. Thus, the finding here that LM11A-31 increased levels of hippocampal CTF and ICD in AβPPL/S mice raises the possibility that p75NTR ligands might promote neurite integrity, in part, by increasing neuritogenic signaling. Alternatively, increased p75NTR proteolytic processing could lead to increased internalization and removal of surface p75NTR , thereby reducing potentially degenerative signaling stimulated by secreted proNGF [38, 108, 109]. It is also of interest that LM11A-31 did not increase levels of p75NTR fragments in wt mice, indicating that p75NTR ligand responses might be enhanced in the neurodegenerative state, a characteristic potentially favorable to therapeutic applications. The mechanistic details underlying these observations remain to be determined. At the ages (6.5–7.5 months) examined in the present study, p75NTR levels are not found to be increased in AβPPL/S mice. It is of interest to note, however, that in subsequent studies in our laboratory assessing AβPPL/S mice at age 13–14 months, a 48 ± 11% increase in p75NTR levels has been observed (Simmons and Longo, unpublished), an observation consistent with its elevation in AD [26, 27] and other models [34, 35, 42].
LM11A-24 crosses the blood-brain barrier and was therefore selected as a structurally distinct p75NTR ligand for the present studies. Pharmacokinetic studies showed that this compound achieved brain concentrations sufficient for Aβ protective effects according to previous in vitro studies [48]; however, at both doses tested, total brain exposure was more than 10-fold less than that achieved with LM11A-31. The reduced effects of LM11A-24 are likely a result of its much lower brain exposure although a differential effect at the receptor cannot be entirely ruled out. Future studies will determine whether LM11A-31 and -24 differentially affect p75NTR processing or signaling. As previously demonstrated for LM11A-31, LM11A-24 demonstrated no other binding in a broad receptor screen.
Earlier in vitro studies found that LM11A-31 and LM11A-24 prevent Aβ-induced activation of tau-phosphorylating kinases, GSK3β, cdk5, and c-Jun, associated with reduced tau hyperphosphorylation [48]. In the current in vivo study, LM11A-31 decreased AT8 p-tau staining, while LM11A-24 reduced only non-cluster/cell body regions, a smaller but still significant effect. This reduced effect size is consistent with the lower brain levels/exposures of LM11A-24. The ability to partially reduce excess tau phosphorylation in cell body regions but not in clusters of dystrophic neurites might reflect a more advanced degenerative stage in such clusters. Tau phosphorylation at critical sites such as Ser202/Thr205 (AT8 antibody epitopes) induces a misfolded, pre-aggregation conformational state detected by the MC-1 antibody [75]. The ability of LM11A-31 to decrease MC-1 signal indicates that its effect on reducing tau phosphorylation at the AT8 site, and possibly at additional tau sites, may be significant in terms of preventing aberrant folding. Whether this beneficial effect on tau structure is sufficient to prevent the formation of tau aggregates will be a topic of future studies.
Since excess phosphorylation and/or misfolding of tau may constitute a major mechanism leading to tau loss of function or toxic gain of function, the development of strategies to inhibit tau kinases has emerged as an important therapeutic area in AD [21, 22, 110, 111]. There are relatively few examples of orally bioavailable small molecules capable of inhibiting excess tau phosphorylation and/or misfolding in AβPP/Aβ-based AD mouse models. Oral administration of the PDE5 inhibitor tadalafil in the J20 AD mouse model was found to inhibit GSK3β-induced tau phosphorylation [112]. Oral treatment of the 3xTg-AD model with the GSK3β inhibitor MMBO led to decreased tau phosphorylation in AD, as well as in normal control mice [113]. Treatment of 3xTg-AD mice by intraperitoneal (IP) administration with a dual GSK3β-CDL5/p25 inhibitor decreased tau phosphorylation and improved fear conditioning response [114]. In addition, IP administration of the flavonoid morin, a GSK3β inhibitor, in 3XTg-AD mice was also found to inhibit tau phosphorylation [115]. Thus far, the primary approaches to inhibiting excess tau phosphorylation have been comprised of the direct targeting of individual tau kinases. Challenges to this strategy include establishing whether the inhibition of just one kinase would prevent tau misfolding given the multiplicity of kinases phosphorylating tau, and potential interference with the physiological roles of the targeted kinases. A key advantage of the strategy established in the present study is that modulation of p75NTR has been found in earlier studies to inhibit Aβ induction of at least four tau kinases, GSK3, CKD5, JNK [48] and p38 (Supplementary Figure 2). Furthermore, p75NTR modulation had no effect on tau phosphorylation in wt mice, while in AβPPL/S mice returned tau phosphorylation to normal rather than below normal levels, and at the same time led to inhibition of aberrant folding. These findings point to the modulation of Aβ-relevant targets ‘upstream’ of aberrant tau kinase activation as a potential high-yield approach in AD therapeutics (see Fig. 11 for a working model of p75NTR modulation of Aβ-induced pathogenic mechanisms).
Fig. 11.

Working model for p75NTR modulation of Aβ-induced neuronal dysfunction and degeneration. Aβ might promote degeneration, in part, through interaction with p75NTR ; however, our working assumption is that Aβ promotes degeneration primarily through non-p75NTR mechanisms. Non-Aβ etiologies affecting oxidation and other degenerative processes might also be important. Previous work from our group [48] and others [1-3] demonstrates that Aβ can promote signal transduction mechanisms such as activation of calpain and stress kinases (cdk5, GSK3β, c-Jun, p38), and increased tau phosphorylation that leads to synaptic dysfunction, degeneration of neurites, and eventual neuronal death. p75NTR signaling modulates a number of these signaling pathways and our previous in vitro studies demonstrate that small molecule p75NTR ligands inhibit Aβ-induced activation of deleterious signaling and also prevent Aβ-induced inactivation of PI3K/AKT and CREB, leading to decreased tau phosphorylation and decreased neurodegeneration.
In AD mouse models, and possibly in human AD, accumulation of Aβ is likely an important inducer of the chronic inflammatory responses driven by activated microglia and astrocytes [116]. Moreover, elements of neuronal degeneration, including formation of pathological tau are also implicated in the activation of the neuroinflammatory cascade [85]. Microglial and astrocyte activation have previously been described in AβPPL/S mice [51, 56] and were also observed in the present study. Administration of LM11A-31 was associated with attenuation of both microglial and astrocytic activation, while LM11A-24 had an effect only on microglial activation. As in the case of tau effects, the narrower spectrum of LM11A-24 activity is likely related to its significantly lower brain exposure compared to LM11A-31. Since microglia and astrocytes did not appear to express p75NTR in this model, compound effects might have been indirect, via their ability to inhibit tau phosphorylation and/or other elements of neuronal degeneration. Another possibility, although less likely, is that the ligands might have induced a small decrease in Aβ levels, which we were unable to detect but could have nevertheless been sufficient to decrease glial activation. Alternatively, microglia and/or astrocytes might express p75NTR at undetectable levels that are nevertheless sufficient for direct regulation of glial activation by small molecule ligands. In terms of potential indirect mechanisms, it is of interest to note that cholinergic deficiency has been linked to microglial and astrocytic activation [117], hence the ability of p75NTR ligands to inhibit cholinergic neurite degeneration might contribute to suppression of glial activation.
Interestingly, the observed effects of LM11A-31 and LM11A-24 on tau phosphorylation, neurite degeneration, and cognitive status occurred without decreasing amyloid burden. These results are consistent with the findings of reduction of dystrophic neurites with no changes in amyloid deposition or soluble Aβ levels in AβPPL/S mice treated with LM11A-31 [49] and in p75NTR−/− mice [43], suggesting that p75NTR ligands achieve their effects by interfering with deleterious Aβ-mediated degenerative signaling (Fig. 11), as we have previously demonstrated in in vitro studies [48], rather than by the reduction of amyloid.
It is well documented that p75NTR regulates the trophic status of BFCNs and that degeneration of cholinergic neurons and/or their neurites contributes significantly to diminished cognitive function [39, 118, 119]. In AβPPL/S mice [43] and most other AD models [120], no significant loss of BFCNs occurs. In contrast, cholinergic neurite degeneration is evident in both mouse models [121] and in human cases of AD [121-123]. In a prior study, LM11A-31 reduced basal forebrain cholinergic neurite degeneration, the formation of dystrophic cholinergic neurites in the cortex, and the formation of AβPP-positive dystrophic neurites in the hippocampus [49]. In the present study, in addition to preventing degeneration of cholinergic neurites in the basal forebrain, both LM11A-31 and LM11A-24 were found to inhibit degeneration of cholinergic fibers within cortical and hippocampal target regions. The similar efficacy of LM11A-24 relative to LM11A-31, in spite of a much lower level of brain exposure, indicates that prevention of cholinergic neurite degeneration might be a particularly sensitive target for p75NTR ligand effects.
The positive effects of p75NTR ligands in the cognitive testing paradigms of the current study are likely related to their ability to prevent loss of basal forebrain cholinergic innervation of hippocampal and cortical targets, although effects involving other neuronal populations expressing p75NTR might also be involved. The ability of LM11A-24 to restore fear conditioning but not enhance water maze performance effects is likely related to its lower levels of brain exposure, although differential mechanisms between the ligands cannot be ruled out. The behavioral studies described here add two cognitive paradigms to novel object recognition and Y-maze performance [49], for which LM11A-31 has previously demonstrated efficacy.
Therapeutic approaches in the AD field have mainly focused on targeting Aβ levels. The complexity and robust nature of AD and our lack of understanding of its precise underlying mechanisms suggest that effective therapies will consist of parallel strategies such as minimizing the accumulation of toxic Aβ and tau species, reducing neuronal vulnerability to those toxic agents and other causative factors, and promoting recovery of, or compensation for, impaired neuronal function. Along these lines, the p75NTR ligands investigated here exhibit several activities relevant to AD therapy. The ability of an orally administered small molecule to inhibit pathological tau phosphorylation and folding is particularly significant. These small molecule actions provide a much needed parallel and complementary approach to the standard Aβ- and AβPP-focused efforts in AD treatment.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by funding from NIA (U01 AG032225), the Jean Perkins Foundation, and the Horngren Family Alzheimer’s Research Fund to FML. The authors thank Hui Zhang and Simret Beraki for their assistance in the execution of this work.
Footnotes
Authors’ disclosures are available online (http://www.j-alz.com/disclosures/view.php?id=2260).
SUPPLEMENTARY MATERIAL
Supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-140036.
REFERENCES
- 1.Ubhi K, Masliah E. Alzheimer’s disease: Recent advances and future perspectives. J Alzheimers Dis. 2013;33(Suppl 1):S185–S194. doi: 10.3233/JAD-2012-129028. [DOI] [PubMed] [Google Scholar]
- 2.Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9:25–34. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- 3.Weiner MW. Dementia in 2012: Further insights into Alzheimer disease pathogenesis. Nat Rev Neurol. 2013;9:65–66. doi: 10.1038/nrneurol.2012.275. [DOI] [PubMed] [Google Scholar]
- 4.Hof PR, Morrison JH. The cellular basis of cortical disconnection in Alzheimer disease and related dementing conditions. In: Terry RD, Katzman R, Bick KL, editors. In Alzheimer Disease. Raven Press; New York: 1994. pp. 197–230. [Google Scholar]
- 5.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 6.Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terry RD. The cytoskeleton in Alzheimer disease. J Neural Transm Suppl. 1998;53:141–145. doi: 10.1007/978-3-7091-6467-9_12. [DOI] [PubMed] [Google Scholar]
- 8.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurode-generation. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol Aging. 1988;9:339–349. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 11.Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunore-activity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer’s disease. Acta Neuropathol. 1991;83:12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2009;16:378–385. doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Hoi Yu AC. Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2011;8:67–80. doi: 10.2174/156720511794604543. [DOI] [PubMed] [Google Scholar]
- 14.Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia. 1989;2:420–436. doi: 10.1002/glia.440020605. [DOI] [PubMed] [Google Scholar]
- 15.von Bernhardi R. Immunotherapy in Alzheimer’s disease: Where do we stand? Where should we go? J Alzheimers Dis. 2010;19:405–421. doi: 10.3233/JAD-2010-1248. [DOI] [PubMed] [Google Scholar]
- 16.Grundman M, Dibernardo A, Raghavan N, Krams M, Yuen E. 2012: A watershed year for Alzheimer’s disease research. J Nutr Health Aging. 2012;17:51–53. doi: 10.1007/s12603-013-0002-2. [DOI] [PubMed] [Google Scholar]
- 17.Laino C. Anti-amyloid-beta drug modestly slows cognitive decline in mild to moderate AD. Neuology Today. 2012;12:34–38. [Google Scholar]
- 18.Blennow K, Zetterberg H, Rinne JO, Salloway S, Wei J, Black R, Grundman M, Liu E. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol. 2012;69:1002–1010. doi: 10.1001/archneurol.2012.90. [DOI] [PubMed] [Google Scholar]
- 19.Longo FM, Massa SM. Neuroprotective strategies in Alzheimer’s disease. NeuroRx. 2004;1:117–127. doi: 10.1602/neurorx.1.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selkoe DJ. Defining molecular targets to prevent Alzheimer disease. Arch Neurol. 2005;62:192–195. doi: 10.1001/archneur.62.2.192. [DOI] [PubMed] [Google Scholar]
- 21.Gotz J, Ittner A, Ittner LM. Tau-targeted treatment strategies in Alzheimer’s disease. Br J Pharmacol. 2012;165:1246–1259. doi: 10.1111/j.1476-5381.2011.01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Himmelstein DS, Ward SM, Lancia JK, Patterson KR, Binder LI. Tau as a therapeutic target in neurode-generative disease. Pharmacol Ther. 2012;136:8–22. doi: 10.1016/j.pharmthera.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu J, Sigurdsson EM. Immunotherapy for tauopathies. J Mol Neurosci. 2011;45:690–695. doi: 10.1007/s12031-011-9576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE. Selective vulnerability in Alzheimer’s disease: Amyloid precursor protein and p75(NTR) interaction. Ann Neurol. 2009;65:294–303. doi: 10.1002/ana.21578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mufson EJ, Kordower JH. Cortical neurons express nerve growth factor receptors in advanced age and Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89:569–573. doi: 10.1073/pnas.89.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakravarthy B, Menard M, Ito S, Gaudet C, Dal Pra I, Armato U, Whitfield J. Hippocampal membrane-associated p75NTR levels are increased in Alzheimer’s disease. J Alzheimers Dis. 2012;30:675–684. doi: 10.3233/JAD-2012-120115. [DOI] [PubMed] [Google Scholar]
- 27.Hu XY, Zhang HY, Qin S, Xu H, Swaab DF, Zhou JN. Increased p75(NTR) expression in hippocampal neurons containing hyperphosphorylated tau in Alzheimer patients. Exp Neurol. 2002;178:104–111. doi: 10.1006/exnr.2002.8018. [DOI] [PubMed] [Google Scholar]
- 28.Yamuy J, Sampogna S, Chase MH. Neurotrophin-receptor immunoreactive neurons in mesopontine regions involved in the control of behavioral states. Brain Res. 2000;866:1–14. doi: 10.1016/s0006-8993(00)02204-6. [DOI] [PubMed] [Google Scholar]
- 29.Morales I, Jimenez JM, Mancilla M, Maccioni RB. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis. 2013;37:849–856. doi: 10.3233/JAD-131843. [DOI] [PubMed] [Google Scholar]
- 30.Miller MW, Pitts FA. Neurotrophin receptors in the somatosensory cortex of the mature rat: Co-localization of p75, trk, isoforms and c-neu. Brain Res. 2000;852:355–366. doi: 10.1016/s0006-8993(99)02176-9. [DOI] [PubMed] [Google Scholar]
- 31.Zagrebelsky M, Holz A, Dechant G, Barde YA, Bonhoeffer T, Korte M. The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci. 2005;25:9989–9999. doi: 10.1523/JNEUROSCI.2492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salehi A, Ocampo M, Verhaagen J, Swaab DF. P75 neurotrophin receptor in the nucleus basalis of meynert in relation to age, sex, and Alzheimer’s disease. Exp Neurol. 2000;161:245–258. doi: 10.1006/exnr.1999.7252. [DOI] [PubMed] [Google Scholar]
- 33.Treanor JJ, Dawbarn D, Allen SJ, MacGowan SH, Wilcock GK. Low affinity nerve growth factor receptor binding in normal and Alzheimer’s disease basal forebrain. Neurosci Lett. 1991;121:73–76. doi: 10.1016/0304-3940(91)90652-a. [DOI] [PubMed] [Google Scholar]
- 34.Chakravarthy B, Gaudet C, Menard M, Atkinson T, Brown L, Laferla FM, Armato U, Whitfield J. Amyloid-beta peptides stimulate the expression of the p75(NTR) neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. J Alzheimers Dis. 2010;19:915–925. doi: 10.3233/JAD-2010-1288. [DOI] [PubMed] [Google Scholar]
- 35.Perez SE, He B, Muhammad N, Oh KJ, Fahnestock M, Ikonomovic MD, Mufson EJ. Cholinotrophic basal forebrain system alterations in 3xTg-AD transgenic mice. Neurobiol Dis. 2011;41:338–352. doi: 10.1016/j.nbd.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruno MA, Mufson EJ, Wuu J, Cuello AC. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J Neuropathol Exp Neurol. 2009;68:1309–1318. doi: 10.1097/NEN.0b013e3181c22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allard S, Leon WC, Pakavathkumar P, Bruno MA, Ribeiro-da-Silva A, Cuello AC. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J Neurosci. 2012;32:2002–2012. doi: 10.1523/JNEUROSCI.1144-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, Fritz J, Lah J, Ginsberg SD, Wuu J, Scheff SW. Hippocampal proNGF signaling pathways and beta-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:1018–1029. doi: 10.1097/NEN.0b013e318272caab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coulson EJ, May LM, Sykes AM, Hamlin AS. The role of the p75 neurotrophin receptor in cholinergic dys-function in Alzheimer’s disease. Neuroscientist. 2009;15:317–323. doi: 10.1177/1073858408331376. [DOI] [PubMed] [Google Scholar]
- 40.Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008;28:3941–3946. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geetha T, Kenchappa RS, Wooten MW, Carter BD. TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 2005;24:3859–3868. doi: 10.1038/sj.emboj.7600845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ito S, Menard M, Atkinson T, Gaudet C, Brown L, Whitfield J, Chakravarthy B. Involvement of insulin-like growth factor 1 receptor signaling in the amyloid-beta peptide oligomers-induced p75 neurotrophin receptor protein expression in mouse hippocampus. J Alzheimers Dis. 2012;31:493–506. doi: 10.3233/JAD-2012-120046. [DOI] [PubMed] [Google Scholar]
- 43.Knowles JK, Rajadas J, Nguyen TV, Yang T, LeMieux MC, Vander Griend L, Ishikawa C, Massa SM, Wyss-Coray T, Longo FM. The p75 neurotrophin receptor promotes amyloid-beta(1-42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci. 2009;29:10627–10637. doi: 10.1523/JNEUROSCI.0620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diarra A, Geetha T, Potter P, Babu JR. Signaling of the neurotrophin receptor p75 in relation to Alzheimer's disease. Biochem Biophys Res Commun. 2009;390:352–356. doi: 10.1016/j.bbrc.2009.09.116. [DOI] [PubMed] [Google Scholar]
- 45.Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- 46.Longo FM, Massa SM. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat Rev Drug Discov. 2013;12:507–525. doi: 10.1038/nrd4024. [DOI] [PubMed] [Google Scholar]
- 47.Massa SM, Xie Y, Yang T, Harrington AW, Kim ML, Yoon SO, Kraemer R, Moore LA, Hempstead BL, Longo FM. Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J Neurosci. 2006;26:5288–5300. doi: 10.1523/JNEUROSCI.3547-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang T, Knowles JK, Lu Q, Zhang H, Arancio O, Moore LA, Chang T, Wang Q, Andreasson K, Rajadas J, Fuller GG, Xie Y, Massa SM, Longo FM. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS One. 2008;3:e3604. doi: 10.1371/journal.pone.0003604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knowles JK, Simmons DA, Nguyen TV, Vander Griend L, Xie Y, Zhang H, Yang T, Pollak J, Chang T, Arancio O, Buckwalter MS, Wyss-Coray T, Massa SM, Longo FM. A small molecule p75 ligand prevents cognitive deficits and neurite degeneration in an Alzheimer's mouse model. Neurobiol Aging. 2013;34:2052–2063. doi: 10.1016/j.neurobiolaging.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faizi M, Bader PL, Saw N, Nguyen TV, Beraki S, Wyss-Coray T, Longo FM, Shamloo M. Thy1-hAPP(Lond/Swe+) mouse model of Alzheimer's disease displays broad behavioral deficits in sensorimotor, cognitive and social function. Brain Behav. 2012;2:142–154. doi: 10.1002/brb3.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huttunen HJ, Havas D, Peach C, Barren C, Duller S, Xia W, Frosch MP, Hutter-Paier B, Windisch M, Kovacs DM. The acyl-coenzyme A: Cholesterol acyltransferase inhibitor CI-1011 reverses diffuse brain amyloid pathology in aged amyloid precursor protein transgenic mice. J Neuropathol Exp Neurol. 2010;69:777–788. doi: 10.1097/NEN.0b013e3181e77ed9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rockenstein E, Adame A, Mante M, Moessler H, Windisch M, Masliah E. The neuroprotective effects of Cerebrolysin in a transgenic model of Alzheimer's disease are associated with improved behavioral performance. J Neural Transm. 2003;110:1313–1327. doi: 10.1007/s00702-003-0025-7. [DOI] [PubMed] [Google Scholar]
- 53.Havas D, Hutter-Paier B, Ubhi K, Rockenstein E, Crailsheim K, Masliah E, Windisch M. A longitudinal study of behavioral deficits in an AbetaPP transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. 2011;25:231–243. doi: 10.3233/JAD-2011-101866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloid-beta protein deposits in a mutantAPPtransgenic model depends on levels of Abeta(1-42) J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 56.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, Jagla W, Schlenzig D, Lindner C, Rudolph T, Reuter G, Cynis H, Montag D, Demuth HU, Rossner S. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 58.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 59.Franklin KBJ, Paxinos G. The mouse in stereotaxic coordinates. Elsevier Science; Amsterdam: 2008. [Google Scholar]
- 60.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 61.Steele RJ, Morris RG. Delay-dependent impairment of a matching-to-place task with chronic and intrahippocampal infusion of theNMDA-antagonist D-AP5. Hippocampus. 1999;9:118–136. doi: 10.1002/(SICI)1098-1063(1999)9:2<118::AID-HIPO4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 62.Dougherty KD, Milner TA. Cholinergic septal afferent terminals preferentially contact neuropeptide Y-containing interneurons compared to parvalbumincontaining interneurons in the rat dentate gyrus. J Neurosci. 1999;19:10140–10152. doi: 10.1523/JNEUROSCI.19-22-10140.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aronica E, Ozbas-Gerceker F, Redeker S, Ramkema M, Spliet WG, van Rijen PC, Leenstra S, Gorter JA, Troost D. Expression and cellular distribution of high- and lowaffinity neurotrophin receptors in malformations of cortical development. Acta Neuropathol. 2004;108:422–434. doi: 10.1007/s00401-004-0906-3. [DOI] [PubMed] [Google Scholar]
- 64.Ozbas-Gerceker F, Gorter JA, Redeker S, Ramkema M, van der Valk P, Baayen JC, Ozguc M, Saygi S, Soylemezoglu F, Akalin N, Troost D, Aronica E. Neurotrophin receptor immunoreactivity in the hippocampus of patients with mesial temporal lobe epilepsy. Neuropathol Appl Neurobiol. 2004;30:651–664. doi: 10.1111/j.1365-2990.2004.00582.x. [DOI] [PubMed] [Google Scholar]
- 65.Cragnolini AB, Huang Y, Gokina P, Friedman WJ. Nerve growth factor attenuates proliferation of astrocytes via the p75 neurotrophin receptor. Glia. 2009;57:1386–1392. doi: 10.1002/glia.20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee TH, Donegan E, Slichter S, Busch MP. Transient increase in circulating donor leukocytes after allogeneic transfusions in immunocompetent recipients compatible with donor cell proliferation. Blood. 1995;85:1207–1214. [PubMed] [Google Scholar]
- 67.Landman N, Kim TW. Got RIP? Presenilindependent intramembrane proteolysis in growth factor receptor signaling. Cytokine Growth Factor Rev. 2004;15:337–351. doi: 10.1016/j.cytogfr.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 68.Jung KM, Tan S, Landman N, Petrova K, Murray S, Lewis R, Kim PK, Kim DS, Ryu SH, Chao MV, Kim TW. Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J Biol Chem. 2003;278:42161–42169. doi: 10.1074/jbc.M306028200. [DOI] [PubMed] [Google Scholar]
- 69.Skeldal S, Matusica D, Nykjaer A, Coulson EJ. Proteolytic processing of the p75 neurotrophin receptor: A prerequisite for signalling? Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR) Bioessays. 2011;33:614–625. doi: 10.1002/bies.201100036. [DOI] [PubMed] [Google Scholar]
- 70.Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41:14885–14896. doi: 10.1021/bi026469j. [DOI] [PubMed] [Google Scholar]
- 71.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 72.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 73.Ren Y, Sahara N. Characteristics of tau oligomers. Front Neurol. 2013;4:102. doi: 10.3389/fneur.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33(Suppl 1):S123–S139. doi: 10.3233/JAD-2012-129031. [DOI] [PubMed] [Google Scholar]
- 75.Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- 76.Eikelenboom P, Veerhuis R, Scheper W, Rozemuller AJ, van Gool WA, Hoozemans JJ. The significance of neuroinflammation in understanding Alzheimer's disease. J Neural Transm. 2006;113:1685–1695. doi: 10.1007/s00702-006-0575-6. [DOI] [PubMed] [Google Scholar]
- 77.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 78.Coria F, Moreno A, Rubio I, Garcia MA, Morato E, Mayor F., Jr The cellular pathology associated with Alzheimer beta-amyloid deposits in non-demented aged individuals. Neuropathol Appl Neurobiol. 1993;19:261–268. doi: 10.1111/j.1365-2990.1993.tb00436.x. [DOI] [PubMed] [Google Scholar]
- 79.Pike CJ, Cummings BJ, Cotman CW. Early association of reactive astrocytes with senile plaques in Alzheimer's disease. Exp Neurol. 1995;132:172–179. doi: 10.1016/0014-4886(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 80.Sheffield LG, Marquis JG, Berman NE. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer's disease. Neurosci Lett. 2000;285:165–168. doi: 10.1016/s0304-3940(00)01037-5. [DOI] [PubMed] [Google Scholar]
- 81.Cras P, Kawai M, Siedlak S, Perry G. Microglia are associated with the extracellular neurofibrillary tangles of Alzheimer disease. Brain Res. 1991;558:312–314. doi: 10.1016/0006-8993(91)90783-r. [DOI] [PubMed] [Google Scholar]
- 82.DiPatre PL, Gelman BB. Microglial cell activation in aging and Alzheimer disease: Partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol. 1997;56:143–149. doi: 10.1097/00005072-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 83.Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: Progressive association of IL-1alpha+microglia and S100beta+astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997;56:285–290. [PubMed] [Google Scholar]
- 84.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 85.Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, Novak M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J Neuroimmunol. 2009;209:16–25. doi: 10.1016/j.jneuroim.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 86.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lance. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 87.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: Loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 88.Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci. 1997;17:7594–7605. doi: 10.1523/JNEUROSCI.17-20-07594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Greferath U, Bennie A, Kourakis A, Bartlett PF, Murphy M, Barrett GL. Enlarged cholinergic forebrain neurons and improved spatial learning in p75 knockout mice. Eur J Neurosci. 2000;12:885–893. doi: 10.1046/j.1460-9568.2000.00976.x. [DOI] [PubMed] [Google Scholar]
- 90.Lamour Y, Dutar P, Jobert A. Topographic organization of basal forebrain neurons projecting to the rat cerebral cortex. Neurosci Lett. 1982;34:117–122. doi: 10.1016/0304-3940(82)90162-8. [DOI] [PubMed] [Google Scholar]
- 91.Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: Cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 92.Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer's disease: Therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Masliah E, Alford M, Adame A, Rockenstein E, Galasko D, Salmon D, Hansen LA, Thal LJ. Abeta1-42 promotes cholinergic sprouting in patients with AD and Lewy body variant of AD. Neurology. 2003;61:206–211. doi: 10.1212/01.wnl.0000073987.79060.4b. [DOI] [PubMed] [Google Scholar]
- 94.Benzing WC, Mufson EJ, Armstrong DM. Alzheimer's disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid betaprotein deposition is present. Brain Res. 1993;606:10–18. doi: 10.1016/0006-8993(93)91563-8. [DOI] [PubMed] [Google Scholar]
- 95.Heredia L, Helguera P, de Olmos S, Kedikian G, Sola Vigo F, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Caceres A, Lorenzo A. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: A potential mechanism of neuronal dystrophy in Alzheimer's disease. J Neurosci. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer diseaseassociated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grace EA, Rabiner CA, Busciglio J. Characterization of neuronal dystrophy induced by fibrillar amyloid beta: Implications for Alzheimer's disease. Neuroscience. 2002;114:265–273. doi: 10.1016/s0306-4522(02)00241-5. [DOI] [PubMed] [Google Scholar]
- 98.Knowles JK, Simmons DA, Nguyen TV, Vander Griend L, Xie Y, Zhang H, Yang T, Pollak J, Chang T, Arancio O, Buckwalter MS, Wyss-Coray T, Massa SM, Longo FM. Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer's mouse model. Neurobiol Aging. 2013;34:2052–2063. doi: 10.1016/j.neurobiolaging.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fanselow MS. Pavlovian conditioning, negative feedback, and blocking: Mechanisms that regulate association formation. Neuron. 1998;20:625–627. doi: 10.1016/s0896-6273(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 100.Solomon PR, Levine E, Bein T, Pendlebury WW. Disruption of classical conditioning in patients with Alzheimer's disease. Neurobiol Aging. 1991;12:283–287. doi: 10.1016/0197-4580(91)90004-4. [DOI] [PubMed] [Google Scholar]
- 101.Rudge JS, Li Y, Pasnikowski EM, Mattsson K, Pan L, Yancopoulos GD, Wiegand SJ, Lindsay RM, Ip NY. Neurotrophic factor receptors and their signal transduction capabilities in rat astrocytes. Eur J Neurosci. 1994;6:693–705. doi: 10.1111/j.1460-9568.1994.tb00981.x. [DOI] [PubMed] [Google Scholar]
- 102.Hanbury R, Charles V, Chen EY, Leventhal L, Rosenstein JM, Mufson EJ, Kordower JH. Excitotoxic and metabolic damage to the rodent striatum: Role of the P75 neurotrophin receptor and glial progenitors. J Comp Neurol. 2002;444:291–305. doi: 10.1002/cne.10104. [DOI] [PubMed] [Google Scholar]
- 103.Dougherty KD, Milner TA. p75NTR immunoreactivity in the rat dentate gyrus is mostly within presynaptic profiles but is also found in some astrocytic and postsynaptic profiles. J Comp Neurol. 1999;407:77–91. doi: 10.1002/(sici)1096-9861(19990428)407:1<77::aid-cne6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 104.Oderfeld-Nowak B, Orzylowska-Sliwinska O, Soltys Z, Zaremba M, Januszewski S, Janeczko K, Mossakowski M. Concomitant up-regulation of astroglial high and low affinity nerve growth factor receptors in the CA1 hippocampal area following global transient cerebral ischemia in rat. Neuroscience. 2003;120:31–40. doi: 10.1016/s0306-4522(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 105.Meeker RB, Poulton W, Feng WH, Hudson L, Longo FM. Suppression of immunodeficiency virus-associated neural damage by the p75 neurotrophin receptor ligand, LM11A-31, in an in vitro feline model. J Neuroimmune Pharmacol. 7:388–400. doi: 10.1007/s11481-011-9325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol. 2002;67:203–233. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 107.Matusica D, Skeldal S, Sykes AM, Palstra N, Sharma A, Coulson EJ. An intracellular domain fragment of the p75 neurotrophin receptor (p75NTR) enhances TrkA receptor function. J Biol Chem. 2013;288:11144–11154. doi: 10.1074/jbc.M112.436469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Al-Shawi R, Hafner A, Chun S, Raza S, Crutcher K, Thrasivoulou C, Simons P, Cowen T. ProNGF, sortilin, and age-related neurodegeneration. Ann N Y Acad Sci. 2007;1119:208–215. doi: 10.1196/annals.1404.024. [DOI] [PubMed] [Google Scholar]
- 109.Ibanez CF. Jekyll-Hyde neurotrophins: The story of proNGF. Trends Neurosci. 2002;25:284–286. doi: 10.1016/s0166-2236(02)02169-0. [DOI] [PubMed] [Google Scholar]
- 110.Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alonso E, Fuwa H, Vale C, Suga Y, Goto T, Konno Y, Sasaki M, LaFerla FM, Vieytes MR, Gimenez-Llort L, Botana LM. Design and synthesis of skeletal analogues of gambierol: Attenuation of amyloid-beta and tau pathology with voltage-gated potassium channel and N-methyl-D-aspartate receptor implications. J Am Chem Soc. 2012;134:7467–7479. doi: 10.1021/ja300565t. [DOI] [PubMed] [Google Scholar]
- 112.Garcia-Barroso C, Ricobaraza A, Pascual-Lucas M, Unceta N, Rico AJ, Goicolea MA, Salles J, Lanciego JL, Oyarzabal J, Franco R, Cuadrado-Tejedor M, Garcia-Osta A. Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology. 2013;64:114–123. doi: 10.1016/j.neuropharm.2012.06.052. [DOI] [PubMed] [Google Scholar]
- 113.Onishi T, Iwashita H, Uno Y, Kunitomo J, Saitoh M, Kimura E, Fujita H, Uchiyama N, Kori M, Takizawa M. A novel glycogen synthase kinase -3 inhibitor 2-methyl-5-(3-4-[(S)-methylsulfinyl]phenyl-1-benzofuran-5-yl)-1,3,4-oxadiazole decreases tau phosphorylation and ameliorates cognitive deficits in a transgenic model of Alzheimer's disease. J Neurochem. 2011;119:1330–1340. doi: 10.1111/j.1471-4159.2011.07532.x. [DOI] [PubMed] [Google Scholar]
- 114.Zhang X, Hernandez I, Rei D, Mair W, Laha JK, Cornwell ME, Cuny GD, Tsai LH, Steen JA, Kosik KS. Diaminothiazoles modify Tau phosphorylation and improve the tauopathy in mouse models. J Biol Chem. 2013;288:22042–22056. doi: 10.1074/jbc.M112.436402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gong EJ, Park HR, Kim ME, Piao S, Lee E, Jo DG, Chung HY, Ha NC, Mattson MP, Lee J. Morin attenuates tau hyperphosphorylation by inhibiting GSK3beta. Neurobiol Dis. 2011;44:223–230. doi: 10.1016/j.nbd.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm. 2010;117:919–947. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- 117.Vijayaraghavan S, Karami A, Aeinehband S, Behbahani H, Grandien A, Nilsson B, Ekdahl KN, Lindblom RP, Piehl F, Darreh-Shori T. Regulated extracellular choline acetyltransferase activity-the plausible missing link of the distant action of acetylcholine in the cholinergic antiinflammatory pathway. PLoS One. 2013;8:e65936. doi: 10.1371/journal.pone.0065936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Niewiadomska G, Mietelska-Porowska A, Mazurkiewicz M. The cholinergic system, nerve growth factor and the cytoskeleton. Behav Brain Res. 2011;221:515–526. doi: 10.1016/j.bbr.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 119.Lad SP, Neet KE, Mufson EJ. Nerve growth factor: Structure, function and therapeutic implications for Alzheimer's disease. Curr Drug Targets CNS Neurol Disord. 2003;2:315–334. doi: 10.2174/1568007033482724. [DOI] [PubMed] [Google Scholar]
- 120.McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 121.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 122.Wong TP, Debeir T, Duff K, Cuello AC. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J Neurosci. 1999;19:2706–2716. doi: 10.1523/JNEUROSCI.19-07-02706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.German DC, Yazdani U, Speciale SG, Pasbakhsh P, Games D, Liang CL. Cholinergic neuropathology in a mouse model of Alzheimer's disease. J Comp Neurol. 2003;462:371–381. doi: 10.1002/cne.10737. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.