

A dual in vitro/in vivo approach is used to show that WAVE directly binds Ena/VASP, coordinating its activity with that of the Arp2/3 complex for enhanced actin assembly.
Abstract
The WAVE complex is the main activator of the Arp2/3 complex for actin filament nucleation and assembly in the lamellipodia of moving cells. Other important players in lamellipodial protrusion are Ena/VASP proteins, which enhance actin filament elongation. Here we examine the molecular coordination between the nucleating activity of the Arp2/3 complex and the elongating activity of Ena/VASP proteins for the formation of actin networks. Using an in vitro bead motility assay, we show that WAVE directly binds VASP, resulting in an increase in Arp2/3 complex–based actin assembly. We show that this interaction is important in vivo as well, for the formation of lamellipodia during the ventral enclosure event of Caenorhabditis elegans embryogenesis. Ena/VASP's ability to bind F-actin and profilin-complexed G-actin are important for its effect, whereas Ena/VASP tetramerization is not necessary. Our data are consistent with the idea that binding of Ena/VASP to WAVE potentiates Arp2/3 complex activity and lamellipodial actin assembly.
INTRODUCTION
The assembly of branched actin networks, nucleated by the Arp2/3 complex, is the driving force behind the protrusion of lamellipodia structures at the leading edge of many types of moving cells (Blanchoin et al., 2014). In lamellipodia, the Arp2/3 complex is activated by the WAVE regulatory complex (WRC) downstream of activation by Rac GTPase and acidic phospholipids, whereas the WASP family of Arp2/3 complex activators is implicated in the formation of filopodia and invadopodia downstream of activation by Cdc42 (Yamaguchi et al., 2005; Sarmiento et al., 2008; Derivery et al., 2009; Lebensohn and Kirschner, 2009; Campellone and Welch, 2010). Another important player in actin dynamics and cell migration is Ena/VASP (Krause et al., 2003). Ena/VASP proteins are correlated with increased actin assembly and lamellipodia-based motility in vivo (Grevengoed et al., 2001, 2003; Gates et al., 2007; Kwiatkowski et al., 2007; Tucker et al., 2011) and increased leading edge protrusion of cells in culture (Rottner et al., 1999; Bear et al., 2002; Lacayo et al., 2007). In keeping with this, the various members of the family (Mena, VASP, and EVL) are part of the invasive signature of human cancers, including those of breast and lung, as well as being associated with other pathologies (Dertsiz et al., 2005; Hu et al., 2008; Philippar et al., 2008; Pula and Krause, 2008). However, these proteins are not actin polymerization nucleators/activators at physiological salt concentrations but instead have anticapping and barbed-end elongation enhancement activity (Barzik et al., 2005; Breitsprecher et al., 2008, 2011; Hansen and Mullins, 2010; Winkleman et al., 2014).
It is not entirely clear how Ena/VASP exercises its effect on actin assembly. In addition to an N-terminal EVH1 domain that binds proline-rich repeats, Ena/VASP proteins possess a central polyproline domain that binds profilin and a C-terminal EVH2 domain that harbors G- and F-actin binding sites and a tetramerization domain (Krause et al., 2003). Several studies of various developmental processes in Drosophila and Caenorhabditis elegans indicated that removal of the tetramerization domain reduced but did not eliminate activity, whereas mutations in the EVH1 domain interfered with localization and gave reduced activity (Shakir et al., 2006; Gates et al., 2007, 2009; Homem and Peifer, 2009; Fleming et al., 2010). On the other hand, removal of the entire EVH2 domain was equivalent to complete lack of protein. However, the EVH2 domain has not been dissected in vivo in model organisms to evaluate the relative contributions of the F- and G-actin binding domains and the importance of the profilin-binding site to Ena/VASP activity. In cells in culture, a study of cell protrusion and Listeria motility in the presence of different VASP deletion mutants gave conflicting results. For example, the form of VASP lacking its F-actin binding site impeded cell protrusion, whereas it enhanced Listeria motility (Geese et al., 2002; Loureiro et al., 2002).
It is also not known how Ena/VASP activity is coordinated with that of the bona fide actin polymerization nucleator, the Arp2/3 complex, at the leading edge of moving cells. Speaking to this point, two Arp2/3 complex activators, ActA protein from the Listeria bacteria and human WASP, bind Ena/VASP's EVH1 domain, leading to enhanced motility (Niebuhr et al., 1997; Castellano et al., 2001; Lin et al., 2010). Regarding WAVE, several studies point to possible interactions between the WAVE complex and Ena/VASP proteins (Tani et al., 2003; Hirao et al., 2006; Dittrich et al., 2010; Maruoka et al., 2012; Okada et al., 2012). Most of these studies identify the Abi subunit of the complex as the site of interaction between Ena/VASP and the WAVE complex, including one recent work that defines the exact amino acids involved in the Abi-Ena/VASP interaction (Chen et al., 2014). However, another study shows that a proline-rich domain (PRD) from the WAVE polypeptide itself pulls down Ena/VASP from cell extracts (Okada et al., 2012). A WAVE-Ena/VASP interaction might explain how Ena/VASP is targeted to the leading edge of moving cells. Lamellipodin was previously believed to fill this role, but in a recent study, removing lamellipodin's Ena/VASP- binding sites did not affect lamellipodia formation (Law et al., 2013).
Here we investigate the idea that there is a conserved mechanism by which Arp2/3 complex activators additionally bind Ena/VASP to maximize actin assembly. We show that this is true for WAVE and test the functional significance of the Ena/VASP-WAVE polypeptide interaction. We further define what functional domains of Ena/VASP proteins are necessary for its effect on WAVE-based actin polymerization. For this study, we use a dual in vitro bead system/in vivo embryogenesis approach. In the in vitro system, cellular actin polymerization is reproduced on the surface of a bead in the form of an actin comet tail capable of propelling the bead forward, similar to the pushing out of the plasma membrane at the front of a moving cell (Wiesner et al., 2002; Plastino and Sykes, 2005). By changing what form of WAVE we absorb to the bead surface and what form of VASP we add to the motility mix, we address the functional consequences of the putative WAVE-VASP interaction and, in addition, which domains of VASP are required for its activity. In parallel, we ask the same questions in the ventral enclosure event of the developing C. elegans embryo. Enclosure involves the formation of actin-filled protrusions by the ventral epidermal cells and their migration to the ventral midline of the embryo to seal the epithelial monolayer (Williams-Masson et al., 1997). As for lamellipodium formation in mammalian cells, WAVE and VASP (WVE-1 and UNC-34, respectively, in C. elegans vocabulary) are major players in ventral enclosure, with WAVE being the essential factor: when WAVE is removed, enclosure fails due to lack of migration of the epidermal cells (Patel et al., 2008).
In both the C. elegans embryo and using the comet assay, we show evidence for a direct interaction between WAVE and VASP, observe that VASP reinforces Arp2/3 complex–based actin assembly when recruited by WAVE, and determine that the G- and F-actin and profilin-binding domains are critical for VASP function but not its tetramerization domain. We propose that WAVE brings the Arp2/3 complex and VASP together for cooperative enhancement of actin assembly.
RESULTS
Ena/VASP interacts with the proline-rich domain of WAVE to enhance actin-based motility in vitro
We first looked for a direct WAVE-VASP interaction using pure proteins, since the previous studies mentioned in the Introduction were done with cell extracts. We coated polystyrene beads with PRD-VCA-WAVE, a form of WAVE comprising both the proline-rich domain and the VCA domain, which is the part that activates the Arp2/3 complex (Figure 1a). When these beads were incubated in purified VASP (Supplemental Figure S1) and then immunostained for VASP, they showed bright staining (Figure 1b). As a positive control for VASP binding, we coated beads with the PRD-VCA construct of human WASP, previously shown to bind VASP (Castellano et al., 2001). These beads showed bright staining, comparable to PRD-VCA-WAVE beads. On the other hand, VCA-coated beads showed dim VASP staining, comparable to that observed when all three types of beads were incubated in ΔEVH1-VASP, a form of VASP lacking the capacity to bind proline-rich domains (Figure 1b). Overall this experiment showed that there was a direct interaction between the EVH1 domain of VASP and the PRD of WAVE.
FIGURE 1:
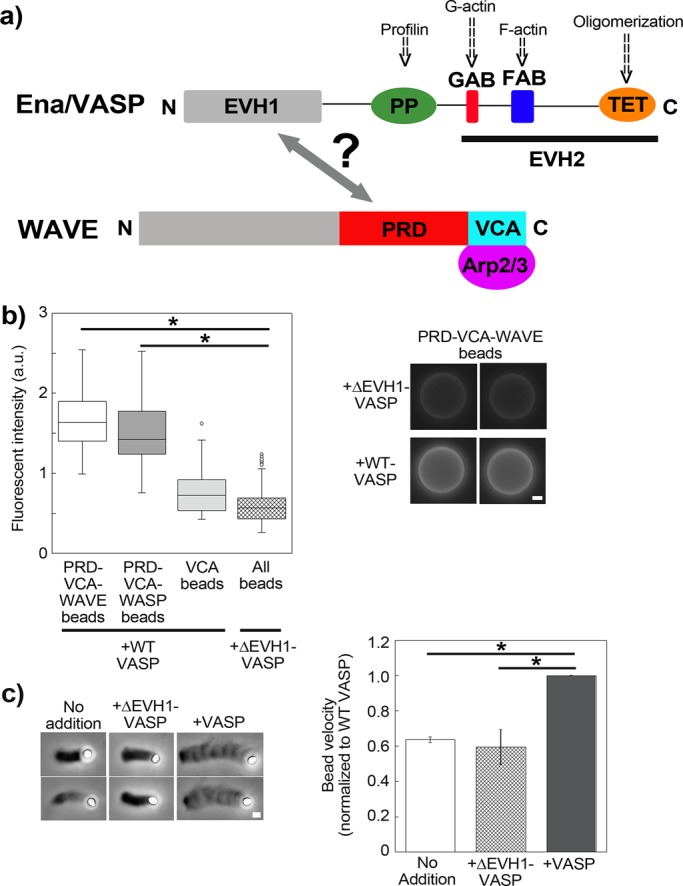
WAVE binds Ena/VASP for increased motility in vitro. (a) Scheme of general Ena/VASP and WAVE domain organization, with the putative interaction between the two marked by a double arrow. (b) Immunolabeling of beads coated with different PRD-VCA and VCA constructs incubated in either full-length VASP or ΔEVH1-VASP (lacking the putative site for interaction with WAVE). Only beads carrying the PRD domain light up and only when incubated in VASP possessing its EVH1 domain; p < 0.0001. PRD-VCA-WAVE and PRD-VCA-WASP beads in VASP are also significantly higher than VCA in VASP, p < 0.0001, not marked on the graph for clarity. Left, fluorescence intensity measurements; right, representative images. From 20 to 50 beads were analyzed per condition. Epifluorescence microscopy. (c) Comets on PRD-VCA-WAVE beads in the presence of wild-type VASP and ΔEVH1-VASP and with no addition. Actin comets appear as darker streaks behind the beads, which appear white. All pictures were taken at ∼10- to 15-min reaction time. In the graph, speeds for PRD-VCA-WAVE beads are represented normalized to wild-type VASP addition to account for day-to-day variations. No addition and addition of ΔEVH1-VASP give speeds that are 60% that of wild type, p = 0.004 and 0.003, respectively. PRD-VCA-WAVE beads moved at speeds of 0.3–1.4 μm/min, depending on the day and the additive. Phase contrast microscopy. All data are represented as averages ± SD. p values calculated with the Student's t test. Bars, 1 μm.
We next sought to determine whether and how this interaction affected WAVE-based motility. To evaluate this, we turned to the actin comet assay. Beads were coated with PRD-VCA-WAVE and incubated in a reconstituted motility mix containing the Arp2/3 complex, capping protein, and profilin/G-actin (Achard et al., 2010). This mix mimicked the high concentration of monomeric actin complexed with profilin in cellular cytosol and also minimized F-actin formation in the bulk solution, targeting actin assembly to the bead surface.
Addition of VASP to the motility mix containing PRD-VCA-WAVE–coated beads gave bead displacement that was 1.7-fold that produced in the presence of ΔEVH1-VASP or with no addition, indicating that surface recruitment of VASP by WAVE had an enhancing effect on motility (Figure 1c). In fact, adding ΔEVH1-VASP gave identical speeds to the control, no-addition case, meaning that VASP in the bulk had no effect on PRD-VCA-WAVE bead motility. As an additional negative control, we prepared VCA-WAVE–coated beads, but they did not form comets, probably due to low Arp2/3 complex activation in the profilin–actin motility mix without the PRD to recruit profilin–actin. Overall these results suggested that VASP was exercising its enhancing effect on motility via direct binding to the PRD domain of WAVE.
Assessing WAVE and Ena/VASP interaction in vivo
Our tests on beads were done with the recombinant WAVE polypeptide in isolation, not taking into account the fact that this polypeptide is part of the WRC in vivo, regulated by Rac GTPase, phospholipids, and phosphorylation. Indeed, although the native WRC has been successfully recruited to membrane-coated glass beads to form actin comets in cell extract, this approach is not adaptable to our pure-protein mix conditions (Koronakis et al., 2011). However, the PRD of WAVE is a disordered domain that is exposed on the surface of the WAVE complex, so access of VASP to this site should not be hampered in vivo (Chen et al., 2010). Given this, we turned to a cell motility event that was known to depend on the WAVE complex—ventral enclosure during C. elegans embryogenesis—and tested whether the PRD of WAVE in the WRC interacted with VASP and increased actin dynamics in vivo as we saw on beads.
To evaluate actin dynamics during ventral closure, we expressed Lifeact–green fluorescent protein (GFP) under an epidermal-specific promoter. We observed the presence of dynamic F-actin structures at the protruding edge of the epidermal cells, especially in the anteriormost “leader cells” (Figure 2, a and b, and Supplemental Video S1), as previously reported using a fluorescently tagged actin-binding domain from VAB-10 (Patel et al., 2008; Gally et al., 2009; Bernadskaya et al., 2012). Also as previously observed, in a VASP-null strain, ventral enclosure still occurred, but the lamellipodia of the leader cells were blunted and less dynamic (Figure 2a and Supplemental Video S2; Sheffield et al., 2007). Somewhat counterintuitively, we observed that the pocket area at the moment of contact of the leader cells in the VASP-null worms was half that of wild type, largely due to the fact that the VASP-null pocket was smaller along its vertical axis, as evidenced by a larger aspect ratio (Figure 2, a and b).
FIGURE 2:

VASP affects lamellipodial actin dynamics during ventral enclosure. (a) Imaging of Lifeact-GFP expressed exclusively in epidermal cells during ventral enclosure for wild-type embryos and for embryos lacking VASP. Times are indicated in minutes in relation to leader cell touch (LCT). The lamellipodia of the lower leader cells are indicated by arrows, and zooms of the boxed red area are shown on the right. In the absence of VASP, leader cell protrusions are blunted and only slightly in advance of adjacent pocket cells. z-stack projections over several micrometers. Spinning disk fluorescence microscopy; ventral view, anterior is to the left. See also Supplemental Videos S1 and S2. (b) Cartoon of the embryo and measurement of the size of the ventral pocket at the moment of leader cell touching. Pocket sizes are represented as percentages: area of pocket/total area of embryo visible by fluorescence. The pocket in the VASP null case is significantly smaller than in the wild-type case (left, p < 0.0001). This is largely due to the fact that the height (h) of the VASP-null pocket is smaller, whereas pocket widths (w) are identical for wild-type and VASP-null embryos, giving a higher pocket aspect ratio for VASP-null embryos (right, p < 0.001). Between 5 and 10 embryos/condition. (c) Migration speeds of leader cells and pocket cells during ventral enclosure. Kymographs are taken as indicated (left) to measure the speed of the first leader cell (LC) and the first pocket cell (PC). Middle, representative kymographs of wild-type and VASP-null embryos (slopes of kymograph in blue; lower cells only for clarity; for PC speeds, only the first, fast phase of enclosure was quantified). Right, LC and PC speeds from several kymographs (6–14). VASP-null leader cells move significantly more slowly than wild type (p = 0.0006). The first LCs in VASP-null embryos move essentially at the same speed as pocket cells in both wild-type and VASP-null conditions. All data are represented as averages ± SD. p values calculated with the Student's t test. Bars, (a) 15 μm, zoom 7.5 μm; (c) 15 μm; kymographs: horizontal bars, 10 min; vertical bar, 15 μm.
To understand this difference in pocket area, we quantified the speeds of leader cells as compared with pocket cells for wild-type and VASP-null embryos using kymograph analysis. The leader cells in the wild-type embryos migrated almost 1.7-fold faster than those of the VASP-null embryo, whereas the speeds of pocket cell movement were identical (Figure 2c). The difference in pocket area upon leader cell contact in the VASP-null mutant versus the wild type therefore seemed to result from the fact that leader cells and pocket cells moved with similar slow speeds in the VASP-null case, whereas in the wild-type case, leader cells were more dynamic and ran ahead of the sheet. Pocket area at the moment of leader cell touch provided a robust visual readout of the dynamics of the leader cells, and we therefore use this measurement, along with cell migration speeds, to quantify the effects of our different mutants.
Mimicking what we had done on beads, we removed the putative Ena/VASP binding site, the PRD of WAVE. This deletion form of WAVE had been studied in vitro and shown to be correctly incorporated into the mammalian and Drosophila WAVE complex (Ismail et al., 2009). We introduced ΔPRD-WAVE and wild-type WAVE as a positive control into a WAVE-null, Lifeact-GFP–positive background, and filmed ventral enclosure events. We observed that reintroduced wild-type WAVE restored leader cell speeds and pocket areas to normal levels, whereas ΔPRD-WAVE gave results that were identical to the VASP-null case shown in Figure 2, even though wild-type VASP was still present in these embryos (Figure 3, a–c, and Supplemental Videos S3 and S4). Other ligands for the PRD domain of WAVE in C. elegans are not known. In vertebrates, the PRD of WAVE2 strongly binds IRSp53, a protein implicated in enhancing WAVE activity (Miki et al., 2000). However C. elegans WAVE is a WAVE1-type protein, and vertebrate WAVE1 proteins have been shown to have a very weak interaction with IRSp53 (Miki et al., 2000; Kurisu and Takenawa, 2009).
FIGURE 3:

WAVE recruits VASP for enhanced actin-based motility in vivo. (a) Lifeact-GFP imaging (WT WAVE and ΔPRD-WAVE) or Lifeact-mCherry imaging (ΔEVH1-VASP) of ventral enclosure in embryos with reintroduced wild-type WAVE or with mutant WAVE and VASP lacking putative interaction sites. Going from left to right, images are shown just before, at the moment of, and just after leader cell touch. Right, zooms of the boxed red areas. Reintroduced wild-type WAVE looks normal (see Figure 2), but introduction of either of the mutants gives leader cell protrusions that are blunted and only slightly in advance of adjacent pocket cells, as if VASP is not present (Figure 2). z-stack projections over several micrometers. Spinning disk fluorescence microscopy; ventral view, anterior is to the left. See also Supplemental Videos S3, S4, and S6. These differences are confirmed by pocket area measurements (b) and leader cell speed measurements (c). ΔPRD-WAVE and ΔEVH1-VASP have significantly smaller pocket sizes than reintroduced wild-type (p < 0.0001 and p = 0.049, respectively) and slower leader cell motility (p = 0.0015 and 0.01, respectively), although pocket cell speeds are unchanged with respect to wild type. (d) Synthetic lethal assay with embryonic survival represented as percentage of total eggs laid. On RNAi against WASP, most VASP-null embryos do not survive. ΔEVH1-VASP and ΔPRD-WAVE have much reduced survival compared with reintroduced wild-type proteins (p < 0.0001 for both), although both mutants are about as viable as reintroduced wild-type in absence of RNAi treatment (unpublished data). All data are represented as averages ± SD. p values calculated with the Student's t test. Bar, 15 μm; zoom, 7.5 μm.
We also performed the converse experiment, removing the putative WAVE binding site, the EVH1 domain, of C. elegans VASP. This ΔEVH1-VASP construct was introduced as a GFP fusion into a VASP-null background, and a wild-type, GFP-tagged VASP transgenic was also prepared as a control. Wild-type VASP-GFP and ΔEVH1-VASP-GFP were localized at cell borders, although cytoplasmic diffuse staining was present for ΔEVH1-VASP-GFP (Supplemental Figure S2a). The bright puncta throughout the cells may have resulted from GFP labeling, since these were not apparent for native VASP observed by immunostaining (Sheffield et al., 2007). Puncta had also been observed upon GFP-Ena expression during dorsal closure in Drosophila, so this seemed to be a general observation for Ena/VASP-GFP overexpression in vivo and did not appear to disrupt cell function (Gates et al., 2007).
The GFP-tagged strains were additionally crossed with a Lifeact-mCherry strain in order to visualize leader cell dynamics and pocket morphology. The double labeling made it clear that VASP was very faint at the leading edge of leader cells, although bright at cell–cell borders, as also observed in Drosophila dorsal closure (Gates et al., 2007; Supplemental Video S5). It seemed probable that the lamellipodia were too thin and dynamic to reliably observe VASP at the leading edge of leader cells. However, observation of F-actin dynamics in the red channel for reintroduced WT-VASP-GFP and ΔEVH1-VASP-GFP embryos revealed blunted leader cells with reduced protrusion speeds and reduced pocket areas in the latter case, meaning that this form of VASP was unable to rescue leader cell dynamics (Figure 3, a–c, and Supplemental Videos S5 and S6).
Taken together these results showed that interfering with domains that ensure the WAVE-VASP interaction gave ventral enclosure events that resembled the VASP-null case. To confirm this result for a whole population of worms, we turned to a synthetic lethal assay consisting of RNA interference (RNAi) against WASP (WSP-1). WASP knockdown is known to sensitize the embryo, making the absence of VASP embryonic lethal due to ventral enclosure failure (Withee et al., 2004; Sheffield et al., 2007), even though WASP removal on its own has no effect on ventral enclosure (Supplemental Figure S3 and Supplemental Video S7). In the following, we use WASP RNAi as a tool to expose deficiencies in VASP activity. The advantage of using this assay is the ability to evaluate hundreds of embryos by a high-throughput visual assessment of embryonic survival.
We first reproduced previous results showing ∼0% survival upon RNAi against WASP in a VASP-null scenario (Figure 3d). Reintroduced wild-type VASP increased survival to 74%, <100%, perhaps due to less efficient expression from extrachromosomal arrays (Stinchcomb et al., 1985). On the other hand, reintroduced ΔEVH1-VASP rescued embryo survival to only 29%, confirming what we had observed concerning leader cell dynamics, that this mutant was much attenuated in its ability to play the role of VASP in ventral enclosure. Its residual activity (not 0% survival like VASP null) indicated that ΔEVH1-VASP was still performing some of its functions. Similarly, we subjected ΔPRD-WAVE transgenic worms to WASP RNAi. As a positive control, we did the same experiment with worms carrying reintroduced wild-type WAVE. Embryonic survival was 25% in the positive control, again perhaps due to inefficient expression from extrachromosomal arrays. However, when ΔPRD-WAVE worms were treated with RNAi against WASP, survival was a solid 0%, phenocopying a Ena/VASP-null phenotype and confirming what we had observed concerning leader cell dynamics and pocket morphology.
Overall these results taken together indicated that when Ena/VASP was present in the cells but not recruited by WAVE, it was inactive to enhance motility, which is what we had also observed with pure proteins in vitro.
Ena/VASP's binding to F-actin and profilin/G-actin are important for its function in vivo
We next wanted to define which domain(s) of VASP, in addition to its WAVE-binding site, were essential to its function of increasing WAVE-based actin dynamics. Into the VASP-null background, we introduced GFP-tagged C. elegans VASP constructs lacking individually the F-actin binding site, the G-actin binding site, the tetramerization site, and the profilin-binding region, ΔFAB-VASP, ΔGAB-VASP, ΔTET-VASP, and ΔPP-VASP, respectively (Figure 1a). We also introduced a mutant composed of just the EVH1 domain and thus lacking both the PP and EVH2 regions, called EVH1-VASP. The GAB site is ill defined in C. elegans VASP, but by sequence alignments, we identified a site that contained a Leu residue adjacent to basic amino acids, which we mutated to acidic amino acids to make our ΔGAB-VASP construct as per Walders-Harbeck et al. (2002) and Barzik et al. (2005). All constructs localized to cell borders as observed for wild type, whereas ΔTET-VASP displayed additional cytoplasmic staining, and EVH1-VASP was also present in the cytoplasm and the nucleus as previously observed in fibroblasts for EVH1-EGFP of Mena (Bear et al., 2000; Supplemental Figure S2b).
ΔPP-VASP, ΔFAB-VASP, ΔGAB-VASP, and ΔTET-VASP GFP-tagged mutant strains were crossed with a Lifeact-mCherry strain, and we observed that leader cell dynamics and the pocket area at the moment of leader cell touch were reduced for ΔPP-VASP and ΔFAB-VASP, identical to that of VASP-null embryos shown earlier, indicating that VASP required its F-actin and profilin-binding sites to exert its function in vivo (Figure 4, a and b, and Supplemental Video S8). On the other hand, ΔTET-VASP and ΔGAB-VASP embryos had dynamic leader cell lamellipodia and resembled the wild-type situation, with pocket areas similar to wild type, indicating that these domains were not essential for VASP function in vivo (Figure 4, a and b, and Supplemental Video S9). In all mutants, pocket cell speeds were identical to each other, so differences in pocket area resulted from differences in leader cell dynamics only (Supplemental Figure S4).
FIGURE 4:

VASP's F-actin and profilin/G-actin binding activities are important for its effect on WAVE-based motility. (a) Lifeact-mCherry imaging of ventral enclosure in embryos carrying GFP-tagged VASP proteins mutant for profilin binding, F- and G-actin binding, and tetramerization (ΔPP, ΔFAB, ΔGAB, and ΔTET, respectively). Left image is just before leader cell touch, and right image is at the moment of contact. The leader cell protrusion is rounded and less in advance of the adjacent pocket cells in the ΔPP and ΔFAB cases as compared with the two others. (b) This gives correspondingly smaller pocket areas for ΔPP and ΔFAB (p = 0.016), whereas ΔGAB and ΔTET are identical to reintroduced wild-type protein (unpublished data; p = 0.79 and 0.87, respectively). See also Supplemental Videos S8 and S9. (c) Embryonic survival of mutant VASP embryos subjected to the synthetic lethal RNAi treatment. ΔTET had a level of survival like wild-type (Figure 3d), whereas ΔGAB was reduced (p = 0.04 as compared with reintroduced wild type), although not as much as ΔFAB and ΔPP, which were identical to the negative control EVH1 (p = 0.97 and 0.12, respectively). (d) PRD-VCA-WAVE–coated beads incubated in the motility mix with different forms of VASP. Left, representative comets at 10- to 15-min reaction time. See Figure 1 for pictures of wild-type, ΔEVH1-VASP, and no-addition comets. Phase contrast microscopy. Right, bead speeds normalized to the wild-type speed for each day, which was on average ∼0.8 μm/min. Two to four independent experiments were averaged for each condition. Wild type and no addition are replotted from Figure 1c for comparison. ΔTET-VASP addition is the same as wild type (p = 0.3), whereas ΔFAB-VASP gives identical speeds to no addition (p = 0.12). ΔGAB-VASP and ΔPP-VASP inhibit motility. All data are represented as averages ± SD. p values calculated with the Student's t test. Bars, 15 μm (a), 1 μm (d).
However, the synthetic lethality assay of these mutants revealed a slight difference between ΔTET-VASP and ΔGAB-VASP. Indeed, when the ΔTET-VASP worms were subjected to WASP RNAi, the lethality was low, identical to wild type shown in Figure 3d, whereas ΔGAB-VASP was mid way between wild type and ΔFAB-VASP (Figure 4c). We performed the synthetic lethality assay on two additional constructs—EVH1-VASP as a negative control, lacking all VASP functional domains for interaction with actin, and ΔPP-VASP. Embryonic lethality of 50–70% was observed in worms carrying ΔPP-VASP, statistically identical to ΔFAB-VASP and to the negative control EVH1-VASP (Figure 4c). We concluded from this that the necessary domains for VASP function in vivo were the F-actin and profilin-binding domains, whereas the tetramerization domain was dispensable. In addition, it appeared that we had correctly identified the G-actin binding domain, and although its removal was not blatantly deleterious to leader cell dynamics, it did appear to play a minor role, as evidenced by the enhanced mortality observed in the RNAi assay.
Ena/VASP's binding to F-actin and profilin/G-actin is important for its function in vitro
In parallel with the ventral enclosure study of the VASP mutants, we used the bead assay to determine which VASP domains were essential for its enhancement of WAVE-based movement in vitro. We applied the different mutants to PRD-VCA-WAVE–coated beads. Addition of VASP lacking the F-actin binding site (ΔFAB-VASP) gave speeds that were 60% that of wild-type protein addition and identical to no addition (Figure 4d). The addition of monomeric VASP (ΔTET-VASP), on the other hand, gave speeds identical to wild type (Figure 4d). Addition of VASP mutants lacking the capacity to interact with G-actin and G-actin/profilin complexes (ΔGAB-VASP and ΔPP-VASP) decreased bead motility, giving split and deformed comets that propelled beads at reduced speeds as compared with no addition (Figure 4d). These results confirmed our in vivo results showing that monomeric VASP was active for motility enhancement and the importance of F-actin and profilin–actin binding. The bead assay further confirmed that the G-actin binding site was important, although it was less essential in vivo.
DISCUSSION
Taken together, our in vivo and in vitro results indicate that the proline-rich domain of WAVE in both C. elegans and human protein interacts with VASP and that this association leads to enhanced actin assembly dynamics and increased motility. When VASP is present in the cytosol/in solution but not recruited to the leading edge/bead surface because WAVE is lacking the proline-rich domain or because VASP lacks its EVH1 domain, actin dynamics resembles that of the no-VASP case. Motility enhancement is only observed when VASP is recruited by WAVE to the membrane or bead surface where Arp2/3 complex branches are being formed.
In both embryo and bead systems, monomeric VASP is just as effective in increasing motility as tetrameric (wild-type) protein, so tetramerization appears to be dispensable for lamellipodial-type actin-based protrusion. Tetramerization may be important for other situations, such as in filopodia formation, where bundling is required (Applewhite et al., 2007). On the other hand, interfering with VASP's F-actin or profilin/G-actin binding abolishes the enhancing effect on actin assembly. This result extends to actin networks in vivo and on beads what has already been observed in single filament in vitro assays: Ena/VASP protein binds filaments via its F-actin binding site and delivers monomers from the G-actin and/or profilin-actin binding site to the barbed end (Chereau and Dominguez, 2006; Ferron et al., 2007; Breitsprecher et al., 2008, 2011; Hansen and Mullins, 2010). The lesser effect observed in vivo for the G-actin binding site deletion may reflect the fact that at in vivo salt concentrations, the main polymerization entity is profilin–actin. This has been shown by in vitro measurements of single filament elongation, where it was hypothesized that the mainly electrostatic interaction of the G-actin binding site with G-actin is not favorable under physiological conditions, whereas the hydrophobic interaction of profilin–actin to proline-rich domains is favored (Hansen and Mullins, 2010).
Our results are consistent with a teamwork mechanism between two different actin polymerization machineries, the Arp2/3 complex and VASP, facilitated by mutual binding to WAVE (Figure 5). The WAVE-activated Arp2/3 complex creates a new branch on the side of an existing filament, and this branch is handed off directly to a molecule of VASP, localized at the bead or membrane surface by its association with the proline-rich domain of WAVE. This point is particularly important in light of recent results showing that Arp2/3 complex activators must dissociate from the Arp2/3 complex in order to allow the new branch to grow (Smith et al., 2013). Another candidate for barbed-end capturing at the surface is the WH2 domain of WASP/WAVE, which binds barbed ends (Co et al., 2007). However, this interaction depends on an intervening molecule of monomeric actin, and WH2 domains are not able to bind profilin–actin (Ferron et al., 2007), so the relevance of this barbed-end capture mechanism is not clear in the high-profilin conditions of in vivo polymerization. We propose therefore that WAVE-bound VASP may act as the link between the surface and the actin network at the same time that it enhances barbed-end growth via the profilin–actin loading mechanism. Together this would enhance polymerization at the surface, which not only would increase protrusion on its own, but also provide more filament primers for further Arp2/3 branching events (Figure 5; Achard et al., 2010).
FIGURE 5:

Teamwork between the Arp2/3 complex and VASP via mutual binding to WAVE. Membrane/bead-bound WAVE activates the Arp2/3 complex with its VCA domain, which then dissociates from the activated Arp2/3 complex to allow the new branch to grow, giving the scenario at the top, where the nascent branch could diffuse away from the surface. When WAVE recruits VASP in addition to binding and activating the Arp2/3 complex, a hand-off of the nascent branch could happen (bottom). VASP provides the link between the surface and the network at the same time that it enhances growth of new barbed ends. This could not only increase surface-directed polymerization on its own, but it could also contribute to providing new filament primers for subsequent rounds of Arp2/3 complex–based branching (bottom, right vignette).
In the bead system, eliminating VASP's ability to interact with either G-actin or profilin/G-actin inhibits bead motility: movement is slower than with no addition. This implies that when VASP is localized at the barbed end via its FAB domain but unable to add actin monomers via its G-actin or profilin-actin binding sites, it slows barbed-end elongation. This result is surprising because for single filaments, interfering with VASP's G-actin binding or with the VASP-profilin/G-actin interaction does not reduce polymerization below that observed for virgin filaments, although it does decrease VASP's capacity to enhance barbed-end elongation (Breitsprecher et al., 2008; Hansen and Mullins, 2010). However, in single-filament assays, VASP does not continue to localize to the barbed end when G-actin binding is abrogated (Hansen and Mullins, 2010). Our observation of motility inhibition may be a reflection of the more complex dynamics of actin network growth confined at a surface where components do not diffuse away as they do from a single filament.
Overall our in vivo and in vitro results allow us to propose a teamwork-type mechanism between the Arp2/3 complex and VASP that leads to enhanced protrusion and motility probably as a result of localized barbed-end elongation enhancement and/or anticapping activity via VASP's capacity to bind profilin, G-actin, and F-actin. Our results ride the wave of similar studies that have brought to light the collaboration of other actin machineries that were previously considered as distinct and independent—for example, the Arp2/3 complex and the formin FMNL2, and the nucleator APC and the formin mDia1 (Block et al., 2012; Breitsprecher et al., 2012). In the light of recent results concerning the direct interaction of the WAVE complex subunit Abi and Ena/VASP proteins (Chen et al., 2014), it seems probable that WAVE coordinates this molecular collaboration between the Arp2/3 complex and Ena/VASP via multiple, perhaps complementary interactions. This mechanism explains why VASP is present in dynamic WAVE-based protrusions in moving cells and gives a first characterization of how VASP activity synergizes with Arp2/3 complex nucleation.
MATERIALS AND METHODS
Worm strains and handling
Worms were maintained and handled using standard techniques (Brenner, 1974). The VASP-null strain unc-34(gm104) was isolated from PE159 strain [unc-34(gm104) hmp-1 (fe4)/mIs10 V] (a gift of Jonathon Pettitt, University of Aberdeen, Aberdeen, United Kingdom). OX308 strain carrying wve-1(ne350) I/hT2[bli-4(e937) let-?(q782) qIs48](I;III) was a gift of Martha Soto (Rutgers University, Piscataway, NJ). NG324 wsp-1(gm324) and DP38 unc119(ed3) were from the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN). The following strains were generated in the present study: JUP30 unc119(ed3); Is[Plin-26::Lifeact::GFP::unc54 3′UTR; Cb-unc119], JUP38 unc119(ed3); Is[Plin-26::Lifeact::mCherry::unc54 3′UTR; Cb-unc119], JUP22 unc34(gm104); Ex[Plin-26::unc-34(WT(full-length cDNA))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP24 unc34(gm104); Ex[Plin-26::unc-34(ΔTET(Δ415-468aa))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP26 unc34(gm104); Ex[Plin-26::unc-34(ΔFAB(Δ301-318aa))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP29 unc34(gm104); Ex[Plin-26::unc-34(ΔPP(Δ196-256aa))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP32 unc34(gm104); Ex[Plin-26::unc-34(ΔEVH1(Δ3-195aa))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP34 unc34(gm104); Ex[Plin-26::unc-34(ΔGAB(LK273MR275->LEME)))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP36 unc34(gm104); Ex[Plin-26::unc-34(EVH1(1-195aa))::GFP::unc54 3′UTR; pRF4; pCFJ90], JUP40 wve-1(ne350); Ex[wve-1; pRF4; pCFJ90], JUP44 wve-1(ne350); Ex[wve-1(ΔPRD(Δ200-390aa)); pRF4; pCFJ90]. pRF4 encodes the dominant rol-6(su1006) cotransformation marker. pCFJ90 encodes Pmyo-2::mCherry cotransformation marker. Crossing of JUP30 with unc34(gm104) and NG324 gave JUP46 and JUP47, respectively. JUP48–JUP53 were issued from crossing of JUP38 with JUP22, JUP24, JUP26, JUP29, JUP32, and JUP34, respectively. JUP54 and JUP55 were issued from crossing of JUP30 with JUP40 and JUP44, respectively.
Constructions
C. elegans expression vectors generated in this study and primers used for their construction are summarized in Supplemental Tables S1 and S2. The pAW5 plasmid, carrying nucleotide sequences for C. elegans lin-26 promoter, unc-34 cDNA (VASP), and unc-54 3′UTR, was a gift of J. Pettitt (Sheffield et al., 2007). Domain boundaries for C. elegans VASP (UNC-34) were predicted by alignment with human and mouse VASP. Constructs coding for ΔPP-VASP (lacking residues 196–256, inclusive numbering), ΔEVH1-VASP (lacking residues 3–195), ΔFAB-VASP (lacking residues 301–318), ΔTET-VASP (lacking residues 415–468) mutants of VASP, or its EVH1 domain (first 195 residues only) were prepared by Splicing by Overlapping Extension PCR (SOEing) using oligonucleotides 1–15 (see Supplemental Tables S2 and S3 for details), followed by digestion/ligation into KasI-BstZ17I fragment of pAW5. Constructs coding for ΔEVH1-VASP and ΔGAB-VASP (K273E, R275E; primers 16–19) were prepared similarly, except that SgrAI–NotI or KasI–NotI sites were used for religation, respectively.
The wve-1 rescuing fragment was prepared as described previously (Patel et al., 2008). Briefly, the wve-1 gene was amplified from genomic DNA using attB-tailed oligonucleotides 20 and 21 and recombined with pDONR201 via Gateway BP reaction (Invitrogen), giving pENTR201/wve-1. The ΔPRD mutant (lacking amino acids 201–390) was prepared by SOEing mutagenesis using primers 20–25 and religation after BglII/EcoRI double digestion into pENTR201/wve-1. As for previous studies (Ismail et al., 2009; Chen et al., 2010), a (Gly-Gly-Ser)6 linker was inserted in place of the PRD to link the N- and C-terminal parts of the molecule.
Sequence for Lifeact and linker was taken as in Riedl et al. (2008) but with C. elegans codon usage and used to amplify GFP from the vector pID3.01B (gift of Geraldine Seydoux, Johns Hopkins University, Baltimore, MD) with attB-tailed oligonucleotides 26 and 27. The product was recombined into pDONR221 and then fused with lin-26 promoter sequence (from pAW5) and the unc-54 3′UTR (gift of G. Seydoux; Addgene plasmid 17253: pCM5.37) in the destination vector pCFJ210 (gift of Erik Jorgensen; Addgene plasmid 30538) using the Multisite Gateway System (Invitrogen). pCFJ210/Plin-26::Lifeact::mCherry::unc543′UTR was prepared in the same way, except that Lifeact::mCherry was prepared by amplifying mCherry from pGH8 (gift of Erik Jorgensen; Addgene plasmid 19359) and fusing it by PCR to the Lifeact sequence of pENTR[1,2]Lifeact-GFP to avoid integrating the long Lifeact sequence on a single oligo (primers 28–33).
Human WAVE-2 cDNA was a gift of Alexis Gautreau (Laboratoire d'Enzymologie et Biochimie Structurales, Gif-sur-Yvette, France). The PRD-VCA domain of WAVE-2, Lys195– Asp498 (full-length protein numbering), was equipped with an N-terminal glutathione S-transferase tag by inserting it between the BamHI and NotI sites of pGEX-4T1 (GE Healthcare). A C-terminal Gly linker and octahistidine tag were added before the stop codon. The VCA domain was prepared in the same way and consisted of Thr424–Asp498. All mouse VASP constructs were from Dorothy Schafer (University of Virginia, Charlottesville, VA) and carried an N-terminal hexahistidine tag (Barzik et al., 2005).
Protein purification
The Arp2/3 complex was purified from bovine thymus using the method described for human leukocytes (Higgs et al., 1999). Bovine brain Arp2/3 complex purchased from Cytoskeleton was not used, as it was found to give very fast PRD-VCA-WAVE bead motility (2–3 μm/min) as compared with home-made Arp2/3 complex, and VASP addition in this situation gave motility inhibition (speeds <1 μm/min). VCA protein (from human N-WASP) and rabbit muscle actin were purchased from Cytoskeleton. The mouse α1β2 capping protein construct was a gift of D. Schafer and was purified as in Palmgren et al. (2001). Untagged human profilin was expressed in Escherichia coli strain Rosetta 2(DE3) pLysS (Novagen) and purified as in Carvalho et al. (2013). Mouse VASP protein and mutants were purified as previously described (Barzik et al., 2005). VASP proteins were further purified via fast protein liquid chromatography using a Superdex 200 10/300GL column (GE Healthcare). Mouse VASP constructs were the following: ΔEVH1-VASP, lacking residues 1–114; ΔPP-VASP, lacking residues 156–207; ΔGAB-VASP double point mutation R232E, K233E; ΔFAB-VASP, lacking residues 255–273; and ΔTET-VASP, lacking residues 331–375.
PRD-VCA-WAVE was expressed in BL21-CodonPlus(DE3)-RIPL (Stratagene) overnight at 30°C with 1 mM isopropyl-β-d-thiogalactoside (IPTG) in 2YT medium containing 50 μg/μl ampicillin and 17 μg/μl chloramphenicol. Cells were lysed in 20 mM Tris, pH 8.0, 200 mM NaCl, 1 mM ETDA, 0.1% Triton X-100, and 1 mM dithiothreitol (DTT) supplemented with complete EDTA-free protease inhibitor cocktail (Roche) then purified using glutathione Sepharose (GE Healthcare). Proteins were eluted with 20 mM Tris, pH 8.0, 200 mM NaCl, 1 mM ETDA, 1 mM DTT, and 25 mM reduced glutathione and then supplemented to 20 mM imidazole. Proteins were then bound to Ni Sepharose High Performance column (GE Healthcare) and eluted in 20 mM Tris, pH 8.0, 200 mM NaCl, 1 mM DTT, and 300 mM imidazole. Proteins were further purified over the Superdex 200 10/300GL column (GE Healthcare) in 20 mM Tris, pH 8.0, 200 mM NaCl, 0.5 mM ETDA, and 1 mM DTT. Protein was dialyzed into 20 mM Tris, pH 8.0, 200 mM NaCl, 0.5 mM ETDA, 1 mM DTT, and 5% glycerol and stored at −80°C. VCA-WAVE was purified essentially in the same way, except that Rosetta 2(DE3) pLysS (Novagen) were used and the Superdex step was omitted. The PRD-VCA-WASP protein was likewise expressed in Rosetta 2(DE3) pLysS but with an overnight expression at 20°C instead of 30°C with 1 mM IPTG. In addition, eluate from the glutathione Sepharose was supplemented to 40 mM imidazole instead of 20 mM before application to the Ni column.
C. elegans transgenesis and imaging
To create wve-1 transgenics, wve-1(ne350) I/hT2[bli-4(e937) let-?(q782) qIs48](I;III) heterozygous animals were injected with DNA coding for either wild-type or ΔPRD mutant versions of wve-1 and the injection markers pRF4 (Mello et al., 1991) and pCFJ90 (Pmyo-2::mCherry; Frokjaer-Jensen et al., 2008). Noninjected homozygous wve-1 animals show Egl (egg-laying defective) and Mel (maternal embryonic lethal) phenotypes. Homozygous wve-1 animals from established transgenic lines, identified as GFP(-) mCherry (+) rollers, were assayed for rescue of these phenotypes. Wild-type (WT) and ΔPRD mutants of wve-1 effectively restored laying of eggs (brood size 278 ± 19 for WT vs. 210 ± 26 for ΔPRD) and abated embryonic lethality of their progeny (72 and 82% eggs dead for WT vs. ΔPRD). The assay was done in triplicate, and 12 animals/strain were assayed.
Lifeact-GFP and Lifeact-mCherry animals were generated by microparticle bombardment (Bio-Rad) as described previously (Praitis et al., 2001). To create VASP transgenic animals, VASP-null hermaphrodites were injected with pAW5 (coding for WT-VASP-GFP) or derived plasmids (coding for GFP-tagged forms of ΔEVH1-VASP, ΔFAB-VASP, ΔGAB-VASP, ΔTET-VASP, ΔPP-VASP, or EVH1 domain) along with pCFJ90 (Pmyo-2::mCherry) and pRF4 injection markers. For ventral enclosure imaging, embryos were extruded from transgenic adults by cutting them in a drop of M9 solution and mounted on a 2% agarose pad. Image acquisition was performed at 22°C. Spinning disk confocal fluorescence images were acquired at a Nikon Eclipse TE2000-E microscope equipped with an oil immersion objective, 100×/1.40 numerical aperture, a piezo stage (Nanoscan Prior), a Yokogawa CSU22 confocal head, a HQ2 charge-coupled device camera (Roper Scientific), and a 491-nm diode laser controlled by MetaMorph software 7.5 (Molecular Devices). The 10- to 20-μm z-stacks were acquired with 0.5-μm distance between planes. For time-lapse imaging of Lifeact-GFP and Lifeact-mCherry during ventral enclosure, z-stacks were acquired at 60- to 90-s intervals on the spinning disk. Owing to low signal, Lifeact-mCherry single-channel images were denoised with the program Safir (Boulanger et al., 2010).
C. elegans RNAi and analysis
Standard RNAi feeding techniques were used (Kamath and Ahringer, 2003). To create wsp-1 RNAi feeding vector, a full-length wsp-1a cDNA was PCR amplified from yk184g1 cDNA clone (gift of Yuji Kohara, National Institute of Genetics, Mishima, Japan) using 5′-GGGCCATGGATGTCGGTATATCCTCCCACGCCGAC and 5′-GGGCTCGAGCTAATCTGACCATTCATTTTTGTCA oligonucleotides and cloned into XhoI–NcoI sites of L4440 plasmid. C. elegans animals were synchronized by hypochlorite treatment. Feeding was carried out at 20°C. A triplicate of Pmyo-2::mCherry(+) embryos issued from 10–20 Pmyo-2::mCherry(+) adult hermaphrodites/condition was assayed for ability to complete embryonic development. Embryos unable to hatch 24 h postlaying were scored as dead. In case of transgenic lines, only mCherry(+) progeny were taken into account. Data are the average of two experiments.
Bead preparation
Carboxylated polystyrene beads of both 1- and 4.5-μm diameter (Polysciences) were coated in Xb (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.5, 0.1 M KCl, 1 mM MgCl2, and 0.1 mM CaCl2) with 4.5 μM coating protein at 20 min in a thermomixer (Eppendorf) at 18°C, 1000 rpm. The amount of beads in 40 μl of protein solution was adjusted to a total surface area of 3 cm2. After coating, the beads were washed twice in 1% bovine serum albumin (BSA)/Xb, resuspended in 120 μl 1% BSA/Xb, and stored on ice for 1 d for bead motility assays.
Immunolabeling of beads
A 0.2-μl amount of coated beads was mixed with 4 μl of 500 nM VASP or ΔEVH1-VASP in Xb/1% BSA, and the reaction was sandwiched between two 12-mm-round coverslips separated by a Parafilm spacer. The reactions were incubated 1 h in a moist chamber at room temperature, and then the sandwiches were floated apart and simultaneously fixed by submersion in a 2% glutaraldehyde/phosphate-buffered saline (PBS) solution. Fixation was continued for 1 h at room temperature, and then the coverslips were neutralized for 10 min in 2 mg/ml NaBH4 in PBS. Coverslips were labeled with a VASP antibody that recognized the C-terminus to detect both wild-type and ΔEVH1-VASP protein (Thermo Scientific) and counterstained with a secondary antibody (goat anti-rabbit) coupled to Alexa 488 (Invitrogen).
Motility assay
The motility medium contained 95 nM Arp2/3 complex, 50 nM capping protein, 5.5 μM profilin, and 5.5 μM G-actin. Actin was diluted to 23 μM in G-buffer (2 mM Tris, 0.2 mM CaCl2, 0.2 mM DTT, pH 8.0) and allowed to depolymerize at 4°C for at least 2 d and used for several weeks. Proteins were diluted in MB13 (10 mM HEPES, 1.5 mM ATP, 3 mM DTT, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid [EGTA], 1% BSA, and 50 mM KCl, pH 7.5, with 0.1–0.2% methylcellulose [M0512, 4000 cP; Sigma-Aldrich]). We added 150 nM VASP proteins (calculated using the tetramer molecular weight, even for the ΔTET mutant) or the equivalent in VASP buffer (20 mM imidazole, 200 mM KCl, 1 mM EGTA, 2 mM MgCl2, and 1 mM DTT, pH 7.0). The final KCl concentration was brought up to 86 mM by addition of KCl in MB13. Owing to dilution by VASP buffer and G-actin solution, final reaction conditions were ∼1 mM ATP, 2 mM DTT, 0.7 mM EGTA, 0.6% BSA, and 0.6–1.2% methylcellulose. For a final reaction volume of 8.4 μl, 0.2 μl of coated beads was added, and the entire volume was placed between a glass slide and coverslip (18 × 18 mm) and sealed with Vaseline/lanolin/paraffin (1:1:1).
Bead observation and data processing
Phase contrast (for motility assay) and epifluorescence (for immunolabeling) microscopy were performed on an Olympus BX51 upright microscope or an Olympus IX70 inverted microscope with a 100× oil-immersion objective and CoolSnap charge-coupled device camera (Photometrics). Phase contrast and fluorescence quantification was done using MetaMorph software (Universal Imaging). Bead velocities were calculated by measuring lengths of the whole population of comets (pictures taken at random over the entire sample) over time. The slope of comet length versus time gave the average velocity of the entire population. This approach meant that at least 50 comets went into each measurement. The measurement was repeated on different days, and reported speeds are the average 2–4 different days, representing the measurement of hundreds of comets.
Supplementary Material
Acknowledgments
We thank Jean-Louis Bessereau for use of his worm injection setup for initial experiments and Vincent Galy and Marie-Anne Felix for use of their microparticle bombardment equipment. For microscope use and support, we acknowledge the imaging facility PICT-IBiSA, Institut Curie, Paris, France, a member of the France BioImaging national research infrastructure. We thank Dorothy Schafer and Kevin Carvalho for discussions and Cécile Sykes and Renaud Legouis for discussions and reading of the manuscript. This work was funded by a Fondation Pierre-Gilles de Gennes grant to S.H., a Human Frontiers Science Program Organization Young Investigator's Grant to J.P., and the Fondation pour la Recherche Médicale (Grant DEQ20120323737) to J.P., Cécile Sykes, and Timo Betz. Some strains were provided by the CGC, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Abbreviations used:
- FAB
F-actin binding domain
- GAB
G-actin binding domain
- GFP
green fluorescent protein
- LC
leader cell
- LCT
leader cell touch
- PC
pocket cell
- PP
profilin binding domain
- PRD
proline-rich domain
- RNAi
RNA interference
- TET
tetramerization domain
- WRC
WAVE regulatory complex.
Footnotes
*These authors contributed equally to this work and are listed in alphabetical order.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1200) on October 29, 2014.
REFERENCES
- Achard V, Martiel J-L, Michelot A, Guérin C, Reymann A-C, Blanchoin L, Boujemaa-Paterski R. A “primer”-based mechanism underlies branched actin filament network formation and motility. Curr Biol. 2010;20:423–428. doi: 10.1016/j.cub.2009.12.056. [DOI] [PubMed] [Google Scholar]
- Applewhite DA, Barzik M, Kojima S-i, Svitkina TM, Gertler FB, Borisy GG. Ena/VASP proteins have an anti-capping independent function in filopodia formation. Mol Biol Cell. 2007;18:2579–2591. doi: 10.1091/mbc.E06-11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280:28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fässler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Bernadskaya YY, Wallace A, Nguyen J, Mohler WA, Soto MC. UNC-40/DCC, SAX-3/Robo, and VAB-1/Eph polarize F-actin during embryonic morphogenesis by regulating the WAVE/SCAR actin nucleation complex. PLoS Genet. 2012;8:e1002863. doi: 10.1371/journal.pgen.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture and mechanics in cell motility. Physiol Rev. 2014;94:235–263. doi: 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- Block J, Breitsprecher D, Kühn S, Winterhoff M, Kage F, Geffers R, Duwe P, Rohn JL, Baum B, Brakebusch C, et al. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr Biol. 2012;22:1005–1012. doi: 10.1016/j.cub.2012.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger J, Kervrann C, Bouthemy P, Elbau P, Sibarita JB, Salamero J. Patch-based nonlocal functional for denoising fluorescence microscopy image sequences. IEEE Trans Med Imaging. 2010;29:442–454. doi: 10.1109/TMI.2009.2033991. [DOI] [PubMed] [Google Scholar]
- Breitsprecher D, Jaiswal R, Bombardier JP, Gould CJ, Gelles J, Goode BL. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 2012;336:1164–1168. doi: 10.1126/science.1218062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Kiesewetter AK, Linkner J, Urbanke C, Resch GP, Small JV, Faix J. Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J. 2008;27:2943–2954. doi: 10.1038/emboj.2008.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Kiesewetter AK, Linkner J, Vinzenz M, Stradal T, Small JV, Curth U, Dickinson RB, Faix J. Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J. 2011;30:456–467. doi: 10.1038/emboj.2010.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho K, Lemière J, Faqir F, Manzi J, Blanchoin L, Plastino J, Betz T, Sykes C. Actin polymerization or myosin contraction: two ways to build up cortical tension for symmetry breaking. Philos Trans R Soc. 2013;B 368:20130005. doi: 10.1098/rstb.2013.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano F, Le Clainche C, Patin D, Carlier M-F, Chavrier P. A WASP-VASP complex regulates actin polymerization at the plasma membrane. EMBO J. 2001;20:5603–5614. doi: 10.1093/emboj/20.20.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Squarr AJ, Stephan R, Chen B, Higgins TE, Barry DJ, Martin MC, Rosen MK, Bogdan S, Way M. Ena/VASP proteins cooperate with the WAVE complex to regulate the actin cytoskeleton. Dev Cell. 2014;30:569–584. doi: 10.1016/j.devcel.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereau D, Dominguez R. Understanding the role of the G-actin-binding domain of Ena/VASP in actin assembly. J Struct Biol. 2006;155:195. doi: 10.1016/j.jsb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Co C, Wong DT, Gierke S, Change V, Taunton J. Mechanism of actin network attachment to moving membranes: barbed end capture by N-WASP WH2 domains. Cell. 2007;128:901–913. doi: 10.1016/j.cell.2006.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E, Lombard B, Loew D, Gautreau A. The Wave complex is intrinsically inactive. Cell Motil Cytoskel. 2009;66:777–790. doi: 10.1002/cm.20342. [DOI] [PubMed] [Google Scholar]
- Dertsiz L, Ozbilim G, Kayisli Y, Gokhan GA, Demircan A, Kayisli UA. Differential expression of VASP in normal lung tissue and lung adenocarcinomas. Thorax. 2005;60:576–581. doi: 10.1136/thx.2004.037622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich M, Strassberger V, Fackler M, Tas P, Lewandrowski U, Sickmann A, Walter U, Dandekar T, Birschmann I. Characterization of a novel interaction between vasodilator-stimulated phosphoprotein and Abelson Interactor 1 in human platelets: a concerted computational and experimental approach. Arterioscler Thromb Vasc Biol. 2010;30:843–850. doi: 10.1161/ATVBAHA.109.200683. [DOI] [PubMed] [Google Scholar]
- Ferron F, Rebowski G, Lee SH, Dominguez R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007;26:4597–4606. doi: 10.1038/sj.emboj.7601874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming T, Chien S-C, Vanderzalm PJ, Dell M, Gavin MK, Forrester WC, Garriga G. The role of C. elegans Ena/VASP homolog UNC-34 in neuronal polarity and motility. Dev Biol. 2010;344:94–106. doi: 10.1016/j.ydbio.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gally C, Wissler F, Zahreddine H, Quintin S, Landmann F, Labouesse M. Myosin II regulation during C. elegans embryonic elongation: LET-502/ROCK, MRCK-1 and PAK-1, three kinases with different roles. Development. 2009;136:3109–3119. doi: 10.1242/dev.039412. [DOI] [PubMed] [Google Scholar]
- Gates J, Mahaffey JP, Rogers SL, Emerson M, Rogers EM, Sottile SL, Van Vactor D, Gertler FB, Peifer M. Enabled plays key roles in embryonic epithelial morphogenesis in Drosophila. Development. 2007;134:2027–2039. doi: 10.1242/dev.02849. [DOI] [PubMed] [Google Scholar]
- Gates J, Nowotarski SH, Yin H, Mahaffey JP, Bridges T, Herrera C, Homem CCF, Janody F, Montell DJ, Peifer M. Enabled and Capping protein play important roles in shaping cell behavior during Drosophila oogenesis. Dev Biol. 2009;333:90–107. doi: 10.1016/j.ydbio.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geese M, Loureiro JJ, Bear JE, Wehland J, Gertler FB, Sechi AS. Contribution of Ena/VASP proteins to intracellular motility of Listeria requires phosphorylation and proline-rich core but not F-actin binding or multimerization. Mol Biol Cell. 2002;13:2383–2396. doi: 10.1091/mbc.E02-01-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevengoed EE, Fox DT, Gates J, Peifer M. Balancing different types of actin polymerization at distinct sites: roles for Abelson kinase and Enabled. J Cell Biol. 2003;163:1267–1279. doi: 10.1083/jcb.200307026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevengoed EE, Loureiro JJ, Jesse TL, Peifer M. Abelson kinase regulates epithelial morphogenesis in Drosophila. J Cell Biol. 2001;155:1185–1197. doi: 10.1083/jcb.200105102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SD, Mullins RD. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol. 2010;191:571–584. doi: 10.1083/jcb.201003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs HN, Blanchoin L, Pollard TD. Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry. 1999;38:15212–15222. doi: 10.1021/bi991843+. [DOI] [PubMed] [Google Scholar]
- Hirao N, Sato S, Gotoh T, Maruoka M, Suzuki J, Matsuda S, Shishido T, Tani K. NESH (Abi-3) is present in the Abi/WAVE complex but does not promote c-Abl-mediated phosphorylation. FEBS Lett. 2006;580:6464–6470. doi: 10.1016/j.febslet.2006.10.065. [DOI] [PubMed] [Google Scholar]
- Homem CCF, Peifer M. Exploring the roles of Diaphanous and Enabled activity in shaping the balance between filopodia and lamellipodia. Mol Biol Cell. 2009;20:5138–5155. doi: 10.1091/mbc.E09-02-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L-D, Zou H-F, Zhan S-X, Cao K-M. EVL (Ena/VASP-like) expression is up-regulated in human breast cancer and its relative expression level is correlated with clinical stages. Oncol Rep. 2008;19:1015–1020. [PubMed] [Google Scholar]
- Ismail AM, Padrick SB, Chen B, Umetani J, Rosen MK. The WAVE regulatory complex is inhibited. Nat Struct Mol Biol. 2009;16:561–563. doi: 10.1038/nsmb.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Koronakis V, Hume PJ, Humphreys D, Liu T, Horning O, Jensen ON, McGhie EJ. WAVE regulatory complex activation by cooperating GTPases Arf and Rac1. Proc Natl Acad Sci USA. 2011;108:14449–14454. doi: 10.1073/pnas.1107666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Kurisu S, Takenawa T. The WASP and WAVE family proteins. Genome Biol. 2009;10:226. doi: 10.1186/gb-2009-10-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, van Veen JE, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, et al. Ena/VASP is required for neuritogenesis in the developing cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Lacayo CI, Pincus Z, VanDuijn MM, Wilson CA, Fletcher DA, Gertler FB, Mogilner A, Theriot JA. Emergence of large-scale cell morphology and movement from local actin filament growth dynamics. PLoS Biol. 2007;5:2035–2052. doi: 10.1371/journal.pbio.0050233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law A-L, Vehlow A, Kotini M, Dodgson L, Soong D, Theveneau E, Bodo C, Taylor E, Navarro C, Perera U, et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J Cell Biol. 2013;203:673–689. doi: 10.1083/jcb.201304051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W-H, Nelson SE, Hollingsworth RJ, Chung CY. Functional roles of VASP phosphorylation in the regulation of chemotaxis and osmotic stress response. Cytoskeleton. 2010;67:259–271. doi: 10.1002/cm.20443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro JJ, Rubinson DA, Bear JE, Baltus GA, Kwiatkowski AV, Gertler FB. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/Vasodilator-stimulated Phosphoprotein (VASP) function during cell migration. Mol Biol Cell. 2002;13:2533–2546. doi: 10.1091/mbc.E01-10-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruoka M, Sato M, Yuan Y, Ichiba M, Fuji R, Ogawa T, Ishida-Kitagawa N, Takeya T, Watanabe N. Abi-1-bridged tyrosine phosphorylation of VASP by Abelson kinase impairs association of VASP to focal adhesions and regulates leukaemic cell adhesion. Biochem J. 2012;441:889–899. doi: 10.1042/BJ20110951. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000;408:732–735. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- Niebuhr K, Ebel F, Frank R, Reinhard M, Domann E, Carl UD, Walter U, Gertler FB, Wehland J, Chakraborty T. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J. 1997;16:5433–5444. doi: 10.1093/emboj/16.17.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Uezu A, Soderblom EJ, Moseley III MA, Gertler FB, Soderling SH. Peptide Array X-linking (PAX): a new peptide-protein identification approach. PLoS One. 2012;7:e37035. doi: 10.1371/journal.pone.0037035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren S, Ojala PJ, Wear MA, Cooper JA, Lappalainen P. Interactions with PIP2, ADP-actin monomers, and capping protein regulate the activity and localization of yeast twinfilin. J Cell Biol. 2001;155:251–260. doi: 10.1083/jcb.200106157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel FB, Bernadskaya YY, Chen E, Jobanputra A, Pooladi Z, Freeman KL, Gally C, Mohler WA, Soto MC. The WAVE/SCAR complex promotes polarized cell movements and actin enrichment in epithelia during C. elegans embryogenesis. Dev Biol. 2008;324:297–309. doi: 10.1016/j.ydbio.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippar U, Roussos ET, Oser M, Yamaguchi H, Kim H-D, Giampieri S, Wang Y, Goswami S, Wyckoff JB, Lauffenburger DA, et al. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev Cell. 2008;15:813–828. doi: 10.1016/j.devcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plastino J, Sykes C. The actin slingshot. Curr Opin Cell Biol. 2005;17:62–66. doi: 10.1016/j.ceb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pula G, Krause M. Role of Ena/VASP proteins in homeostasis and disease. Handb Exp Pharmacol. 2008;186:39–65. doi: 10.1007/978-3-540-72843-6_3. [DOI] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner K, Behrendt B, Small JV, Wehland J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1999;1:321–322. doi: 10.1038/13040. [DOI] [PubMed] [Google Scholar]
- Sarmiento C, Wang W, Dovas A, Yamaguchi H, Sidani M, El-Sibai M, DesMarais V, Holman HA, Kitchen S, Backer JM, et al. WASP family members and formin proteins coordinate regulation of cell protrusions in carcinoma cells. J Cell Biol. 2008;180:1245–1260. doi: 10.1083/jcb.200708123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakir MA, Gill JS, Lundquist EA. Interactions of UNC-34 Enabled with Rac GTPases and the NIK Kinase MIG-15 in Caenorhabditis elegans axon pathfinding and neuronal migration. Genetics. 2006;172:893–913. doi: 10.1534/genetics.105.046359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield M, Loveless T, Hardin J, Pettitt J. C. elegans enabled exhibits novel interactions with N-WASP, Abl and cell-cell junctions. Curr Biol. 2007;17:1791–1796. doi: 10.1016/j.cub.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BA, Padrick SB, Doolittle LK, Daugherty-Clarke K, Correa IR, Xu M-Q, Goode BL, Rosen MK, Gelles J. Three-color single molecule imaging shows WASP detachment from Arp2/3 complex triggers actin filament branch formation. eLife. 2013;2:e01008. doi: 10.7554/eLife.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcomb DT, Shaw JE, Carr SH, Hirsh D. Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol Cell Biol. 1985;5:3484–3496. doi: 10.1128/mcb.5.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani K, Sato S, Sukezane T, Kojima H, Hirose H, Hanafusa H, Shishido T. Abl Interactor 1 promotes tyrosine 296 phosphorylation of mammalian Enabled (Mena) by c-Abl kinase. J Biol Chem. 2003;278:21685–21692. doi: 10.1074/jbc.M301447200. [DOI] [PubMed] [Google Scholar]
- Tucker PK, Evans IR, Wood W. Ena drives invasive macrophage migration in Drosophila embryos. Dis Model Mech. 2011;4:126–134. doi: 10.1242/dmm.005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walders-Harbeck B, Khaitlina SY, Hinssen H, Jockusch BM, Illenberger S. The vasodilator-stimulated phosphoprotein promotes actin polymerisation through direct binding to monomeric actin. FEBS Lett. 2002;529:275–280. doi: 10.1016/s0014-5793(02)03356-2. [DOI] [PubMed] [Google Scholar]
- Wiesner S, Boujemaa-Paterski R, Carlier M-F. Actin-based motility of Listeria monocytogenes and Shigella flexneri. Methods Microbiol. 2002;31:245–262. [Google Scholar]
- Williams-Masson EM, Malik AN, Hardin J. An actin-mediated two-step mechanism is required for ventral enclosure of the C. elegans hypodermis. Development. 1997;124:2889–2901. doi: 10.1242/dev.124.15.2889. [DOI] [PubMed] [Google Scholar]
- Winkleman JD, Bilancia CG, Peifer M, Kovar DR. Ena/VASP Enabled is a highly processive actin polymerase tailored to self-assemble parallel-bundled F-actin networks with Fascin. Proc Natl Acad Sci USA. 2014;111:4121–4126. doi: 10.1073/pnas.1322093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withee J, Galligan B, Hawkins N, Garriga G. Caenorhabditis elegans WASP and Ena/VASP proteins play compensatory roles in morphogenesis and neuronal cell migration. Genetics. 2004;167:1165–1176. doi: 10.1534/genetics.103.025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP–Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.