

Abstract
A disintegrin and metalloproteinase domain 10 (Adam10), a member of the ADAM family of cell membrane–anchored proteins, has been linked to the regulation of the Notch, EGF, E-cadherin, and other signaling pathways. However, it is unclear what role Adam10 has in the kidney in vivo. In this study, we showed that Adam10 deficiency in ureteric bud (UB) derivatives leads to a decrease in urinary concentrating ability, polyuria, and hydronephrosis in mice. Furthermore, Adam10 deficiency led to a reduction in the percentage of aquaporin 2 (Aqp2)+ principal cells (PCs) in the collecting ducts that was accompanied by a proportional increase in the percentage of intercalated cells (ICs). This increase was more prominent in type A ICs than in type B ICs. Foxi1, a transcription factor important for the differentiation of ICs, was upregulated in the Adam10 mutants. The observed reduction of Notch activity in Adam10 mutant collecting duct epithelium and the similar reduction of PC/IC ratios in the collecting ducts in mice deficient for mindbomb E3 ubiquitin protein ligase 1, a key regulator of the Notch and Wnt/receptor-like tyrosine kinase signaling pathways, suggest that Adam10 regulates cell fate determination through the activation of Notch signaling, probably through the regulation of Foxi1 expression. However, phenotypic differences between the Adam10 mutants, the Mib1 mutants, and the Foxi1 mutants suggest that the functions of Adam10 in determining the fate of collecting duct cells are more complex than those of a simple upstream factor in a linear pathway involving Notch and Foxi1.
Keywords: diabetes insipidus, genetics and development, renal tubular epithelial cells
During nephrogenesis, the ureteric bud (UB) invades the metanephric mesenchyme and undergoes dichotomous branching. The distal branches of the UB fuse with the metanephric mesenchyme–derived portions of the nephrons and become collecting duct epithelia.1 When renal collecting ducts mature, the uniform UB epithelial cells give rise to two distinct cell types: principal cells (PCs) and intercalated cells (ICs). The PCs express several water channel proteins, including aquaporin (Aqp)-2 on the apical membrane, Aqp3/4 on the basolateral membrane, and many ion channels and transporters. The PCs are primarily responsible for water and sodium reabsorption. The ICs are primarily responsible for acid-base homeostasis through proton and bicarbonate secretion.2 The ICs have two canonical types, A and B, although other ICs with dual characteristics or transitional phenotypes have been reported.3 Type A ICs have an apically located H+-ATPase (ATPase) and an anion exchanger, AE1 (Slc4a1), on their basolateral membrane. Such arrangement allows type A ICs to secrete protons into the collecting duct lumen and to have HCO3 transported across the basolateral membrane. Type B ICs have an apical Cl:HCO3 exchanger, Pendrin (Slc26a4), and a basolaterally located H+-ATPase, facilitating the secretion of HCO3 into the urine from these cells.
The relative number and activity of type A and type B ICs can influence the acid-base balance and are regulated through several mechanisms that may include the switching of IC subtypes.2,3 Deletion of Hensin/DMBT1 blocks the conversion of type B to type A ICs and induces distal renal tubular acidosis.4 The apparent ability of ICs to switch types highlights the commonalities between these cells despite their opposite functions. Both types of ICs express carbonic anhydrase II (CAII), an enzyme important for HCO3 and CO2 handling. Deficiency of CAII leads to the depletion of both types of ICs and the proportional increase of Aqp2+ PCs.5 The ratio of PCs and ICs can be altered by genetic mutations and chemical treatment, such as lithium.6,7 Although it is still unclear how lithium treatment induces the increase of ICs, the study of Mib1, a regulator of Notch signaling, suggested that Notch signaling confers PC fate and the absence of Notch signaling results in IC fate.7 Similar Notch role in cell fate choice, between multiciliated versus transporter cell, was also observed in zebrafish pronephros.8 The forkhead transcription factor Foxi1 mediates the differentiation of ICs from epithelial precursors in the UB and acts upstream of IC-specific proteins, such as AE1 and Pendrin.9
The ADAMs (a disintegrin and metalloproteinase) belong to the zinc protease superfamily and are best known for their sheddase activities.10 A member of the ADAMs family, Adam10, is best known for its ability to mediate the S2 cleavage of Notch ligands.11–14 In addition to Notch, Adam10 is capable of proteolytic cleavage of a wide range of substrates, including collagen IV, epidermal growth factor, Ephrin, and chemokines.15 High level of Adam10 expression was observed in the developing UB,16 although its functional role in UB and UB derivatives has not been directly studied. Here, we generated mice with UB-specific deletion of Adam10 and found that Adam10 helps regulate cell fates in the renal collecting ducts.
Results
Deletion of Adam10 in UB Derivatives Led to Polyuria and Hydronephrosis
To investigate the role of Adam10 in the collecting duct, we used a Hoxb7-Cre transgene to drive Cre expression and subsequent deletion of Adam10 from a floxed-Adam10 (Adam10flox) allele specifically in the UB derivatives, including the ureteral and collecting duct epithelium (Figure 1, A–E). Adam10 is present in the collecting duct epithelial cells in the controls with no Adam10 deletion but is absent in these cells in mice with homozygous UB-specific deletion of Adam10 (mutants) (Figure 1, F–I). Deletion of Adam10 can also be detected by PCR in the mutant kidneys (Figure 1J). Hydronephrosis was observed postnatally in about 30% of mice with homozygous deletion of Adam10 in the UB (Figure 1, K–N). Mice with heterozygous deletion showed no hydronephrosis or any other phenotypes described below. Thus, “mutants” in this report refer only to mice with homozygous deletion of Adam10. Severe loss of renal parenchyma, especially in the medullary region, was observed in many mutants with hydronephrosis. We measured water intake and urine output in adult mice and found that the mutants have polydipsia and polyuria. The mutants drank more water and produced more urine than their littermate controls (Figure 1, O and P), resembling diabetes insipidus in humans. Accordingly, urine osmolality was decreased in these mutant mice compared with their littermate controls (Figure 1Q). Kidneys of newborn mutants showed no hydronephrosis or any physical obstruction of the urinary path (data not shown). We thus suspect that functional defects in handling urine transport or polyuria may be the primary cause of hydronephrosis, although other causes cannot be excluded at this point.17–19
Figure 1.

Deletion of Adam10 in UB derivatives cause hydronephrosis and polyuria. Whole-mount β-galactosidase assay showed the expression of the Hoxb7-Cre transgene in the UB derivatives in mice that are Hoxb7-Cre; ROSALacZ/+ (A–C). Cre expression is detected in almost all collecting duct epithelial cells (D). We crossed the Hoxb7-Cre mice with mice carrying an Adam10flox allele to induce Adam10 deletion in UB derivatives, including the collecting duct system (E). Sections from control (F and G) and mutant (H and I) newborn kidneys were immunostained for Aqp2 and Adam10. (F and H) Signals from both antibodies are shown to highlight the collecting ducts that contain Aqp2+ cells. (G and I) Only the Adam10 staining is shown for better appreciation of the changes in the mutants. For more accurate tracing of the collecting ducts, segments of the collecting ducts lined by easily identifiable Aqp2+ cells are shown. The dashed lines in G and I follow the contour of the collecting ducts. In the controls, Adam10 is located primarily on the membrane in the collecting duct cells (arrows in G) and some other nephron segments (asterisks). Although the signal in other nephron segments did not change in the mutants (asterisks), the membrane signal in the collecting ducts was largely gone in the mutant collecting ducts. (J) Adam10 deletion in the mutant kidneys can be detected by PCR. Lane 1 is kidney DNA from a control (Adam10flox/+, no Cre). Lanes 2 and 3 are from mutant kidneys. The PCR primers used are different from the genotyping primers and are designed to reveal the deletion mediated by Cre-loxP. The upper bands represent the floxed-allele and the wild-type allele (very close in size). The lower bands (asterisk) represent the deletion allele created after Cre-mediated loxP recombination. The deletion band is present only in the mutant samples, not in the controls. The upper bands in the mutant samples are less robust because of amplification competition between upper and lower bands. About a third of the mice with Adam10 deletion in UB derivatives developed hydronephrosis at the time of examination (K–N). (M and N) Hematoxylin and eosin–stained sections from kidneys of control and mutant mice after weaning. Mutant mice have higher water intake (3.85±2.41 ml/d versus 1.51±0.86 ml/d) (O) and urine output (1.29±0.87 ml/d versus 0.60±0.39 ml/d) (P). (Q) Urine osmolality decreased in the mutants (1317±465 mmol/kg) compared with controls (2800±596 mmol/kg). n=16 for each group. *P<0.05; **P<0.01. CT, controls; MT, mutants. Original magnification, ×3 (stereoscope) in A and B; ×1 (stereoscope) in C, and K–N; ×20 in D; ×40 in F–I.
Absence of Adam10 in Collecting Duct Progenitor Cells Results in an Altered Ratio of PCs to ICs
To distinguish the two main cell types in the collecting duct, namely the PCs and the ICs, we performed immunofluorescence staining on kidney sections with Aqp2 (a PC marker), and ATPase (an IC marker) (Figure 2, A–H). The proportion of ATPase+ ICs was significantly higher in the mutants than in the controls (Figure 2, A–F and I). The percentage of ATPase+ collecting duct cells in the inner medulla was 17.40%±4.79% in the controls and 38.04%±12.62% in the mutants (Data are presented as mean±SDs). In the outer medulla, this percentage was 26.34%±5.57% in the controls and 47.86%±5.95% in the mutants. In the cortex, it was 38.55%±5.61% in the controls and 46.51%±4.38% in the mutants. In all three areas, the differences between control and mutant samples were significant (P<0.01). Correspondingly, the percentage of Aqp2+ PCs was significantly lower in the mutants (Figure 2, A–F and I). Besides the differences in the relative proportion of PC and IC cells, the mutant samples also showed a general decrease in total number of cells in the collecting duct, which was statistically significant in the inner medulla (Figure 2J). Such a decrease is caused by the decrease in Aqp2+ cells. The average number of Aqp2+ cells within a 40× field in the controls was 83.3±29.38 in the inner medulla, 57.19±26.70 in the outer medulla, and 41.09±18.31 in the cortex. The corresponding set of numbers in the mutants (52.82±22.13, 38.53±21.14, and 30.05±14.05) was statistically significantly smaller than those of the controls (P<0.001 in all three areas). In fact, the IC numbers within a 40× field in the inner medulla (30.59±11.64) and outer medulla (34.14±16.84) of the mutants were higher than those of the controls (16.95±6.46 and 19.81±8.30; P<0.001). This increase in IC numbers partially offset the decrease in number of PCs in the mutant medulla, making the differences in total numbers of collecting duct epithelial cells less dramatic in the medullary area. The decrease in the relative abundance and the absolute numbers of Aqp2+ PCs can affect the ability of the collecting duct to absorb water from the prourine, leading to deficient urinary concentrating ability, lower urinary osmolality, and polyuria. Polyuria has the potential to eventually overwhelm the pyeloureteral peristaltic machinery for urine transfer, leading to hydronephrosis and renal failure.17–19 Because the mice with hydronephrosis had various degrees of erosion of the renal parenchyma starting from the inner medulla, we analyzed these mice separately and focused mostly on the cortex, where structures were still relatively intact and recognizable. We found that the percentage of ATPase+ ICs in the cortex was similar between mutants with hydronephrosis (46.51%±4.38%) and mutants without hydronephrosis (46.91%±5.49%) (P=0.68). The percentage of Aqp2+ cells is thus also similar. It is still unclear why some mutants have more severe phenotypes than others. It is worth noting that these mutants were in a mixed outbred background and the genetic background differences among individual mutants may at least partially account for the phenotypic variations.
Figure 2.
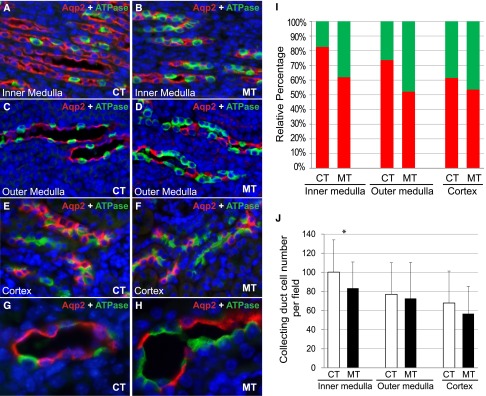
Absence of Adam10 in collecting duct progenitor cells altered the ratio of PCs and ICs. Immunofluorescence staining showed PCs (Aqp2+) and ICs (ATPase+) in inner medulla (A and B), outer medulla (C and D), and cortex (E and F) of adult controls (A, C, E) and mutants (B, D, F). (G and H) Higher-magnification images of control and mutant collecting ducts immunostained for Aqp2 and ATPase. The percentage of ATPase+ IC cells was increased while the percentage of Aqp2+ PC cells was decreased in the mutant inner medulla (61.90%±12.62% for PCs, 38.04%±12.62% for ICs) compared with controls (82.60%±4.79% for PCs, 17.40%±4.79% for ICs); in the outer medulla (52.14%±5.95% for PCs, 47.86%%±5.95% for ICs) compared with controls (73.66%±5.57% for PCs, 26.34%±5.57% for ICs); and in the cortex (53.49%±4.38% for PCs, 46.51%±4.38% for ICs) compared with controls (61.45%±5.61% for PCs, 38.55%±5.61% for ICs) (I). P<0.01 for all comparisons between control and mutant samples in all regions. Total number of cells per field deceased in the inner medulla, but such a decrease did not reach statistical significance in outer medulla and cortex (J). CT, controls; MT, mutants. For inner medulla, our analysis focused on the more proximal regions away from the papilla. Original magnification, ×20 in A–F; ×60 in G and H.
Adam10 Inactivation Affected Type A ICs More Than Type B ICs but Did Not Affect Urine pH
Although both A and B type ICs produce ATPase and CAII, they express distinct sets of transporters and enzymes for accomplishing their different, and mostly opposite, functional roles. To determine whether Adam10 deletion alters the differentiation of the IC subtypes, we quantified type A and type B ICs by immunostaining with AE1 and Pendrin, respectively. The number of AE1+ type A ICs was significantly higher in the mutant kidneys (Figure 3, A–H). The ratio of type A ICs/PCs increased significantly in mutants (Figure 3I). There was a general increase of AE1+ cells in the mutants with the most prominent increase in the medullary region. In adult kidneys, Pendrin+ type B ICs were detectable in the cortex but not the medulla (Figure 3, K–P). Thus, the medullary collecting ducts are primarily composed of AE1+ type A ICs and Aqp2+ PCs. The cortical collecting ducts have both types of ICs in addition to PCs. The relative ratio of Pendrin+ type B ICs/PCs was only moderately higher in mutants than in their control littermates (Figure 3J). As such, the differences in ICs between the mutants and controls appear to be greater in the type A ICs than in the type B ICs. We measured the pH of urine samples from adult mice and found no significant differences between control and mutant mice (Figure 3Q). This is not surprising because both types of ICs are present in sufficient numbers in the absence of Adam10. It is still unclear whether this reflects the functional plasticity for the ICs.2,3 It appears, however, that the major physiologic consequence of the alterations of cell fates in the collecting ducts is caused by the reduction of PCs, but not the increase of ICs or the relative abundance of the two subtypes. Both ATPase and Pendrin appeared to maintain their proper intracellular localization in the mutant ICs (Figure 3, D, H, M, P).
Figure 3.

Adam10 inactivation affected type A intercalated cells more than type B intercalated cells but did not affect urine pH. In adult controls (A–D) and mutants (E–H), immunofluorescence staining showed PCs (Aqp2+) and type A ICs (AE1+) in inner medulla (A and E), outer medulla (B and F), and cortex (C and G). (D and H) Higher-magnification images of medullary collecting ducts. The ratio of type A ICs/PCs increased significantly in the inner medulla, the outer medulla, and the cortex in mutants (0.82±0.41, 0.75±0.15, and 0.57±0.23, respectively) compared with the controls (0.33±0.07, 0.31±0.06, and 0.31±0.10, respectively; P<0.001 in all three regions) (I). Immunofluorescence staining of Aqp2 and Pendrin in the adult medulla (K and N) showed essentially no Pendrin+ cells in either genotype. In the cortex of adult controls (L and M) and mutants (O and P), the relative ratio of Pendrin+ type B ICs/PCs was only moderately higher in mutants (0.52±0.09) than in their control littermates (0.49±0.10) (P=0.02) (J). The urine pH did not significantly differ between mutants (6.0±0.6) and controls (6.1±0.7) (Q). CT, controls; MT, mutants. Original magnification, ×20 in A–C, E–G, K, L, N, and O; ×60 in D, H, M, and P.
Adam10 Inactivation Results in Deficient Notch Signaling but Does Not Affect Cell Proliferation, Apoptosis, or Polarity
To better understand the mechanism by which Adam10 deletion caused altered cell fate determination in the collecting duct, we examined the activity of Notch signaling, a known target for Adam10. Although Notch1, Notch2, and Notch3 receptors are expressed in the developing collecting ducts,20 we could reliably detect activated Notch1 only by immunohistochemistry. The activated form of Notch1, the Notch intracellular domain (NICD), was detected predominantly in the nascent nephrogenic bodies (arrows) in both control and mutant kidneys (Figure 4, A–D). Whereas nuclear presence of NICD was evident in many collecting duct cells that express Aqp2 in control kidneys (arrowheads in Figure 4C), it was essentially absent in the mutant collecting duct cells (Figure 4, B and D). This suggests that Adam10 is required for Notch1 activation in the collecting ducts.
Figure 4.

Adam10 deficiency results in deficient Notch signaling but does not affect cell proliferation, apoptosis, or polarity. A and B are low magnification images of cortical and outer medullary regions in which the Notch intracellular domain (NICD) for activated Notch1 (N1 Val1744) was detected predominantly in the nascent nephrogenic bodies (arrows) in both control and mutant kidneys at E17.5. Red membrane/cytoplasmic signal in some of the tubules in A and B is non-specific background. Whereas we detected nuclear staining of NICD in many collecting duct cells expressing Aqp2 in control kidneys (arrowheads in C), very few, if any, such collecting duct cells showed nuclear staining for NICD in mutant kidneys (B and D). C and D are higher magnification images. No significant difference in apoptosis (by terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling assay, E and F) or proliferation (by PHH3 immunostaining, G and H) was detected in the newborn controls (E and G) and mutants (F and H). Furthermore, Aqp3 and Aqp4 in the mutant collecting duct (J and L) were basolaterally located, similar to the controls (I and K). CT: Controls; MT: Mutants. Original magnification, ×20 in A, B, and E–H; ×40 in C and D; ×60 in I–L.
Postnatal distribution of ICs in rat kidneys was reported to be partially regulated by the loss of ICs via apoptosis or shedding.21 In light of this finding and other reports that apoptosis and proliferation may play a role in the distribution of ICs and PCs,21,22 we set out to examine whether Adam10 deficiency affects apoptosis or proliferation of the collecting duct cells. Terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling assay and phospho-histone 3 (PHH3) staining on newborn kidneys showed no significant difference in apoptosis and proliferation between controls (Figure 4, E and G) and mutants (Figure 4, F and H). In addition, Aqp3 and 4 have a normal basolateral distribution in the mutants (Figure 4, I–L), hinting that the main difference in the mutant collecting ducts is in the composition of the cells and probably not altered cellular characteristics. This is consistent with the findings that the key markers of ICs (ATPase, AE1, and Pendrin, as shown in Figure 2H and Figure 3, H and P, respectively) have a normal intracellular distribution in the Adam10 mutants.
Foxi1 Is a Potential Downstream Effector for Adam10 in the Regulation of Collecting Cell Fate
Because Foxi1 regulates key transporters in ICs,9 we asked whether deletion of Adam10 affects cell fate determination in the collecting duct through differences of Foxi1 expression. We observed more Foxi1+ cells in the renal medulla and cortex in mutants than in controls (Figure 5, A–F). The percentage of Foxi1+ cells in the collecting duct was higher in mutants than controls (Figure 5G). The percentage of Aqp2+ cells decreased correspondingly in mutants. The difference in Foxi1 expression appears to be associated with, and probably mediates, the effects of Adam10 deletion on determination of collecting duct cell fate.
Figure 5.

Foxi1 is a potential downstream effector for Adam10 regulation of collecting duct cell fate. Immunofluorescence staining showed Aqp2+ PCs and Foxi1+ cells in inner medulla (A and B), outer medulla (C and D), and cortex (E and F) of adult controls (A, C, E) and mutants (B, D, F). The percentage of Foxi1+ cells in the collecting ducts was 8.6%±1.6% in controls and 33.2%±6.1% in mutants in the inner medulla (P<0.001), 20.8%±4.7% in controls and 42.5%±9.8% in mutants in the outer medulla (P<0.001), and 42.2%±6.0% in controls and 45.2%±3.8% in mutants in the cortex (P<0.01) (G). CT, controls; MT, mutants. Original magnification, ×40 in A–F.
The Roles of Adam10 in Determination of Collecting Duct Cell Fate Appear to Be Largely Fulfilled Prenatally
To determine the timing of cell fate determination, we examined the PCs and ICs in the collecting ducts at E17.5, when markers for both cell types can be reliably visualized and quantified. At E17.5, we observed higher percentage of ATPase+ cells in the inner medulla in mutants than in controls (Figure 6, A–C). The relative ratios of ICs to PCs in newborn controls and mutants were similar to those in their adult counterparts (Figures 2 and 6), suggesting that cell-fate determination in the collecting ducts is largely completed by late gestation and that key effects of Adam10 in cell fate determination were fully evident by E17.5. The increase in the density of ICs at this early stage was accompanied by an increase in Foxi1 expression (Figure 6, D and E).
Figure 6.
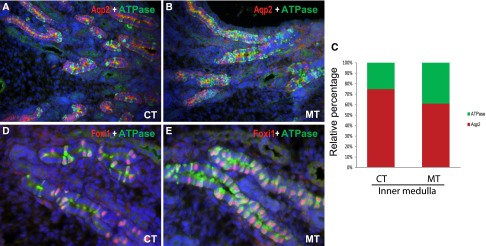
The roles of Adam10 in determination of collecting duct cell fate appear to be largely fulfilled prenatally. As early as E17.5, there were more ATPase+ ICs in the mutants (60.7%±7.5% for PCs, 39.3%±7.1% for ICs) (B) than in controls (74.8%±4.7% for PCs versus 25.2%±4.6% for ICs) (A) in the kidney inner medulla. The ratio of ATPase+ ICs to Aqp2+ PCs was increased in the E17.5 mutants (C). An increase in the number of Foxi1+ cells in the mutants was also observed as early as E17.5 (D and E). CT, controls; MT, mutants. Original magnification, ×20 in A and B; ×40 in D and E.
Discussion
This study showed that mice deficient for Adam10, specifically in the collecting ducts, have a urinary concentrating defect and polyuria that might have caused hydronephrosis in some mice (Figure 1). These defects originated from abnormal cell-fate determination in the mutant collecting duct, where disproportionally more IC cells and fewer PC cells were found (Figure 2). The abnormal PC/IC ratio was evident as early as E17.5, suggesting that the process of cell-fate determination in the collecting duct occurs early and is largely completed during embryogenesis (Figure 6). All of these results indicate an indispensable role for Adam10 in the normal development of the renal collecting ducts.
Considering the known role of Adam10 in regulating Notch signaling, the reduced activation of Notch1 in Adam10 mutant collecting ducts (Figure 4), and the similar reduction in PC to IC ratio observed in mice separately deficient for Adam10 and Mib17 (an upstream regulator of Notch) in collecting ducts, we conclude that Adam10-mediated Notch signaling ensures that sufficient number of collecting duct cells select the principal cell fate during collecting duct development in mammals. Foxi1, a transcription factor essential for the differentiation of ICs, was upregulated in the Adam10 mutants (Figure 5). One possible mechanism for the Adam10 regulation of principal cell-fate selection in the collecting duct is by the suppression of Foxi1 expression through Notch signaling. However, the phenotypic differences between the Adam10 mutants, the Mib1 mutants, and the Foxi1 mutants, suggest a more complex picture of Adam functions in collecting duct cell-fate determination. Because Adam10 has many known substrates besides Notch, it is possible that the pathway through Adam10, Notch, and Foxi1 is not linear. Although results from transgenic models suggest that Adam10 serves as the key S2 cleavage enzyme for Notch in vivo, it is still possible that other enzymes, especially other Adams (such as Adam17),23 may provide some compensatory function in the absence of Adam10, leading to the milder phenotypes in Adam10 mutants than expected. In addition, in the Xenopus skin, where Notch signaling inhibition increased the proton secreting intercalated-like cell types, this increase was primarily in type B IC-like cells and not as much in the type A IC-like cells.24 The involvement of Notch signaling in the development of type A IC-like cells in the Xenopus skin24 is not conserved in mammalian type A IC differentiation in the collecting ducts, or Adam10 and Mib1 are compensated for by other gene products that mediate Notch-dependent type A IC differentiation.
The normal differentiation of renal collecting duct epithelial cells into proper numbers of PCs and ICs ensures the functions of the collecting system both in urine concentration and in maintaining acid-base balance. Although the proportional decrease of PCs and proportional increase of ICs were both dramatic in appearance, the effects of the decrease in PCs appeared to be far more devastating, probably as a result of an absence of any compensatory mechanism for the reduced capacity in water reabsorption through the Aqp2 water channels in the collecting ducts. In addition, a recent study indicates that Aqp2 is not just a water channel and can interact with integrins to regulate cell behavior and kidney development.25 Thus, it is possible that some of the Adam10 phenotypes may be mediated through altered Aqp2–integrin interactions. In the case of ICs, although the number of acid-producing type A ICs was increased more dramatically compared with the base-producing type B ICs, sufficient acid-producing and base-producing capabilities among the two types of IC cells develop in the Adam10 mutants. Unlike Foxi1,9 Adam10 deletion appears to affect the number, but not the characteristics, of the ICs on a large scale (Figure 2H and Figure 3, G and P); thus, the excess acid-producing capacity may be held in check by the intact physiologic regulation of these cells. Thus, it may not be a surprise that we have not detected any overt defects associated with the increase of IC under normal dietary and housing conditions. Whether these mice will develop acid-base imbalance when challenged remains to be determined.
When we examined the papilla region, we found an almost complete absence of ICs in the controls (Supplemental Figure 1). Because the Hoxb7-Cre transgene has uniform expression in the UB derivatives (Figure 1), collecting ducts in this region would have Adam10 deletion, just as in other parts of the collecting duct system. However, although Adam10 deletion results in an increase of ICs in other regions of the kidney, there is still an absence of IC cells in the papilla. At least in the case of Adam10 deletion, there appears to be segmental differences in the ability of the UB derivatives to differentiate into IC cells. Thus, switching off Adam10 is enough for collecting duct cells outside of the papillary region to adapt IC traits but not enough to do so in the papillary collecting duct epithelial cells that may be under more intrinsic or extrinsic restrictions.
Plasticity of cells in the collecting ducts has been observed long before any mechanism underlying their cell fate determination was revealed. In 1992, researchers reported that type B ICs can differentiate into type A ICs and then progress to PCs in culture.26 If normal collecting duct cell differentiation were to involve type B ICs differentiating into type A ICs and then PCs, then the phenotype of Adam10 deficiency may be explained by a requirement for Adam10 in the conversion of type A ICs into PCs. Another in vitro study also suggested the possibility of ICs originating from PCs.27 Key molecular switches for cell fate determination in the collecting duct have started to come to light in recent years. Mice deficient for Foxi1 showed no distinct cells, only one cell type with expression of both Aqp2 and CAII.9 The removal of this key regulator required for the expression of IC-specific genes, such as AE1 and Pendrin, appears to cause progenitors to arrest at an ambiguous state between PCs and ICs. Recent findings in mice with a deficiency for a histone methyltransferase suggest the presence of a group of Aqp2+ “plastic PCs” that can differentiate into both PCs and ICs.28 In the Adam10 mutants, we have observed a small population of cells that are positive for both Aqp2 and Foxi1 (Figure 5). It is still unclear whether this population is, or overlaps with, the group of Aqp2+ “plastic PCs”. Besides the possible existence of multipotent progenitor cells, conversion between differentiated PCs and ICs and between the IC subtypes may result from genetic mutations or biochemical changes. As such, the molecular circuitry responsible for the initial differentiation of the primary UB epithelial cells into specialized PCs and IC subtypes may also be regulating the plasticity of these cells later in response to environmental or physiologic alterations. In fact, published studies and our recent results have shown that treatment with lithium (the classic mood stabilizer widely used in psychiatry clinics) results in alterations in collecting duct composition that may be at least partially responsible for the adverse effects of lithium in the kidney.6 Better understanding of cell fate determination in the collecting ducts may help to design new strategies to manage undesirable alterations of the cell composition in the collecting ducts in the future.
Concise Methods
Mouse (Mus musculus) Strains
All animal studies were approved by the Washington University Animal Studies Committee and were conducted according to relevant National Institutes of Health guidelines. The Adam10flox/flox mice were purchased from the The Jackson Laboratory (Bar Harbor, ME) and crossed with Hoxb7-Cre mice.29 Hoxb7-Cre; Adam10flox/flox mice were mutants, and Hoxb7-Cre; Adam10+/+ or Hoxb7-Cre; Adam10flox/+ were used as controls. All experiments were repeated at least three times. For detection of Adam10 deletion in the mutant kidneys, the following PCR primers were used, KOF: ACCTCTTAGCGATACCACAAGCC and KOB: CCAAGCGTCAAAGCGTTAC.30
Histology and Immunostaining
Tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Sections of 5 µm were collected and stained with hematoxylin and eosin. Xgal staining and immunostaining on cryostat sections were performed as described elsewhere.17,18 Antibodies used were anti-Aqp2 antibody (Santa Cruz Biotechnologies, 1:200); anti–V-ATPase antibody (Santa Cruz Biotechnologies, 1:200), anti-CAII antibody (Santa Cruz Biotechnologies, 1;200), anti-AE1 antibody (EMD Millipore, 1:500), anti-Pendrin antibody (a gift from Dr. Aronson,31 1:500), anti-PHH3 antibody (EMD Millipore, 1:500), anti-Adam10 antibody (Abcam, Inc., 1:400), anti-Notch1 ICD (Cell signaling Technology, 1:300), and anti-Foxi1 antibody (Abcam, Inc., 1:300). Appropriate AlexaFlour488 or 555-conjugated secondary antibodies (Molecular Probe, 1:500) and NL557-conjugated secondary antibodies (R&D Systems, 1:300) were used for detection. For micrographs of control and mutant samples being compared, the camera settings were identical. Cell numbers were quantified on micrographs without genotype labels. For the quantification of the absolute number of cells, counting was done for each 40× field. Data analyses were performed in Excel (Microsoft Corp., Redmond, WA). For statistical analyses, P<0.05 was considered to represent statistically significant differences in two-tailed t tests. For analyses of the inner medulla, we focused on the proximal portion of the inner medulla away from the papilla. While the medullary regions have collecting ducts in all images taken systematically from one side of the section to the other, the cortex has areas completely void of colleting ducts. To avoid huge variations, we excluded such collecting duct–free areas for the analyses. As such, we effectively compared areas rich in collecting ducts in the cortex between controls and mutants.
Serologic Analysis and Urinalysis
Mice were housed in metabolic cages (Nalgene, Rochester, NY) and given free access to water and powdered chow (Rodent Diet 5001; LabDiet, St. Louis, MO). Urine samples were collected for 24 hours from the metabolic cage. A vapor pressure osmometer (Wescor, Logan, UT) was used to determine urine osmolality. Urine and serum chemistry was analyzed by the Renal Chemistry Laboratory at Washington University School of Medicine.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Dr. Carlton Bates for the Hoxb7-Cre mice. We also thank the George M. O’Brien Center for Kidney Disease Research at Washington University (P30-DK079333) for core services and Drs. Peter Aronson and Zhirong Jiang for the kind gift of the Pendrin antibody. F.C. is supported in part by institutional funds from the Department of Medicine/Renal Division at Washington University School of Medicine and National Institutes of Health (NIH) grants (DK81592 and DK87960). M.M. and K.S. are supported by funds from Sanford Research/University of South Dakota and NIH (P20_GM103620-01A1).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013070764/-/DCSupplemental.
References
- 1.Dressler GR: The cellular basis of kidney development. Annu Rev Cell Dev Biol 22: 509–529, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Al-Awqati Q, Gao XB: Differentiation of intercalated cells in the kidney. Physiology (Bethesda) 26: 266–272, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Wall SM: Recent advances in our understanding of intercalated cells. Curr Opin Nephrol Hypertens 14: 480–484, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Gao X, Eladari D, Leviel F, Tew BY, Miró-Julià C, Cheema FH, Miller L, Nelson R, Paunescu TG, McKee M, Brown D, Al-Awqati Q: Deletion of hensin/DMBT1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosis. Proc Natl Acad Sci U S A 107: 21872–21877, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breton S, Alper SL, Gluck SL, Sly WS, Barker JE, Brown D: Depletion of intercalated cells from collecting ducts of carbonic anhydrase II-deficient (CAR2 null) mice. Am J Physiol 269: F761–F774, 1995 [DOI] [PubMed] [Google Scholar]
- 6.Christensen BM, Marples D, Kim YH, Wang W, Frøkiaer J, Nielsen S: Changes in cellular composition of kidney collecting duct cells in rats with lithium-induced NDI. Am J Physiol Cell Physiol 286: C952–C964, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Jeong HW, Jeon US, Koo BK, Kim WY, Im SK, Shin J, Cho Y, Kim J, Kong YY: Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J Clin Invest 119: 3290–3300, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma M, Jiang YJ: Jagged2a-notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genet 3: e18, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergström G G, Enerbäck S: Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 113: 1560–1570, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seals DF, Courtneidge SA: The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev 17: 7–30, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lübke T, Lena Illert A, von Figura K, Saftig P: The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet 11: 2615–2624, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P: The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci 30: 4833–4844, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber S, Niessen MT, Prox J, Lüllmann-Rauch R, Schmitz A, Schwanbeck R, Blobel CP, Jorissen E, de Strooper B, Niessen CM, Saftig P: The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development 138: 495–505, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieber T, Kidd S, Young MW: kuzbanian-mediated cleavage of Drosophila Notch. Genes Dev 16: 209–221, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein T, Bischoff R: Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. J Proteome Res 10: 17–33, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Stuart RO, Bush KT, Nigam SK: Changes in gene expression patterns in the ureteric bud and metanephric mesenchyme in models of kidney development. Kidney Int 64: 1997–2008, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Chang CP, McDill BW, Neilson JR, Joist HE, Epstein JA, Crabtree GR, Chen F: Calcineurin is required in urinary tract mesenchyme for the development of the pyeloureteral peristaltic machinery. J Clin Invest 113: 1051–1058, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDill BW, Li SZ, Kovach PA, Ding L, Chen F: Congenital progressive hydronephrosis (cph) is caused by an S256L mutation in aquaporin-2 that affects its phosphorylation and apical membrane accumulation. Proc Natl Acad Sci U S A 103: 6952–6957, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen F: Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 24: 1621–1632, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Al-Awqati Q: Segmental expression of Notch and Hairy genes in nephrogenesis. Am J Physiol Renal Physiol 288: F939–F952, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Cha JH, Tisher CC, Madsen KM: Role of apoptotic and nonapoptotic cell death in removal of intercalated cells from developing rat kidney. Am J Physiol 270: F575–F592, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Christensen BM, Kim YH, Kwon TH, Nielsen S: Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol 291: F39–F48, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Zolkiewska A: ADAM proteases: Ligand processing and modulation of the Notch pathway. Cell Mol Life Sci 65: 2056–2068, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quigley IK, Stubbs JL, Kintner C: Specification of ion transport cells in the Xenopus larval skin. Development 138: 705–714, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Rice W, Gu Z, Li J, Huang J, Brenner MB, Van Hoek A, Xiong J, Gundersen GG, Norman JC, Hsu VW, Fenton RA, Brown D, Lu HA: Aquaporin 2 promotes cell migration and epithelial morphogenesis. J Am Soc Nephrol 23: 1506–1517, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fejes-Tóth G, Náray-Fejes-Tóth A: Differentiation of renal beta-intercalated cells to alpha-intercalated and principal cells in culture. Proc Natl Acad Sci U S A 89: 5487–5491, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jamous M, Bidet M, Tauc M, Koechlin N, Gastineau M, Wanstok F, Poujeol P: In young primary cultures of rabbit kidney cortical collecting ducts intercalated cells originate from principal or undifferentiated cells. Eur J Cell Biol 66: 192–199, 1995 [PubMed] [Google Scholar]
- 28.Wu H, Chen L, Zhou Q, Zhang X, Berger S, Bi J, Lewis DE, Xia Y, Zhang W: Aqp2-expressing cells give rise to renal intercalated cells. J Am Soc Nephrol 24: 243–252, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, Baum M, Bates CM: Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276: 403–415, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian L, Wu X, Chi C, Han M, Xu T, Zhuang Y: ADAM10 is essential for proteolytic activation of Notch during thymocyte development. Int Immunol 20: 1181–1187, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS: Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci U S A 98: 9425–9430, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.