

Abstract
Chronic mountain sickness (CMS) is a disease that affects many high-altitude dwellers, particularly in the Andean Mountains in South America. The hallmark symptom of CMS is polycythemia, which causes increased risk of pulmonary hypertension and stroke (among other symptoms). A prevailing hypothesis in high-altitude medicine is that CMS results from a population-specific “maladaptation” to the hypoxic conditions at high altitude. In contrast, the prevalence of CMS is very low in other high-altitude populations (e.g., Tibetans and Ethiopians), which are seemingly well adapted to hypoxia. In recent years, concurrent with the advent of genomic technologies, several studies have investigated the genetic basis of adaptation to altitude. These studies have identified several candidate genes that may underlie the adaptation, or maladaptation. Interestingly, some of these genes are targeted by known drugs, raising the possibility of new treatments for CMS and other ischemic diseases. We review recent discoveries, alongside the methodologies used to obtain them, and outline some of the challenges remaining in the field.
More than 140 million people around the globe reside at high altitude (≥3,000 m) in locations such as the Ethiopian Highlands in Africa, the Himalaya Mountains in Asia, and the Andes Mountain Range in South America. Although there is no doubt that the elevation in these regions represents stressful environmental conditions, chiefly due to low environmental O2, adaptation of high-altitude dwellers has varied qualitatively and quantitatively. For example, a higher hemoglobin concentration and lower oxygen saturation have been observed in Andean highlanders compared with Tibetans or Ethiopians at similar altitude (3, 5). Furthermore, infants of Tibetans have a higher birth weight and arterial O2 saturation compared with infants of other populations (Han Chinese) at similar altitudes (3). In fact, a sizeable percentage of individuals in these populations (as much as 16% in certain regions, especially males) are maladapted and are thus threatened by the low levels of inspired O2 to this day (31). The primary manifestation of this maladaptation to high altitudes is chronic mountain sickness (CMS) or Monge's disease, first described by Carlos Monge in the Andes in 1925 (29). It is characterized by polycythemia (hematocrit > 65%) and hypoxemia (O2 < 85%), both of which improve upon descent from altitude. The most frequent symptoms and signs of CMS are headache, dizziness, breathlessness, palpitations, sleep disturbance, mental fatigue, and confusion (30). People affected by CMS often suffer from stroke and myocardial infarction in early adulthood, mostly due to increased blood viscosity and tissue hypoxia. Why some are affected by this disease and not others has so far been a mystery. Differences in adaptation patterns between the high-altitude populations suggest that there are, at least in part, distinct genetic mechanisms underlying the adaptations.
Despite this genetic basis having been proposed for many years, it is only in the past few years that our understanding of human adaptation to high altitude has accelerated (1, 6, 22, 34, 37, 39, 42, 46, 47, 51). These rapid discoveries have mostly resulted from the advent of genomic technologies, particularly deep sequencing, as well as the concurrent developments in computational genetics. Understanding the molecular basis of high-altitude adaptation and maladaptation will also provide us with a unique handle on genes that are important for human health and disease. This is particularly true of conditions where oxygen deprivation plays a major etiological role, such as arterial pulmonary hypertension, myocardial ischemia and injury, stroke, and polycythemia (to name a few). It is possible that learning about regions of the human genome that evolved over many thousands of years to allow adaptation to hypoxia will lead to a better understanding of adaptation in humans subjected to other stresses.
In this review, we highlight several recent studies (including ours) dealing with the genetic underpinnings of high-altitude adaptation or mal-adaptation. CMS is a maladaptation to high altitude, and any understanding of CMS will likely shed light on relevant genetic and physiological mechanisms. We evaluate the methods used in previous studies and the results obtained. In addition, we highlight some of the remaining open questions in the field.
A Genomic Approach to Understanding Chronic Mountain Sickness
Adaptation to hypoxia (as well as the diagnosis of CMS) is often measured using related phenotypes, such as blood oxygen saturation, Hb, or hematocrit levels (5, 30). From available data, we can surmise that the different populations all adapted separately: their phenotypic values show distinct inherited traits. It is possible that a founder effect may have been at work when the populations first migrated, and standing genetic variation in the founders was subjected to selective constraints leading to this variable adaptation. Although most high-altitude populations in the world are well-adapted, in some regions (e.g., the Andes), 10–20% of the male population is threatened by the CMS syndrome (31). Such populations provide us with a unique opportunity to study the nature of adaptation by contrasting the genetics of well adapted individuals and those with CMS (51). CMS is believed to arise, at least partly, due to an excessive production of red blood cells (RBC). By increasing RBC production, humans attempt to mitigate the effect of low environmental oxygen via increased oxygen carrying capacity in the blood. Hence, CMS is considered an adaptation to life under chronic hypoxia at altitude. Indeed, sea-level-dwelling humans visiting high altitudes show a similar, albeit more modest, response. However, a long-term consequence of this increase in RBC production is increased blood viscosity, leading to vascular sludging and increasing the likelihood of vascular occlusion, stroke, myocardial ischemia, and infarcts in early adulthood. It also results in uneven blood flow through the lungs, increasing the ventilation-perfusion mismatch and leading to hypoxemia, contrary effects to what is desired. In trying to adapt, the organism has effectively responded in a way that is adverse to its survival and well being (mal-adaptation) at high altitude (51). Therefore, the genetic basis for CMS can be investigated in the context of adaptation to hypoxic environments at high altitudes, and our survey keeps that perspective. FIGURE 1A describes a generic strategy that is implicit in many highlander studies. The first steps of this strategy, correlating genotypes and phenotypes and/or scanning the genome for signatures of natural selection, reveal candidate genes. Due to many factors that may confound this step, candidate genes must then be validated using various approaches. Our review focuses on these two steps, where current studies suggest a complex, multigenic adaptation.
FIGURE 1.

Proposed workflow for hypoxia-related therapeutics and schematic genealogical tree
A: a proposed workflow for hypoxia related therapeutics, starting with genetic samples and ending with candidate therapeutic targets. B: schematic genealogical tree illustrating the evolution of a non-recombining genomic fragment across three populations, one of which migrates to high altitude (HA population) and undergoes genetic adaptation, whereas the others remain at low altitudes (LA and Outgroup populations). The bottom of the tree (leaves) represents individuals sampled from the current generation, whereas the upper sections reflect the past genealogy. In the HA population, hypoxia imposes positive natural selection on the beneficial allele (blue star), increasing its frequency (in the non-CMS group) at the expense of individuals carrying the maladapted allele (CMS). As long as phenotypic variation persists in the adaptive trait (e.g., Hb levels are still variable in the HA population, meaning the selective sweep is ongoing), genetic association may find variants associated with the trait. However, after the trait reaches fixation or given small effect sizes and/or smaller cohorts, genome-wide association (GWA) is unlikely to reveal the adaptive genes. Neutrality tests can be used to pinpoint genomic regions under selection in both settings (i.e., pre- and postfixation, and given a smaller sample). These tests utilize properties of the genealogical tree. The LSBL/PBS tests approximate the branch length leading to the MRCA of the HA population, which is unusually high in regions under selection (see long branch with blue SNPs). Tajima's π uses the mean allelic heterogeneity, which is unusually low in regions under selection (since HA individuals are genetically similar given their relatively recent MRCA). The iHS/EHH tests use haplotype homozygosity, which is unusually high and spans longer regions under selection (most variation in HA individuals, shown as SNPs on the path from MRCA to the present HA individuals, is common to the entire HA sample). Common practice is to genotype a population sample, followed by imputation from a nearby, and densely sequenced, reference population (e.g., the LA population). Because imputation relies on conserved linkage disequilibrium (LD) between target and reference populations, and LD is strongly altered by selective sweeps, imputation will be inaccurate in regions evolving under strong selection. This further illustrates the importance of WGS. MCRA, most recent common ancestor.
Genome-Wide Association Studies and Disease Traits
Association tests measure correlation between genotype segregation and a phenotype, and can be applied to any of the many hypoxia-related phenotypes (e.g., Hb levels). Different studies have explored this with different designs involving the choice of phenotype, population (mainly Tibetan, Ethiopian, and Andean highlanders), genotyping technology, and statistical methods. It was recognized early on that there are population-specific differences in highlander phenotypes. For instance, Tibetan highlanders have lower Hb levels but also lower O2 saturation levels compared with Andeans (3). Hanaoka et al. (18) showed that serum erythropoietin (EPO) levels in Sherpas at 3,440 m was equal to that in non-Sherpas at a much lower altitude, indicating that Sherpas have a resistance response to EPO levels. Moore (30) and Beall (4, 5, 7) review many relevant traits across highlander populations, including hematocrit and hemoglobin levels, O2 saturation, arterial O2 content, ventilatory response to hypoxia, exhaled NO, and pulmonary vasoconstriction. In our context, individuals with and without CMS can be used in a case-control association study, with the caveat that there may be significant population substructure [see Figure S3 of Zhou et al. (51)]. Most association studies to date have focused on Hb levels due to ease of measurement and its correlation with other traits of interest. In a typical genome-wide association study (GWAS), the association between sampled genotypes and a specific trait is measured in case-control cohorts using statistical tests (see Table 1). The merits of different statistical tests have been previously reviewed (23, 45) and will not be discussed here.
Table 1.
Genes identified via association with a hypoxia-tolerance phenotype
| Study | Phenotype | Population(s) | Assay | Gene Candidates |
|---|---|---|---|---|
| Simonson et al. (39) | Hb | Tibet (n = 31) | Genotyping array (Affymetrix 6.0) | EPAS1, PPARA |
| Yi et al. (47) | Hb, erythrocyte | Tibet (n = 50) | Exome sequence (NimbleGen) | EPAS1 |
| Beall et al. (6) | Hb | Tibet (n = 70)Tibet (n = 91) | Genotyping array(Illumina 0.5M) | EPAS1 |
| Scheinfeldt et al. (37) | Hb | Ethiopia (n = 42) | Genotyping array(Illumina 1M) | ARNT2, THRB |
| Alkorta-Aranburu et al. (1) | Hb, O2 saturation | Ethiopia (n = 260)Amhara (102H, 60L)Oromo (63H, 35L) | Genotyping array (Illumina) | RORA*COL6A1*SLC30A9* HGF* |
Not genome-wide significant after multiple testing correction. H and L, high- and low-altitude individuals, respectively.
In the domain of genomic technologies, we now have a multitude of options, including candidate gene sequencing, SNP genotyping arrays, whole exome sequencing (WES), and whole genome sequencing (WGS). Each of these options has difficult trade-offs, arguably making this the single most important decision in the study-design process. For example, genotyping arrays sample a large corpus (up to several million) of genomic loci but suffer from a serious problem of ascertainment bias [recently summarized by Lachance and Tishkoff (24) and illustrated schematically in FIGURE 1B]. Normally, conserved haplotype structure between populations implies that, even if only a small collection of SNPs is sampled (e.g., by array), most alleles can be inferred from a previously sequenced reference population (10). Although generally accurate, this strategy (genotype imputation) may fail in genomic regions affected by strong selection (blue dots in FIGURE 1B), since the haplotype structure is highly sensitive to selective sweeps and is expected to diverge in such regions. Although some [e.g., Yi et al. (47)] have used whole exome sequencing to overcome such issues, this technology does not sample the vast noncoding portions of the genome, including regulatory and many noncoding RNA regions. Indeed, one of the most important sites discovered to date was in an intron of the EPAS1 gene not specifically targeted by the exon array.
A second problem, unrelated to the technology domain, is the reduced power stemming from the multigenic response to hypoxia. As with many other complex traits, most published GWA studies to date have failed to achieve genome-wide significance after multiple testing correction. Finally, adaptive alleles fix in the population, reducing phenotypic variation in their respective traits, and consequently reducing the power of association testing. Therefore, researchers have focused on candidate genes with known physiology (mainly HIF pathway genes; see Table 1). Simonson et al. (39) used a short list of five regions and zoomed in on EPAS1. Yi et al. (47) focused on genes annotated by the ontology term “response to hypoxia.” Beall et al. (6) looked carefully at sites around the EPAS1 gene and validated them through independent cohorts. Scheinfeldt et al. (37) and Alkorta-Aranburu et al. (1) studied Ethiopian highlanders and reported many interesting candidate genes; however, their results do not achieve genome-wide significance after Bonferroni correction for any SNPs located near genes. Yet, they validated through secondary means and identified a number of novel genes, suggesting a very different adaptive response in Ethiopians compared with Tibetan highlanders. In summary, association tests (particularly with ascertained SNPs) must be applied to larger populations, validated on independent cohorts, or focused on a reduced set of candidate regions. A viable approach to identifying an unbiased list of candidate genes is through searching for genomic signatures of natural selection, discussed next.
Natural Selection and Disease Pathogenesis
Given the strong selective constraints stemming from low environmental oxygen, identifying genetic signatures of natural selection in highlander populations provides us with an alternative approach to genotype/phenotype association for candidate gene detection. FIGURE 1B provides a schematic of the evolutionary history of a short (non-recombining) chromosomal segment under positive selection. The bottom (“leaves”) of the evolutionary tree corresponds to the genomic region in extant individuals of different subpopulations, whereas the top (“root”) of the tree represents the region in the most recent common ancestor (MRCA). Mutations (green or blue circles) on a specific lineage are inherited by all its descendants. The highlander (HA) migration exerts a selective constraint. Consequently, individuals carrying a beneficial allele (blue star) rise rapidly in frequency in the population, outcompeting other individuals. These individuals 1) present the non-CMS phenotype; 2) have a recent common ancestor, with all other non-CMS individuals sharing a longer than average branch of common mutations (i.e., blue circles); and 3) have had limited time to differentiate (i.e., few mutation and recombination events), leading to a lack of allelic diversity and long homogenous haplotypes in the region. Statistical tests capturing these characteristics have been used to identify regions under selection (Table 2). For example, LSBL/PBS (38, 47) and FST (21) were used by Yi et al. (47), Bigham et al. (9), Alkorta-Aranburu et al. (1), Zhou et al. (51), and Udpa et al. (42) to approximate the branch length leading to MRCA. Zhou et al. (51) and Udpa et al. (42) used multiple tests to measure allelic diversity (or lack thereof). Simonson et al. (39) and Scheinfeldt et al. (37) used the iHS test (36, 44) to compute the decay of haplotype similarity. These tests may be confounded by recent admixture of populations, which can also lead to lack of allelic diversity in some cases. Recently, an admixture-correction was proposed by Huerta-Sanchez et al. (22) to refine the analysis.
Table 2.
Genes identified via tests of selection
| Study | Population(s) | Assay | Gene Candidates |
|---|---|---|---|
| Bigham et al.* (9) | Tibet (n = 49) Andes (n = 49) | Genotyping array (Affymetrix 6.0) | Tibet: EGLN1, EPAS1 Andes: EGLN1, TH, NOS2A, PRKAA1 |
| Simonson et al. (39) | Tibet (n = 31) | Genotyping array (Affymetrix 6.0) | EPAS1, EGLN1, CYP2E1, EDNRA, ANGPTL4, CAMK2D, HMOX2, CYP17A1, PPARA, PTEN |
| Yi et al.* (47) | Tibet (n = 50) | Exome sequence (NimbleGen) | EPAS1, HBB, HBG2, FANCA, PKLR, HFE |
| Beall et al. (6) | Tibet (n = 35) | Illumina Quad (0.5M) | EPAS1 |
| Xu et al. (46) | Tibet (n = 46) | Genotyping array (Affymetrix 6.0) | EPAS1, EGLN1 |
| Peng et al.* (34) | Tibet (n = 1334) | Genotyping array (Affymetrix 6.0) | EPAS1, EGLN1 |
| Scheinfeldt et al.* (37) | Ethiopia Amhara HA (n = 28) Aari/Hamer LA (n = 19) | Genotyping array (Illumina 1M) | CBARA1, ARHGAP15, RNF216, SYNJ2, NAT2, AIMP1, VAV3, ARNT2, THRB |
| Alkorta-Aranburu et al.* (1) | Ethiopia (n = 260) Amhara (102H, 60L) Oromo (63H, 35L) | Genotyping array (Illumina) | CUL3, ADRBK1, CORO1B, ASF1B, MAPKAPK2, ADH6, SLC30A9, TMEM33 |
| Huerta-Sanchez et al.* (22) | Ethiopia Tigray-Amhara (n = 47) Oromo (n =21) | Genotyping array (Illumina Omni 1M) | BHLHE41, CASP1, SMURF2 |
| Zhou et al. (51) | Andes CMS (n = 10) non-CMS (n = 10) | Whole genome sequencing (Illumina) | SENP1, ANP32D |
| Udpa et al. (42) | Ethiopia Amhara (n = 7) Oromo (n = 6) | Whole genome sequencing (Illumina) | CIC, LIPE, PAFAH1B3, EDNRB |
Complete gene set not listed due to space limitations.
Taken together, these studies reveal the complexity of understanding hypoxia adaptation. On one hand, small sample sizes, low effect, and reduced phenotypic diversity (after fixation of a beneficial allele) make it difficult to achieve genome-wide significance in association studies. Indeed, with the exception of the EPAS1 genes in Tibetans, few genes have been identified with genome-wide significance. On the other hand, recent population admixture and complex demographic histories may confound tests of selection. We believe that, with further refinement, both methodologies will likely yield additional insights.
Another major issue is the choice of genetic assaying technology. Genotype arrays are designed to exploit haplotype blocks by directly assaying only select SNPs from the existing variation (so-called “tag SNPs”), and then using imputation (10) to fill in missing genotypes. Yet, Udpa et al. (42) and Zhou et al. (51) demonstrated that highlander populations may have a different haplotype structure specifically in areas under selection, and that many regions with strong signature of selection would be missed by genotype arrays or exome sequencing (FIGURE 2). Instead, these studies used WGS from small population samples of highlanders. In a study of Andean highlanders with CMS and non-CMS phenotypes, Zhou et al. (51) identified 11 regions genome-wide with significant haplotype frequency differentials between the CMS and non-CMS individuals, which are consistent with selective sweeps. Two distinct regions contained genes that had fly orthologs and could be validated in a model organism system (an erythropoiesis regulator, SENP1, and an oncogene, ANP32D). These studies illustrate the potential of whole genome sequencing in identifying the genetic basis of CMS and long-term hypoxia adaptation in general. Although the studies by Zhou et al. (51) and Udpa et al. (42) illustrate the advantages of WGS, it is currently too expensive for use on large cohorts. Therefore, a cost-effective design may be created using tests of selection on WGS of a smaller cohort, followed by genotyping and association tests on a larger cohort. The identified genes would be candidates for secondary validation, followed by functional testing.
FIGURE 2.
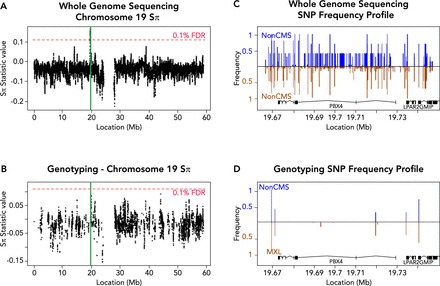
The effects of sequence assay on genome-wide scans for selection
A and B: test statistic values on chromosome 19, when taking into account all variants discovered by WGS (A) or only the subset found in a common ∼1M SNP genotyping array (B) (1% FDR computed separately based on the genome-wide distribution of test statistic values). Highlighted in green is 1 of the 11 significant peaks reported in our laboratory's study (51), which does not exceed the 1% FDR using only genotype data. C and D: SNP frequency profiles of the highlighted (green) region in non-CMS (blue) compared with MXL (brown, inverted) showing all variants from WGS (C) or only the subset present in genotyping (D). WGS reveals many variants in the region, allowing a robust estimate of the allele frequency distribution, whereas genotyping detects only a handful of alleles, making inference of adaptive evolution difficult. However, genotyping studies in large populations can be used to validate the frequency differences obtained via WGS of smaller cohorts. Figure adapted from Zhou et al. (51), with permission from Elsevier.
From Association to Causative Genes and Pathways
Independent Cohorts
Most highlander studies to date are not powered to achieve genome-wide significance for association, even when large population cohorts have been used [e.g., Peng et al. (34)]. Therefore, many studies seek to replicate their findings on independent cohorts from the same population. Good examples of this are the EPAS1 and EGLN1 genes, which have been shown repeatedly as important for hypoxia adaptation in Tibetans, pointing to the essential role of the HIF pathway. Interestingly, these genes appear to play a less substantial role in Andeans and have not been observed in Ethiopians (see Tables 1 and 2). In general, even genes that appear repeatedly in multiple highlander cohorts require further investigation to elucidate their specific biological role.
Model Organisms
To validate the effects of genes found via genomic scans for association or selection, animal models may be of great value (42, 51). In the context of CMS, the idea is to determine the functional impact of genetic variants identified as potentially significant in both affected (CMS) and unaffected (non-CMS) individuals. Drosophila melanogaster provides a powerful in vivo model to dissect the genetic mechanisms that contribute to human disease (8, 15, 33), including aging (17, 28), neurological and cardiac disease (13, 25, 27), cancer (35, 43), and the mechanisms underlying hypoxia tolerance or susceptibility (2, 50, 52). Several genes obtained from human studies of high-altitude adaptation (and maladaptation) have orthologs in the Drosophila genome and could thus be tested for effects on hypoxia tolerance. For example, Zhou et al. (51) observed a dramatic increase in survival when the expression of candidate genes SENP1 and ANP32D (obtained from genomic tests of selection) was reduced in flies under hypoxic conditions, indicating a likely role for these genes in human adaptation to high altitude (see FIGURE 3). Indeed, SENP1 is known to regulate erythropoiesis, and Senp1−/− mice die of anemia in early life. If CMS pathogenesis is even partially caused by abnormal polycythemia, then SENP1 may be of prime importance, potentially linking erythropoiesis to the pathogenesis of CMS.
FIGURE 3.
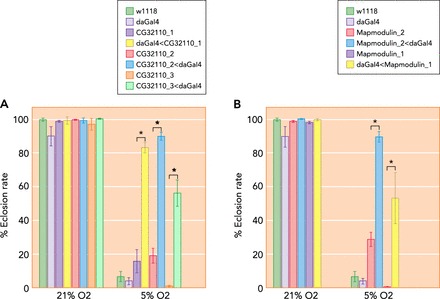
Experimental validation in a model system of candidate genes for human high-altitude adaptation
Downregulation of human SENP1 and ANP32D orthologs in Drosophila enhances survival under hypoxia. The da-Gal4 driver was used to ubiquitously knock down the individual candidate genes by crossing with respective UAS-RNAi lines. Eclosion rates were then measured at 21% and 5% O2. A: significant increase in eclosion rate under 5% O2 in three RNAi lines targeting the same human SENP1 ortholog (CG32110). B: the differences in eclosion rates were also significant in the two lines targeting the human ANP32D ortholog (Mapmodulin). Each bar represents mean 5 SE of eclosion rate. The w1118 and da-Gal4 stocks were tested and used as background controls. Figure adapted from Zhou et al. (51), with permission from Elsevier.
Although to the best of our knowledge the model organism work done by Zhou et al. (51) and Udpa et al. (42) represents the only such work to validate results from studies of genetic variation in high-altitude human populations, other models have also been used in studying acute or chronic hypoxic stress (20, 40, 49). Zebrafish and C. elegans have been used, mostly to dissect the genetic basis of response to hypoxic stress and genetic predisposition. These model systems could be very useful to corroborate findings from human studies where cellular and molecular studies are difficult to perform.
In Vitro Models
Another possible approach for validation of candidate genes obtained from statistical tests of association or selection is to study the relevant phenotypic effects in vitro. Zhou et al. (51) determined the expression levels of candidate genes in fibroblasts (obtained from skin biopsies) placed under decreasing levels of O2. Furthermore, a similar approach can be applied to reprogrammed iPS cells. This can be very useful toward replicating the disease in a dish, as has been done before in other cases (11). As shown by Zhou et al. (51), such in vitro models are able to capture aspects of CMS. In this way, we may better understand the effect of genetic variants on phenotype in the condition when the disease is manifested.
Functional Networks
Naively, one might expect that similar adaptive genes should surface among populations experiencing similar selective stresses. To some extent this is true, as is the case for EGLN1 that exhibits a strong signature of selection in both the Andean and the Tibetan populations (see Table 2). Yet, a striking observation from Tables 1 and 2 is the relative lack of overlap in candidate adaptive genes among the different populations. Partially, this may result from technical limitations in the respective studies (meaning the true overlap may be greater than currently observed). Nevertheless, we believe the small overlap stems chiefly from the structure and connectivity of the underlying genetic networks. Differently put, although different genes are involved across different populations, it is plausible that similar mechanisms and/or pathways are at play. For instance, different loss of function (LOF) mutations in critical genes in a pathway, or different mutations disrupting regulatory sites of such genes, may suffice to mediate the chronic hypoxia response.
In FIGURE 4, we show a genetic network constructed by GeneMania (32) from the genes reported in the studies appearing in Tables 1 and 2. The network includes connections representing physical (protein-protein) interactions as well as known pathways. GeneMania reports a statistically significant enrichment of several relevant biological processes, including response to hypoxia (FDR = 2.28 × 10−6), blood circulation (FDR = 2.06 × 10−3), endothelial cell proliferation (FDR = 4.76 × 10−3), and response to oxidative stress (FDR = 9.98 × 10−3). In addition, we note that to a certain extent the network segregates into components that correspond to underlying physiological processes. Importantly, genes observed across different populations are often present in the same component, further supporting the hypothesis of similarity at the process level.
FIGURE 4.

GeneMania (32) network constructed from candidate genes for adaptation to hypoxia
The network contains two types of edges: physical interaction (red) and known pathways (blue), and includes genes from Tables 1 and 2 (green, blue, and yellow circles), and additional genes with direct connections (gray circles). Genes with no connecting edges are not shown. Genes are shaded according to the geographical region in which they were identified. Note that many genes from the hypoxia response pathway are directly implicated in multiple populations. The hypoxia response directly affects metabolism. Specifically, the transcription factor HIF1A also upregulates Angiopoietin-like protein 4 (26), which in turn regulates the PPAR-dependent expression of LIPE. Other genes impacted by hypoxia involve the vascular system, such as the vasoconstrictor EDNRB, and MAP kinase 2, which influences pulmonary vascular permeability (12). The Fanconi anemia complex genes [which also complex with Spectrin (SPTA)] are key members of a DNA repair pathway that are regulated by hypoxic stress. In addition, the FANCG gene interacts with cytochrome P450 protein CYP2E1 (14). Together, the studies demonstrate the complex, multi-locus adaptation to hypoxia achieved by different populations.
Future Directions
Although there is little doubt that genetic factors underlie human adaptation to high altitude, there is a paucity of investigations into the role of epigenetics in these adaptations. There is reason to suspect that the harsh environmental stress at high altitudes may cause changes in DNA methylation or histone modification. Indeed, a study by Hartley et al. (19) showed experimentally that major epigenetic changes occur in a culture system using primary neurons. Moreover, preliminary experiments from our laboratory have recently demonstrated that even a short period of hypoxia induces many changes in DNA methylation that last for weeks after exposure (Zhou D, Haddad GG, unpublished observations).
Another important area is proving causation when genes and pathways are identified as candidates. Zhou et al. (42, 51) recently used fibroblasts from skin biopsies in conjunction with reprogramming strategies to study the role of candidate genes in hypoxia tolerance. In addition, Zhau et al. (48) studied signaling pathways that may underlie causation in human (in vitro) models of adaptation and maladaptation to hypoxia.
The accelerated rate of discovery using newer technologies and the potential for rapid functional validation using reprogrammed iPSC underscore the opportunities in genetics-based approaches. We stress that the methodology is not limited to hypoxia tolerance and can be applied to any condition caused by evolutionary constraints, including adaptation to parasitic infections, and (through a study of somatic variation in individual cells) to cancer progression. In this domain, we may therefore be reaching the stage for studies that aim at understanding the cell physiology and biology of adaptation.
Finally, it is important to highlight here that a number of available drugs target some of the gene products reported by the studies on high-altitude adaptation. For example, PPAR(alpha), EDNRB, EPAS1, and EGLN1, among others, are targeted by existing drugs or compounds (16). It remains an intriguing possibility that some of these drugs could direct new treatments for CMS and other ischemic diseases.
Acknowledgments
The authors thank Efrat Golan, Matan Hofree, and Patricia Spindler for technical assistance, as well as two anonymous reviewers for insightful comments.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grant 1P01 HL-098053 to G.G.H., NSF grant CCF-1115206 to V.B., and NIH grants U54-HL-108460 and 1P01 HD-070494 to V.B. V.B. was also supported in part by NIH Grant 5RO1-HG-004962.
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: R.R. and D.Z. performed experiments; R.R., D.Z., V.B., and G.G.H. analyzed data; R.R., D.Z., V.B., and G.G.H. interpreted results of experiments; R.R., D.Z., and V.B. prepared figures; R.R., D.Z., V.B., and G.G.H. drafted manuscript; R.R., D.Z., V.B., and G.G.H. edited and revised manuscript; R.R., D.Z., V.B., and G.G.H. approved final version of manuscript; D.Z., V.B., and G.G.H. conception and design of research.
References
- 1.Alkorta-Aranburu G, Beall CM, Witonsky DB, Gebremedhin A, Pritchard JK, Di Rienzo A. The genetic architecture of adaptations to high altitude in Ethiopia. PLos Genet 8: e1003110, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azad P, Zhou D, Zarndt R, Haddad GG. Identification of genes underlying hypoxia tolerance in Drosophila by a P-element screen. G3 (Bethesda) 2: 1169–1178, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beall CM. Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxia. Integr Comp Biol 46: 18–24, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Beall CM. Tibetan and Andean contrasts in adaptation to high-altitude hypoxia. Adv Exp Med Biol 475: 63–74, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Beall CM, Brittenham GM, Strohl KP, Blangero J, Williams-Blangero S, Goldstein MC, Decker MJ, Vargas E, Villena M, Soria R, Alarcon AM, Gonzales C. Hemoglobin concentration of high-altitude Tibetans and Bolivian Aymara. Am J Phys Anthropol 106: 385–400, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, Li JC, Liang Y, McCormack M, Montgomery HE, Pan H, Robbins PA, Shianna KV, Tam SC, Tsering N, Veeramah KR, Wang W, Wangdui P, Weale ME, Xu Y, Xu Z, Yang L, Zaman MJ, Zeng C, Zhang L, Zhang X, Zhaxi P, Zheng YT. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA 107: 11459–11464, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beall CM, Song K, Elston RC, Goldstein MC. Higher offspring survival among Tibetan women with high oxygen saturation genotypes residing at 4,000 m. Proc Natl Acad Sci USA 101: 14300–14304, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet 6: 9–23, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Bigham A, Bauchet M, Pinto D, Mao X, Akey JM, Mei R, Scherer SW, Julian CG, Wilson MJ, Lopez Herraez D, Brutsaert T, Parra EJ, Moore LG, Shriver MD. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLos Genet 6: e1001116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 84: 210–223, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chailangkarn T, Acab A, Muotri AR. Modeling neurodevelopmental disorders using human neurons. Curr Opin Neurobiol 22: 785–790, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damarla M, Hasan E, Boueiz A, Le A, Pae HH, Montouchet C, Kolb T, Simms T, Myers A, Kayyali US, Gaestel M, Peng X, Reddy SP, Damico R, Hassoun PM. Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLos One 4: e4600, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diop SB, Bodmer R. Drosophila as a model to study the genetic mechanisms of obesity-associated heart dysfunction. J Cell Mol Med 16: 966–971, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Futaki M, Igarashi T, Watanabe S, Kajigaya S, Tatsuguchi A, Wang J, Liu JM. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis 23: 67–72, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert LI. Drosophila is an inclusive model for human diseases, growth and development. Mol Cell Endocrinol 293: 25–31, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, Miller CA, Subramanian J, Govindan R, Kumar RD, Bose R, Ding L, Walker JR, Larson DE, Dooling DJ, Smith SM, Ley TJ, Mardis ER, Wilson RK. DGIdb: mining the druggable genome. Nat Methods 10: 1209–1210, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grotewiel MS, Martin I, Bhandari P, Cook-Wiens E. Functional senescence in Drosophila melanogaster. Ageing Res Rev 4: 372–397, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Hanaoka M, Droma Y, Basnyat B, Ito M, Kobayashi N, Katsuyama Y, Kubo K, Ota M. Genetic variants in EPAS1 contribute to adaptation to high-altitude hypoxia in Sherpas. PLos One 7: e50566, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartley I, Elkhoury FF, Heon Shin J, Xie B, Gu X, Gao Y, Zhou D, Haddad GG. Long-lasting changes in DNA methylation following short-term hypoxic exposure in primary hippocampal neuronal cultures. PLos One 8: e77859, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu T. Complex cellular functions of the von Hippel-Lindau tumor suppressor gene: insights from model organisms. Oncogene 31: 2247–2257, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics 132: 583–589, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huerta-Sanchez E, Degiorgio M, Pagani L, Tarekegn A, Ekong R, Antao T, Cardona A, Montgomery HE, Cavalleri GL, Robbins PA, Weale ME, Bradman N, Bekele E, Kivisild T, Tyler-Smith C, Nielsen R. Genetic signatures reveal high-altitude adaptation in a set of ethiopian populations. Mol Biol Evol 30: 1877–1888, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42: 348–354, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lachance J, Tishkoff SA. SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. Bioessays 35: 780–786, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lessing D, Bonini NM. Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat Rev Genet 10: 359–370, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Ge C, Zhao F, Yan M, Hu C, Jia D, Tian H, Zhu M, Chen T, Jiang G, Xie H, Cui Y, Gu J, Tu H, He X, Yao M, Liu Y, Li J. Hypoxia-inducible factor 1 alpha-activated angiopoietin-like protein 4 contributes to tumor metastasis via vascular cell adhesion molecule-1/integrin beta1 signaling in human hepatocellular carcinoma. Hepatology 54: 910–919, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Lu B. Recent advances in using Drosophila to model neurodegenerative diseases. Apoptosis 14: 1008–1020, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michno K, van de Hoef D, Wu H, Boulianne GL. Modeling age-related diseases in Drosophila: can this fly? Curr Topics Dev Biol 71: 199–223, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Monge CC, Whittembury J. Chronic mountain sickness. Johns Hopkins Med J 139, Suppl: 87–89, 1976. [PubMed] [Google Scholar]
- 30.Moore LG. Human genetic adaptation to high altitude. High Alt Med Biol 2: 257–279, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Moore LG, Niermeyer S, Zamudio S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol Suppl 27: 25–64, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol 9, Suppl 1: S4, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev 63: 411–436, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng Y, Yang Z, Zhang H, Cui C, Qi X, Luo X, Tao X, Wu T, Ouzhuluobu Basang Ciwangsangbu Danzengduojie Chen H, Shi H, Su B. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol Biol Evol 28: 1075–1081, 2011. [DOI] [PubMed] [Google Scholar]
- 35.Polesello C, Roch F, Gobert V, Haenlin M, Waltzer L. Modeling cancers in Drosophila. Prog Mol Biol Trans Sci 100: 51–82, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Scheinfeldt LB, Soi S, Thompson S, Ranciaro A, Woldemeskel D, Beggs W, Lambert C, Jarvis JP, Abate D, Belay G, Tishkoff SA. Genetic adaptation to high altitude in the Ethiopian highlands. Genome Biol 13: R1, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, Akey JM, Jones KW. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum Genomics 1: 274–286, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, Bai Z, Lorenzo FR, Xing J, Jorde LB, Prchal JT, Ge R. Genetic evidence for high-altitude adaptation in Tibet. Science 329: 72–75, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Stevenson TJ, Trinh T, Kogelschatz C, Fujimoto E, Lush ME, Piotrowski T, Brimley CJ, Bonkowsky JL. Hypoxia disruption of vertebrate CNS pathfinding through ephrinB2 Is rescued by magnesium. PLos Genet 8: e1002638, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Udpa N, Ronen R, Zhou D, Liang J, Stobdan T, Appenzeller O, Yin Y, Du Y, Guo L, Cao R, Wang Y, Jin X, Huang C, Jia W, Cao D, Guo G, Claydon VE, Hainsworth R, Gamboa JL, Zibenigus M, Zenebe G, Xue J, Liu S, Frazer KA, Li Y, Bafna V, Haddad GG. Whole genome sequencing of Ethiopian Highlanders reveals conserved hypoxia tolerance genes. Genome Biol 15: R36, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vidal M, Cagan RL. Drosophila models for cancer research. Curr Opin Genet Dev 16: 10–16, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol 4: e72, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet 14: 507–515, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu S, Li S, Yang Y, Tan J, Lou H, Jin W, Yang L, Pan X, Wang J, Shen Y, Wu B, Wang H, Jin L. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol Biol Evol 28: 1003–1011, 2011. [DOI] [PubMed] [Google Scholar]
- 47.Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng H, Liu T, He W, Li K, Luo R, Nie X, Wu H, Zhao M, Cao H, Zou J, Shan Y, Li S, Yang Q, Asan Ni P, Tian G, Xu J, Liu X, Jiang T, Wu R, Zhou G, Tang M, Qin J, Wang T, Feng S, Li G, Huasang Luosang J, Wang W, Chen F, Wang Y, Zheng X, Li Z, Bianba Z, Yang G, Wang X, Tang S, Gao G, Chen Y, Luo Z, Gusang L, Cao Z, Zhang Q, Ouyang W, Ren X, Liang H, Huang Y, Li J, Bolund L, Kristiansen K, Li Y, Zhang Y, Zhang X, Li R, Yang H, Nielsen R, Wang J. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329: 75–78, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao H, Gu X, Chailangkarn T, Appenzeller O, Poulsen O, Zhou D, Muotri A, Haddad G. Study of chronic mountain sickness using inducible pluripotent stem cells. Soc Neurosci Abstract: 2013. [Google Scholar]
- 49.Zhou D, Haddad GG. Genetic analysis of hypoxia tolerance and susceptibility in Drosophila and humans. Annu Rev Genomics Hum Genet 14: 25–43, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou D, Udpa N, Gersten M, Visk DW, Bashir A, Xue J, Frazer KA, Posakony JW, Subramaniam S, Bafna V, Haddad GG. Experimental selection of hypoxia-tolerant Drosophila melanogaster. Proc Natl Acad Sci USA 108: 2349–2354, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou D, Udpa N, Ronen R, Stobdan T, Liang J, Appenzeller O, Zhao HW, Yin Y, Du Y, Guo L, Cao R, Wang Y, Jin X, Huang C, Jia W, Cao D, Guo G, Gamboa JL, Villafuerte F, Callacondo D, Xue J, Liu S, Frazer KA, Li Y, Bafna V, Haddad GG. Whole-genome sequencing uncovers the genetic basis of chronic mountain sickness in Andean highlanders. Am J Hum Genet 93: 452–462, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou D, Visk DW, Haddad GG. Drosophila, a golden bug, for the dissection of the genetic basis of tolerance and susceptibility to hypoxia. Pediatr Res 66: 239–247, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]