

Background: Hdac7 is a transcriptional co-repressor in skeletal tissues, but its role in chondrocytes is undefined.
Results: Proteasomal degradation reduced Hdac7 levels during chondrogenic maturation and conditional deletion of Hdac7 in chondrocytes increased proliferation and β-catenin levels.
Conclusion: Hdac7 degradation enhances β-catenin transcriptional activity in growth plate chondrocytes.
Significance: Suppressing Hdac7 levels increases chondrocyte proliferation and promotes cartilage formation and regeneration.
Keywords: Beta-catenin (β-catenin), Cartilage, Growth Plate, Histone Deacetylase (HDAC), Insulin, ATDC5 Cells, Histone Deacetylase 7 (Hdac7)
Abstract
Histone deacetylases (Hdacs) regulate endochondral ossification by suppressing gene transcription and modulating cellular responses to growth factors and cytokines. We previously showed that Hdac7 suppresses Runx2 activity and osteoblast differentiation. In this study, we examined the role of Hdac7 in postnatal chondrocytes. Hdac7 was highly expressed in proliferating cells within the growth plate. Postnatal tissue-specific ablation of Hdac7 with a tamoxifen-inducible collagen type 2a1-driven Cre recombinase increased proliferation and β-catenin levels in growth plate chondrocytes and expanded the proliferative zone. Similar results were obtained in primary chondrocyte cultures where Hdac7 was deleted with adenoviral-Cre. Hdac7 bound β-catenin in proliferating chondrocytes, but stimulation of chondrocyte maturation promoted the translocation of Hdac7 to the cytoplasm where it was degraded by the proteasome. As a result, β-catenin levels and transcription activity increased in the nucleus. These data demonstrate that Hdac7 suppresses proliferation and β-catenin activity in chondrocytes. Reducing Hdac7 levels in early chondrocytes may promote the expansion and regeneration of cartilage tissues.
Introduction
Endochondral ossification is a developmental process of bone formation that is recapitulated during fracture repair, heterotopic ossification, and osteoarthritis (1). Proceeding through a defined program, endochondral ossification begins with mesenchymal condensations that give rise to the cartilaginous skeletal template that eventually becomes mineralized (2). Coordination of each step, including proper maturation of chondrocytes within the cartilaginous phase, is essential for skeletal formation and longitudinal growth. Disruption of any phase of chondrocyte maturation within the growth plate alters bone length, shape and/or strength. Many growth factors, including insulin, insulin-like growth factor (Igf),2 bone morphogenic proteins (BMPs), transforming growth factor β (Tgfβ), and Wnts regulate the endochondral transitions between chondrogenesis, chondrocyte proliferation, chondrocyte hypertrophy, osteoblast recruitment, and chondrocyte apoptosis, all of which are required for optimal bone formation.
Histone deacetylases (Hdacs) remove acetyl groups from lysine residues to control a number of cellular processes, including chromatin compaction and gene expression. Numerous Hdacs control chondrocyte differentiation during endochondral ossification (3–6). For example, Hdac4 suppresses chondrocyte hypertrophy by repressing Runx2 activity and facilitating PTHrP signaling (6, 7) and Hdac5/Hdac9 double mutant mice exhibit severe growth retardation (8). Hdac7 has structural and functional similarity to Hdacs 4, 5, and 9. Class IIa Hdacs are phosphorylated by calcium/calmodulin dependent-kinase (CaMK) and protein kinase D (PKD) in response to kinase-dependent signaling such as that elicited by vascular endothelial growth factor (Vegf) or BMP2 (9–13). This promotes interactions between Hdacs and 14-3-3 proteins and cytosolic localization (14). Hdac7 possesses little intrinsic deacetylase activity and requires association with the class I Hdac, Hdac3, to suppress gene expression (15). Germline deletion of Hdac7 causes early embryonic lethality prior to skeletal formation (16), and its role in skeletal development and endochondral ossification is unknown.
β-Catenin-dependent Wnt signaling (commonly referred to as the canonical pathway) is an important, ubiquitously utilized, developmental pathway. β-Catenin levels are strictly controlled through growth factor-elicited degradation. In cartilage, β-catenin has different functions during chondrocyte commitment, proliferation and hypertrophy. Genetic experiments show that β-catenin inhibits commitment of mesenchymal progenitors to the chondrocyte linage and promotes osteoblast differentiation (17, 18). In contrast, in committed chondrocytes β-catenin promotes proliferation and differentiation (19). Transgenic expression of the inhibitor of β-catenin (ICAT) suppresses chondrocyte proliferation and delays ossification (20). In addition, β-catenin activity in chondrocytes may also orchestrate osteoclast differentiation (21). Thus, β-catenin plays crucial roles in cartilage tissue.
To define the role of Hdac7 during endochondral ossification we conditionally ablated Hdac7 expression in proliferating chondrocytes using tamoxifen-inducible collagen type 2a1-driven Cre recombinase. Postnatal deletion of Hdac7 increased β-catenin levels in growth plate cartilage to promote chondrocyte proliferation, but delayed type X collagen synthesis to prevent peak bone mass accrual. Similar results were observed in chondrocyte micromass cultures in vitro. These data indicate that Hdac7 slows chondrocyte proliferation to control cartilage tissue maturation and endochondral ossification.
MATERIALS AND METHODS
Generation of Hdac7 Conditional Knock-out Mice
Mice harboring two copies of the Hdac7 allele with loxP sites flanking a 300-kp region encompassing exons 2 to 5 (Hdac7fl/fl) (18) were crossed with Col2a1Cre mice (16, 22). Hdac7fl/fl animals were also crossed with mice homozygous for the tamoxifen-inducible Col2a1CreERT transgene to obtain Hdac7fl/fl:Col2CreERT mice (16, 23). These animals are referred to as Hdac7Col2ERT mice in this report. Animals were housed in an accredited facility under a 12-hour light/dark cycle and provided water and food (PicoLab® Rodent Diet 20, LabDiet) ad libitum. Tamoxifen (1 mg, Sigma Aldrich) was dissolved in corn oil and injected at postnatal day 5 (P5) as described by Nakamura et al. (23). For BrdU labeling studies, vehicle- (n = 4) or tamoxifen (n = 5)-treated mice were injected when they were 9 days old (P9) with 50 μg of 5-bromo-2deoxyuridine (BrdU) labeling reagent (Invitrogen) per kg body weight two hours prior to euthanasia. All animal research was performed in adherence with the National Institutes of Health and the Institute of Laboratory Animal Resources, National Research Council guidelines. The Mayo Clinic Institutional Animal Care and Use Committee preapproved all animal studies.
Isolation and Culture of IMAC Cells
Immature mouse articular chondrocytes (IMACs) were extracted from P5 mice as previously described by Gosset et al. (24). Briefly, cartilage from the femoral head and tibial plateau were digested twice in 3 mg/ml collagenase for 1 h and then overnight in 0.5 mg/ml collagenase. The resulting cell suspension was placed into micromasses consisting of 2 × 105 cells in a 10 μl drop of DMEM. After 1 h, chondrogenic medium (DMEM, supplemented with 5% FBS, 1% antimycotic/antibiotic and 1× mixture of insulin, transferrin, and selenium (ITS) (Invitrogen, Carlsbad, CA)) was added to the cultures for the indicated periods of time (3, 25, 26). Cells were infected with adenoviral (Ad)-GFP or Ad-Cre (MOI = 10) on day 0 and harvested for analysis on day 3.
ATDC5 Cell Culture and Transfection
ATDC5 cells were cultured at a density of 6 × 103cells/cm2 in DMEM, 5% FBS, 1% antimycotic/antibiotic. DNA plasmid mixtures 100 ng TOPFLASH (luciferase reporter), 10 ng of pRL-null (Renilla luciferase reporter vector and internal control), 150 ng pcDNA3-β-catenin, and 150 ng pcDNA3 or pcDNA-Hdac7 were transfected into cells with Lipofectamine (Invitrogen) at a 3:1 ratio within 24 h of seeding the cells. All transfections were done in triplicate. Following transfection, cells were cultured in 5% FBS, 1% antimycotic/antibiotic, and 1× ITS. Luciferase activities were assayed 48 h later with the Dual Luciferase Reporter System (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity and fold changes relative to control transfections were calculated.
For immunoprecipitation experiments, cells were transfected with Flag-tagged Hdac7 and Myc-tagged β-catenin. For fractionation and proteasome inhibitor experiments, cells were cultured at a density of 6 × 103 cells/cm2 in DMEM, 5% FBS, 1% antimycotic/antibiotic. After 3 days, cells were cultured for 2 h in DMEM lacking FBS and antimycotic/antibiotics. For proteasome inhibitor experiments, 5 nm MG132 (Millipore) or vehicle (DMSO) was included. After this serum starvation, 1× ITS solution or 10 ng/ml Igf1 was added to the culture medium, and cells were incubated for an additional 24 h.
Cytosolic and Nuclear Fractionation
ATDC5 cells were treated as per experimental design, washed, and harvested in ice-cold PBS. Cells were homogenized and pelleted in ice-cold PBS. Each pellet was then resuspended in 10 mm Tris-HCl (pH 7.84), 140 mm NaCl, 1.5 mm MgCl2, 0.5% Nonidet P-40, and 1× Inhibitor mixture (Roche, Mannheim, Germany). The resulting lysate was cleared by centrifugation at 2,000 rpm for 5 min at 4 C. The supernatant containing the cytoplasmic extract was retained for Western blotting. The nuclear pellet was then washed twice with ice-cold PBS and suspended in 20 mm HEPES (pH 7.9), 1 m NaCl, 0.2 mm EDTA, 0.1 mm EGTA, 1.5 mm MgCl2, 1.2 mm PMSF, 20% glycerol, and 1× Inhibitor mixture (Roche) and incubated on ice for 5 min. The nuclear lysate was cleared by centrifugation at 5,000 rpm for 5 min at 4 °C. The resulting supernatant was collected as a nuclear extract and used for Western blotting.
RNA Isolation and Real-Time PCR
Total RNA was extracted from IMAC cultures using TRIzol (Invitrogen) and chloroform. mRNA (2 μg) was reverse transcribed using the SuperScript III first-strand synthesis system (Invitrogen). The resulting cDNAs were used to assay gene expression via real-time PCR using the following gene-specific primers: Aggrecan (F: 5′-CCGCTTGCCAGGGGGAGTTG-3′, R: 5′-GATGATGGGCGCACGCCGTA-3′), Axin2 (F: 5′-CGCCACCAAGACCTACATACG-3′, R: 5′-ACATGACCGAGCCGATCTGT-3′), Cyclin D3 (F: 5′-CGTCGCTTCTCCTAGGACTC-3′, R: 5′-AACACAGCAGCTCCATCCTG-3′), Gapdh (F: 5′-GGGAAGCCCATCACCATCTT-3′, R: 5′-GCCTCACCCCATTTGATGTT-3′), Hdac7 (F: 5′-AACGGCACGTGCTCCTCTAC-3′, R: 5′-TCCTGTAAGCTTCCCGTTATCC-3′), Ihh (F: 5′-GCTTTCCTGCCGGAGCCCAG-3′, R: 5′-GGTGGGGGTCCCATCCTCCC-3′), Lef1 (F; 5′-GATCCCCTTCAAGGACGAAG-3′, R: 5′-GGCTTGTCTGACCACCTCAT-3′), p21 (F: 5′-GAACATCTCAGGGCCGAAAA-3′, R: 5′-TGCGCTTGGAGTGATAGAAATC-3′), Runx2 (F: 5′-GGCACAGACAGAAGCTTGATG-3′, R: 5′-GAATGCGCCCTAAATCACTGA-3′) Sox9 (F: 5′-AGGAAGCTGGCAGACCAGTA-3′, R: 5′-CGTTCTTCACCGACTTCCTC-3′), Type 2a1 collagen (F: 5′-ACTGGTAAGTGGGGCAAGAC-3′, R: 5′-CCACACCAAATTCCTGTTCA-3′), Type X collagen (F: 5′-CTTTGTGTGCCTTTCAATCG-3′, R: 5′-GTGAGGTACAGCCTACCAGTTTT-3′), Vegf (F: 5′-AGAGGCTTGGGGCAGCCGAG-3′, R: 5′-ACTCCCGGGCTGGTGAGTCC-3′) (27). Fold changes in gene expression for each sample were calculated using the 2(−ΔΔCt) method relative to control after normalization of gene-specific Ct values to Gapdh Ct values. Each experiment was performed in triplicate. The average of three experiments is reported.
Immunoprecipitation and Western Blotting
For immunoprecipitation experiments, lysates were collected in radioimmune precipitation assay (RIPA) buffer and incubated overnight with 2 μg of each indicated antibody and in the presence of 20 μl protein G dynabeads (Invitrogen). Beads were collected magnetically, washed, and eluted with SDS sample buffer (0.1% glycerol, 0.01% SDS, 0.1 m Tris, pH 6.8). For Western blotting, cell lysates were collected in SDS sample buffer on ice. After determining total protein concentration using the Bio-Rad DC assay (Bio-Rad), 20 μg of total protein per sample was resolved by SDS-PAGE and transferred to nitrocellulose. Western blotting was performed with antibodies (1:2000 dilution) for p-Thr202/Tyr204 ERK1/2, total ERK, Lef1, Cre, Myc (Cell Signaling Technologies, Boston, MA), Hdac7, (AbCam, Cambridge, MA), histone 3 (H3), total β-catenin (BD Transduction Laboratories, San Jose, CA), active β-catenin (unphosphorylated at Ser37 or Thr41) (Millipore), Flag (Sigma-Aldrich) or actin (Santa Cruz Biotechnology, Santa Cruz, CA), and corresponding secondary antibodies conjugated to horseradish peroxidase (HRP) (Santa Cruz Biotechnology). Protein detection was accomplished using the Supersignal West Femto Chemiluminescent Substrate (Pierce). When necessary blots were stripped using Restore Western blot Stripping Buffer (Pierce). Experiments were repeated as detailed in the figure legends with representative data shown.
Alcian Blue Staining
IMAC cultures were fixed with 10% neutral buffered formalin and stained with Alcian blue (1% Alcian blue, 3% acetic acid) for 2 h. Tibias were fixed in 10% neutral buffered formalin, decalcified in 15% EDTA for 7 days, paraffin embedded, sectioned, and stained with Alcian blue and Eosin.
Immunohistochemical Staining
Tibias from P9 or 4-week-old mice were fixed in 10% neutral buffered formalin, decalcified in 15% EDTA for 5 days, and embedded in paraffin. Immunohistochemical staining was performed with antibodies directed to BrdU, Hdac7, Ki67 (Cell Signaling Technologies), p21 (Santa Cruz Biotechnology), unphosphorylated active β-catenin, or IgG isotype control. Detection was accomplished using the Mouse and Rabbit Specific HRP (ABC) Detection IHC Kit (AbCam) using 3,3′-diaminobenzidine as a chromogen (Sigma Aldrich). Sections were counterstained with Alcian blue.
In Situ Hybridization
Tibias from P9 or 4 week-old mice were fixed in 10% neutral buffered formalin, decalcified in 15% EDTA for 5 days, and embedded in paraffin. In situ hybridization (ISH) was performed using digoxigenin (DIG)-labeled type X collagen alpha 1 (ColXa1) probes as previously described (28).
Micro-computed Tomography
Bone mineral density and architecture of the secondary spongiosa in the distal femurs of male mice was evaluated using ex vivo micro-computed tomography (μCT) imaging (μCT35 scanner; Scanco Medical AG, Basserdorf, Switzerland). Trabecular bone scans were performed with 7 μm voxel size using an energy setting of 70 kVp and an integration time of 300 ms.
Dual-energy X-ray Absorptiometry Analysis
Cortical bone mineral density (BMD) was assessed via dual-energy x-ray absorptiometry (DXA) scanning (PIXImus, GE Healthcare) of the femoral midshaft of 8-week-old mice.
Image Quantitation
Images were digitally scanned or collected using phase contrast microscopy. Densitometric values of Western blots were obtained by measuring the mean gray values using Image J software. Total plate depths were determined by taking the average of 10 measurements across the imaged growth plate from each mouse (vehicle: n = 3, tamoxifen: n = 3) using Image J software. Proliferative and hypertrophic cells were distinguished using type X collagen ISH. Proliferative cells were defined as type X collagen-negative cells and hypertrophic chondrocytes were defined as type X collagen-positive cells. The average of 10 measurements per zone across the imaged growth plate from each mouse (vehicle: n = 3, tamoxifen: n = 3) using Image J software was determined.
Statistical Analysis
Experiments were repeated a minimum of three times. Data obtained are presented as the means ± S.E. of the mean (S.E.). p values were determined with the Student's t test.
RESULTS
Hdac7 Deficiency Promotes Chondrocyte Proliferation
To understand the function of Hdac7 during endochondral bone development, its expression pattern was determined in growth plate chondrocytes by IHC. Hdac7 was highly expressed in resting and proliferating chondrocytes, but noticeably lower in hypertrophic cells (Fig. 1A).
FIGURE 1.

Hdac7 deletion expands the growth plate proliferative zone and increases chondrocyte numbers. A, 4-week-old Hdac7Col2ERT mice were injected with vehicle (n = 4) or tamoxifen (n = 5). Paraffin-embedded sections of tibias mice were incubated with an IgG isotype control antibody (upper row) or an anti-Hdac7 antibody (lower row). Shown are representative 20× images of the growth plates. The proliferative (P) and hypertrophic (H) zones are designated in select panels. B–F, 9-day-old Hdac7Col2ERT mice injected with vehicle (n = 4) or tamoxifen (n = 5) and BrdU. B, paraffin-embedded sections (10×) of tibias from BrdU-injected Hdac7Col2ERT mice that were incubated with an IgG isotype control antibody or an anti-BrdU antibody. C, in situ hybridization using DIG-labeled type X collagen-specific probes was performed on paraffin-embedded tibia sections. Probes were detected using an anti-DIG antibody or corresponding IgG control. Shown are representative 40× images. D, average total (E) proliferative and hypertrophic zone depths within the growth plate were determined using image J software. F, average number of proliferative cells per tissue area was determined using Image J software. *, p < 0.05 compared with control. G, average femur lengths of 4 week-old vehicle or tamoxifen-injected animals.
Since type 2a1 collagen is expressed in the regions of high Hdac7 expression, we crossed Hdac7fl/fl mice to animals expressing Col2a1-Cre to determine if Hdac7 is required for growth plate cartilage maintenance and endochondral ossification. Hdac7 conditional mutants were born at lower than expected ratios, indicating partially penetrant lethality (data not shown). Therefore, we deleted it postnatally by crossing the Hdac7fl/fl mice to animals expressing a tamoxifen-inducible Col2a1-Cre. Tamoxifen injection on P5 substantially reduced Hdac7 expression in all growth plate chondrocytes (Fig. 1A). More BrdU-positive (Fig. 1B) and Ki67-positive (data not shown) cells were observed after tamoxifen injection, but type X collagen expression was reduced in the hypertrophic zone (Fig. 1C). Based on these molecular markers, the proliferative zone occupied 8% more of the growth plate and the size of the hypertrophic zone was reduced in Hdac7Col2ERT mice (Fig. 1, E and F). There was no significant change in overall growth plate depth (Fig. 1D). Tamoxifen injection did not change growth plate depth or zone percentages in control C57Bl6 animals (data not shown). In addition to the expansion of the proliferative zone, the number of cells per tissue area in this zone was significantly increased in tamoxifen injected Hdac7Col2ERT animals by 18%. One consequence of these disruptions in the endochondral ossification program was a 15% reduction in femur length in Hdac7Col2ERT mice (Fig. 1G).
Developmental growth plate disruptions can also impact cancellous bone density because the cartilage matrix acts as a scaffold for osteoblasts and osteoclasts. To determine if Hdac7 deficiency affected bone density, Hdac7Col2ERT mice were injected at P5 with tamoxifen or vehicle (corn oil) and bone density and architecture were measured at 8 weeks of age by μCT imaging (Fig. 2). Bone volume density (BV/TV) was decreased by 37% in tamoxifen-treated animals. Trabecular numbers (Tb N) were reduced 53%, resulting in increased trabecular spacing (Tb Sp). Trabecular thickness (Tb Th) was not affected. No changes in cortical bone density were observed. These data demonstrate that promotion of chondrocyte proliferation by Hdac7 deficiency negatively impacts bone accrual and lengthening.
FIGURE 2.
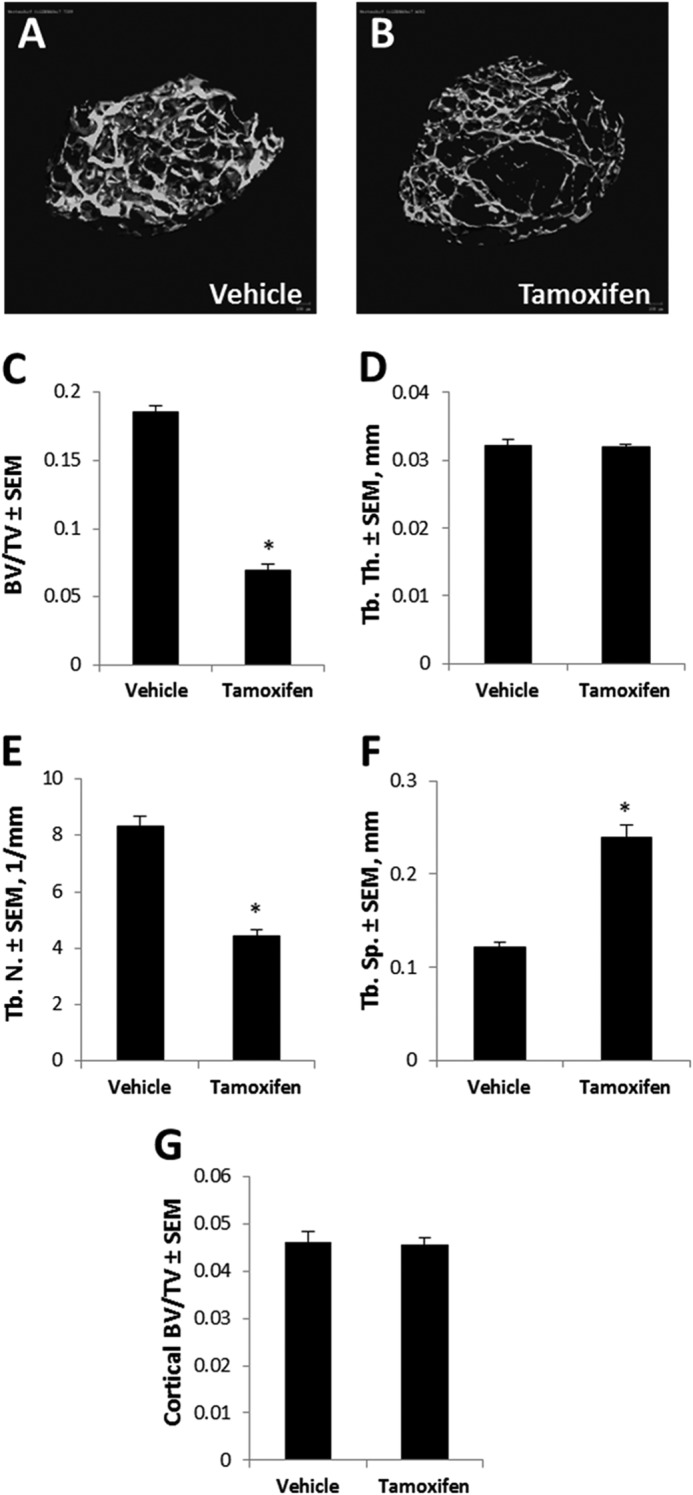
Cartilage-specific deletion of Hdac7 decreases bone accrual. A and B, representative three-dimensional reconstructions of trabecular bone from the distal femur of Hdac7Col2ERT mice treated with vehicle or tamoxifen. C, bone volume per total tissue volume (BV/TV), D, trabecular thickness (Tb. Th.), E, trabecular number (Tb. N.), and F, trabecular spacing (Tb. Sp.) from the distal femur are also shown (n = 5 per group). G, cortical bone density was measure at the femoral diaphysis. *, p < 0.05 compared with control.
Hdac7 Deficiency Enhances β-Catenin Activity
To further understand the effects of Hdac7 deficiency on chondrocytes, we collected primary immature mouse articular chondrocytes (IMACs) from Hdac7Col2ERT mice and placed them into micromass cultures. Cells were infected with adenoviruses expressing GFP (Ad-GFP) or Cre recombinase (Ad-Cre). Hdac7 protein and mRNA levels were substantially reduced in cells infected with Ad-Cre (Fig. 3, A and C). Three-day-old micromasses of Hdac7-depleted IMACs had stronger Alcian blue staining, suggesting the presence of more matrix-producing chondrocytes (Fig. 3B). To determine if this phenotype was related to increased cell proliferation, as observed in vivo, or enhanced differentiation to hypertrophy, the expression of many chondrocyte differentiation genes was assessed in these micromasses by qPCR. Multiple chondrogenic genes (e.g. Sox9, Col2a1, Ihh, Runx2, and Vegf) were not altered in Hdac7-depleted cells (Fig. 3, D–H). Aggrecan (Acan) message was modestly increased 1.8-fold, but type X collagen transcripts were reduced by ∼40% (Fig. 3, I and J). In contrast, transcripts for the cell cycle regulator, cyclin D3, were 3-fold more abundant in Hdac7-depleted micromasses while transcript levels for p21, a cyclin-dependent kinase inhibitor, were reduced 40% (Fig. 3, K and L). Protein levels of p21 were also suppressed in tamoxifen-treated animals (Fig. 3O). Together these data demonstrate that Hdac7 depletion has a profound effect on chondrocyte proliferation.
FIGURE 3.

Hdac7 deficiency increases active β-catenin levels and chondrocyte proliferation. A–N, immature mouse articular chondrocytes from Hdac7Col2ERT mice were cultured in micromass and infected with Ad-GFP or Ad-Cre on day 0. A, Western blotting for the indicated proteins was performed on day 3. This experiment was repeated three times, and data from a representative experiment are shown. B, micromasses were fixed on day 3 and stained with Alcian blue. Relative expression levels of (C) Hdac7, (D) Sox9, (E) Type 2a1 collagen, (F) Ihh, (G) Runx2, (H) Vegf, (I) Aggrecan, (J) Type X collagen, (K) Cyclin D3, (L) p21, (M) Lef1, and (N) Axin2 were assayed by qPCR. *, p < 0.05 compared with control. These data are the average of three independent experiments. O and P, 4-week-old Hdac7Col2ERT mice were injected with vehicle (n = 4) or tamoxifen (n = 5). O, paraffin-embedded sections of tibias were incubated with an IgG isotype control antibody (left column) or an antibody that recognizes p21 (right column). Shown are representative 40× images of the growth plate. P, paraffin-embedded sections of tibias were incubated with an IgG isotype control antibody (left column) or an antibody that recognizes unphosphorylated (active) β-catenin (right column). Shown are representative 40× images of the growth plate.
β-Catenin is a well-known inducer of chondrocyte proliferation. Thus, we sought to determine if altering Hdac7 levels affected β-catenin activity in chondrocytes. Levels of unphosphorylated and active β-catenin levels were higher in Hdac7-depleted micromasses, whereas total levels of β-catenin remained constant (Figs. 3A). Immunohistochemistry for unphosphorylated (active) β-catenin showed that tamoxifen-treated, Hdac7-deficient animals exhibited higher levels of active β-catenin within proliferative cells of the growth plate than control animals (Fig. 3P). Transcripts of Lef1 and Axin2, downstream β-catenin target genes, were also elevated in Hdac7-deficient cells (Fig. 3, M and N). Thus, Hdac7 deletion increases β-catenin activity in chondrocytes.
Hdac7 Levels Are Reduced during Chondrogenesis by Proteasomal Degradation
We next sought to determine if Hdac7 and β-catenin formed complexes in chondrogenic ATDC5 cells. Co-immunoprecipitation experiments revealed the association of endogenous Hdac7 with β-catenin (Fig. 4A). Similar complexes were observed in ATDC5 cells transfected with Flag-tagged Hdac7 and Myc-tagged β-catenin (Fig. 4B).
FIGURE 4.
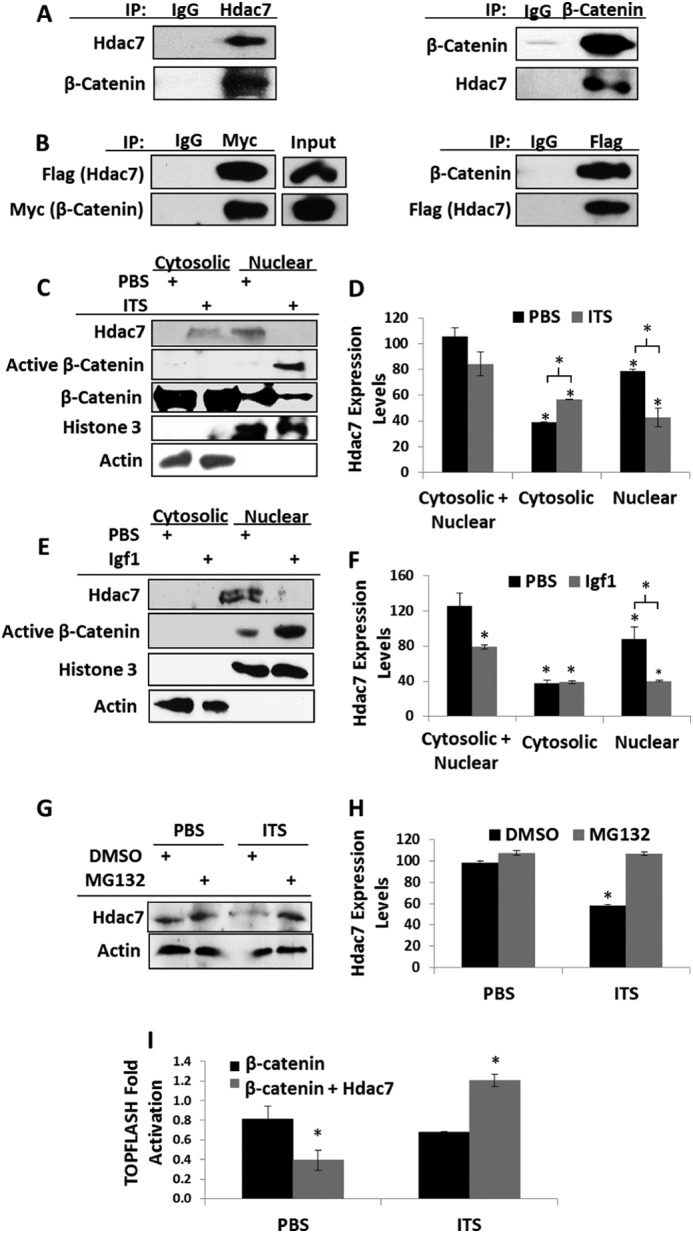
Chondrogenic stimuli promote Hdac7 degradation and β-catenin activity. A, co-immunoprecipitations of Hdac7 and β-catenin endogenous to ATDC5 cells. B, ATDC5 cells were transfected with Flag-tagged Hdac7 and Myc-tagged β-catenin. Co-immunoprecipitations were performed as shown. C and D, ATDC5 cells were serum starved, treated with 1× ITS for 24 h, and then biochemically fractionated into cytosolic and nuclear extracts. C, Western blotting for the indicated proteins was performed and D, the expression levels of Hdac7 in each compartment were quantified, n = 3. E and F, ATDC5 cells were serum starved, treated with 10 ng/ml Igf1 for 24 h, and then biochemically fractionated into cytosolic and nuclear extracts. E, Western blotting for the indicated proteins was performed and F, the expression levels of Hdac7 in each compartment were quantified, n = 3. G and H, ATDC5 cells were serum starved in the presence of 5 nm MG132 or vehicle control. Cells were then treated with 1× ITS for 24 h. G, Western blotting was then performed as indicated and H, the expression levels of Hdac7 were quantified, n = 3. I, ATDC5 cells were transfected with the TOPFLASH reporter, a constitutively active form of β-catenin and pcDNA3 or pcDNA3-Hdac7 expression constructs as indicated. After transfection, cells were exposed to 1× ITS for 48 h. *, p < 0.05 compared with control, n = 3.
To determine if Hdac7 affected β-catenin function during chondrogenesis, ATDC5 cells were cultured in the presence or absence of ITS or Igf1, two potent inducers of chondrogenesis, for 24 h. Both ITS and Igf1 reduced overall cellular Hdac7 levels. Cell fractionation experiments revealed that the remaining Hdac7 in ITS-treated cultures was predominantly in the cytosol (Fig. 4, C–F). Since Hdac7 was predominantly nuclear in untreated cells, we hypothesized that it was translocated to the cytosol where it would be degraded. Indeed, the proteasome inhibitor MG132 attenuated the ITS-induced reductions in Hdac7 levels (Fig. 4, G and H).
The translocation of Hdac7 to the cytosol coincided with increased β-catenin levels in the nucleus (Fig. 4, C and E). To ascertain if Hdac7 attenuated β-catenin transcriptional activity, we co-transfected an Hdac7 expression construct with a constitutively active form of β-catenin and the β-catenin-responsive TOPFLASH reporter into ATDC5 cells and treated cells with ITS. Hdac7 reduced β-catenin-induced TOPFLASH reporter activity in the absence but not the presence of ITS (Fig. 4I). Together, these data demonstrate that Hdac7 suppresses β-catenin driven proliferation of growth plate chondrocytes.
DISCUSSION
Hdacs are emerging as crucial regulators of skeletal development and endochondral ossification. In this study we examined the function of Hdac7 in growth plate chondrocyte differentiation. Hdac7 is robustly expressed in proliferating chondrocytes. Using a tamoxifen-inducible collagen type 2a1-driven Cre recombinase to ablate Hdac7 expression post-natally, we observed greater numbers of proliferating chondrocytes, decreased chondrocyte hypertrophy, increased active β-catenin levels within the growth plate, and decreased bone mass accrual. This observation was recapitulated with in vitro cultured primary chondrocytes using adenoviral Cre-mediated excision of Hdac7. Exposure of ATDC cells to ITS or Igf-1 induced proteasome-mediated degradation of Hdac7 and increased active β-catenin levels (Fig. 5). These data are the first demonstration that Hdac7 has a non-redundant role in regulating chondrocyte differentiation via suppression of chondrocyte proliferation and β-catenin activity.
FIGURE 5.
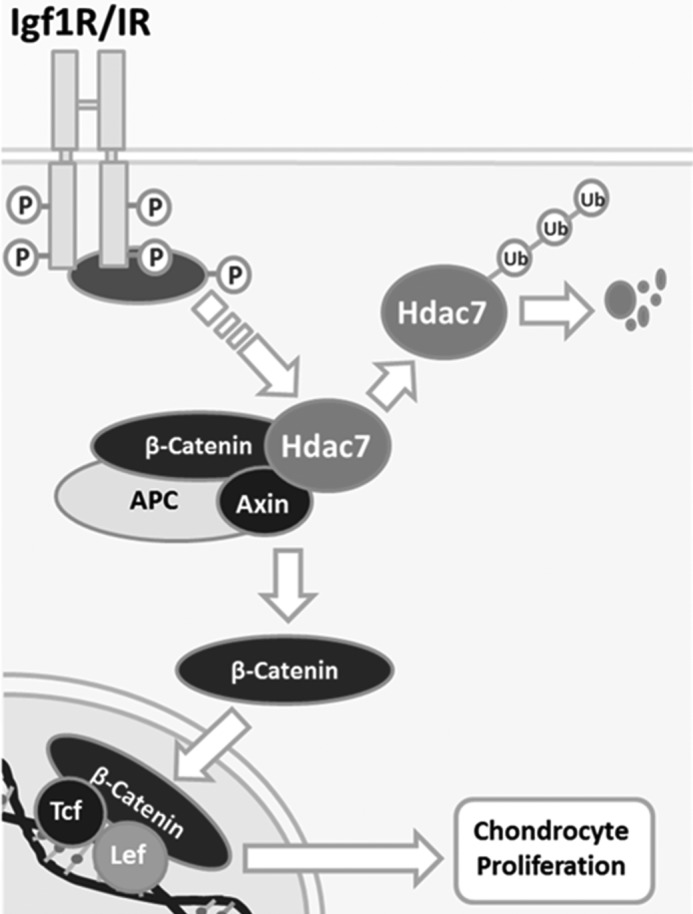
Working model. Igf1/insulin-dependent signaling induces Hdac7 degradation and β-catenin stability.
Hdacs are important modulators of development, cellular differentiation, and responses to growth factors and cytokines. Although Hdacs are critical regulators of gene expression due to their ability to promote chromatin compaction, the non-epigenetic effects of the Hdacs may be equally important. This is especially true of the class IIa Hdacs, like Hdac7, that shuttle between nuclear and cytoplasmic compartments and have little purported intrinsic deacetylase activity. Hdac7 subcellular localization is regulated by extracellular signals and phosphorylation while its promotion of histone deacetylation requires the activity of Hdac3 in the nucleus (15). Importantly, we show that chondrogenic stimuli, including Igf1, regulate Hdac7 function. Thus a role for Hdac7 in modulating signaling via the insulin/Igf1 receptor may also be true of other tissues.
We previously demonstrated that Hdac3 also suppresses chondrocyte hypertrophy (3). Deletion of Hdac3 in osteo/chondroprogenitor cells and committed osteoblasts reduces bone mass (4, 5). Since Hdac7 participates in protein complexes containing Hdac3 and also inhibits the activity of Runx2 (15, 30), we determined if Hdac7 had a role in chondrocyte differentiation. In contrast to Hdacs 3 and 4 that are strongly expressed in prehypertrophic chondrocytes (3, 6), Hdac7 is expressed mainly by proliferating chondrocytes. This suggested that Hdac7 has a function independent of Hdac3 and Hdac4 during chondrocyte differentiation. Indeed, deletion of Hdac7 in type 2al collagen-expressing cells increased chondrocyte proliferation. The difference in the effects of Hdac7 deficiency as compared with either Hdac3 or Hdac4 suggested an alternate mechanism of action that we herein describe. Similar disparities in the function of Hdac3 and Hdac7 on cellular differentiation were observed in osteoclasts, where Hdac3 is required for osteoclast formation, but Hdac7 suppresses osteoclast differentiation by repressing Mitf activity (31). Mitf has no reported functions in chondrocytes. We explored alternative functions of Hdac7 in governing chondrocyte differentiation and demonstrate a novel mechanistic link between Igf1/ITS and Hdac7 that delays chondrocyte hypertrophy.
Germline deletion of Hdac7 causes early embryonic lethality prior to bone development (16). We also observed decreased survival using the Col2a1-Cre to delete Hdac7 during embryonic development (data not shown). To circumvent this lethality, we utilized a tamoxifen-inducible type 2al collagen-driven Cre to inactivate Hdac7 at postnatal day 5. The tamoxifen-inducible type2a1 collagen-driven Cre, when crossed with the Rosa26 reporter mouse, is active in chondrocytes as well as perichondrial cells and derived osteoblasts (32). We observed reductions in bone mass associated with decreased trabecular number and increased trabecular spacing. No changes in cortical bone density were observed. One limitation of this approach is the potential confounding effects of tamoxifen. As an estrogen metabolite, tamoxifen promotes growth plate chondrocyte apoptosis and reduces the proliferation of articular chondrocytes (33, 34). We did not observe any changes in growth plate depth or proliferative growth plate chondrocytes in tamoxifen-injected C57 animals. Thus, the effects of tamoxifen and Hdac7 deficiency are distinct, and any effects of tamoxifen may be acting in opposition to the effects of Hdac7 deficiency.
This work demonstrates that chondrogenesis promotes Hdac7 degradation to increase β-catenin stability and activity. Similar results were observed with both ITS and Igf1, which is one of the most potent growth factors promoting cartilage maintenance and regeneration (35–37). β-Catenin signaling also enhances post-natal chondrocyte proliferation and is required for the maintenance of articular cartilage (20, 38–40). Cartilage-specific knockouts of the Igf1 receptor or transgenic expression of the inhibitor of β-catenin decrease chondrocyte proliferation (20, 41). Our data show that Hdac7 represses β-catenin activity, but signals from the insulin receptor may induce Hdac7 degradation. This is similar to what has been observed in endothelial cells where signaling through another tyrosine kinase receptor, Vegfr, induced Hdac7 degradation and enhanced β-catenin-dependent proliferation (12, 29). In cartilage, Vegf is active in hypertrophic chondrocytes, but not in proliferating cells. Although further study is needed, it is possible that we did not detect any Hdac7 in hypertrophic chondrocytes because Vegf activity is high in this zone. It is tempting to speculate that induction of Hdac7 degradation may be a common function of growth factors employing receptor tyrosine kinases.
In summary, Hdac7 is highly expressed in proliferating chondrocytes. Hdac7 deficiency increases the numbers of proliferating chondrocytes within the growth plate and negatively impacts chondrocyte hypertrophy and cancellous bone mass. Hdac7 associates with β-catenin in chondrocytes, but during chondrogenesis Hdac7 is translocated to the cytosol, where it is degraded. Reducing Hdac7 levels in early chondrocytes may be an effective strategy to promote the expansion and regeneration of chondrocytes.
Acknowledgments
We thank Xiaodong Li, David Razidlo, and Bridget Stensgard for technical assistance and Dr. Meghan McGee-Lawrence for helpful discussions and μCT expertise. We also thank Dr. Rhonda Bassel-Duby for assistance with the Hdac7fl/fl mice. Contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
This work was supported, in whole or in part, by National Institutes of Health Grants AR056950, AR61873, AR65397, and GM055252, Mayo Clinic, and the Mayo Graduate School.
- Igf
- insulin-like growth factor
- Hdacs
- histone deacetylases
- Igf1R
- insulin-like growth factor receptor
- ER
- estrogen receptor
- ITS
- insulin-transferrin-selenium
- HRP
- horseradish peroxidase
- ERK
- extracellular signal-regulated kinase
- DMSO
- dimethyl sulfoxide
- PTHrP
- parathyroid hormone-related peptide.
REFERENCES
- 1. Staines K. A., Pollard A. S., McGonnell I. M., Farquharson C., Pitsillides A. A. (2013) Cartilage to bone transitions in health and disease. J. Endocrinol. 219, R1–R12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kronenberg H. M. (2003) Developmental regulation of the growth plate. Nature 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 3. Bradley E. W., Carpio L. R., Westendorf J. J. (2013) Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J. Biol. Chem. 288, 9572–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGee-Lawrence M. E., Bradley E. W., Dudakovic A., Carlson S. W., Ryan Z. C., Kumar R., Dadsetan M., Yaszemski M. J., Chen Q., An K. N., Westendorf J. J. (2013) Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone 52, 296–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Razidlo D. F., Whitney T. J., Casper M. E., McGee-Lawrence M. E., Stensgard B. A., Li X., Secreto F. J., Knutson S. K., Hiebert S. W., Westendorf J. J. (2010) Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS One 5, e11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vega R. B., Matsuda K., Oh J., Barbosa A. C., Yang X., Meadows E., McAnally J., Pomajzl C., Shelton J. M., Richardson J. A., Karsenty G., Olson E. N. (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119, 555–566 [DOI] [PubMed] [Google Scholar]
- 7. Kozhemyakina E., Cohen T., Yao T. P., Lassar A. B. (2009) Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol. Cell. Biol. 29, 5751–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang S., McKinsey T. A., Zhang C. L., Richardson J. A., Hill J. A., Olson E. N. (2004) Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 24, 8467–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Backs J., Song K., Bezprozvannaya S., Chang S., Olson E. N. (2006) CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 116, 1853–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vega R. B., Harrison B. C., Meadows E., Roberts C. R., Papst P. J., Olson E. N., McKinsey T. A. (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 24, 8374–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S., Li X., Parra M., Verdin E., Bassel-Duby R., Olson E. N. (2008) Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. U.S.A. 105, 7738–7743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jensen E. D., Gopalakrishnan R., Westendorf J. J. (2009) Bone morphogenic protein 2 activates protein kinase D to regulate histone deacetylase 7 localization and repression of Runx2. J. Biol. Chem. 284, 2225–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grozinger C. M., Schreiber S. L. (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. U.S.A. 97, 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischle W., Dequiedt F., Fillion M., Hendzel M. J., Voelter W., Verdin E. (2001) Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J. Biol. Chem. 276, 35826–35835 [DOI] [PubMed] [Google Scholar]
- 16. Chang S., Young B. D., Li S., Qi X., Richardson J. A., Olson E. N. (2006) Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126, 321–334 [DOI] [PubMed] [Google Scholar]
- 17. Day T. F., Guo X., Garrett-Beal L., Yang Y. (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 [DOI] [PubMed] [Google Scholar]
- 18. Hill T. P., Später D., Taketo M. M., Birchmeier W., Hartmann C. (2005) Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738 [DOI] [PubMed] [Google Scholar]
- 19. Akiyama H., Lyons J. P., Mori-Akiyama Y., Yang X., Zhang R., Zhang Z., Deng J. M., Taketo M. M., Nakamura T., Behringer R. R., McCrea P. D., de Crombrugghe B. (2004) Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 18, 1072–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen M., Zhu M., Awad H., Li T. F., Sheu T. J., Boyce B. F., Chen D., O'Keefe R. J. (2008) Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J. Cell Sci. 121, 1455–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang B., Jin H., Zhu M., Li J., Zhao L., Zhang Y., Tang D., Xiao G., Xing L., Boyce B. F., Chen D. (2014) Chondrocyte beta-Catenin Signaling Regulates Postnatal Bone Remodeling Through Modulation of Osteoclast Formation in a Murine Model. Arthritis Rheumatol. 66, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ovchinnikov D. A., Deng J. M., Ogunrinu G., Behringer R. R. (2000) Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 26, 145–146 [PubMed] [Google Scholar]
- 23. Nakamura E., Nguyen M. T., Mackem S. (2006) Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev. Dyn. 235, 2603–2612 [DOI] [PubMed] [Google Scholar]
- 24. Gosset M., Berenbaum F., Thirion S., Jacques C. (2008) Primary culture and phenotyping of murine chondrocytes. Nature Protocols 3, 1253–1260 [DOI] [PubMed] [Google Scholar]
- 25. Soung do Y., Dong Y., Wang Y., Zuscik M. J., Schwarz E. M., O'Keefe R. J., Drissi H. (2007) Runx3/AML2/Cbfa3 regulates early and late chondrocyte differentiation. J Bone Miner Res 22, 1260–1270 [DOI] [PubMed] [Google Scholar]
- 26. Bradley E. W., Drissi M. H. (2010) WNT5A Regulates Chondrocyte Differentiation through Differential Use of the CaN/NFAT and IKK/NF-{kappa}B Pathways. Mol. Endocrinol. 24, 1581–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Razidlo D. F., Whitney T. J., Casper M. E., McGee-Lawrence M. E., Stensgard B. A., Li X., Secreto F. J., Knutson S. K., Hiebert S. W., Westendorf J. J. (2010) Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PloS one 5, e11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dao D. Y., Jonason J. H., Zhang Y., Hsu W., Chen D., Hilton M. J., O'Keefe R. J. (2012) Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J. Bone Miner Res. 27, 1680–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Margariti A., Zampetaki A., Xiao Q., Zhou B., Karamariti E., Martin D., Yin X., Mayr M., Li H., Zhang Z., De Falco E., Hu Y., Cockerill G., Xu Q., Zeng L. (2010) Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ. Res. 106, 1202–1211 [DOI] [PubMed] [Google Scholar]
- 30. Jensen E. D., Schroeder T. M., Bailey J., Gopalakrishnan R., Westendorf J. J. (2008) Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J. Bone Miner. Res. 23, 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pham L., Kaiser B., Romsa A., Schwarz T., Gopalakrishnan R., Jensen E. D., Mansky K. C. (2011) HDAC3 and HDAC7 have opposite effects on osteoclast differentiation. J. Biol. Chem. 286, 12056–12065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maes C., Kobayashi T., Selig M. K., Torrekens S., Roth S. I., Mackem S., Carmeliet G., Kronenberg H. M. (2010) Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chagin A. S., Karimian E., Zaman F., Takigawa M., Chrysis D., Sävendahl L. (2007) Tamoxifen induces permanent growth arrest through selective induction of apoptosis in growth plate chondrocytes in cultured rat metatarsal bones. Bone 40, 1415–1424 [DOI] [PubMed] [Google Scholar]
- 34. Talwar R. M., Wong B. S., Svoboda K., Harper R. P. (2006) Effects of estrogen on chondrocyte proliferation and collagen synthesis in skeletally mature articular cartilage. J. Oral Maxillofac. Surg. 64, 600–609 [DOI] [PubMed] [Google Scholar]
- 35. Kolettas E., Muir H. I., Barrett J. C., Hardingham T. E. (2001) Chondrocyte phenotype and cell survival are regulated by culture conditions and by specific cytokines through the expression of Sox-9 transcription factor. Rheumatology 40, 1146–1156 [DOI] [PubMed] [Google Scholar]
- 36. Nixon A. J., Fortier L. A., Williams J., Mohammed H. (1999) Enhanced repair of extensive articular defects by insulin-like growth factor-I-laden fibrin composites. J. Orthop Res. 17, 475–487 [DOI] [PubMed] [Google Scholar]
- 37. Schmidt M. B., Chen E. H., Lynch S. E. (2006) A review of the effects of insulin-like growth factor and platelet derived growth factor on in vivo cartilage healing and repair. Osteoarthritis Cartilage 14, 403–412 [DOI] [PubMed] [Google Scholar]
- 38. Yuasa T., Kondo N., Yasuhara R., Shimono K., Mackem S., Pacifici M., Iwamoto M., Enomoto-Iwamoto M. (2009) Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am. J. Pathol. 175, 1993–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu M., Chen M., Zuscik M., Wu Q., Wang Y. J., Rosier R. N., O'Keefe R. J., Chen D. (2008) Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum 58, 2053–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu M., Tang D., Wu Q., Hao S., Chen M., Xie C., Rosier R. N., O'Keefe R. J., Zuscik M., Chen D. (2009) Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 24, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y., Cheng Z., Elalieh H. Z., Nakamura E., Nguyen M. T., Mackem S., Clemens T. L., Bikle D. D., Chang W. (2011) IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J. Bone Miner. Res. 26, 1437–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]