

Abstract
Trichloroethylene (TCE) is a widely used organic solvent. Although TCE is classified as carcinogenic to humans, substantial gaps remain in our understanding of inter-individual variability in TCE metabolism and toxicity, especially in the liver. We tested a hypothesis that amounts of oxidative metabolites of TCE in mouse liver are associated with liver-specific toxicity. Oral dosing with TCE was conducted in sub-acute (600 mg/kg/d; 5 days; 7 inbred mouse strains) and sub-chronic (100 or 400 mg/kg/d; 1, 2, or 4 weeks; 2 inbred mouse strains) designs. We evaluated the quantitative relationship between strain-, dose-, and time-dependent formation of TCE metabolites from cytochrome P450-mediated oxidation [trichloroacetic acid (TCA), dichloroacetic acid (DCA), and trichloroethanol] and glutathione conjugation [S-(1,2-dichlorovinyl)-L-cysteine and S-(1,2-dichlorovinyl)glutathione] in serum and liver, and various liver toxicity phenotypes. In sub-acute study, inter-strain variability in TCE metabolite amounts was observed in serum and liver. No induction of Cyp2e1 protein levels in liver was detected. Serum and liver levels of TCA and DCA were correlated with increased transcription of peroxisome proliferator-marker genes Cyp4a10 and Acox1, but not with degree of induction in hepatocellular proliferation. In sub-chronic study, serum and liver levels of oxidative metabolites gradually decreased over time despite continuous dosing. Liver protein levels of Cyp2e1, Adh and Aldh2 were unaffected by treatment with TCE. While the magnitude of induction of peroxisome proliferator-marker genes also declined, hepatocellular proliferation increased. This study offers a unique opportunity to provide a scientific data-driven rationale for some of the major assumptions in human health assessment of TCE.
Introduction
Trichloroethylene (TCE) is a chlorinated organic solvent widely used as a feedstock material in chemical manufacturing, as well as in various industrial applications including dry cleaning and vapor degreasing. Since the beginning of its wide-spread production dating back to the 1920s (NICNAS 2000), it has been recognized as an occupational and environmental health concern due to high human exposure and its potential to be a health hazard (NRC 2006). The number of workers in occupations with likely TCE exposure has declined in the developed countries in the past 20 years (von Grote et al. 2003). Still, TCE is one of the major contaminants found in the Superfund hazardous waste sites across the United States (U.S. EPA 2011) and is ranked 16th on the Substance Priority List by the US Agency for Toxic Substances and Disease Registry (ATSDR 2011).
TCE poses a potential human health hazard for non-cancer toxicity to the central nervous system, kidney, liver, immune system, male reproductive system, and the developing fetus (Chiu et al. 2013). In addition, TCE is classified as carcinogenic to humans by IARC (Guha et al. 2012) and the U.S. EPA (U.S. EPA 2011) based on the evidence in humans of a causal relationship between kidney cancer and exposure to TCE. Positive, although less consistent, associations with TCE were reported studies of liver cancer and non-Hodgkin lymphoma (Scott and Jinot 2011).
Liver toxicity and carcinogenicity remain largely unresolved with regard to the human health hazard of TCE (Rusyn et al. 2014). There is clear evidence that TCE is a liver carcinogen in rodents (National Toxicology Program 1990; Anna et al. 1994); however, the relationship between occupational TCE exposure and risk of liver cancer in humans is still inconclusive, given that epidemiologic studies have observed both positive (Hansen et al. 2013) and negative associations (Vlaanderen et al. 2013; Radican et al. 2008). A recent meta-analysis reported a meta-relative risk of 1.3 (95% CI 1.1–1.6) for the overall TCE exposure and liver cancer based on nine human cohorts (Scott and Jinot 2011).
There are also uncertainties regarding the potential mode of action for TCE tumorigenesis in liver (Rusyn et al. 2014). TCE is metabolized through both cytochrome P450-dependent oxidation and glutathione conjugation to form a number of toxic species (Lash et al. 2014). Metabolites of the oxidative pathway, trichloroacetic (TCA) and dichloroacetic (DCA) acids, are widely considered to be primary mediators of the toxicity and carcinogenicity of TCE in the liver via peroxisome proliferator-activated receptor (PPARα) activation. The latter is one of the most studied mechanisms of action for TCE-induced liver cancer in rodents (Klaunig et al. 2003; Keshava and Caldwell 2006). Studies in vitro have shown that human PPARα is activated by TCA and DCA, but relatively inactive to TCE itself (Maloney and Waxman 1999; Zhou and Waxman 1998). In addition, human hepatocytes transfected with mouse PPARα displayed increased expression of PPARα, and increased peroxisome proliferator response element-reporter activity when treated with TCA and DCA (Walgren et al. 2000).
Even though it is widely assumed that tissue-specific formation of TCE metabolites is one of the critical determinants for the plethora of its adverse health effects, most studies of TCE toxicokinetics were performed in blood. Few studies evaluated TCE metabolism in tissues, however one study in rats did not find a dose-response relationship in formation of TCE metabolites in liver and kidney (Lash et al. 2006). Thus, we tested a hypothesis that formation of oxidative metabolites of TCE in mouse liver is associated with liver-specific toxicity by evaluating the quantitative relationship between strain-, dose-, and time-dependent formation of TCA and DCA in serum and liver, and various liver toxicity phenotypes in a panel of mouse inbred strains.
Materials and Methods
Animals and treatments
Male mice (aged 6–7 weeks) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in polycarbonate cages on Sani-Chips (P.J. Murphy Forest Products Corp., Montville, NJ) irradiated hardwood bedding. Animals were fed an NTP-2000 (Zeigler Brothers, Inc., Gardners, PA) wafer diet and water ad libitum on a 12 h light-dark cycle. All studies were approved by the UNC Institutional Animal Care and Use Committee.
Two study designs were utilized in this work. First, we performed a sub-acute study where vehicle (10 mL/kg, 5% Alkamuls EL-620 in saline) or TCE (600 mg/kg/d, in vehicle) was administered by gavage to mice from 7 inbred strains (129S1/SvImJ, A/J, BTBR T+tf/J, C57BL/6J, CAST/EiJ, NOD/ShiLtJ, and NZW/LacJ) for 5 consecutive days. These strains were selected to maximize inter-strain differences in metabolism of TCE based on the previous study of TCE metabolism in a panel of inbred strains (Bradford et al. 2011) and the results of the statistical modeling of the effect of time and strain on TCE metabolite concentrations which supports the sample size used in this study (Chiu et al. 2014). Second, based on the data from the sub-acute study, we selected two inbred strains (C57BL/6J and NZW/LacJ) that represented widely varying degrees of formation of oxidative metabolites of TCE for a subsequent sub-chronic study. Specifically, animals of each strain were randomly assigned to one of the three groups (5% Alkamuls EL-620 in saline vehicle, 100, or 400 mg/kg/d of TCE) and were dosed by oral gavage daily for 5 days, 2 weeks, or 4 weeks (the latter two were dosed for 5 days/week).
In all studies, mice were given drinking water containing 0.2 g/L of 5-bromo-2′-deoxyuridine (BrdU) for 72 h prior to sacrifice. Blood, tissues (liver, kidney, etc.) and a section of a duodenum were collected 2 hrs after the last treatment. The timing of sacrifice was selected based on the toxicokinetic studies of TCE in the mouse (Bradford et al. 2011; Kim et al. 2009b) showing that concentrations of both oxidative and glutathione conjugation metabolites of TCE peak around 2 hrs after dosing. Blood was drawn from vena cava and centrifuged to prepare serum using Z-gel tubes (Sarstedt, Germany) according to the manufacturer’s instructions. Body and organ weights were recorded. Liver and duodenum sections were fixed in neutral buffered formalin for 24 hrs, and the remainder of the liver tissue was frozen in liquid nitrogen. All serum and tissue samples were stored at −80°C until analyzed.
Quantification of TCE metabolites
Concentrations of TCA, DCA, S-(1,2-dichlorovinyl)-L-cysteine (DCVC), and S-(1,2-dichlorovinyl) glutathione (DCVG) in serum and liver were determined using HPLC-ESI-MS/MS as detailed elsewhere (Kim et al. 2009a) with slight modifications. Quantification of trichloroethanol (TCOH) in liver was performed using the method of (Song and Ho 2003). The configuration of the instruments was identical to that in the above mentioned references, but the extraction methods were optimized for each tissue (liver or serum) and metabolite as follows.
TCA and DCA
Serum (50 μL) was mixed with 100 μL of the internal standards (difluoroacetic acid (DFA) and trifluoroacetic acid (TFA), 50 nmol/mL each). Serum proteins were then removed by filter-centrifugation (Microcon YM-10; Millipore, Billerica, MA) at 14,000×g for 1 hr. Liver samples (100 mg) were homogenized in 500 μL of 0.01 M PBS (pH 7.4) with 20 μL of internal standards (DFA and TFA, 20 nmol/mL each) using Tissuelyser (Qiagen, Valencia, CA) for 1 min. The homogenates were filter-centrifuged (Amicon Ultra Centrifugal Filters 10K; Millipore) at 14,000×g for 1 hr. After the filtrate was acidified with 100 μL of 3% (v/v) sulfuric acid, 2 mL of diethyl ether was added and solutions were vortexed vigorously for 1 min. The upper ether layer was transferred to another vial, reduced in volume to less than 300 μL under continuous stream of N2, then transferred to a glass vial insert containing 5 μl of double-distilled water and dried completely. The residue was reconstituted in 20 μL of HPLC mobile phase consisting of 70% acetonitrile, 30% 1 mM ammonium citrate in double-distilled water. The lower limits of quantification (LLOQ) were: 0.04 nmol/mL in serum and 0.1 nmol/g in liver for DCA, 5 nmol/mL in serum and 8 nmol/g in liver for TCA.
DCVG and DCVC
Serum (50 μL) was mixed with 100 μL of the internal standard solution ([13C2,15N]DCVG and [13C3,15N]DCVC, 5 nmol/mL each). Serum proteins were then removed by filter-centrifugation (Microcon YM-10; Millipore) at 14,000×g for 1 hr. Liver tissue (100 mg) was homogenized in 500 μL of 0.01 M PBS (pH 7.4) with 20 μL of internal standards ([13C2,15N]DCVG and [13C3,15N]DCVC, 25 nmol/mL each) using Tissuelyser (Qiagen) for 1 min. The homogenates were filter-centrifuged (Amicon Ultra Centrifugal Filters 10K; Millipore) at 14,000×g for 1 hr. From each prepared sample, DCVG and DCVC were extracted using a solid phase extraction cartridge (StrataTM X-AW, 30mg 96-well plate; Phenomenex, Torrance, CA). The cartridges were conditioned with 300 μL of methanol, followed by equilibration with 300 μL of water. Samples were loaded, washed with 300 μL of water, and eluted with 250 μL of basic methanol (pH adjusted to 10.8 by 29.1% NH4OH). The final eluent was collected in 300 μL glass vial inserts and dried in a SpeedVac Concentrator before reconstitution with 20 μL of 4:1 water/methanol containing 0.1% acetic acid. The LLOQ were: 0.5 pmol/mL in serum and 2 pmol/g in liver for DCVG, 1 pmol/mL in serum and 20 pmol/g in liver for DCVC.
TCOH
Liver tissue (30 mg) was homogenized in 500 μL of acetate buffer (pH 4.6) with 1,000 units of β-glucuronidase (Sigma [G0751], St. Louis, MO) using Tissuelyser (Qiagen) for 1 min, followed by overnight incubation at 37°C. After centrifugation at 14,000×g for 5 min, the supernatant was transferred to a new tube, then mixed with 20 μL internal standard (DCA, 10 mM in methanol) and 550 μL of water/0.1 M sulfuric acid/methanol (6:5:1). The mixture was heated at 70°C for 20 min. After cooling to room temperature, 2.5 mL hexane was added, the mixture vortexed for 10 min and centrifuged at 2,500×g for 2 min. The upper layer was concentrated under a stream of N2 to less than 20 μL and used for GC-MS analysis as detailed in (Song and Ho 2003). The LLOQ was 5 nmol/g in liver.
Gene expression analysis by real-time PCR
Total RNA was isolated from liver samples using an RNeasy kit (Qiagen) according to the manufacturer’s instructions. RNA concentration and quality were determined using an ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE) and Agilent 2000 Bioanalyser, respectively. Total RNA was reverse transcribed using random primers and the high capacity complementary DNA archive kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. The following gene expression assays (Applied Biosystems) were used for quantitative real-time PCR: PPARα (Mm00440939_m1); palmitoyl acyl-Coenzyme A oxidase 1 (Acox1, Mm01246831_m1); cytochrome P450, family 4, subfamily a, polypeptide 10 (Cyp4a10, Mm01188913_g1); and beta glucuronidase (Gusb, Mm00446953_m1). Reactions were performed in a 96-well plate, and all samples were plated in duplicate using a LightCycler® 480 instrument (Roche Applied Science, Indianapolis, IN). The cycle threshold (Ct) for each sample was determined from the linear region of the amplification plot. The ∆Ct values for all genes relative to the control gene Gusb were determined. The ∆∆Ct were calculated using treated group means relative to strain-matched control group means. Fold change data were calculated from the ∆∆Ct values.
Protein level measurements
Proteins were extracted from 20 mg of frozen liver samples using T-PER® Tissue Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL) and Halt™ Protease Inhibitor Cocktail (Pierce Biotechnology) according to the manufacturer’s instructions. Protein concentration was measured using Pierce® BCA Protein Assay Kit (Pierce Biotechnology) and a DTX 880 Multimode Detector (Beckman Coulter, Brea, CA). Extracts containing 30 μg of protein were resolved on 12% polyacrylamide sodium dodecyl sulfate-containing gels and transferred to polyvinylidene difluoride membranes. Membranes were blocked in Odyssey® Blocking Buffer (LI-COR, Lincoln, NE) and probed with 1:5000 diluted anti-Cytochrome P450 2e1 antibody (Abcam, Cambridge, MA), anti-Aldh2 antibody (Abcam), or anti-Adh antibody (Abcam) overnight at 4°C. Blots were washed in 0.1% Tween20 in 0.01 M PBS, incubated with 1:20,000 IRDye® 680LT donkey anti-rabbit IgG (LI-COR), and detected using an Odyssey Infrared Imaging System (LI-COR). Equal protein loading was confirmed by total protein staining with 0.1% (w/v) naphthol blue black in 7% (v/v) acetic acid in water for 10 min. The signal intensity was analyzed by Image Studio Software (LI-COR).
Statistical analysis
The significance of inter-strain effect on metabolism was assessed by ANOVA modeling. Given the small sample size, the exact permutation test was used to determine significant differences between control and TCE-treated groups (α=0.05). The Spearman (rank) correlation analysis across all variables was conducted to account for the difference in scale of the variables. In correlation analyses, false discover rate correction (Storey and Tibshirani 2003) was applied to all p-values to correct for multiple comparisons. The resultant significant (<0.1) q-values are reported in the Supplemental Tables. All statistical analyses were performed using SAS software ver. 9.3 (SAS Institute, Cary, NC).
Data availability
Individual animal-level data for all quantitative endpoints is available publicly at the Mouse Phenome Database (http://phenome.jax.org/; accession ID: Rusyn6).
Results
Sub-acute (5 days) studies of strain-dependent effects of TCE in mouse liver
We evaluated serum and liver levels of TCE metabolites from P450-mediated oxidation (TCA, DCA, TCOH) and glutathione conjugation (DCVC and DCVG) in seven mouse inbred strains (Figure 1). Mice were treated for 5 days by oral gavage of 600 mg/kg of TCE in aqueous vehicle. Across all strains, serum levels (Figure 1, left panel) of TCA were on average 1,000-fold greater than those for DCA, and the amounts of DCA were about 100-fold higher than those of either DCVC or DCVG. Higher levels of TCA (about 2-fold) and DCA (about 10-fold) were found in liver (Figure 1, right panel) than in serum, but the relative ratio of TCA to DCA was similar. The levels of TCOH, also a major TCE metabolite in liver, were comparable to those of TCA. Levels of DCVG were much higher in liver than in serum (about 100-fold) and only about 10-fold lower than those of DCA. We observed a substantial difference between the concentrations of DCVG and DCVC in liver. While the amounts of DCVG and DCVC in serum were comparable (<2-fold difference), the concentration of DCVG in liver was at least 50 to 100-fold higher than that of DCVC, which was below the LLOQ in most samples. We found a significant strain effect (p<0.05) in both serum and liver for every metabolite examined (TCA, DCA, TCOH, DCVG, and DCVC).
Figure 1. Inter-strain variability in TCE metabolism in the mouse in a sub-acute study.
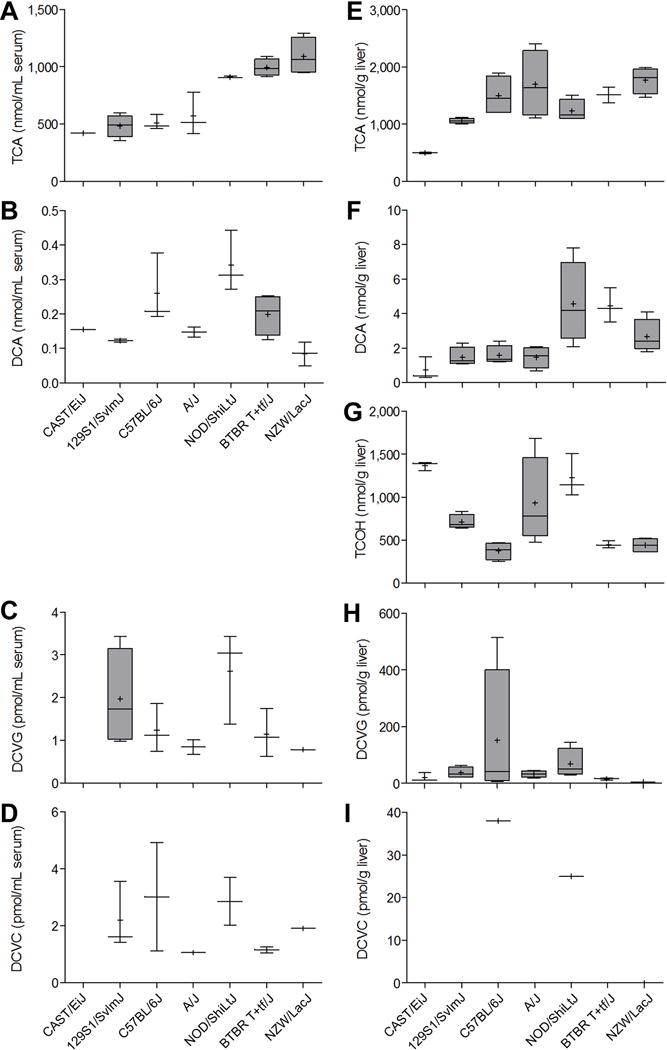
Serum (A-D) and liver (E-I) levels of metabolites were assessed 2 h following the last of 5 daily doses (600 mg/kg) of TCE. Box and whisker plots are shown (+, mean; line, median; box, inter-quartile range; whiskers, min to max). When box is shown, 4 animals per group were available. Otherwise, there were 3 animals per group.
We examined the relationships between TCE metabolites in either serum or liver (Supplemental Table 1). In serum, we found no significant correlations among 5 metabolites. In liver, levels of TCA and DCA were significantly correlated (ρ=0.74, q=0.001). Although both TCA and TCOH are major oxidative metabolites, we found no correlation between their levels in liver. Inter-tissue (liver vs serum) correlation of TCE metabolites was also evaluated. We found significant correlation between liver and serum levels for TCA (ρ=0.62, q=0.020), serum TCA and liver DCA (ρ=0.78, q=0.001), and serum DCVC and liver DCVG (ρ=0.67, q=0.017).
To determine whether inter-strain differences in TCE metabolism may be due to strain-dependent variability in expression of Cyp2e1, we measured the levels of liver Cyp2e1 protein in vehicle- and TCE-treated animals (Figure 2). A significant strain effect (p<0.05) was found in baseline liver Cyp2e1 levels. However, treatment with TCE was without effect on liver Cyp2e1 across all strains. Interestingly, the background liver levels of Cyp2e1 were not correlated (ρ=0.74, p=0.055) with the amount of TCA in liver of TCE-treated animals.
Figure 2. Relationships between TCE metabolism in a sub-acute study and liver Cyp2e1 protein levels.
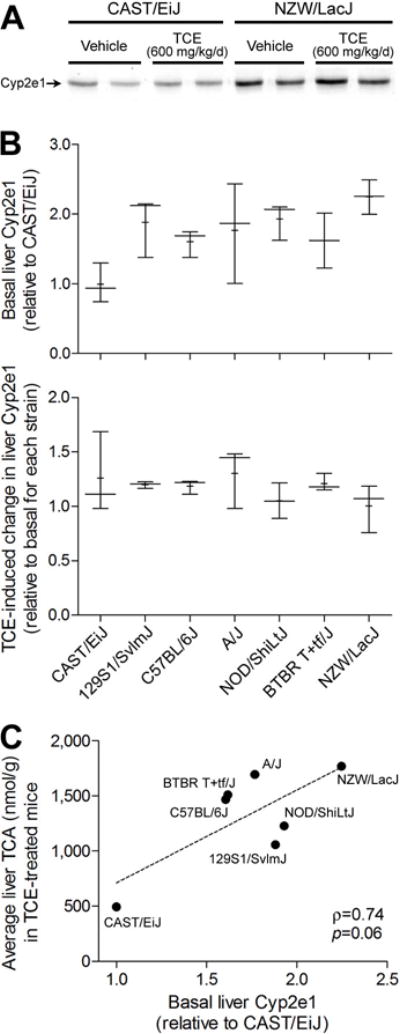
(A) A representative Western blot of Cyp2e1 protein expression in liver of vehicle- and TCE-treated mice from CAST/EiJ and NZW/LacJ strains. (B) Inter-strain differences in basal and TCE-induced liver expression of Cyp2e1. Box and whiskers plots are shown (+, mean; line, median; box, inter-quartile range; whiskers, min to max). There were 3 animals per group. (C) Correlation between basal liver expression of Cyp2e1 and liver TCA amounts in TCE-treated mice of 7 strains. Each dot represents a mouse strain. Spearman rank (ρ) correlation is shown.
Liver size (relative to body weight) and hepatocellular proliferation were examined in vehicle- and TCE-treated mice (Figures 3A–B). We observed significant effects of sub-acute treatment with TCE on liver enlargement in 129S1/SvlmJ, NOD/ShiLtJ and BTBR T+tf/J, and on hepatocyte proliferation in NOD/ShiLtJ, BTBR T+tf/J, and NZW/LacJ strains. Because peroxisome proliferation has been suggested as contributing to hepatomegaly in rodents (Marsman et al. 1988), we evaluated expression of Cyp4a10 and Acox1, marker genes for this mechanistic event, in mouse liver. Expression of the transcription factor Ppara, was not affected by TCE (data not shown); however, expression of Cyp4a10 and Acox1 was markedly elevated in all strains, except for CAST/EiJ (Figures 3C–D), with TCE-induced effect being greater for Cyp4a10 than for Acox1.
Figure 3. Inter-strain differences in liver toxicity of TCE in a sub-acute study.
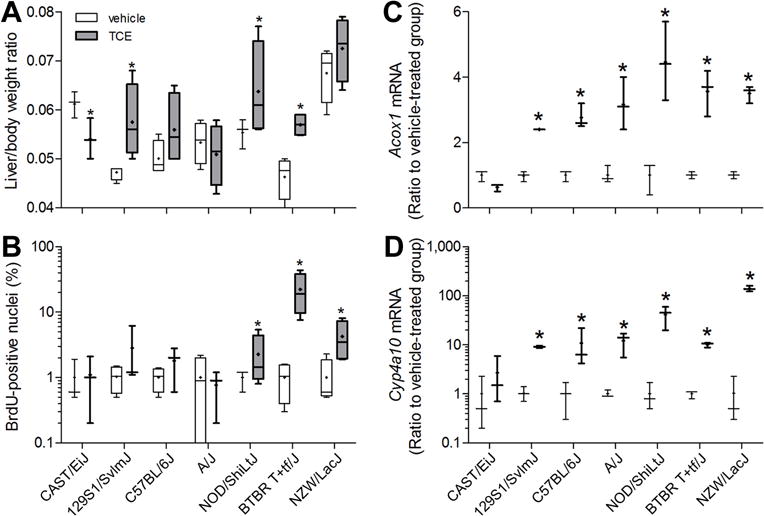
Liver to body weight ratios (A), percent BrDU-positive hepatocyte nuclei (B), and liver expression of peroxisome proliferation marker genes Acox1 (C) and Cyp4a10 (D) were evaluated in mice treated with vehicle (white) or TCE (black; 600 mg/kg) for 5 days. Box and whiskers plots are shown (+, mean; line, median; box, inter-quartile range; whiskers, min to max). When box is shown, 4 animals per group were available. Otherwise, there were 3 animals per group. Asterisk (*) denotes a significant difference (p<0.05) compared to vehicle-treated group within same strain.
Because inter-strain differences in both TCE metabolism and liver effects were observed in this study, we examined the associations among these endpoints in liver (Supplemental Table 2). Levels of TCA and DCA were strongly correlated with expression of Acox1 and Cyp4a10 (Figures 4A–B). However, neither the expression of Acox1 and Cyp4a10, nor the level of TCA, was correlated with hepatocellular proliferation (Figures 4C and 4D, respectively).
Figure 4. Correlation between TCE metabolites and liver toxicity phenotypes.
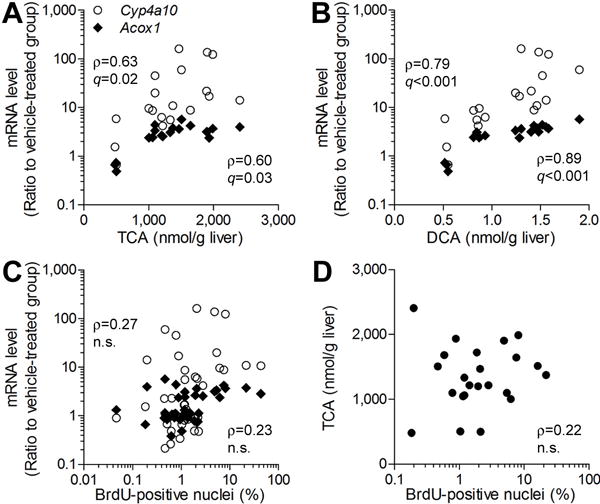
TCE-induced increase in Cyp4a10 and Acox1 expression in the mouse liver was significantly correlated with liver levels of TCA (A) and DCA (B), and hepatocellular proliferation (C). Weak correlation was observed between TCA concentration in the liver and hepatocellular proliferation (D). Each symbol represents an individual animal in the study. Spearman rank (ρ) correlations and false discovery rate-corrected significance values (q<0.1) are shown.
Sub-chronic (up to 4 weeks) studies of strain-dependent effects of TCE in mouse liver
Based on the differences in TCE metabolism observed in the sub-acute (5 days) TCE exposure studies, we selected C57BL/6J and NZW/LacJ strains to further test our hypothesis that inter-strain differences in amounts of oxidative metabolites of TCE in the liver are associated with liver-specific toxicity. In these studies, we examined the time-dependent (1, 2 and 4 weeks) and dose-dependent (100 and 400 mg/kg/day, i.g.) effects of TCE.
As expected, the difference in TCE metabolism between two strains was also observed at lower doses and in longer-term studies (Figure 5). Serum and liver levels of TCA and DCA were higher in NZW/LacJ mice, and liver levels of DCVG were higher in C57BL/6J mice. Similar to the findings in the sub-acute study, levels of TCA and TCOH in liver were 100 to 1,000-fold greater than those of DCA, and levels of DCA were 10 to 100-fold higher than those of DCVG. Levels of DCVC in serum and liver were below the LLOQ. Amounts of all measured TCE metabolites in liver and serum generally trended downward over time. Specifically, the levels of TCA in serum and liver decreased over time in both strains, while the levels of TCOH in liver decreased over time only in C57BL/6J strain. Likewise, the level of DCVG in liver was highest after 5 days of treatment.
Figure 5. Time-course analysis of TCE metabolism in C57BL/6J and NZW/LacJ mice in a sub-chronic study.
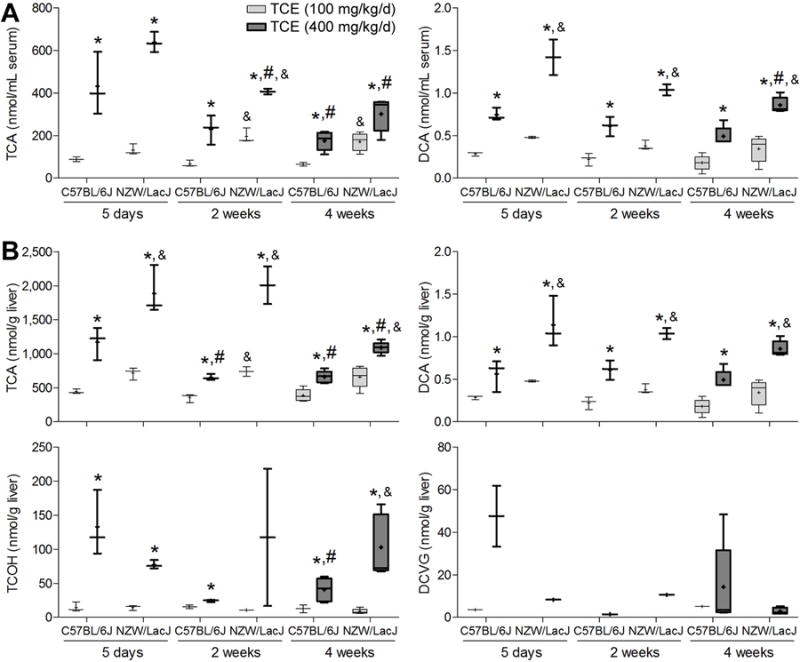
Serum (A) and liver (B) levels of metabolites were assessed 2 h following the last dose after 1, 2 or 4 wks (100 or 400 mg/kg/d) of TCE. Box and whiskers plots are shown (+, mean; line, median; box, inter-quartile range; whiskers, min to max). Light-gray, 100 mg/kg/d groups; dark gray, 400 mg/kg/d groups. When box is shown, 4 animals per group were available. Otherwise, there were 3 animals per group. Asterisks denote a significant (p<0.05) difference as compared to (*) the group dosed with 100 mg/kg/d (same strain and time point), (#) the 5 day treatment group (same strain and dose), or (&) the values in C57BL/6J strain (same dose and time point).
Among TCE metabolites that were measured in this study, serum and liver levels of TCA and DCA were highly correlated (q<0.001) across all animals in the study (Supplemental Table 3). Liver levels of TCOH exhibited similar strong correlation with the levels of TCA and DCA in either serum or liver. No correlation was found among the levels of DCVG in liver and other TCE metabolites.
To examine whether strain-dependent changes in TCE-metabolizing enzymes could account for the observed decrease in metabolite formation over time, we examined the levels of Cyp2e1, Aldh2, and Adh proteins in liver, and found that expression of these enzymes was not affected by TCE treatment (Figure 6).
Figure 6. The effect of a sub-chronic treatment with TCE on liver Cyp2e1 (A), Aldh2 (B) and Adh (C) protein levels in C57BL/6J and NZW/LacJ mice.
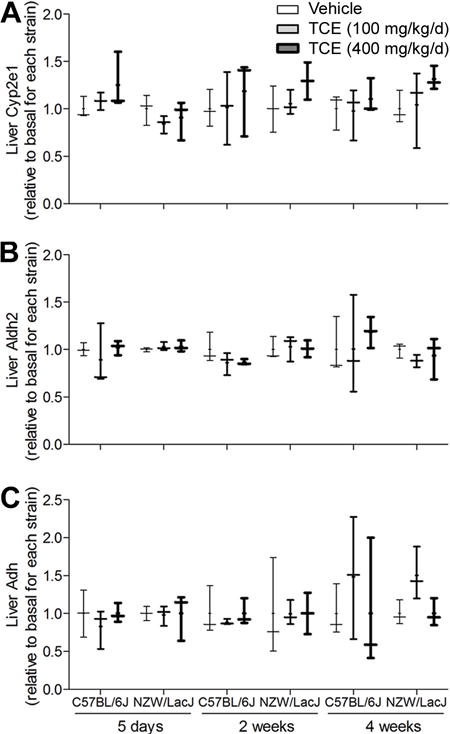
Box and whiskers plots are shown (+, mean; line, median; box, inter-quartile range; whiskers, min to max). Thickness of the line corresponds to the vehicle, 100 and 400 mg/kg/d groups. There were 3 animals per group.
We also examined the effects of sub-chronic treatment with TCE on liver weight and the marker genes of hepatocellular proliferation and peroxisome proliferator expression. Even though the amount of oxidative metabolites of TCE trended downward over time, the increased liver to body weight ratio was still observed in mice treated for 2 week or 4 week in a strain-dependent manner. In the case of hepatocellular proliferation, most prominent effects were observed at the 4 week time-point (Figures 7A–B). Histopathological evaluation of the liver sections revealed concordant increases in relative size of the hepatocytes, as well as hemosiderin deposits in the high-dose 4 week treatment groups of both strains (Supplemental Figure 1). A prominent effect of TCE on peroxisome proliferation in the liver was observed in the sub-chronic study. Although TCE did not affect expression of Ppara (data not shown), expression of Acox1 and Cyp4a10 was strongly induced in a dose-dependent manner in both strains of mice treated with TCE for 5 days (Figures 7C–D). Interestingly, the magnitude of upregulation of Acox1 and Cyp4a10 by TCE diminished over time. In the 2 and 4 week treatment groups, there was either no difference in gene expression between vehicle and TCE-treated animals, or differences were less prominent.
Figure 7. Differences in liver toxicity of TCE in C57BL/6J and NZW/LacJ mice in a sub-chronic study.
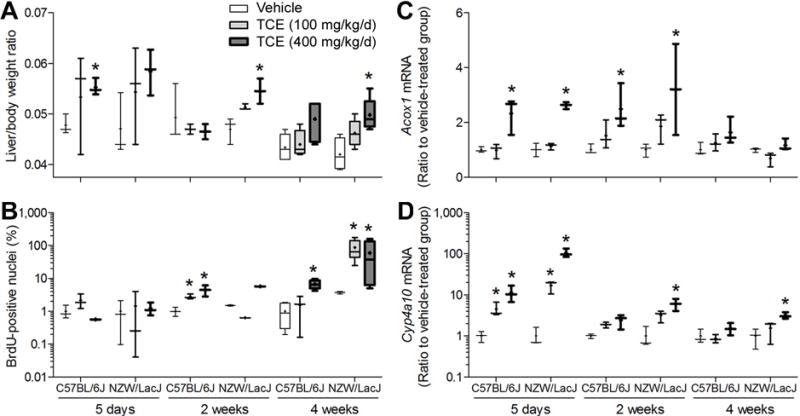
Liver to body weight ratios (A), percent BrDU-positive hepatocyte nuclei (B), and liver expression of peroxisome proliferation marker genes Acox1 (C) and Cyp4a10 (D) were evaluated in mice treated with TCE (100 or 400 mg/kg) for 1, 2 or 4 wks. Box and whiskers plots are shown (+, mean; line, median; box, inter-quartile range; whiskers, min to max). White, vehicle-treated groups; light-gray, 100 mg/kg/d groups; dark gray, 400 mg/kg/d groups. When box is shown, 4 animals per group were available. Otherwise, there were 3 animals per group. Asterisk (*) denotes a significant (p<0.05) difference as compared to vehicle-treated group (same strain and time point).
Similar to the result of the sub-acute study, the levels of TCA and DCA were significantly correlated with Acox1 and Cyp4a10 expression in liver regardless of TCE dose or treatment duration (Figures 8A–B). The Cyp4a10 expression was significantly correlated with liver-to-body weight ratio (ρ=0.68, q<0.001, Figure 8C); however, hepatocellular proliferation did not correlate with other variables (Figure 8D, Supplemental Table 4).
Figure 8. Correlation between TCE metabolites and liver toxicity phenotypes in C57BL/6J and NZW/LacJ mice in a sub-chronic study.
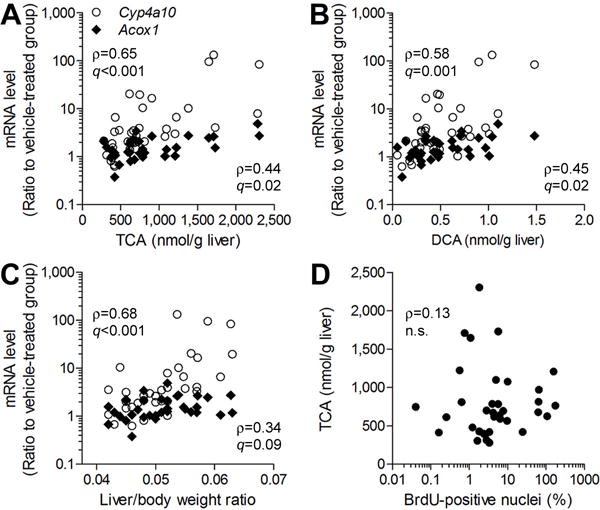
TCE-induced increase in Cyp4a10 and Acox1 expression in the mouse liver was significantly correlated with liver levels of TCA (A) and DCA (B), and liver to body weight ratio (C). (D) No correlation was observed between TCA concentration in the liver and hepatocellular proliferation. Each symbol represents an individual animal in the study. Spearman rank (ρ) correlations and false discovery rate-corrected significance values (q<0.1) are shown.
Discussion
The challenge of addressing the variability in susceptibility to environmental exposures is frequently one of the most contentious issues in human health assessments. Because of the heterogeneity of the human population, it is generally expected that there will be a broad range of toxicokinetic and toxicodynamic responses to chemicals or drugs (Zeise et al. 2013). Traditionally, the inter-individual differences in the toxicokinetics are accounted for by default assumptions and only in rare cases, are based on human toxicokinetic data. Seldom is there sufficient data to evaluate the extent of variability in toxicodynamics.
Because TCE metabolism to form chloroacetic acids and GSH-conjugates is widely accepted as the mechanism leading to toxicity in various organs (Lash et al. 2001), the interplay between toxicokinetics and toxicodynamics is a critically important consideration in the evaluation of human health hazard of TCE. Some understanding of the human population variability in toxicokinetics of TCE is available based on the limited data from clinical studies and Bayesian modeling (Chiu et al. 2009). The metabolism of TCE across species (e.g., rodents and humans) is qualitatively similar (Lash et al. 2014), and thus inter-species and –individual variability in TCE toxicity is likely due to the variability in TCE metabolism (Chiu et al. 2013; Green 1990). TCE toxicity is also dose-dependent, which suggests the link between toxicokinetics and toxicodynamics (Chiu et al. 2013). It is yet to be experimentally demonstrated, however, that inter-individual variability in TCE metabolism, not exposure (i.e., dose), will result in quantitative differences in its toxicity. In this regard, an examination of the relationship between the variability of metabolism and the variability in toxicity may shed light on our understanding of human health hazard of TCE. Therefore, the purpose of this study was to use a population-based approach to test the hypothesis that TCE-induced toxicity in liver is associated with the liver-specific formation of oxidative TCE metabolites.
In our previous work, we demonstrated that both oxidative metabolism and GSH conjugation of TCE vary considerably among inbred mouse strains (Bradford et al. 2011), and that such variability was associated with strain-specific differences in gene expression in mouse liver. While these results allowed for a quantitative evaluation of the relationship between metabolism and TCE-induced gene expression in the liver, the focus of the study was on toxicokinetic profiling over a 24 hour time period and the use of one high dose of TCE (2.1 g/kg, i.g.). To further explore the time- and dose-relationships between TCE metabolism and toxicity in the context of inter-strain variability, we conducted a series of studies that aimed to quantitate the levels of TCE metabolites in serum and liver. We also evaluated cell proliferation and peroxisome proliferation, two widely accepted liver toxicity phenotypes reflective of the major mechanistic events considered to play a role in liver carcinogenesis of TCE in rodents (Rusyn et al. 2014).
The first important and novel outcome of this study is quantitative data on a broad range of metabolites produced via oxidative metabolism and GSH conjugation of TCE in serum and liver in the context of inter-strain variability in TCE metabolism. While many published reports provide quantitative information on serum levels of TCA along with a few reports of DCA after treatment with TCE in rodents and humans, little information exists on the formation of these metabolites in the liver. Similar to our findings in the single-dose studies in mouse serum (Bradford et al. 2011; Kim et al. 2009b), there is up to 5 orders of magnitude difference in the relative flux of TCE through CYP450-dependent oxidation (primarily TCA) compared to GSH conjugation. The similarities in the levels of TCE metabolites formed through oxidation and GSH conjugation in this and our previous studies confirm that GSH conjugation of TCE is a minor pathway in mice, regardless of the dose or duration of treatment.
In the mouse liver, TCA was also the predominant metabolite formed (about 2 orders of magnitude greater than DCA); however, while the difference in relative flux of TCE through CYP450-dependent oxidation compared to GSH conjugation was still large, it was smaller than that in serum, about 3 orders of magnitude. In the rat liver, TCA and TCOH are also predominant TCE metabolites and there is about 100-fold difference between CYP450-dependent oxidative metabolism and GSH-conjugation (Lash et al. 2006). TCE metabolism by CYP450-dependent pathway is known to occur at a faster rate in mice than in rats (Prout et al. 1985), and our data are in line with these observations.
Serum levels of TCA and DCA were not correlated with each other, which is consistent with our previous study (Kim et al. 2009b) that postulated that DCA formation is not occurring exclusively from TCA. However, we found that liver levels of TCA and DCA did correlate significantly, which supports a hypothesis that TCA is the major precursor for the formation of DCA in this organ (Ketcha et al. 1996). A detailed liver toxicokinetic study of oxidative metabolites of TCE may be necessary to further characterize the metabolic fate of TCA and DCA.
Our finding that the concentration of DCVG in liver was much higher than that in serum and higher than the concentration of DCVC in either serum or liver further demonstrates that TCE conjugation with GSH to form DCVG occurs predominantly in the liver (Lash et al. 2014). Given that DCVG is rapidly excreted into the bile (van Bladeren 2000), the relative flux of TCE through GSH conjugation in the liver may be even greater than that observed in our study. As it has been suggested that GSH conjugation may be much greater in humans than in rodents (Lash et al. 1999), careful estimation of the biliary excretion of GSH conjugates of TCE may be needed to completely understand the kinetics of this metabolic pathway.
Based on the levels of TCA and DCA observed in the sub-acute study (Figure 1), two strains were chosen, C57BL/6J and NZW/LacJ, which represent low or high levels of CYP450-dependent oxidation of TCE, respectively. Time-course analysis of TCE metabolites in liver showed a decreasing trend of TCA and TCOH concentration, especially in the high dose (400 mg/kg/day) group. Previous studies of TCE metabolism showed that under conditions of acute exposure in mice, the induction of the monooxygenase system results in greater liver metabolism of TCE (Dekant et al. 1986) and no apparent saturation of metabolism is observed (Prout et al. 1985). However, saturation of TCE metabolism does occur in the rat liver at high doses (Prout et al. 1985; Dekant et al. 1986). A long-term study with TCE in rats and mice showed that daily dosing for 180 days did not induce the overall metabolism of TCE, but did double the urinary excretion of TCA (Green and Prout 1985).
The time-dependent change in kinetics may be explained by either auto-induction (Chaudhry et al. 2010) or depletion of co-substrates such as NADPH, NADH, and NAD+ (Lipscomb et al. 1996). Auto-induction may not explain our findings because Cyp2e1, Aldh2, and Adh, major enzymes involved in oxidative metabolism of TCE, were not affected by TCE dose or by the duration of exposure. In addition, it is possible that a decrease in TCE metabolism may be due to insufficient availability of co-substrates that are affected by not only by the extent of metabolism, but also by the redox status of the liver. Oxidative stress, as a secondary event that follows cytotoxicity and peroxisome proliferation in the liver, is one such mechanistic event. Several studies that examined TCA- and DCA-induced hepatic oxidative stress demonstrated small, albeit significant, increases in lipid peroxidation and oxidative DNA damage (Austin et al. 1996; Parrish et al. 1996). While we reason that the time-dependent decrease in TCE metabolism observed herein may plausibly result from the saturation of co-substrate supply or from oxidative stress, these mechanisms need further study.
The second important and novel outcome of our work is the investigation of the quantitative relationships among TCE metabolite levels and liver toxicity phenotypes. Because significant inter-strain variability was observed in TCE metabolism and liver toxicity, this study offers a unique opportunity to provide a scientific data-driven rationale for some of the major assumptions in human health assessment of TCE and other chlorinated solvents. Specifically, we examined major metabolizing enzymes responsible for oxidative biotransformation of TCE, as well as markers of cell proliferation and peroxisome proliferation in liver.
Inter- and intra-species differences in oxidative metabolism of TCE have been examined in relation to the expression levels of key xenobiotic metabolism enzymes, primarily CYP2E1 (Lash et al. 2014). We observed a correlation between the amounts of TCA produced in liver and a baseline Cyp2e1 protein level across strains. This finding may be attributable to the previously reported observation that not only is CYP2E1 involved in the first step of TCE oxidation, but CYP2E1 also catalyzes the transformation of TCOH to TCA (Stenner et al. 1997). CYP2E1 is not the only CYP450 enzyme that may act on TCE. For example, liver Cyp2e1 content was found to be similar in rats and mice (Nakajima et al. 1993) even though major differences in TCE metabolism are known to exist between mice and rats. Human CYP1A1/2, CYP2A6, and CYP3A4 are also known to oxidize TCE, with CYP1A2 being the major alternative to CYP2E1 (Lash et al. 2014).
A recent study using a mouse model of the human population showed that TCE metabolism was strongly associated with induction of PPARα-mediated lipid and nucleic acid metabolism pathways in mouse liver regardless of the genetic background (Bradford et al. 2011). In our study, we found that under conditions of sub-acute and sub-chronic treatment with TCE, significant up-regulation of Acox1 and Cyp4a10 expression occurred in 6 out of 7 strains and the extent of gene expression was correlated with the liver levels of TCA and DCA. Strong correlation between TCA and DCA levels in liver makes it difficult to interpret their relative contributions to the induction of peroxisome proliferation response. However, DCA is widely regarded as a weaker ligand for PPARα activation compared to TCA (Corton 2008), and the amount of DCA detected in this study in the liver is orders of magnitude lower than that of TCA.
Similar to an observed time-dependent decrease in TCE metabolite formation, the expression levels in Acox1 and Cyp4a10 decreased over time in the sub-chronic study, which indicates a close association between peroxisome proliferation and the oxidative metabolism of TCE. Contrary to the decrease in the oxidative metabolism and peroxisome proliferation over time, liver enlargement and hepatocellular proliferation effects were most prominent in mice treated with TCE for 4 weeks. Neither TCA nor DCA in liver was correlated with hepatocellular proliferation in the sub-chronic study, which may suggest that multiple metabolites or pathways are likely to be involved in liver toxicity due to TCE (Rusyn et al. 2014).
Supplementary Material
Acknowledgments
Funding
This work was supported, in part, by grants from NIEHS (P42 ES005948 and P30 ES010126) and AstraZeneca (Bioscience PhD Studentship to Hong Sik Yoo). The authors are grateful to Dr. Gary Boorman for his contribution to pathologic considerations in this study. The authors also thank Dr. Sungkyoon Kim (Seoul National University) for useful discussions of the study design and results.
List of Abbreviations
- BrdU
5-bromo-2′-deoxyuridine
- DCA
Dichloroacetic acid
- DCVC
S-(1,2-dichlorovinyl)-L-cysteine
- DCVG
S-(1,2-dichlorovinyl)glutathione
- DFA
Difluoroacetic acid
- GSH
Glutathione
- LLOQ
Lower limit of quantification
- NADPH
Nicotinamide adenine dinucleotide phosphate
- PPARα
Peroxisome proliferator-activated receptor alpha
- TCA
Trichloroacetic acid
- TCE
Trichloroethylene
- TCOH
Trichloroethanol
- TFA
Trifluoroacetic acid
Footnotes
Previous publication
Partially presented at the Society of Toxicology annual meeting, San Francisco, CA, March 2012, and at the International Congress of Toxicology, Seoul, South Korea, July 2013.
References
- Anna CH, Maronpot RR, Pereira MA, Foley JF, Malarkey DE, Anderson MW. Ras proto-oncogene activation in dichloroacetic acid-, trichloroethylene- and tetrachloroethylene-induced liver tumors in B6C3F1 mice. Carcinogenesis. 1994;15(10):2255–61. doi: 10.1093/carcin/15.10.2255. [DOI] [PubMed] [Google Scholar]
- ATSDR. Priority List of Hazardous Substances. 2013 Oct. 25th, 2011 [cited July 26th 2013]. Available from http://www.atsdr.cdc.gov/SPL/index.html.
- Austin EW, Parrish JM, Kinder DH, Bull RJ. Lipid peroxidation and formation of 8-hydroxydeoxyguanosine from acute doses of halogenated acetic acids. Fundam Appl Toxicol. 1996;31(1):77–82. doi: 10.1006/faat.1996.0078. [DOI] [PubMed] [Google Scholar]
- Bradford BU, Lock EF, Kosyk O, Kim S, Uehara T, Harbourt D, DeSimone M, Threadgill DW, Tryndyak V, Pogribny IP, Bleyle L, Koop DR, Rusyn I. Interstrain differences in the liver effects of trichloroethylene in a multistrain panel of inbred mice. Toxicol Sci. 2011;120(1):206–17. doi: 10.1093/toxsci/kfq362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry AS, Urban TJ, Lamba JK, Birnbaum AK, Remmel RP, Subramanian M, Strom S, You JH, Kasperaviciute D, Catarino CB, Radtke RA, Sisodiya SM, Goldstein DB, Schuetz EG. CYP2C9*1B promoter polymorphisms, in linkage with CYP2C19*2, affect phenytoin autoinduction of clearance and maintenance dose. J Pharmacol Exp Ther. 2010;332(2):599–611. doi: 10.1124/jpet.109.161026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WA, Campbell JL, Clewell HJ, Zhou YH, Wright FA, Guyton KZ, Rusyn I. Physiologically-Based Pharmacokinetic (PBPK) Modeling of Inter-strain Variability in Trichloroethylene Metabolism in the Mouse. Environ Health Perspect. 2014;122(5):456–463. doi: 10.1289/ehp.1307623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WA, Jinot J, Scott CS, Makris SL, Cooper GS, Dzubow RC, Bale AS, Evans MV, Guyton KZ, Keshava N, Lipscomb JC, Barone S, Fox JF, Gwinn MR, Schaum J, Caldwell JC. Human health effects of trichloroethylene: key findings and scientific issues. Environ Health Perspect. 2013;121(3):303–11. doi: 10.1289/ehp.1205879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WA, Okino MS, Evans MV. Characterizing uncertainty and population variability in the toxicokinetics of trichloroethylene and metabolites in mice, rats, and humans using an updated database, physiologically based pharmacokinetic (PBPK) model, and Bayesian approach. Toxicol Appl Pharmacol. 2009;241(1):36–60. doi: 10.1016/j.taap.2009.07.032. [DOI] [PubMed] [Google Scholar]
- Corton JC. Evaluation of the role of peroxisome proliferator-activated receptor alpha (PPARalpha) in mouse liver tumor induction by trichloroethylene and metabolites. Crit Rev Toxicol. 2008;38(10):857–875. doi: 10.1080/10408440802209796. [DOI] [PubMed] [Google Scholar]
- Dekant W, Schulz A, Metzler M, Henschler D. Absorption, elimination and metabolism of trichloroethylene: a quantitative comparison between rats and mice. Xenobiotica. 1986;16(2):143–52. doi: 10.3109/00498258609043517. [DOI] [PubMed] [Google Scholar]
- Green T. Species differences in carcinogenicity: the role of metabolism in human risk evaluation. Teratog Carcinog Mutagen. 1990;10(2):103–13. doi: 10.1002/tcm.1770100206. [DOI] [PubMed] [Google Scholar]
- Green T, Prout MS. Species differences in response to trichloroethylene. II. Biotransformation in rats and mice. Toxicol Appl Pharmacol. 1985;79(3):401–11. doi: 10.1016/0041-008x(85)90138-3. [DOI] [PubMed] [Google Scholar]
- Guha N, Loomis D, Grosse Y, Lauby-Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Baan R, Mattock H, Straif K, International Agency for Research on Cancer Monograph Working Group Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. Lancet Oncol. 2012;13(12):1192–3. doi: 10.1016/s1470-2045(12)70485-0. [DOI] [PubMed] [Google Scholar]
- Hansen J, Sallmen M, Selden AI, Anttila A, Pukkala E, Andersson K, Bryngelsson IL, Raaschou-Nielsen O, Olsen JH, McLaughlin JK. Risk of cancer among workers exposed to trichloroethylene: analysis of three nordic cohort studies. J Natl Cancer Inst. 2013;105(12):869–77. doi: 10.1093/jnci/djt107. [DOI] [PubMed] [Google Scholar]
- Keshava N, Caldwell JC. Key issues in the role of peroxisome proliferator-activated receptor agonism and cell signaling in trichloroethylene toxicity. Environ Health Perspect. 2006;114(9):1464–70. doi: 10.1289/ehp.8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketcha MM, Stevens DK, Warren DA, Bishop CT, Brashear WT. Conversion of trichloroacetic acid to dichloroacetic acid in biological samples. J Anal Toxicol. 1996;20(4):236–241. doi: 10.1093/jat/20.4.236. [DOI] [PubMed] [Google Scholar]
- Kim S, Collins LB, Boysen G, Swenberg JA, Gold A, Ball LM, Bradford BU, Rusyn I. Liquid chromatography electrospray ionization tandem mass spectrometry analysis method for simultaneous detection of trichloroacetic acid, dichloroacetic acid, S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine. Toxicology. 2009a;262(3):230–8. doi: 10.1016/j.tox.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Pollack GM, Collins LB, Rusyn I. Pharmacokinetic analysis of trichloroethylene metabolism in male B6C3F1 mice: Formation and disposition of trichloroacetic acid, dichloroacetic acid, S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine. Toxicol Appl Pharmacol. 2009b;238(1):90–9. doi: 10.1016/j.taap.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, Deluca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33(6):655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Lash LH, Chiu WA, Guyton KZ, Rusyn I. Trichloroethylene biotransformation and its role in mutagenicity, carcinogenicity and target organ toxicity. Mutat Res. 2014 doi: 10.1016/j.mrrev.2014.04.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Brashear WT, Abbas R, Parker JC, Fisher JW. Identification of S-(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene. J Toxicol Environ Health A. 1999;56(1):1–21. doi: 10.1080/009841099158204. [DOI] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Parker JC. Metabolism and tissue distribution of orally administered trichloroethylene in male and female rats: identification of glutathione- and cytochrome P-450-derived metabolites in liver, kidney, blood, and urine. J Toxicol Environ Health A. 2006;69(13):1285–1309. doi: 10.1080/15287390500360133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Qian W, Putt DA, Hueni SE, Elfarra AA, Krause RJ, Parker JC. Renal and hepatic toxicity of trichloroethylene and its glutathione-derived metabolites in rats and mice: sex-, species-, and tissue-dependent differences. J Pharmacol Exp Ther. 2001;297(1):155–164. [PubMed] [Google Scholar]
- Lipscomb JC, Mahle DA, Brashear WT, Garrett CM. A species comparison of chloral hydrate metabolism in blood and liver. Biochem Biophys Res Commun. 1996;227(2):340–50. doi: 10.1006/bbrc.1996.1511. [DOI] [PubMed] [Google Scholar]
- Maloney EK, Waxman DJ. Trans-activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol Appl Pharmacol. 1999;161(2):209–218. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- Marsman DS, Cattley RC, Conway JG, Popp JA. Relationship of hepatic peroxisome proliferation and replicative DNA synthesis to the hepatocarcinogenicity of the peroxisome proliferators di(2-ethylhexyl)phthalate and [4-chloro-6-(2,3-xylidino)-2- pyrimidinylthio]acetic acid (Wy-14,643) in rats. Cancer Res. 1988;48(23):6739–6744. [PubMed] [Google Scholar]
- Nakajima T, Wang RS, Elovaara E, Park SS, Gelboin HV, Vainio H. Cytochrome P450-related differences between rats and mice in the metabolism of benzene, toluene, and trichloroethylene in liver microsomes. Biochemical Pharmacology. 1993;45(5):1079–1085. doi: 10.1016/0006-2952(93)90252-r. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program. Carcinogenesis Studies of Trichloroethylene (Without Epichlorohydrin) (CAS No. 79-01-6) in F344/N Rats and B6C3F1 Mice (Gavage Studies) Natl Toxicol Program Tech Rep Ser. 1990;243:1–174. [PubMed] [Google Scholar]
- NICNAS. Trichloroethylene: Priority Existing Chemical Assessment Report. Australia: National Industrial Chemicals Notification and Assessment Scheme; 2000. [Google Scholar]
- NRC. Assessing the human health risks of trichloroethylene: Key scientific issues. Washington, D.C.: The National Academies Press; 2006. [Google Scholar]
- Parrish JM, Austin EW, Stevens DK, Kinder DH, Bull RJ. Haloacetate-induced oxidative damage to DNA in the liver of male B6C3F1 mice. Toxicology. 1996;110(1–3):103–11. doi: 10.1016/0300-483x(96)03342-2. [DOI] [PubMed] [Google Scholar]
- Prout MS, Provan WM, Green T. Species differences in response to trichloroethylene. I. Pharmacokinetics in rats and mice. Toxicol Appl Pharmacol. 1985;79(3):389–400. doi: 10.1016/0041-008x(85)90137-1. [DOI] [PubMed] [Google Scholar]
- Radican L, Blair A, Stewart P, Wartenberg D. Mortality of aircraft maintenance workers exposed to trichloroethylene and other hydrocarbons and chemicals: extended follow-up. J Occup Environ Med. 2008;50(11):1306–19. doi: 10.1097/JOM.0b013e3181845f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Chiu WA, Lash LH, Kromhout H, Hansen J, Guyton KZ. Trichloroethylene: Mechanistic, epidemiologic and other supporting evidence of carcinogenic hazard. Pharmacol Ther. 2014;141:55–68. doi: 10.1016/j.pharmthera.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CS, Jinot J. Trichloroethylene and cancer: systematic and quantitative review of epidemiologic evidence for identifying hazards. Int J Environ Res Public Health. 2011;8(11):4238–72. doi: 10.3390/ijerph8114238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JZ, Ho JW. Simultaneous detection of trichloroethylene alcohol and acetate in rat urine by gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;789(2):303–9. doi: 10.1016/s1570-0232(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Stenner RD, Merdink JL, Stevens DK, Springer DL, Bull RJ. Enterohepatic recirculation of trichloroethanol glucuronide as a significant source of trichloroacetic acid. Metabolites of trichloroethylene. Drug Metab Dispos. 1997;25(5):529–35. [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA. Toxicological Review of Trichloroethylene (CAS No. 79-01-6): In Support of Summary Information on the Integrated Risk Information System (IRIS) Washington, DC: National Center for Environmental Assessment; 2011. [Google Scholar]
- van Bladeren PJ. Glutathione conjugation as a bioactivation reaction. Chem Biol Interact. 2000;129(1–2):61–76. doi: 10.1016/s0009-2797(00)00214-3. [DOI] [PubMed] [Google Scholar]
- Vlaanderen J, Straif K, Pukkala E, Kauppinen T, Kyyronen P, Martinsen JI, Kjaerheim K, Tryggvadottir L, Hansen J, Sparen P, Weiderpass E. Occupational exposure to trichloroethylene and perchloroethylene and the risk of lymphoma, liver, and kidney cancer in four Nordic countries. Occup Environ Med. 2013;70(6):393–401. doi: 10.1136/oemed-2012-101188. [DOI] [PubMed] [Google Scholar]
- von Grote J, Hurlimann C, Scheringer M, Hungerbuhler K. Reduction of occupational exposure to perchloroethylene and trichloroethylene in metal degreasing over the last 30 years: influences of technology innovation and legislation. J Expo Anal Environ Epidemiol. 2003;13(5):325–40. doi: 10.1038/sj.jea.7500288. [DOI] [PubMed] [Google Scholar]
- Walgren JE, Kurtz DT, McMillan JM. Expression of PPAR(alpha) in human hepatocytes and activation by trichloroacetate and dichloroacetate. Res Commun Mol Pathol Parmacol. 2000;108(1–2):116–132. [PubMed] [Google Scholar]
- Zeise L, Bois FY, Chiu WA, Hattis D, Rusyn I, Guyton KZ. Addressing human variability in next-generation human health risk assessments of environmental chemicals. Environ Health Perspect. 2013;121(1):23–31. doi: 10.1289/ehp.1205687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YC, Waxman DJ. Activation of peroxisome proliferator-activated receptors by chlorinated hydrocarbons and endogenous steroids. Environ Health Perspect. 1998;106(Suppl 4):983–988. doi: 10.1289/ehp.98106s4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual animal-level data for all quantitative endpoints is available publicly at the Mouse Phenome Database (http://phenome.jax.org/; accession ID: Rusyn6).