

SUMMARY
Self-renewal and proliferation of nephron progenitor cells and the decision to initiate nephrogenesis are crucial events directing kidney development. Despite recent advancements in defining lineage and regulators for the progenitors, fundamental questions about mechanisms driving expansion of the progenitors remain unanswered. Here we show that Eya1 interacts with Six2 and Myc to control self-renewing cell activity. Cell fate tracing reveals a developmental restriction of the Eya1+ population within the intermediate mesoderm to nephron-forming cell fates and a common origin shared between caudal mesonephric and metanephric nephrons. Conditional inactivation of Eya1 leads to loss of Six2 expression and premature epithelialization of the progenitors. Six2 mediates translocation of Eya1 to the nucleus, where Eya1 uses its threonine phosphatase activity to control Myc phosphorylation/dephosphorylation and function in the progenitor cells. Our results reveal a functional link between Eya1, Six2, and Myc in driving the expansion and maintenance of the multipotent progenitors during nephrogenesis.
INTRODUCTION
Kidney tissue is derived from the intermediate mesoderm (IM), a strip of tissue located adjacent to the axial mesoderm in the developing embryo (Saxén and Sariola, 1987). The IM gives rise to three types of kidney tissue in an anterior-to-posterior sequence: the pronephros, a transient embryonic structure; the mesonephros, the functional embryonic kidney; and the metanephros, the permanent adult kidney. Formation of the permanent kidney requires the generation of distinct precursor cells that differentiate into more than 30 different cell types within a mature kidney. Elucidating how these cell types are derived and how coordinated morphogenesis of these distinct cell types leads to the formation of a functional organ is essential for understanding cellular hierarchies in development and disease.
In mice, kidney development initiates at approximately embryonic day 10.5 (E10.5) via inductive interaction between the metanephric mesenchyme (MM) and the ureteric bud (UB) epithelium. MM formation at the caudal end of the nephrogenic cord is a critical step in kidney organogenesis because this tissue secretes signals inducing UB outgrowth and its branching morphogenesis to form the collecting duct system of the mature kidney (Davies and Fisher, 2002; Saxén and Sariola, 1987). The UB induces the MM to condense to form a precursor cell population that either self-renews to maintain the progenitor pool at the UB tips (cap mesenchyme [CM]) or undergoes epithelialization from pretubular aggregate (PA) to form the renal vesicle (RV), the precursor of the nephron. The balance between self-renewal and differentiation of the progenitor cells is essential for generation of a sufficient number of nephrons in a mature kidney.
Previous cell fate marking suggested that the UB and MM are both derived from a common Osr1+ IM, which appears at E8.5 (Mugford et al., 2008). A more recent study suggested that the MM might be derived from the caudal T (Brachyury)+/Osr1− mesoderm based on the observations that the MM precursors are maintained in the T+ caudal population until E8.5 and that the caudal T+ mesoderm can be induced to form nephrons in vitro (Taguchi et al., 2014). However, how the caudal T+ mesoderm is induced to adopt a nephron fate and how the MM and UB lineages are specified and segregated from each other are still unclear.
Among the regulatory genes identified in the MM, only Eya1 and Osr1 are found to be required for the initial formation of the MM, whereas all other genes are instead required for its subsequent differentiation. Six2 is essential for maintaining the renal progenitor population because Six2−/− MM undergoes premature epithelialization (Self et al., 2006). More recently, studies have shown that the Six2+ CM is compartmentalized into molecularly distinct subdomains and that signaling molecules such as Wnt, Fgfs, and Bmps play crucial roles in compartmentalization and promotion of progenitor maintenance and nephrogenesis (Brown et al., 2013; Karner et al., 2011). However, despite the importance of these factors in the maintenance of the progenitor pool and nephrogenesis, how Six2 activity is regulated and what intrinsic mechanisms drive the progenitors to expand is unclear.
The Eya family proteins are transcriptional coactivators and interact with the homeodomain So/Six proteins (Chen et al., 1997; Pignoni et al., 1997; Xu et al., 1997). Eya also possesses a phosphatase catalytic motif (Rebay et al., 2005). However, whether Eya’s phosphatase activity is necessary for maintaining the multipotency of the progenitor pool during nephrogenesis is not understood.
Among the Eya and Six family genes, Eya1, Six1, and Six2 are coexpressed in the MM at E10.5. Although Six1 expression in the MM disappears after the initial “T” stage (Nie et al., 2011), Eya1 and Six2 expression persists in the CM throughout nephrogenesis. However, whether the Eya1+ IM represents the earliest MM-committed population, how Eya1 drives MM formation and whether it interacts with Six2 to regulate the maintenance of the nephron progenitors remains to be elucidated.
Here we addressed the lineage of Eya1-expressing cells and the role of Eya1 in regulating nephrogenesis. Cell fate tracing reveals a developmental restriction of the Eya1+ IM at E8.5 to nephron-forming cell fates and a common origin shared between the caudal mesonephric and metanephric nephron. Eya1+ progenitors represent a multipotent progenitor population throughout nephrogenesis. Temporal deletion of Eya1 leads to loss of Six2 and premature epithelialization of the progenitors. Eya1 requires Six2 for its nuclear localization, and its nuclear activity regulates postphosphorylation modification of Myc. Our findings indicate a functional link between Eya1, Six2, and Myc in driving the expansion and maintenance of the multipotent progenitor population during nephrogenesis.
RESULTS
Eya1 Is Expressed in Caudal Mesonephric Tubules and Metanephric Progenitors
We performed X-gal staining for the Eya1LacZ knockin allele. Like Eya1 mRNA expression (Sajithlal et al., 2005), LacZ activity was detected in the IM from E8.5 (data not shown). Eya1+ (LacZ+) cells extended caudally and became restricted to the caudal region where UB forms (Figures 1A–1C and 1E). LacZ+ cells were also found in the caudal mesonephric tubules but not in the rostral mesonephric tubules that are fused with the nephric duct at the level of the forelimb (Figure 1D). By E11.5, its expression was confined to the MM and had disappeared in the anterior region (Figures 1E and 1F). Within the induced MM at E11.5, LacZ activity appeared uneven, and shorter staining only detected a subset of cells that were LacZ+ (Figures 1G), whereas all cells were LacZ+, as revealed by longer staining (Figure S1 available online). Throughout nephrogenesis, high LacZ activity was maintained in the CM, whereas low activity was also detectable in the PA, RV, and S-shaped body but not in its later derivatives (Figure 1H; Figures S1C and S1D). The Eya1+ CM can be divided into a high Eya1 subdomain directly opposed to the branching tip and a low Eya1 compartment next to the PA (Figures 1H and 1I) that overlaps with that of Six2 (Figure 1J). No expression was detected in the nephric duct and its derivatives. Together, the spatiotemporal pattern of Eya1 expression suggests that it may have a critical role in specifying and maintaining nephron progenitors.
Figure 1. The Eya1+ IM Contributes to Caudal Mesonephric and Metanephric Nephrons.
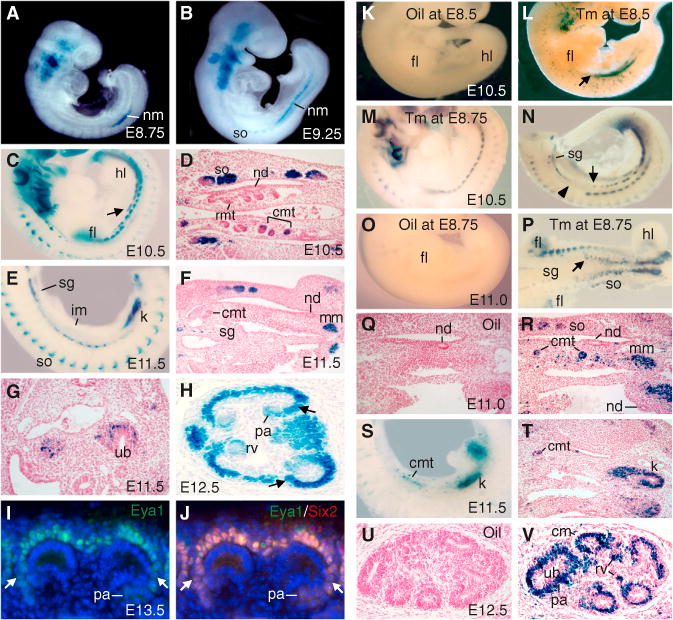
(A–C and E) β-gal staining of an Eya1LacZ/+ embryo at E8.75 (A), E9.25 (B), E10.5 (C), and E11.5 (E). The arrow points to the Eya1+ IM.
(D and F) Sections of β-gal+ embryos at E10.5 (D) and E11.5 (F).
(G and H) β-gal staining on E11.5 (shorter staining) (G) and E12.5 (H) kidney sections. The arrows in (H) point to weaker activity of the LacZ+ subregion. (I and J) Immunostaining for Eya1/Six2on anE13.5 kidney section, showing Eya1 alone (I) and a merged image for both Eya1 (green) and Six1 (red) (J). The arrows point to lower levels of the Eya1/Six2 subregion.
(K–V) Fate mapping of Eya1+ cells in Eya1CreERT2/+; R26RLacZ/+ embryos at E10.5–E12.5 after injection of oil (K, O, Q, and U) or 2–3 mg Tm (L–N, P, R–T, and V) at E8.5–E8.75. (K–O and S) show whole-mount lateral or (P) ventral views. The arrows point to the anterior limit of the Eya1+ IM. The arrowhead points to the forelimb region. (Q, R, and T–V) show sections counterstained with hematoxylin.
cmt, caudal mesonephric tubule; fl, forelimb; hl, hindlimb; k, kidney; nd, nephric duct; nm, nephrogenic mesoderm; rmt, rostral mesonephric tubule; sg, sympathetic ganglion; so, somite. See also Figure S1.
Caudal Mesonephric and Metanephric Nephrons Share a Common Developmental Origin of the Eya1+ IM
Because Eya1 is broadly expressed in the IM, it is possible that only a subpopulation of the Eya1+ IM at the caudal end takes a metanephric fate and maintains Eya1 expression, whereas the anterior Eya1+ IM may take other cell fates in which Eya1 expression is suppressed. To determine the fate map of the Eya1+ IM between E8.5 and E9.5, we generated the Eya1-CreERT2 knockin mouse line. Instead of the LacZ transgene, a tamoxifen (Tm)-regulated Cre recombinase (CreERT2) transgene was similarly introduced into the Eya1 locus at the position of the Eya1 initiation codon (Figures S1F and S1G). Eya1CreERT2 mice were intercrossed with Cre reporter R26RLacZ mice to permanently label descendant cells from the Eya1+ population. Without Tm, we essentially noted no leakiness of Cre activity (Figures 1K, 1O, 1Q, and 1U; Figure S2A), demonstrating that the Cre recombinase activity is absolutely dependent on drug administration. Single Tm (2 mg) injections facilitated genetic tracing of Eya1+ cells and their offspring. We first marked Eya1+ cells by injecting Tm at E8.5–E8.75, and subsequent LacZ staining was performed at various phases of kidney development.
When analyzed at E10.5–E11.5, during which the mesonephros achieves its maximum volume, marked cells were not detected in the rostral mesonephric vesicles but were restricted to the IM caudal to the forelimb (Figures 1L, 1N, and 1P). Although a subset of Eya1+ cells within this region contributed to the caudal mesonephric tubules, scattered LacZ+ cells were also present but disappeared from this region by E11.5 (Figures 1R–1T). The majority of marked cells condensed at the caudal end were confined to the MM at E11.5 (Figures 1S and 1T; Figure S2B). When analyzed at E12.5–postnatal day 0 (P0), marked cells contributed to the CM, PA, RV, and nephron tubules (Figures 1V and 2). No labeled cells contributed to the nephric duct and its derivatives (Figures 1R, 1T, 1V, 2C–2E, and 2K). Together, these results indicate that the caudal mesonephric and metanephric nephron share a common developmental origin from the Eya1+ IM.
Figure 2. Eya1+ Cells Contribute Continuously to Nephron Tubules throughout Kidney Development.
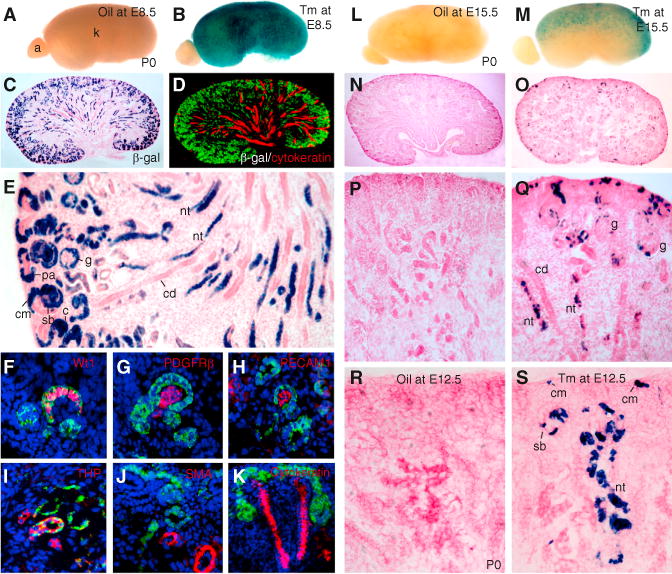
(A–E) β-gal-stained kidneys from Eya1CreERT2/+; R26RLacZ/+ embryos at E18.5 (oil or 2 mg Tm at E8.5). (A and B) show a whole-mount view. (C) shows a section view. (D) shows a kidney section costained for β-gal/cytokeratin. (E) shows a higher magnification of (C).
(F–K) Sections costained for β-gal/Wt1 (F), PDGFRb (G), PECAM1 (H), uromodulin (I), αSMA (J), or cytokeratin (K).
(L–Q) β-gal-stained kidneys at P0 after injection of oil (L, N, and P) or 1.5 mg Tm (M, O, and Q) at E15.5. (L and M) show a whole-mount view. (N–Q) show a section view. (P and Q) show a higher magnification of (N and O).
(R and S) β-gal-stained kidney sections at P0 injected with oil (R) or 0.5 mg Tm (S) at E12.5. a, adrenal gland; c, comma-shaped body; cd, collecting duct; cm, cap mesenchyme; g, glomerulus; nt, nephron tubule; pa, pretubular aggregate; sb, S-shaped body. See also Figure S2.
We next analyzed kidney at P0 to investigate which cell types were populated by the Eya1+-descendant lineage. Marked cells contributed to all MM-derived structures of the nephron (Figure 2C–2E). Immunostaining revealed that Bowman’s capsule and podocytes marked by Wt1 were β-galactosidase+ (β-gal+) (Figure 2F), confirming that Eya1+ progenitors are the cellular source of these components. In contrast, the glomerular mesangial cells marked by platelet-derived growth factor receptor β (PDGFRβ) and the glomerular capillary system marked by PECAM1 were β-gal−(Figures 2G and 2H), indicating that these cells are clonally distinct from the Eya1+ population. In the medullary region, uromodulin+ (THP+) (loop of Henle), Phaseolus vulgaris (PHA-E) lectin+ (proximal tubule), and peanut agglutinin (PNA) lectin+ (distal tubule) cells of the nephron were also β-gal+ (Figure 2I and data not shown), indicating that these cells are linearly related. Smooth muscle actin (SMA)+ stromal mesenchymal (SM) cells (Figure 2J) and cytokeratin+ cells in the collecting duct (Figure 2K) were β-gal− confirming that there is no contribution of the Eya1+ IM to those structures. Our observation that marked cells were specifically detected within the epithelial body of the nephron indicates that the MM progenitors originate from the Eya1+ IM and confirms that the metanephric nephron lineage is specified and segregated from the nephric duct and SM from E8.5.
Eya1+ CM Continues to Contribute to All Cell Types of the Nephron Tubule throughout Kidney Development
We further determined whether Eya1+ cells at later stages similarly contribute to nephron formation in kidney development by injecting Tm at E12.5–15.5 and analyzing labeled cells in Eya1CreET2;R26RLacZ/+ kidneys at P0. No labeled cells were detected in the absence of Tm administration (Figures 2L, 2N, 2P, and 2R). Labeled cells were observed in the CM and nephron tubules of the embryos from dams injected with 2 mg Tm at E15.5 (Figures 2M, 2O, and 2Q). We then reduced the dose of Tm by injecting a single dose of 0.5 mg into dams carrying Eya1-CreERT2/+;R26RlacZ/+ embryos at E12.5. Analysis of serial sections from six kidneys for each injection further demonstrated that multiple LacZ+ cells were observed within the CM and that they contributed to PA, RV, podocytes, and proximal and distal tubules at P0 (Figure 2S and data not shown). Therefore, the Eya1+ population is capable of continuously contributing to nephron formation throughout kidney development.
Clonal Tracing of Individual Eya1+ Cells
To address the multipotency of individual Eya1+ cells, limiting doses of Tm were used, and serial kidney sections with a single cluster of marked cells (putative clone) or a few (two to six) well isolated clusters were characterized. When Tm was injected at E12.5 at 0.1–0.2 mg, very few dispersed clusters of marked cells at P0 (two to six clusters per section at 10 μm), indicative of clonal events (Figure 3B), were observed. We analyzed 65 clusters, and all gave similar results. Multiple labeled cells were observed within the CM (Figures 3C–3E), confirming that Eya1+ cells undergo self-renewal. As expected, the majority of PA cells were labeled (Figures 3D). Within a single mature nephron tubule, serial section analysis demonstrated that labeled cells contributed to podocytes and proximal and distal tubules (Figures 3C–3E). Coimmunostaining further confirmed that β-gal+ cells contribute to Wt1+, PHA-E lectin+, PNA lectin+, and uromodulin+ cells in a clone (Figure S2C–S2E and data not shown). These results suggest that descendants of a single Eya1+ cell can differentiate into multiple cell types within the nephron.
Figure 3. Clonal Tracing of Individually Labeled Eya1+ Cells.
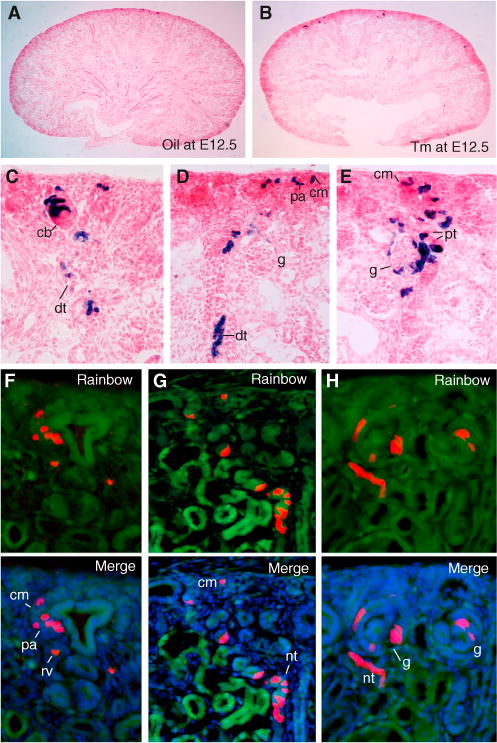
(A and B) β-gal-stained kidney sections from P0 Eya1CreERT2;R26RLacZ/+ mice injected with oil (A) or 0.1–0.2 mg Tm (B) at E12.5).
(C–H) Higher magnification showing three different clusters on serial sections (C–E) or three clones (red) on serial sections (F–H) from P0 Eya1CerERT2, R26RRainbow mice (0.2 mg Tm at E12.5).
cb, Comma-shaped body; cm, cap mesenchyme; dt, distal tubule; pt, proximal tubule. See also Figures S2C–S2H.
Because all labeled descendants are LacZ+, the early observations leave open questions of whether the distinct LacZ-labeled descendant cells are derived from a common progenitor or separate progenitors. To further confirm the clonality of labeled clusters, we analyzed individually labeled cells using a multicolor Cre-dependent reporter “Rainbow” mouse line that harbors a four-color reporter transgene (red, yellow, green, and blue) (Rinkevich et al., 2011). Tm injection induces single Eya1+ cells to randomly adopt one of the fluorescent colors, allowing discrimination between the clonal progeny of neighboring cells within the same Eya1+ pool. If different cell types of the entire nephron segment are derived from a common Eya1+ progenitor, they should appear in same color. We analyzed a total of 68 clusters at P0 treated with low dose of Tm, and all clones were single-colored and spanned the entire nephron axis, including the CM, PA, RV, podocytes, and proximal and distal tubules (Figures 3F–3H; Figures S2F–S2H), thus confirming that a single labeled Eya1+ cell can be programmed to form all cell types of the entire nephron.
Temporal Deletion of Eya1 in the CM Leads to Premature Differentiation and Depletion of the Nephron Progenitors
To address the requirement of Eya1 in the maintenance of MM progenitors, we used Eya1CreERT2 mice and crossed them with conditional Eya1flox mice (Figure S3) to delete Eya1 in a temporally controlled fashion during kidney development. Tm was injected as a single dose at E10.75–E11.0 after UB outgrowth to transiently delete Eya1 in MM progenitors, and kidneys were analyzed at E12.5–E17.5. Eya1Eya1CreCko/Cko (CkO) kidney was ~70% ± 3% (n = 8, p = 0.0208) at E17.5 and ~40% ± 2% (n = 8, p = 0.0315) at E14.5 shorter than that of the wild-type littermate (Figures 4A, 4A′, 4B, and 4B′). Histological analysis revealed CM formation surrounding branching UB tips in the wild-type embryos at E11.75 (Figure 4C). However, in Eya1Cko/Cko littermates, the mesenchymal cells surrounding the UB tips appeared as clustered aggregates or vesicle-like structures (Figure 4C′). At E12.5, the second round of UB branching and epithelial RVs were evident in controls (Figure 4D). However, the mutant UB development arrested at the initial T stage, and the ectopic RVs on the peripheral side and the absence of condensing MMs were already apparent (Figure 4D′). At E14.5, the wild-type kidney exhibited condensing mesenchyme and growing UB branches in the cortical nephrogenic zone (Figure 4E). In contrast, Eya1Eya1CreCko/Cko kidneys lacked condensing mesenchyme in the outmost region (Figure 4E′), which was confirmed by Wt1 staining (Figures 4F and F′). This phenotype was fully penetrant in all analyzed mutants (n = 8). A similar phenotype was obtained with the Six2Cre deletor (data not shown). Therefore, Eya1 is necessary for maintaining the nephron progenitor pool and preventing it from undergoing premature epithelialization.
Figure 4. Temporal Deletion of Eya1 in the MM Progenitors Results in Depletion and Premature Differentiation of the Progenitors.
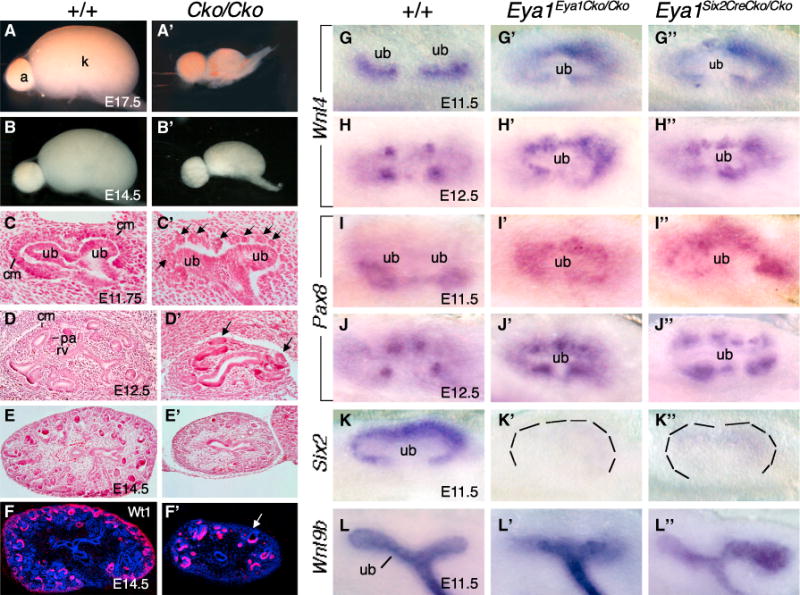
(A, A′, B, and B′) Kidneys at E17.5 (A and A′) and E14.5 (B and B′) of wild-type and Eya1CreERT2/+;Eya1Flox/Flox (Cko/Cko) embryos (Tm at ~E10.75).
(C and C′) Hematoxylin and eosin (H&E)-stained kidney section of the wild-type (C) and Eya1CreERT2/+;Eya1Flox/Flox (C′) at E11.75. The arrows point to ectopic vesicles.
(D, D′, E and E′) H&E-stained section of wild-type (D and E) and Eya1CreERT2/+;Eya1Flox/Flox (D′ and E′) kidneys at E12.5 (D and D′) and E14.5 (E and E′). The arrows point to ectopic vesicles.
(F and F′) Immunostaining with anti-Wt1 on wild-type (F) and Eya1CreERT2/+;Eya1Flox/Flox (F′) kidney sections at E14.5. The arrows point to the depletion of nephron progenitors.
(G–J″) In situ hybridization showing Wnt4 (G–G″ and H–H″) and Pax8 expression (I–I″ and J–J″) in PAs at E11.5 (G–G″ and I–I″) and RVs at E12.5 (H–H″ and J–J″) in wild-type and CKO embryos.
(K–K″) Six2 expression in the MM in E11.5 wild-type and CKO embryos.
(L–L″) Wnt9b expression in the UB in wild-type and CKO embryos.
See also Figures S3 and S4.
Eya1 Acts Upstream of and Interacts with Six2 to Maintain Nephron Progenitors
We next examined molecular markers expressed by the mesenchyme and epithelium to address the basis of the defects. Wnt4 is expressed first in mesenchymal aggregates on the medullary side of the branching UB at E11.5 and in RVs at E12.5 (Figures 4G and 4H). In Eya1Cko/Cko mutants, ectopic Wnt4 expression was observed on the peripheral side of the UB at E11.5 and E12.5 (Figures 4G′, 4G″, 4H′, and 4H″). Similar results were obtained for Pax8 (Figures 4I–4I″ and 4J–4J″).
Similar to Eya1, Six2 expression is high in nephron progenitors (Figure 4K), and loss of Six2 also causes premature differentiation and depletion of nephron progenitors (Self et al., 2006). In contrast to the presence of Eya1 expression in the Six2−/− MM at E10.5–E11.5 (Figure S4J), Six2 expression in the MM was almost undetectable in Eya1Cko/Cko mutants (Figures 4K′ and 4K″). However, Wnt9b was expressed in the branching UB tips in the CKO mutants, which often exhibited abnormal morphology because of incomplete or abnormal branching (Figures 4L–L″). Therefore, Eya1 is required for Six2 expression in the MM, and loss of Six2 most likely plays a significant role in causing the CKO mutant phenotype.
We next tested whether these two genes interact during nephrogenesis by examining kidney development in the Eya1;Six2 compound mutant. Consistent with previous observations (Self et al., 2006), Six2−/− kidneys were ~50% smaller in length than wild-type littermate controls at E14.5 (Figure S4A). Although kidneys of Eya1+/−;Six2+/− embryos at E14.5 were ~21% smaller than wild-type controls, no nephron structures were detectable in Eya1+/−; Six2−/− kidneys (Figure S4A). At E12.5, only very few Wt1+ cells were present in Eya1+/−; Six2−/− kidneys (Figures S4C and S4F) compared with Six2−/− (Figures S4D and S4G) and wild-type controls (Figures S4B and S4E). In Eya1+/−; Six2−/− kidneys, first branching occurred but appeared incomplete, and the MM progenitors were largely depleted and had disappeared completely by E11.5 as labeled by Eya1 (Figure S4I). Although ectopic Pax8+ cells were detectable in the peripheral mesenchyme, fewer Pax8+ or Wnt4+ cells were observed in the compound mutant (Figures S4K–S4Q). The enhancement of the kidney phenotype observed in the compound mutants suggests that Eya1 and Six2 interact genetically to synergistically mediate the expansion of the progenitors.
Eya1 Modulates Phosphorylation Modification of Myc in the CM to Maintain Its Multipotency
Our results identify an essential role of Eya1 in the expansion and maintenance of nephron progenitors. The Myc family of proto-oncogenes is also expressed in the MM progenitors and is essential for progenitor cell proliferation and kidney growth (Bates, 2000; Couillard and Trudel, 2009). We isolated Myc as Eya1’s binding partner via a yeast two-hybrid screen from a mouse E11.5 cDNA library (Figure S5A). We then examined whether Eya1 interacts with Myc to regulate self-renewal and proliferation of the progenitors. Although coimmunoprecipitation (coIP) analysis using E13.5 kidney extracts confirmed the physical interaction between Eya1 and Six2, they also interact with N- or C-Myc (Figure 5A). Therefore, Myc proteins appear to form a complex or complexes with Eya1 and Six2 in nephron progenitors.
Figure 5. Eya1 Interacts with Six2 and Myc and Regulates Myc Postphosphorylation Modification in Nephron Progenitors.
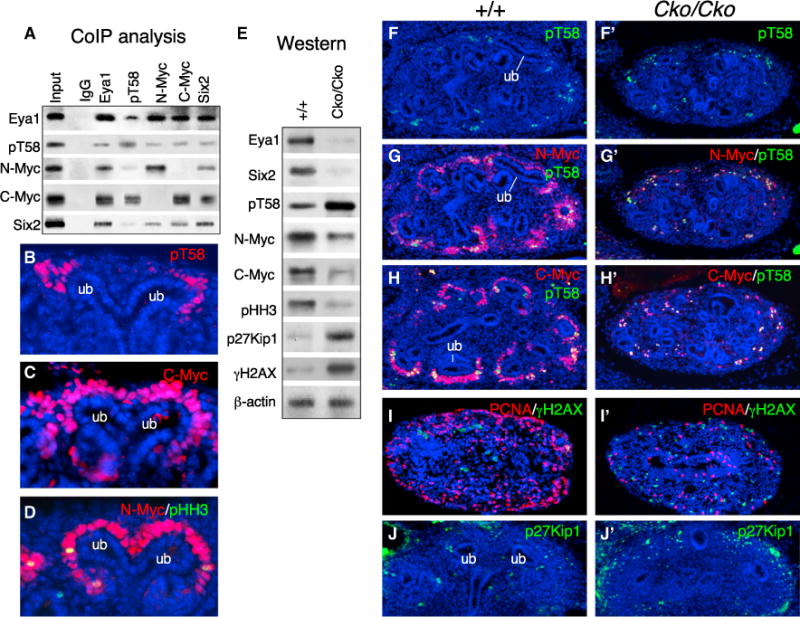
(A) CoIP analysis. Antibodies for IP and western blot are indicated.
(B–D) Immunostaining of E13.5 kidney sections for pT58 (B), C-Myc (C), and N-Myc/pHH3 (D).
(E) Western blot of cell extracts from E12.5 wild-type or CKO kidneys (Tm at E11.0) with the indicated antibodies. The membrane was stripped and reprobed.
(F–J′) Immunostaining of E12.5 kidney sections for pT58 (Fand F′), N-Myc/pT58(G and G′), C-Myc/pT58 (Hand H′), PCNA/γH2AX (I and I′), and p27Kip1 (J and J′) in the wild-type (F–J) and mutant (F′–J′).
See also Figure S5.
Myc proteins are subjected to posttranslational modifications such as phosphorylation and ubiquitination, and cells lacking C- or N-Myc cease to proliferate and exit the cell cycle (de Alboran et al., 2001; Domínguez-Frutos et al., 2011; Trumpp et al., 2001). Because Eya1 possesses phosphatase activity, we asked whether it regulates Myc phosphorylation modification. Phosphorylation at T58 and S62 within the highly conserved N-terminal Myc box1 in all mammalian Myc family proteins is known to play a critical role in Myc protein turnover. Phosphorylation at S62 stabilizes Myc and is required for its subsequent phosphorylation of T58, which is associated with Myc protein degradation targeted by ubiquitin ligases (Sjostrom et al., 2005; Welcker and Clurman, 2008; Welcker et al., 2004). An antibody against phospho-T58 C-Myc (pT58), which detects the phosphorylated Myc on T58, also coprecipitated Eya1 (Figure 5A). The weaker intensity of Eya1 precipitated by the anti-pT58 might be due to lower levels of pT58 in the progenitors. Indeed, immunostaining revealed that pT58 only accumulated in a subset of the CM close to the branching tip (Figure 5B), whereas C- and N-Myc were co-localized in the CM (Figures 5C and 5D; Figure S5B). Together, these data indicate physical interactions between Eya1, Six2, and Myc in nephron progenitors.
Western blot analysis of cell extracts from E12.5 kidneys (Tm at E10.75–E11.0) revealed a reduction in Myc levels but an increase in pT58 levels in the Eya1Cko/Cko mutant compared with littermate control (Figure 5E; Figure S5E). Double immunostaining confirmed that only a subset of Myc+ cells next to the branching tips were pT58+ in controls (Figures 5F–5H). In Eya1Cko/Cko kidneys, Myc+ cells were largely reduced compared with those in wild-type controls, and the majority of them were pT58+ (Figures 5F′–5H′). pHH3+, PCNA+, and 5-ethynyl-2′-deoxyuridine (EdU)+ cells were decreased markedly in the peripheral region of Eya1Cko/Cko kidneys (Figures 5E, 5I, and 5I′; Figure S5C). In contrast, the number of γH2AX+ cells was increased throughout the kidney in the mutant (Figures 5E, 5I, and 5I′), indicating a general increase in DNA double-strand breaks in the mutant. A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay confirmed that apoptotic cells were increased in the mutant (Figure S5D).
Western blot analysis revealed an elevation of cell cycle inhibitor p27Kip1 levels in the mutant compared with the littermate control (Figure 5E). Immunostaining confirmed numerous p27Kip1+ cells in the peripheral region of the mutant kidney (Figure 5J′) compared with only a few sporadic p27Kip1+ cells present in the peripheral region of the wild-type controls (Figure 5J). Quantitative real-time RT-PCR with total RNA isolated from ~E12.5 kidneys (Tm at E11.0) confirmed that C- or N-Myc mRNA levels were relatively unaffected in the mutant (Figure S5F). The relatively unchanged Myc mRNA and elevation of pT58 in Eya1 CKO suggest that the reduction of Myc might be caused by the dysregulation of posttranslational modification. Our observation of a strong interaction between Eya1, Six2, and Myc also suggests that these factors might act together to regulate the expansion of the nephron progenitors.
Reduction of Myc Levels in the Nephron Progenitors Leads to Upregulation of p27Kip1
We further tested the hypothesis that Myc dosage is critical for proper cell division to take place and that a reduction in its dosage causes premature cell cycle exit/arrest. Because mice lacking N-Myc or C-Myc die at E11.5 or E10.5, respectively, we analyzed kidneys from embryos homozygous for a hypomorphic mutation (N-Myc9a/9a) that express ~25%–30% of wild-type levels of N-Myc (Moens et al., 1993). Consistent with previous observations (Bates, 2000), the N-Myc9a/9a kidneys were ~19% ± 1.5% and ~27% ± 2.3% smaller than wild-type littermates at E14.5 and E18.5, respectively (n = 6; Figures S6A and S6B). At E14.5, while the CMs in N-Myc9a/9a kidneys were reduced noticeably as labeled by anti-Six2 (Figures 6A and 6A′), PCNA+ cells within the CM were also largely decreased (Figure 6B′) compared with those in the wild-type control (Figure 6B). However, p27Kip1+ cells were increased in the mesenchyme surrounding the UB tips in the mutant (Figures 6B, 6B′, and 6C). Because elevation of p27Kip1 is known to cause cell cycle exit/arrest, normal levels of N-Myc appear to be critical for cell cycle progression.
Figure 6. Eya1 Requires Six2 for Its Nuclear Localization, and Reduction of N-Myc Causes Elevation of p27Kip1.
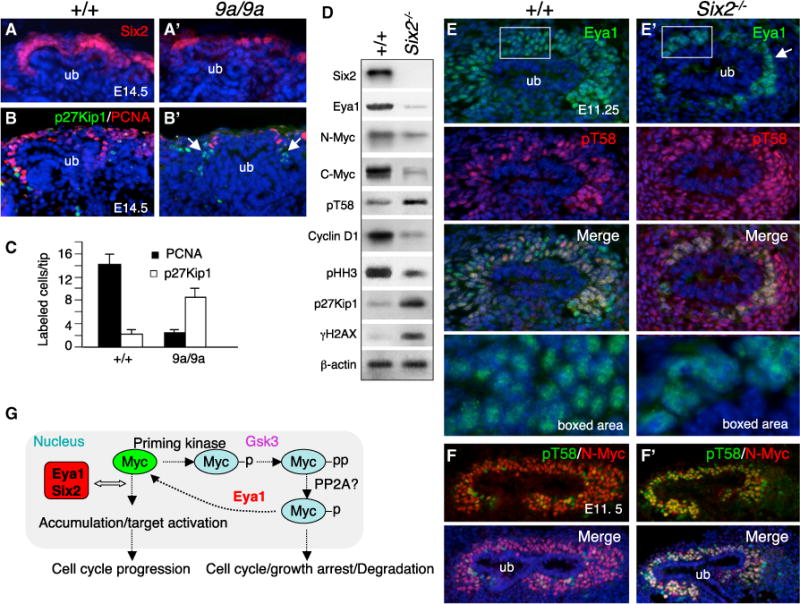
(A–B′) Immunostaining for Six2 (A and A′) and p27Kip1/PCNA (B and B′) on kidney sections of E14.5 wild-type and N-Myc9a/9a embryos. The arrows point to increased p27Kip1+ cells.
(C) Quantification of p27Kip1+ and PCNA+ cells per UB tip. P27Kip1+ or PCNA+ cells were counted in the mesenchyme surrounding the UB tip from a total of 25 tips on 10 μm sections and quantified using a StatView t test. Error bars indicate SD. p Values were between 0.0297 and 0.0318.
(D) Immunoblot of cell extracts from wild-type or Six2−/− kidneys at E11.75–E12.0 with the indicated antibodies. The membrane was stripped and reprobed.
(E and E′) Immunostaining for Eya1/pT58 on sections of E11.25 wild-type (E) and Six2−/− (E′) kidneys. The bottom panels show a higher magnification of the boxed areas.
(F and F′) Coimmunostaining for N-Myc/pT58 on sections of E11.5 wild-type (F) and Six2−/− (F′) kidneys.
(G) Model of the combined effects of Eya1-Six2-Myc on nephron progenitor cell proliferation. After receiving a growth-stimulatory signal, Myc gene transcription is induced, and newly synthesized Myc is phosphorylated on S62, which is necessary for the subsequent Gsk3-mediated phosphorylation at T58. Previous work demonstrated that S62 phosphate is removed by PP2A in the process of Myc ubiquitination. Our findings indicate that the maintenance of Myc protein is regulated in an Eya1/Six2-dependent manner. In the absence of Eya1 or Six2, pT58 levels are accumulated and targeted for degradation, which causes cell cycle/growth arrest as well as cell death. Eya1-Six2-Myc may also form a complex to activate target genes to control the timing of cell cycle exit. p, phosphorylation at S62; pp subsequent phosphorylation at T58.
See also Figure S6.
Six2 Is Necessary for Nuclear Localization of Eya1 in Nephron Progenitors
Because Six2, Eya1, and Myc interact and show nuclear colocalization in MM progenitors, we further investigated whether Six2 acts in the Eya1-Myc pathway to regulate progenitor cell division. Analysis of kidneys in Eya1;N-Myc;Six2 double- or triple-compound mutant embryos at E14.5 revealed a genetic interaction between these three genes in the developing kidney (Figures S6A and S6B). Western blot analysis of ~E11.75–E12.0 control and Six2−/− kidneys revealed that Myc was detectable in Six2−/− kidneys but was reduced (Figures 6D, 6F, and 6F′; Figure S6C). This reduction is likely due to depletion of the progenitors because Eya1 levels were also reduced (Figure 6D; Figures S4J and S6C). Interestingly, however, pT58 levels were elevated in the Six2−/− mutant (Figures 6D–6F′), further suggesting that regulation of Myc postphosphorylation is a critical event for maintaining MM progenitors. However, according to our hypothesis, pT58 should not accumulate in the presence of Eya1 in Six2−/− MM progenitors at E10.5–E11.5 (Figure S4J; Self et al., 2006). One likely explanation is that the subcellular localization of Eya1 might be altered in Six2−/− progenitors. Indeed, Eya1, which normally colocalizes with Six2 in the nuclear compartment of MM progenitors (Figures 1J and 6E), predominantly accumulated in the cytoplasmic compartment of Six2−/− progenitors (Figure 6E′). Furthermore, unlike its normal expression in the multilayered MM progenitors at this stage, Eya1 was only detectable in the innermost MM cells located on the surface of the UB epithelium in the Six2−/− mutant (Figure 6E′). In contrast, pT58 accumulation was observed in the multilayered Six2−/− MMs, whereas Myc+ cells were largely decreased in the mutant (Figures 6E′ and 6F′), similar to what was observed in the Eya1 CKO mutant (Figures 5F′–5H′). Therefore, this result provides in vivo evidence that Eya1 requires Six2 for its nuclear localization, thereby interacting with and dephosphorylating Myc to maintain the progenitors in the cell cycle to divide (Figure 6G).
Eya1 Dephosphorylates Myc from T58 to Prevent Degradation
To further test our model, we investigated the dependence of Six2 for nuclear localization of Eya1 and its interaction with Myc in cell lines. Western blot of cytoplasmic and nuclear extracts revealed that, when transfected into 293 cells, both cytoplasmic and nuclear FLAG-Eya1 was detected (Figure 7A). When cotransfected with His-Six2, FLAG-Eya1 was predominantly distributed in the nucleus (Figure 7A). Immunostaining confirmed this observation (Figure 7B and data not shown). Incontrast, cotransfection with FLAG-Myc did not obviously alter the subcellular distribution of FLAG-Eya1 (Figure 7A), suggesting that Myc is insufficient to mediate this process. CoIP analysis using nuclear extracts from 293 cells transfected with Eya1/Myc, Six2/Myc, or Eya1/Six2/Myc confirmed the physical interaction between Eya1 and Myc in the presence or absence of Six2 in 293 cells (Figure 7C). However, no interaction or a very weak physical interaction was observed between Myc and Six2 without Eya1 (Figure 7C). These results confirm that coexpression with Six2 leads to nuclear translocation of Eya1 and that Eya1 interacts with Myc when exogenously expressed in 293 cells.
Figure 7. Eya1 Stabilizes Myc.
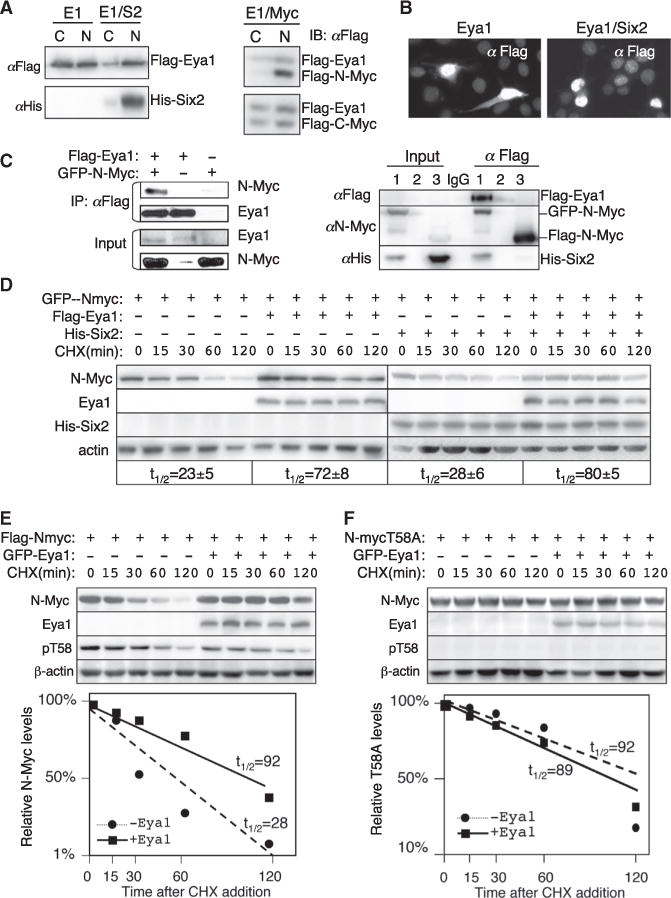
(A) Immunoblots (IB) of cytoplasmic (C) and nuclear (N) extracts from 293 cells transfected with FLAG-Eya1, FLAG-Eya1/His-Six2, or FLAG-Eya1/FLAG-Myc.
(B) Immunostaining with anti-FLAG of 293 cells transfected with FLAG-Eya1 or FLAG-Eya1/His-Six2.
(C) CoIP analysis. 293 cells were transfected with the plasmids indicated on the left or for lanes (lane 1, FLAG-Eya1/His-Six2/GFP-N-Myc; lane 2, empty lane; lane 3, His-Six2/FLAG-N-Myc; lane IgG for IP. Anti-FLAG was used for IP. Input was ~5% of the amount of proteins used for IP.
(D–F) Immunoblots with anti-N-Myc (D), anti-FLAG (D–F), anti-His (D), anti-Eya1 (E and F), anti-pT58 (E and F), or β-actin (loading control). Twenty-four hours posttransfection with the indicated plasmids, the cells were treated with CHX and lysed at the indicated times. Experiments were performed in triplicate, and graphs show quantification of the average results (t1/2, half-life). Myc levels were normalized to β-actin.
See also Figure S7.
Next we examined whether coexpression of Eya1/Myc in 293 cells can prevent Myc degradation by dephosphorylating it at T58. When transfected alone, only low levels of N-Myc protein were detected (Figure S7A). However, N-Myc was accumulated when cotransfected with FLAG-Eya1, or its accumulation was largely increased in a 293/FLAG-Eya1 stable line that constitutively expresses FLAG-Eya1 (Figure S7A). We measured the N-Myc half-life by treating the transfected cells with cycloheximide (CHX) to block new protein synthesis, and cells were harvested at different time points afterward. When expressed alone, N-Myc protein was degraded rapidly and showed a half-life of ~20–28 min after CHX treatment (Figures 7D and 7E), consistent with its half-life of ~20–30 min (Hann, 2006; Slamon et al., 1986). Coexpression of Eya1 markedly increased the levels of Myc protein and extended its half-life to ~72–92 min (Figures 7D and 7E; Figure S7B) but had no effect on Myc mRNA levels (Figure S7C). Addition of Six2 without or with Eya1 did not appear to have a significant effect on the half-life of Myc in 293 cells (Figure 7D).
To further evaluate whether the accumulation of Myc in the presence of Eya1 is a result of postphosphorylation modification regulated by Eya1, we used the Myc phosphorylation-dead mutants T58A and S62A as controls. Previous studies have shown that mutation of T58 to alanine (T58A) results in a stable and more oncogenic Myc protein that is no longer a substrate for ubiquitination (Gregory and Hann, 2000; Sears et al., 2000; Sears, 2004). Consistent with a half-life ranging from 50–110 min, T58A protein showed a half-life of ~89 min after CHX treatment (Figure 7F). Coexpression of Eya1 did not affect the levels of T58A, and its half-life was ~92 min (Figure 7F). Similarly, coexpression of Eya1 had no effect on the levels of S62A (Figure S7D), which had a half-life similar to the wildtype protein (Hann, 2006). FLAG-N-Myc-transfected cells revealed higher levels of pT58 detected with anti-pT58 (Figure 7E) than the negative control T58A (Figure 7F), confirming the specificity of the antibody. In contrast, pT58 levels in cells cotransfected with Myc/Eya1 were lower compared with those in cells transfected with N-Myc alone (Figure 7E). These results suggest a role of Eya1 in stabilizing the Myc protein by blocking its degradation through the regulation of postphosphorylation modification.
We then performed an in vitro phosphatase assay to directly examine the role of Eya1 in targeting the T58 phosphate. The in vitro assay, using purified FLAG-N-Myc (Figure S8), revealed that, after phosphorylation of Myc by GSK3β, the 32P-labeled protein disappeared from the substrates when incubated with purified FLAG-Eya1 (Figure 8A; Figure S8). Next we used Myc protein immunoprecipitated from transfected 293 cells, incubated with buffer only or purified FLAG-Eya1, and assessed T58 phosphorylation by western blotting with the pT58 antibody. Eya1 removed T58 phosphate from N- or C-Myc without reducing S62 phosphate (Figure 8B). Consistent with previous reports, phosphorylation at S62 is not affected in T58A, but T58 phosphorylation is blocked in the S62A mutant (Figure 8B) because phosphorylation at S62 is required for subsequent phosphorylation at T58. Therefore, T58, but not S62 phosphate, is a substrate for Eya1. These results support our model in which Eya1 interacts with and dephosphorylates Myc to maintain its levels in the progenitors to promote cell cycle progression for self-renewal and expansion (Figure 6G).
Figure 8. Eya1 Targets the T58 Phosphate of Myc.
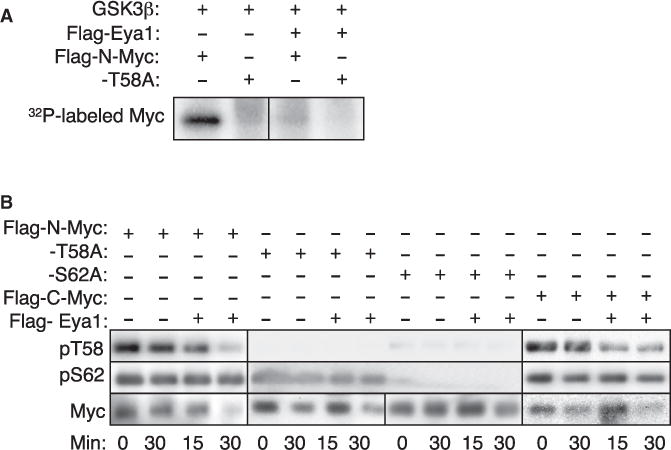
(A) In vitro phosphatase assay. Purified Myc protein was phosphorylated by GSK3β with 32P-γATP. The phosphorylated proteins were incubated with FLAG-Eya1, analyzed on SDS-PAGE, and exposed by phosphorimager.
(B) Eya1 dephosphorylates Myc at T58 but not S62. Purified Myc was incubated with either buffer (−) or FLAG-Eya1 at 30°C for 15 or 30 min. Samples were run in triplicate and immunoblotted with anti-pT58, anti-pS62, or anti-FLAG as indicated.
See also Figure S8.a
DISCUSSION
The Eya1+ Population within the IM at E8.5 Represents Nephron-Committed Progenitors
In mice, the nephric duct differentiates from the IM within the pronephric region at the level of somite 5, and it extends caudally toward the cloaca and induces adjacent IM to differentiate into two sets of mesonephric tubules (18–26 on each side): rostral tubules (four to six pairs at the level of the forelimb) fused with the nephric duct and caudal tubules with no connection to the nephric duct (Sainio et al., 1997). All except the most rostral tubules, which form the epididymal ducts in males, regress by E15.0. Our genetic lineage tracing indicates that Eya1+ cells contribute to caudal mesonephric and metanephric nephron-forming cell fates. No contribution is seen to the nephric duct, pronephros, rostral mesonephros, or other cell types of the kidney. Our finding that the rostral mesonephros has a distinct developmental origin from the caudal mesonephros is consistent with previous observations that the rostral and caudal mesonephros are structurally and functionally distinct and have different regulatory mechanisms (Sainio et al., 1997).
How is the nephron lineage specified and segregated from the UB lineage? It has been suggested previously that the MM and UB lineages might be derived from a common Osr1+ IM because the Osr1+ IM at E8.5 gives rise to the majority of cell types in the kidney (Mugford et al., 2008). Eya1 is also activated in the IM from E8.5, but Eya1+ cells show nephron-restricted cell fates. Therefore, the nephron lineage is specified and segregated from the UB lineage from E8.5. This is in agreement with a recent lineage tracing study showing that the MM, but not the UB, is derived from the caudal T+ population at E8.5 (Taguchi et al., 2014). T is a marker for the primitive streak and posterior nascent mesoderm, and its inactivation leads to a truncation of caudal structures, including the kidney (Herrmann et al., 1990). Based on the common nephron-restricted fate of the Eya1+ IM and T+ posterior population, we speculate that Eya1 may play a crucial role in a very early event specifying a subset of T+ mesodermal cells to adopt an Eya1+ nephron fate. When the nephron fate is specified, T is no longer needed. This explains why the nephron precursors are maintained in the caudal T+ mesoderm until E8.5 and why T+ or Eya1+ cells do not contribute to the nephric duct-derived structures. Osr1 might also collaborate with Eya1 during MM formation because deletion of either one leads to absence of the MM. Although detailed in vivo characterization of the contribution of the Osr1+ and T+ populations is needed to clarify the developmental origins of distinct kidney tissues, our data suggest that the T+/Eya1−/Osr1− mesoderm might be induced to become T−/Eya1+/Osr1− nephron precursors, which then give rise to T−/Eya1+/Osr1+ MM precursors. Therefore, Eya1 may provide a critical link from caudal T+ mesodermal cells to Osr1+ MM progenitors during metanephric specification.
How is the MM formed? We found that the uncondensed Eya1+ descendants are still present in the mesonephric region at E11.0 but disappear by E11.5. In contrast to its marked descendants, Eya1 is not expressed in those scattered anterior populations, and its expression is already restricted to the MM at E11.0–E11.5. This suggests that the disappearance of the marked Eya1+ descendants in the mesonephric region is unlikely to be a result of their caudal active migration to form the MM because, if those scattered cells were still actively migrating to contribute to the MM, Eya1 would still be expressed in those cells because the whole MM is Eya1+. However, it is possible that all Eya1+ descendants within the mesonephric region undergo regression after kidney organogenesis initiates, which leads to the disappearance of those scattered Eya1+ descendants. Therefore, shortly before the nephric duct reaches the cloaca, the nephrogenic precursors condense at the caudal end of the nephrogenic cord to form a functional MM for UB outgrowth, whereas the anterior mesenchyme is induced for mesonephric differentiation that eventually degenerates by apoptosis.
The genetic analysis of Eya1+ cell fates following low-dose Tm induction and using a multicolor reporter in this study provides additional in vivo evidence in support of the conclusion that nephron progenitors are multipotent and capable of giving rise to all segments of the nephron by early fate tracing studies. Our results are in general agreement with recent cell fate studies of Kobayashi et al. (2008) with Six2-Cre. However, unlike Eya1, Six2 is expressed in the MM from ~E10.5. Tm injection at E9.5 or earlier failed to induce labeled cells in the kidney (Kobayashi et al., 2008; Taguchi et al., 2014), further indicating that Six2 is not activated in MM precursors at those early stages. In contrast, Tm injection at E8.75 induced labeled cells in the mesonephros (Taguchi et al., 2014). Because our results demonstrate that the caudal mesonephric nephron shares a common origin with the metanephric nephron, Six2+ IM cells induced by Tm injection at E8.75 probably contributed to the rostral mesonephros, which explains why the labeled descendants were not observed in the MM (Taguchi et al., 2014). Future studies are necessary to clarify this. Nonetheless, our studies indicate that when Six2 is activated in the MM, it overlaps substantially with Eya1+ cells, and these two factors interact to regulate the maintenance of the progenitor pool.
The Role of Eya1 in the Maintenance and Expansion of the Multipotent Nephron Progenitor Pool
Our analyses indicate that Eya1 functions at multiple levels to regulate the nephron progenitors. Eya1 activity is required for Six2 expression in the mesenchymal progenitors. When Six2 is turned on, its activity is necessary for Eya1’s nuclear localization. The nuclear activity of Eya1 in the progenitors appears to be crucial for postphosphorylation modification and stabilization of Myc. Our results suggest that Myc is a physiological substrate for Eya1’s threonine phosphatase activity during proliferation of the nephron progenitor pool.
The importance of Six2 levels in the maintenance and differentiation of the progenitors via its interaction with Wnt signaling has been highlighted in several recent studies. Wnt9b signaling, which is crucial for promoting proliferation of self-renewing progenitor cells and induction of nephrogenesis, is transmitted in the Six2+ progenitors depending on the levels of Six2 and the coregulatory inputs through Six2 and β-catenin (Brown et al., 2013; Carroll et al., 2005; Karner et al., 2011; Park et al., 2012). Because Eya1 is expressed in the Wnt9b (Karner et al., 2011) and Six2 mutants at E11.5, it clearly has Six2/Wnt9b-independent early role(s) in the mesenchymal progenitors, including its requirement for MM formation and regulation of Six2 expression.
How does Eya1 act to regulate Six2 expression? Eya1 forms a transcriptional complex with Six family proteins to regulate downstream genes (Ahmed et al., 2012a, 2012b; Ohto et al., 1999). Therefore, it may interact with other members of the Six protein family at earlier stages to regulate MM formation, UB branching, and Six2 expression. In support of this, Six1 is coexpressed with Eya1 in the MM, and, in Six1−/− mice, the UB fails to undergo branching morphogenesis, and Six2 expression in the MM is also reduced (Nie et al., 2011; Xu et al., 2003). Because Six1 in the MM has disappeared after the initiation of branching morphogenesis (Nie et al., 2011), Six2 expression becomes necessary for Eya1’s nuclear localization in progenitors during nephrogenesis. Eya1 may have a cooperative role with Six2 in response to Wnt signaling during branching morphogenesis. Indeed, a recent study reported that Eya1 is positively regulated by Wnt signaling (Park et al., 2012).
Our finding of the dysregulation of postphosphorylation modification of Myc in the progenitors in the Eya1 CKO and Six2−/− mutants provides insights into the mechanistic basis of the Six2 and Eya1 mutant kidney phenotype. Myc proteins are known to play a crucial role in the expansion of progenitors. Deletion of C-Myc using Bmp7-Cre results in renal hypoplasia because of depletion of the progenitor cells in the CM, causing a decrease of the Six2+/Cited1+ population and of proliferation that likely impairs self-renewal (Couillard and Trudel, 2009). Because N-Myc is coexpressed in the MM and also regulates cell proliferation, these two members of the Myc family may function synergistically to regulate the proliferation of progenitors during nephrogenesis. An analysis of kidney development in their double mutants should reveal their redundant role in the expansion of the progenitors. However, in contrast to the Six2 and Eya1 CKO mutants, loss of Myc does not induce precocious differentiation of nephron progenitors. This is most likely due to the presence of Six2 in the Myc mutant (Couillard and Trudel, 2009), which could prevent the premature onset of nephrogenesis through its interaction with Wnt signaling. Our observation of upregulation of p27Kip1 in the N-Myc9a/9a kidney suggests that exhaustion of nephron progenitors in the Myc mutants is probably caused by the dysregulation of cell cycle progression during proliferation. This is in agreement with the observation of upregulation of p27Kip1 and premature cell cycle withdrawal of cochlear sensory progenitors in the N-Myc CKO mutant (Domínguez-Frutos et al., 2011).
Previous studies have suggested that C-Myc, cyclin A1, cyclin D1, and p27Kip1, as well as other cell cycle-related genes, might be downstream targets of the Eya-Six complex (Coletta et al., 2004; Li et al., 2003; McCoy et al., 2009; Wu et al., 2013; Xu, 2013). Although these findings highlight the importance of Eya-Six for normal expression of those cell cycle related genes, direct transcriptional regulation by the Eya-Six complex has not been demonstrated for any of those genes. Therefore, the reduction of their expression in Eya or Six mutants might reflect an indirect cause of Eya-Six. In our studies, we did not find evidence that Eya1 is directly involved in the regulation of Myc transcription but is necessary for its posttranslational modification. Because Myc proteins are well known drivers of proliferation of undifferentiated cells and because pT58 is known to be targeted by ubiquitin ligases, we propose a model in which Eya1 interacts with Six2 to translocate into the nucleus, whereby it binds with Myc to dephosphorylate Myc at T58 to prevent it from degradation during cell division to ensure normal cell cycle progression (Figure 6G). This model is supported by our results showing that Eya1 interacts with and stabilizes Myc when coexpressed in 293 cells and that T58 phosphate is a substrate for Eya1. In the absence of Eya1, pT58 is accumulated, but Myc is reduced, therefore causing an upregulation of p27Kip1 and a reduction of cyclin D, which, in turn, leads to cell cycle/growth arrest and degeneration of the progenitors. Because the Eya and Six genes are known to be oncogenic in multiple cancer cells (Xu, 2013), stabilization of Myc via dephosphorylation by Eya1 could be an essential function in regulating cell proliferation during development and tumorigenesis.
We found that pT58 levels are high in a subpopulation of Eya1+/Myc+ progenitors directly opposed to the branching tip. This might reflect a cell type-specific feature controlled by not only cell-intrinsic mechanism(s) but also extrinsic signals. Increasing Gsk3β activity has been reported to result in enhanced Myc turnover (Kenney et al., 2004), whereas inhibition of Gsk3 activity has been shown to enhance cerebellar granule neuron progenitor cell proliferation and endogenous N-Myc stabilization. Our data suggest that dephosphorylation of Myc by Eya1 might be critical in preventing Myc degradation induced by Gsk3β to maintain proper levels of Myc to regulate proliferation during self-renewal and expansion of the nephron progenitors. Too little Myc protein or activity in the absence of Eya1 may severely affect the proper functioning of cells and, consequently, affect their proliferation, differentiation, and apoptosis. Because Myc is a known downstream target for Wnt signaling in other systems during progenitor renewal/proliferation (He et al., 1998; Tetsu and McCormick, 1999), it is possible that Myc may also respond to Wnt signaling during nephrogenesis. In addition to Wnt signaling, Myc is targeted by multiple signal transduction cascades, including the Ras/Raf/mitogen-activated protein kinase, Jak/Stat, transforming growth factor β, and NF-κB pathways in cancer cells (Clevers, 2004; Liu and Levens, 2006) and cerebellar neuronal precursors (Kenney et al., 2004). Therefore, our finding of the molecular linkage between Eya1-Six2-Myc provides insights into the intracellular events that integrate effects of divergent signaling pathways crucial for coordinating nephron precursor proliferation and nephrogenesis. The deregulated proliferative activity conferred by loss of either Myc, Eya1, or Six2 suggests that inactivation of these genes may also impair the lengthening of the cell cycle, which may accompany the shift from proliferation to differentiation as well as cell death. Because Myc proteins function primarily as transcription factors, the physical interaction with Eya1/Six2 suggests that these factors may form complexes to synergistically regulate their targets. Future studies defining the nature of their complex formation and elucidating the intracellular events that coordinate effects of divergent signaling pathways active in the developing kidney are necessary for a comprehensive understanding of growth control in the nephron progenitors.
EXPERIMENTAL PROCEDURES
Mouse Strains
Eya1+/− (Xu et al., 1999), Six2+/− (Self et al., 2006), and N-Myc9a (Moens et al., 1993) mice were maintained on a 129/Sv and C57BL/6J mixed background. Eya1lacZ mice have been reported previously (Zou et al., 2008). The Eya1CreERT2 knockin allele was generated by replacing the LacZ gene with CreERT2. R26RLacZ Cre reporter mice were purchased from The Jackson Laboratory and maintained on C57BL/6J 3 Swiss Webster mixed and 129/Sv inbred backgrounds, respectively. The R26RRainbow mice contain a transgene that constitutively expresses GFP and, in the presence of Cre recombinase, is randomly recombined to express one of three other fluorescent proteins: mCherry, mOrange, or mCerulean (Rinkevich et al., 2011). All animal experiments were approved by the Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai (#06-822). Details regarding tamoxifen treatment and genotyping are provided in the Supplemental Experimental Procedures.
Histology
Dissected kidneys were fixed in 4% paraformaldehyde (PFA) for 1 hr at 4°C, dehydrated, and embedded in wax. Paraffin sections were generated at 10 μm. For cryosections, after fixation, kidneys were soaked in 30% sucrose overnight and embedded in OCT compound (Sakura, catalog no. 4583). Cryosections were generated at 10–13 μm using a Leica CM1900 cryostat.
β-gal Staining
β-gal staining was performed as described previously (Zou et al., 2008). Briefly, whole-mount kidneys were fixed in 4% PFA for 10 min and processed for cryosection. Cryosections were stained with X-gal at 37°C overnight and counterstained with 0.2% Eosin-Y or diluted hematoxylin. For whole-mount staining, kidneys were fixed in 4% PFA for 20 min at room temperature and stained at 37°C overnight for embryonic samples or at 4°C for 1–3 days for neonate samples.
In Situ Hybridization
Whole-mount or section in situ hybridization was performed according to standard procedures.
EdU and TUNEL Assays
The EdU assay was performed using a kit (catalog no. C10640, Life Technologies) following the manufacturer’s instructions. The TUNEL assay was performed using the ApopTag kit for in situ apoptosis fluorescein detection (catalog no. NC9815837, Millipore) following the manufacturer’s instructions.
Yeast Two-Hybrid Screen, Cell Transfection, CoIP, and Western Blot Analysis
For the yeast two-hybrid screen, the MATCHMAKER system and a pretransformed mouse E11 embryonic cDNA library (Clontech) were used, following the manufacturer’s instructions. Cell transfection, coIP analysis, and western blot analysis were performed as described previously (Ahmed et al., 2012b).
Protein Purification
The FLAG-Eya1 protein was purified from the FLAG-Eya1 stable cell line B22 using anti-FLAG M2 affinity gel (Sigma). The FLAG-Myc wild-type or mutant proteins were similarly purified from 293 cells transiently transfected with individual FLAG-Myc wild-type or mutant plasmids, respectively. Details are provided in the Supplemental Experimental Procedures.
In Vitro Phosphatase Assays
Purified FLAG-N-Myc or its mutant proteins as substrates were incubated with purified FLAG-Eya1. Details are provided in the Supplemental Experimental Procedures.
Quantitative Real-Time PCR
Total RNA was isolated from E12.25 kidneys with Trizol reagent (Invitrogen) according to the manufacturer’s instructions. A QuantiTect reverse transcription kit (QIAGEN) was used to synthesize first-strand cDNA from the total RNA template. Quantitative PCR was performed using the StepOnePlus PCR system and SYBR green detector (QIAGEN). Normalization was performed using specific amplification of β-actin, and PCR reactions were performed in triplicate for each biological duplicate experiment. The relative amounts of mRNA were calculated using the comparative Ct (threshold cycle) method (see Supplemental Experimental Procedures) for primers.
Supplementary Material
Acknowledgments
We thank Kevin Kelley at our mouse transgenic facility for helping with the ES work, Irving Weissman at Stanford for providing the Rainbow mice, Yuval Rinkevich at Stanford for technical assistance, and Guillermo Oliver for providing Six2 mutant mice. This work was supported by NIH Grant RO1 DK064640 (to P.X.X.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and eight figures and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2014.10.015.
References
- Ahmed M, Wong EY, Sun J, Xu J, Wang F, Xu PX. Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev Cell. 2012a;22:377–390. doi: 10.1016/j.devcel.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M, Xu J, Xu PX. EYA1 and SIX1 drive the neuronal developmental program in cooperation with the SWI/SNF chromatin-remodeling complex and SOX2 in the mammalian inner ear. Development. 2012b;139:1965–1977. doi: 10.1242/dev.071670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates CM. Kidney development: regulatory molecules crucial to both mice and men. Mol Genet Metab. 2000;71:391–396. doi: 10.1006/mgme.2000.3072. [DOI] [PubMed] [Google Scholar]
- Brown AC, Muthukrishnan SD, Guay JA, Adams DC, Schafer DA, Fetting JL, Oxburgh L. Role for compartmentalization in nephron progenitor differentiation. Proc Natl Acad Sci USA. 2013;110:4640–4645. doi: 10.1073/pnas.1213971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell. 2005;9:283–292. doi: 10.1016/j.devcel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Chen R, Amoui M, Zhang Z, Mardon G. Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell. 1997;91:893–903. doi: 10.1016/s0092-8674(00)80481-x. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt breakers in colon cancer. Cancer Cell. 2004;5:5–6. doi: 10.1016/s1535-6108(03)00339-8. [DOI] [PubMed] [Google Scholar]
- Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L, Wolf DM, Müller-Tidow C, Golub TR, Kawakami K, Ford HL. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci USA. 2004;101:6478–6483. doi: 10.1073/pnas.0401139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillard M, Trudel M. C-myc as a modulator of renal stem/progenitor cell population. Dev Dyn. 2009;238:405–414. doi: 10.1002/dvdy.21841. [DOI] [PubMed] [Google Scholar]
- Davies JA, Fisher CE. Genes and proteins in renal development. Exp Nephrol. 2002;10:102–113. doi: 10.1159/000049905. [DOI] [PubMed] [Google Scholar]
- de Alboran IM, O’Hagan RC, Gärtner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14:45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- Domínguez-Frutos E, López-Hernández I, Vendrell V, Neves J, Gallozzi M, Gutsche K, Quintana L, Sharpe J, Knoepfler PS, Eisenman RN, et al. N-myc controls proliferation, morphogenesis, and patterning of the inner ear. J Neurosci. 2011;31:7178–7189. doi: 10.1523/JNEUROSCI.0785-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-MycinBurkitt’s lymphoma cells. Mol Cell Biol. 2000;20:2423–2435. doi: 10.1128/mcb.20.7.2423-2435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann SR. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin Cancer Biol. 2006;16:288–302. doi: 10.1016/j.semcancer.2006.08.004. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, Carroll TJ. Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development. 2011;138:1247–1257. doi: 10.1242/dev.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426:247–254. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- Liu J, Levens D. Making myc. Curr Top Microbiol Immunol. 2006;302:1–32. doi: 10.1007/3-540-32952-8_1. [DOI] [PubMed] [Google Scholar]
- McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, Welm AL, Ford HL. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest. 2009;119:2663–2677. doi: 10.1172/JCI37691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens CB, Stanton BR, Parada LF, Rossant J. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development. 1993;119:485–499. doi: 10.1242/dev.119.2.485. [DOI] [PubMed] [Google Scholar]
- Mugford JW, Sipilä P, McMahon JA, McMahon AP. Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev Biol. 2008;324:88–98. doi: 10.1016/j.ydbio.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Xu J, El-Hashash A, Xu PX. Six1 regulates Grem1 expression in the metanephric mesenchyme to initiate branching morphogenesis. Dev Biol. 2011;352:141–151. doi: 10.1016/j.ydbio.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto H, Kamada S, Tago K, Tominaga SI, Ozaki H, Sato S, Kawakami K. Cooperation of six and eya in activation of their target genes through nuclear translocation of Eya. Mol Cell Biol. 1999;19:6815–6824. doi: 10.1128/mcb.19.10.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Ma W, O’Brien LL, Chung E, Guo JJ, Cheng JG, Valerius MT, McMahon JA, Wong WH, McMahon AP. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell. 2012;23:637–651. doi: 10.1016/j.devcel.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignoni F, Hu B, Zavitz KH, Xiao J, Garrity PA, Zipursky SL. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell. 1997;91:881–891. doi: 10.1016/s0092-8674(00)80480-8. [DOI] [PubMed] [Google Scholar]
- Rebay I, Silver SJ, Tootle TL. New vision from Eyes absent: transcription factors as enzymes. Trends Genet. 2005;21:163–171. doi: 10.1016/j.tig.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Rinkevich Y, Lindau P, Ueno H, Longaker MT, Weissman IL. Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature. 2011;476:409–413. doi: 10.1038/nature10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainio K, Hellstedt P, Kreidberg JA, Saxén L, Sariola H. Differential regulation of two sets of mesonephric tubules by WT-1. Development. 1997;124:1293–1299. doi: 10.1242/dev.124.7.1293. [DOI] [PubMed] [Google Scholar]
- Sajithlal G, Zou D, Silvius D, Xu PX. Eya 1 acts as a critical regulator for specifying the metanephric mesenchyme. Dev Biol. 2005;284:323–336. doi: 10.1016/j.ydbio.2005.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxén L, Sariola H. Early organogenesis of the kidney. Pediatr Nephrol. 1987;1:385–392. doi: 10.1007/BF00849241. [DOI] [PubMed] [Google Scholar]
- Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004;3:1133–1137. [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006;25:5214–5228. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell. 2005;9:327–338. doi: 10.1016/j.devcel.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Boone TC, Seeger RC, Keith DE, Chazin V, Lee HC, Souza LM. Identification and characterization of the protein encoded by the human N-myc oncogene. Science. 1986;232:768–772. doi: 10.1126/science.3008339. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R. Redefining the invivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2014;14:53–67. doi: 10.1016/j.stem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001;414:768–773. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14:1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- Wu K, Li Z, Cai S, Tian L, Chen K, Wang J, Hu J, Sun Y, Li X, Ertel A, Pestell RG. EYA1 phosphatase function is essential to drive breast cancer cell proliferation through cyclin D1. Cancer Res. 2013;73:4488–4499. doi: 10.1158/0008-5472.CAN-12-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX. The EYA-SO/SIX complex in development and disease. Pediatr Nephrol. 2013;28:843–854. doi: 10.1007/s00467-012-2246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Cheng J, Epstein JA, Maas RL. Mouse Eya genes are expressed during limb tendon development and encode a transcriptional activation function. Proc Natl Acad Sci USA. 1997;94:11974–11979. doi: 10.1073/pnas.94.22.11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–117. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130:3085–3094. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D, Erickson C, Kim EH, Jin D, Fritzsch B, Xu PX. Eya1 gene dosage critically affects the development of sensory epithelia in the mammalian inner ear. Hum Mol Genet. 2008;17:3340–3356. doi: 10.1093/hmg/ddn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.