

Lyme disease, caused by spirochetes of the Borrelia burgdorferi sensu lato complex (hereafter referred to as Lyme borrelia), is the most common tick-borne infection in the northern hemisphere (1). The potential for the infection to cause arthritis led to its recognition as a clinical entity in 1975 (2), and since then, the number of reported cases has steadily increased in the United States and elsewhere. The Centers for Disease Control and Prevention (CDC) estimates the annual case incidence of Lyme disease in the United States is ~300,000, ten-fold more than the number reported by physicians. This review is intended to provide rheumatologists with a current understanding of North American Lyme disease, with emphasis on arthritis pathogenesis and expected outcomes from treatment.
Ticks, Borrelia Burgdorferi and Lyme Disease
Ticks within the Ixodidae family transmit Lyme borrelia and an enzootic cycle maintaining both ticks and spirochetes must be established for Lyme disease to occur in a particular geographic area (1). In the United States, the prevalence of Lyme borrelia-infected ticks and disease incidence are greatest in the Northeast, mid- and south-Atlantic regions, and upper Midwest, with 95% of cases reported from the following 13 states: Connecticut, Delaware, Maine, Maryland, Massachusetts, Minnesota, New Hampshire, New Jersey, New York, Pennsylvania, Vermont, Virginia and Wisconsin.
Different Lyme borrelia genospecies are found in endemic areas worldwide, among which B. burgdorferi sensu stricto (hereafter referred to as B. burgdorferi), B. garinii, and B. afzelii are the most clinically relevant (1). B. burgdorferi is the sole cause of Lyme disease in North America, whereas all three genospecies are associated with European Lyme disease. The prevalence of certain infectious complications reflects the species of Lyme borrelia in the region, with B. burgdorferi more commonly associated with arthritis, B. garinii with neurologic disease, and B. afzelii with the late skin manifestation acrodermatitis chronica atrophicans. The genetic heterogeneity of B. burgdorferi also contributes to disease expression as strains vary in their invasive potential (3, 4). Of the lineages detected in human infections, those found in skin are significantly more diverse than those identified in blood, synovial fluid (SF) or cerebrospinal fluid (CSF), with a limited subgroup predominating in disseminated infections (3, 4).
Clinical Manifestations
Lyme disease presents in phases that reflect the immune response to the spirochete as it multiplies at the inoculation site, disseminates, and establishes foci of infection elsewhere in the skin and other tissues (5, 6). Clinical signs may resolve or overlap with new manifestations as the infection progresses. Most patients present at an early stage of infection, during the nymphal tick feeding period in late spring through early fall. The most common presenting sign is the skin lesion erythema migrans (EM), which appears at the tick bite site 1–2 weeks after the tick has fed (Figure 1A). EM usually begins as a homogeneous erythema that expands over time, occasionally developing a central clearing to form the classic bull’s eye lesion. A vesicular or necrotic center occurs in ~5% of cases. The skin lesion is relatively asymptomatic and may be overlooked; sometimes there is tingling, burning, pain or mild pruritis. Multiple EM lesions (Figure 1B) occur as a result of disseminated infection, not from multiple tick bites. Secondary lesions are often smaller and lack a central punctum (the residua of the tick bite).
Figure 1.
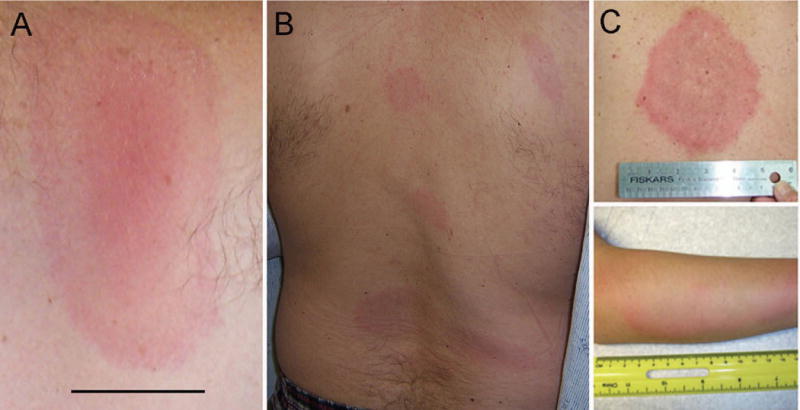
A and B, Erythema migrans presenting as a single lesion (A) (bar = 2 cm) and as multiple lesions (B). C, Skin lesion in southern tick–associated rash illness, with an appearance similar to that of erythema migrans. (Reproduced from http://www.cdc.gov/stari/symptoms/.)
Extracutaneous signs of disseminated B. burgdorferi infection most often involve the musculoskeletal, cardiovascular, and nervous systems. Musculoskeletal symptoms are a central feature of infection at all stages (Table I) (5, 6). Migratory arthralgias and myalgias accompanied by fever, headache and fatigue can be seen early after infection, with or without EM. Brief episodes of muscle, joint and/or periarticular pain, lasting hours to days, may ensue (7) and are often present in the setting of neurologic or cardiac disease (5). Frank arthritis (discussed below) is a late manifestation of infection.
Table I.
Common Musculoskeletal Manifestations of North American Lyme Disease1
| Early Localized Infection | Acute Disseminated Infection | Late Stage Infection1 | |
|---|---|---|---|
| Onset after tick bite | Days to one month | Weeks to a few months | Months |
| Systemic symptoms | None or variable degrees of fever, headache | Fatigue, headache, malaise; Occasionally fever | None or fatigue, malaise Occasionally fever |
| Musculoskeletal signs/symptoms | None or myalgia/arthralgia | Migratory musculoskeletal pain in joints, bursa, tendons, muscles, lasting hours or days in a location | Oligoarticular arthritis, usually involving the knee, lasting weeks to months, with recurrence; other joints include shoulder, ankle, elbow, hip, wrist; temporomandibular joint pain, bursitis, and tendinitis may be present. Children may present with an acute arthritis resembling septic arthritis, but usually are able to bear weight on the affected joint. Inflammation that persists in a single joint for >12 months is an unusual presentation of Lyme arthritis |
Rare late musculoskeletal manifestations relevant to the rheumatologist are clinically apparent myositis, including orbital myositis, and dactylitis; carpal tunnel syndrome has been described but would be unusual in the absence of a polyneuropathy or associated wrist arthritis.
Neurologic manifestations may appear weeks to a few months after a tick bite, most often as a seventh cranial nerve palsy, which may be bilateral (8). Meningitis and sensory and motor radiculoneuropathies (mononeuritis multiplex) may accompany a cranial nerve palsy or occur separately. Encephalomyelitis and axonal polyneuropathy are rare late manifestations. Early studies usually reported these late neurologic signs in patients who had other objective disease manifestations and were either untreated or received inadequate antibiotic therapy relative to current guidelines (8).
Lyme carditis as a presenting manifestation constitutes 1% of cases reported to the CDC. Patients may experience symptoms of shortness of breath, palpitations, lightheadedness and anxiety that result from varying degrees of atrioventricular nodal block; other manifestations of myopericarditis occur less frequently (5). Lyme carditis can progress to complete heart block and may be fatal; sudden death has been reported (9). Chronic myocarditis and cardiomyopathy, reported in Europe, are not recognized consequences of North American Lyme disease.
Arthritis is the most common late manifestation of Lyme disease and was documented in one study to occur in 60% of patients in whom infection was not treated at earlier stages, usually many months (average 6) after infection (7). Most patients present with an oligoarticular inflammatory arthritis affecting one or more large joints, especially the knee (7) (Table II). Arthritis can be migratory, but involvement of more than 5 joints is unusual. Other signs may include bursitis or tendinitis and temporomandibular joint pain. Children often present more acutely than adults, with fever and higher peripheral blood and SF white blood cell counts, suggesting septic arthritis (10, 11). Alternatively, patients with Lyme arthritis may have no other symptoms of systemic infection and present with a large joint effusion with complaints of stiffness more than pain. Baker’s cysts can form and rupture. Without antibiotics, Lyme arthritis may wax and wane, with an episode of synovitis lasting weeks to months; episodes can decrease in frequency and resolve over time (7). A minority (<10%) of patients develop synovitis of a joint that persists despite multiple courses of antibiotics, an entity termed “antibiotic-refractory Lyme arthritis” (7). The genetics of both the host (12, 13) and the strain of B. burgdorferi causing the infection (14) appear to play a role in determining this outcome, which shares features with other forms of non-septic inflammatory arthritis (Table II).
Table II.
Comparison of Lyme Arthritis with Other Common Infectious and Autoimmune Arthritidies1
| Lyme Arthritis | Non-Gonococcal Bacterial Arthritis2 | Gonococcal Arthritis | Reactive Arthritis | Oligoarticular Juvenile Inflammatory Arthritis | Rheumatoid Arthritis | |
|---|---|---|---|---|---|---|
| Onset | Intermediate, months (average 6 months) after EM | Acute (days – 2 weeks) | Acute, within 1 month of initial infection | Acute, 2–4 weeks post initial GI/GU3 infection | Insidious | Insidious |
| Arthritis Features | Oligoarticular, especially knee; may be migratory (<5 joints) Stiffness>Pain | Usually monoarticular, knee in adults; hip in young children Pain, redness | Usually monoarticular, knee Pain, redness | Oligoarticular, lower limb, especially knee Pain, stiffness | Oligoarticular, Lower limb, knee Stiffness>Pain | Classically symmetric, polyarticular Pain, stiffness |
| Systemic signs/symptoms at arthritis onset | 50% with antecedent migratory arthralgia; Fatigue; occasionally fever | Fever in 60% | Migratory polyarthralgia, often fever, rash, tenosynovitis | Enthesitis, Extra-articular features (skin, GI/GU, eye) | Unusual | Fatigue |
| ESR/CRP | Usually modest elevation, but occasionally normal or very elevated | Usually elevated | Usually elevated | Marked elevation in acute phase | Usually elevated | Usually elevated |
| Synovial fluid cell count (white blood cells/mm3) | 500 – >100,000 (average 25,000) 75% PMNs4 | 2000 – >100,000 >90% PMNs | 15,000 – >100,000 >90% PMNs | 2000 – 64,000 variable PMNs | 2000 – 50,000 (ave 20,000) 60% PMNs | 2000 – 50,000 (ave 15,000) variable PMNs |
| Synovial fluid culture | Negative | Positive in 70–90% | Positive in <50% | Negative | Negative | Negative |
| Synovial fluid PCR for bacterial DNA | Positive | Data not available | Positive | Negative | Negative | Negative |
| HLA association | DRB1*0401, −0404, 0101, 1501 in antibiotic-refractory form | None | None | HLA-B27 | DRB1*0801 and 11, DRB1*1301 | DRB1*0401, −0404, −0405, −0101, and −1001 |
| ANA, RF, ACPA5 | Negative or low titer ANA, RF | Negative | Negative | Negative | ANA, occasionally ACPA | RF, ACPA |
Information compiled from Kelley’s Textbook of Rheumatology, 9th Edition
Data refer to arthritis caused by Staphylococcus aureus and common Streptococcal species
GI/GU = gastrointestinal/genitourinary
PMN = polymorphonuclear leukocytes
ANA = anti-nuclear antibodies; RF = rheumatoid factor; ACPA = anti-citrullinated protein antibodies
Pathogenesis
Establishment of infection and dissemination
Onset of tick feeding initiates global changes in B. burgdorferi gene expression that are required for infection of the blood meal host (15). Although spirochetes expand greatly in numbers in the tick, few (estimated to be in the hundreds) ultimately make their way from the midgut to the salivary glands to pass with saliva into the dermis, a process that takes more than 24 hours. A period of continued host adaptation ensues in which spirochetes multiply and eventually disseminate from the skin bite site. With the exception of a recently identified protease in B. burgdorferi with aggrecanase activity (16), Lyme borrelia do not directly degrade extracellular matrix and instead rely on their motility and the expression of adhesins and proteins that bind host proteases to invade tissues (17). Spirochetes also induce the production of matrix metalloproteinases, which render tissues more permissive to pathogen invasion.
The strategies that Lyme borrelia employ for immune evasion are consistent with its genomic characterization as an extracellular pathogen for which antibody, complement and phagocytes are critical for host defense. During transmission, spirochetes bind a tick salivary protein to shield against host antibodies and complement. Lyme borrelia also express proteins that bind host factor H to further inhibit complement-mediated lysis (18). Antigenic variation of VlsE, an outer surface protein required for long-term survival of the pathogen in mammals, subverts specific antibody-mediated clearance of spirochetes (19). As infection progresses, Lyme borrelia may reduce surface lipoprotein expression to further impair immune clearance.
Immune response and clinical disease
Because the B. burgdorferi genome lacks identifiable virulence factors and toxins, the clinical signs of Lyme disease result from the immune response to infection (5, 20). On histopathologic analysis, most involved tissues exhibit a mononuclear-type inflammation without granuloma or giant cell formation (21). Lymphocytes with associated macrophages, dendritic cells, and plasma cells are characteristically seen in all tissues except for SF, where polymorphonuclear leukocytes (PMNs) predominate.
Inflammation begins when dendritic cells and macrophages respond to Lyme borrelia via pattern recognition receptors. The main recognition pathway is the interaction of B. burgdorferi components with Toll-like receptors (TLRs) especially lipoproteins with TLR2; other receptors such as CD14, NOD-like receptors, and scavenger receptors are also engaged (20). TLR stimulation activates MAP kinases and NFκb, resulting in the production of proinflammatory cytokines, including IL-1β, TNFα, IL-6, and type I IFNs. Lyme borrelia and lipoproteins also potently induce the anti-inflammatory cytokine IL-10, which may be a survival strategy (22). The balance between proinflammatory and anti-inflammatory cytokines may determine the efficiency at which immune cells respond to infected sites and the clinical expression of disease.
B and T cell-recruiting chemokines are found in biopsies of skin and synovium and can be detected in blood, CSF and SF (5, 23). The particular chemokine pattern produced and resultant expansion of different B cell and Th cell subsets (Th1, Th2, Th17) is likely determined by host-specific factors, the infecting strain of Lyme borrelia, and the duration of infection. An early Th1 response that promotes phagocyte activation may be important for limiting Lyme borrelia burden (24). Predominant Th17 responses early may result in less efficient clearance of Lyme borrelia as PMNs (recruited by IL-17) ingest spirochetes inefficiently in the absence of specific antibody (25).
Th1 cells are the main T cell subset detected in the blood of patients with disseminated infection (26, 27). In CSF, B cells predominate with fewer CD4+ Th1 cells; despite elevated IL-17 levels, PMNs are rare (28). In contrast to CSF, PMNs abound in SF, where IL-17 and IFN-γ-producing Th17 and Th1 cells, CD8+ T cells and regulatory T cell subsets are also found (26, 27). Synovial biopsies show lymphocytic infiltrates comprised mainly of CD4+ T cells, often in lymphoid aggregates with B cells, some CD8+ T cells, and scattered macrophages, dendritic dells and plasma cells. Angiogenesis and an obliterative vasculopathy have been reported in synovial tissue (21).
Pathogenesis of Lyme arthritis
Unexplained clinical features of Lyme arthritis are the delayed onset after infection and the spontaneous remission of inflammation in one joint with recurrence in the same or a different joint. The association of antibiotic use with resolution of arthritis and elimination of recurrences suggests that viable organisms drive Lyme arthritis throughout much of its course, with the exception of antibiotic-refractory Lyme arthritis as discussed below.
In most cases, Lyme borrelia reach the joint via hematogenous dissemination, a phase only documented in early infection. The presumption is that the joint was infected long before the onset of swelling. The universal presence of high titer antibodies to multiple borrelia antigens at the time of presentation is consistent with a longer duration of infection. In chronically infected mice, B. burgdorferi have a predilection for collagen-rich tissues and have been visualized in small numbers in the highly vascular synovium as well as avascular tendons without associated inflammation (20). Where B. burgdorferi resides in the joint prior to clinically apparent arthritis in humans and what enables its immune recognition to trigger inflammation after a period of quiescence is not known (29). The migratory periarticular pain preceding arthritis and the rare finding of spirochetes in the inflamed synovium during an episode raise questions as to whether the synovium per se is the primary site of B. burgdorferi persistence during the pre-arthritis phase. If synovial resident spirochetes trigger inflammation, they must be altered from their previous host-adapted state to become visible to the immune system (Figure 2). One theory is that spirochetes in protective niches such as tendons, ligaments or even the walls of arterioles in the joint initiate inflammation after transiently seeding the synovium, where they are bound by specific antibody or recognized as foreign by innate immune cells. Joint microtrauma may be one way in which B. burgdorferi in these sites become exposed to synovial cells. Immune clearance or further host adaptation of spirochetes may signal arthritis resolution. Spirochetes in select sites such as blood vessel walls or tendons may elicit host responses that have an effect on these tissues. Obliterative vasculopathy is a feature of Lyme arthritis, but whether the presence of B. burgdorferi in tendons influences tendon remodeling has not been explored. Enthesopathic changes and cartilage calcification have been noted occasionally on imaging studies of long-standing human Lyme arthritis (5), although MRI studies usually reveal inflammation primarily involving the synovium and occasionally adjacent muscles (30).
Figure 2.
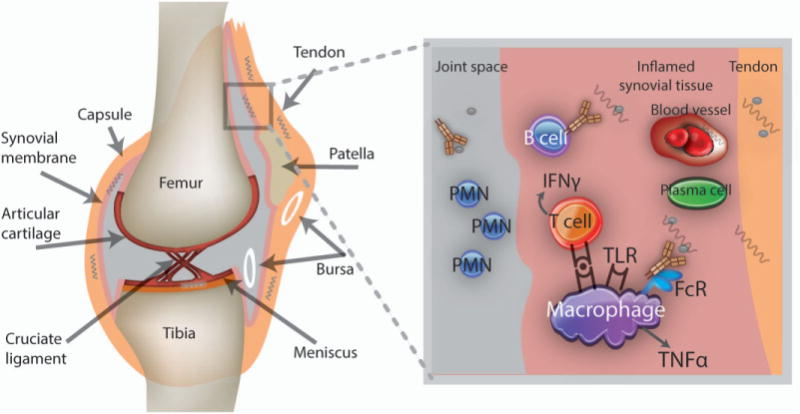
Proposed model for the initiation of Lyme arthritis. Lyme borrelia introduced into the skin disseminate hematogenously and establish infection in the joint. Borrelia in highly vascular areas such as the synovium may host-adapt to elude immune recognition by synovial immune cells and specific antibodies; others may be partially protected against these defenses because of their location in tendons, ligaments, and arteriolar walls. Months later, microdamage may expose Lyme borrelia in these latter sites or synovial resident spirochetes may be altered to suddenly become “visible,” initiating acute inflammation through Toll-like receptors (TLRs) and immune complex formation. Borrelia-specific T and B cells home to the synovium, where they contribute to the inflammatory response; lymphoid follicles may form, and with protracted inflammation, fibrinous exudates appear. These exudates may sequester Lyme borrelia and their remnants, which could also trigger inflammation if later released. In some people, the inflammatory response may become dysregulated, leading to antibiotic-refractory Lyme arthritis. Autoimmunity triggered by infection may be perpetuated when normal mechanisms of immune regulation, such as those mediated by CD25+ Treg cells, γ/δ T cells, or natural killer T cells, are deficient. Arthritis resolution occurs once residual borrelia remnants are contained and the immune response is regulated. PMN = polymorphonuclear leukocyte; IFNγ = interferon- γ; FcR = Fc receptor; TNFα = tumor necrosis factor α.
Prolonged joint inflammation results in the development of fibrinous exudates lining the synovium (21). This fibrinous material may sequester Lyme borrelia in the joint space. Rarely, B. burgdorferi has been observed in SF encased in a meshlike substance, giving rise to the “amber theory” of Lyme arthritis (31). This theory proposes that spirochetes or their remnants become enmeshed in a host-derived fibrinous matrix at some location within the joint. If the material dislodges into the joint space, it may degrade and release borrelia products that drive inflammation directly or via immune complex formation. Presumably viable organisms are present elsewhere in the joint to explain why antibiotics are usually effective in resolving inflammation. Persistence of encased dead spirochetes, however, could explain the occasional recurrence of arthritis in the same or different joint after what initially appeared to be successful antibiotic treatment.
Antibiotic-refractory Lyme arthritis
The rate of arthritis resolution after a course of antibiotics may depend on the extent of synovial pathology present, the clearance of inflammatory B. burgdorferi components, and regulation of the immune response. Analogies have been drawn between pathogenetic mechanisms of RA and antibiotic-refractory Lyme arthritis, stemming initially from the finding of an increased prevalence of the RA-associated alleles HLADRB1*0401, *0101 and others in these patients; however, alleles not linked to RA such as DRB1*1501 have also been reported (12). Alterations in innate immunity may play a role, as a TLR1 polymorphism that reduces expression of the TLR1/2 heterodimer critical for pathogen recognition is found in higher frequency in patients with refractory arthritis and paradoxically is associated with heightened inflammation (13). Autoreactive B and T cell responses can be detected in both antibiotic-responsive and antibiotic-refractory Lyme arthritis and may perpetuate inflammation if not regulated. CD25+FoxP3+ T cells and invariant NKT cells from SF of antibiotic-refractory Lyme arthritis patients are less abundant in comparison to antibiotic-responsive Lyme arthritis, consistent with a deficiency in immune regulation (27, 32, 33). Antibodies to endothelial cell growth factor are more pronounced in patients with antibiotic-refractory Lyme arthritis and have been proposed as a biomarker for this form of arthritis (34). Although autoimmunity may underlie the pathogenesis of antibiotic-refractory Lyme arthritis, the dysregulated immune response affects only a limited number of joints (usually one) and is amenable to permanent regulation because synovectomy can resolve inflammation, as does treatment with intraarticular steroids and/or DMARDs (35, 36).
Diagnosis
Lyme disease should be suspected when patients with potential exposure to Lyme borrelia-infected ticks present with an EM-like skin lesion or other clinical features suggestive of the infection (37). Empiric antibiotic therapy is indicated for EM as culture for Lyme borrelia is not routinely available and spirochetes are rarely seen on histopathology. The lesion, however, is not pathognomonic for Lyme disease. An EM-like lesion is seen in Southern Tick Associated Rash Illness (STARI) (38), which occurs after the bite of the lone star tick Amblyomma americanum (Figure 1C). This tick is found mainly in the south central and southeastern United States, but its geographic distribution extends up to coastal Maine. The etiology of STARI is unknown as is the role of antibiotic therapy for this condition.
In the absence of EM, laboratory testing should be performed to support a diagnosis of Lyme disease (39). Routine laboratory tests cannot distinguish Lyme disease from other entities. Except in the case of coinfection with other tick-borne pathogens, the complete blood count is generally normal. Although ESR and CRP levels may be significantly elevated, these tests usually only exhibit a modest elevation. Mild elevations of liver function tests may be present in early infection. SF cell counts range from 500–110,000 white blood cells/mm3 (average ~25,000 with ~75% PMN); counts of 50,000 or more may be present in children presenting with a septic arthritis picture. In cases of meningitis, CSF reveals a lymphocytic pleocytosis with modestly elevated protein and normal glucose levels; similar findings can be seen in patients with seventh cranial nerve palsy in the absence of meningeal signs. Abnormalities found using imaging techniques such as MRI of the brain or the joint are not sufficiently distinctive to secure a diagnosis of Lyme disease without supporting laboratory tests.
Culture for Lyme borrelia is not routinely performed or available (39). Blood cultures are positive in approximately 50% of patients with EM in the United States, but the organism is rarely cultured from blood in the absence of EM or from the CSF or SF of patients with extracutaneous disease. A novel serum culture assay claiming to detect Lyme borrelia has not been validated and should not be used (40).
PCR testing for B. burgdorferi DNA is positive in up to 80% of SF specimens before treatment if multiple genes are targeted (41); the sensitivity of PCR for CSF specimens tends to be much lower (5% in one US study) (42). A new method for amplifying Lyme borrelia DNA that first enriches for borrelia DNA prior to multiplex PCR of target genes has been reported (43). More data are needed on the sensitivity and specificity of the test. Positive PCR results are not necessarily indicative of active infection. B. burgdorferi DNA can be detected for extended periods in vitro after viable spirochetes have been killed with antibiotics, and PCR positivity of SF specimens months after antibiotics is not predictive of relapse or arthritis persistence (41).
Serologic assays remain the mainstay of Lyme disease diagnostic testing. A two-tier approach is recommended using an enzyme-linked immunosorbent assay (EIA), followed by separate IgM and IgG immunoblots if the EIA is positive or equivocal (39). The two-tier assay has low sensitivity in early infection, but is highly sensitive after 6–8 weeks of untreated infection. The C6 peptide EIA, based on an invariant region of the VlsE protein, is an alternative first-tier test that is more specific than whole sonicate antigen-based EIAs (44). Detection of intrathecal production of Lyme borrelia antibodies using paired serum and CSF samples can be helpful in diagnosing neuroborreliosis (39); CSF antibodies may rarely precede serum antibody. Antibody testing of SF is not recommended because of false positive results. Seronegative late Lyme disease, including seronegative Lyme arthritis, is not known to exist in patients capable of producing antibodies.
The ACR recommends against the routine screening for Lyme disease as a cause of musculoskeletal symptoms without an exposure history and appropriate physical examination findings (45). Polyclonal B cell activation and antibody cross-reactivity with other bacterial antigens are common causes of false positive results, particularly for the IgM EIA and immunoblot. False positive tests occur when the first tier EIA is omitted, when non evidence-based criteria are used to interpret the immunoblot, and when nonspecific weak bands are reported as positive. Serologic tests assess exposure history and are not indicated in routine follow-up because borrelia antibodies may persist for years in successfully treated patients.
Treatment and Expected Outcomes
The Infectious Diseases Society of America published guidelines for the diagnosis and treatment of Lyme disease and all recommendations were upheld after subsequent independent review (Table III) (37). Oral antibiotics (preferably doxycycline, amoxicillin or cefuroxime axetil) are first line therapies for patients who present with single or multiple EM, uncomplicated seventh nerve palsy, or arthritis. Intravenous therapy with ceftriaxone is the treatment of choice for neurologic abnormalities other than seventh nerve palsy, symptomatic cardiac involvement (or those with advanced heart block) or arthritis that does not improve with oral antibiotics. Oral doxycycline appears to be highly effective, however, for all of the early neurologic manifestations of Lyme disease based on studies conducted in Europe. In general, earlier manifestations of the illness respond more rapidly to antibiotics than later ones such as arthritis.
Table III.
| Manifestation | Drug | Adult Dosage | Pediatric Dosage | Duration (Range) |
|---|---|---|---|---|
| Early Disease Erythema migrans |
Doxycycline3 |
100 mg po BID |
<8 years – not recommended ≥8 years – 4 mg/kg/d in two divided doses (maximum 100 mg/dose) |
14 days (10–21 days) |
| Amoxicillin | 500 mg po TID | 50 mg/kg/day in three divided doses (maximum 500 mg/dose) | 14 days (14–21 days) | |
| Cefuroxime axetil | 500 mg po BID | 30 mg/kg/day in two divided doses (maximum 500 mg/dose) | 14 days (14–21 days) | |
| Early Neurologic Disease | ||||
| Cranial nerve palsy4 | Same as for EM | 14 days (14–21 days) | ||
| Meningitis or radiculopathy5 | Ceftriaxone | 2 g IV qd | 50–75 mg/kg IV qd in a single dose (maximum 2 g/day) | 14 days (10–28 days) |
| Cardiac Disease | Same as oral regimen for EM OR use IV regimen as for neurologic disease6 | 2 g IV qd | 50–75 mg/kg IV qd in a single dose (maximum 2 g/day) | 14 days (14–21 days) |
| Late Disease | ||||
| Arthritis without neurologic | Same as for EM | 28 days (28 days) | ||
| Recurrent arthritis after oral regimen | Repeat oral regimen OR use IV regimen as for neurologic disease | 28 days for oral regimens and 14–28 days for IV regimens | ||
| Central or Peripheral Nervous System Disease | IV regimen as for early neurologic disease | 14 days (14–28 days) | ||
A complete list of recommended and alternate therapies can be found in reference 37.
Complete response to treatment may be delayed beyond the treatment period, regardless of the clinical manifestation, and relapse may recur. Patients with objective signs of relapse may need a second course of treatment.
Tetracyclines are relatively contraindicated in pregnant or lactating women and in children < 8 years of age.
Patients without clinical evidence of meningitis may be treated with an oral regimen. The recommendation is based on experience with seventh cranial nerve palsy. Whether oral therapy would be as effective for patients with other cranial neuropathies is unknown; the decision between oral and parenteral therapy should be individualized.
For non-pregnant adult patients intolerant of beta-lactam agents, doxycycline 200–400 mg/day orally (or IV if unable to take oral medications) in two divided doses may be adequate. For children ≥ 8 years of age, the dosage of doxycycline for this indication is 4–8 mg/kg/d in two divided doses (maximum daily dosage of 200–400 mg).
A parenteral antibiotic regimen is recommended at the start of therapy for patients who have been hospitalized for cardiac monitoring; an oral regimen may be substituted to complete a course of therapy or to treat outpatients. A temporary pacemaker may be required for patients with advanced heart block.
Patients with Lyme arthritis are initially treated with a four-week course of oral antibiotics. NSAIDS are often used to ameliorate symptoms during and after treatment. Incomplete resolution of arthritis at 4 weeks has been observed in about 40% of patients and an additional course of oral or intravenous antibiotics may be necessary (37). No controlled trials are available to guide the decision as to whether further antibiotics should be prescribed versus anti-inflammatory medications, including intra-articular corticosteroids, to hasten arthritis resolution. Arthritis may be considered refractory to antibiotics if it persists for more than 2 months after a course of intravenous antibiotics or one month after two 4-week courses of oral antibiotics (if parenteral therapy is not possible) and PCR of SF is negative for borrelia DNA (37). Antibiotic-refractory Lyme arthritis is not likely due to persistent infection because intraarticular steroids (36) and/or DMARD therapies (hydroxychloroquine, sulfasalazine, or methotrexate) and rarely TNFα antagonists have been used successfully without signs of recrudescent infection in the joint or elsewhere (35). Synovectomy can be curative if most of the synovial tissue is removed; excised synovial tissue has been uniformly negative for B. burgdorferi by PCR and culture (41).
Similar to other manifestations of disseminated disease, the outcome from early neurologic Lyme disease is generally favorable, although mild residual deficits can occur in a minority of patients (incompletely resolved facial palsy or mild motor or sensory deficits).
Symptoms after Lyme disease treatment
New or recurrent objective signs of Lyme disease appearing after antibiotics should prompt evaluation for reinfection or incomplete treatment. A recent study in which the causative strain of B. burgdorferi was cultured from EM lesions showed that recurrent EM after treatment was due to reinfection rather than a relapse of the prior treated infection (46).
About 25% of patients with EM resolve clinical disease but continue to experience fatigue, cognitive issues or joint and muscle aches at 3 months after treatment, falling to about 10% at 6 months. Post-treatment Lyme disease syndrome (PTLDS) refers to such symptoms that last for more than 6 months after a documented episode of Lyme disease and are disabling. The frequency with which PTLDS occurs is believed to be substantially less than 10%. PTLDS should not be confused with “chronic Lyme disease”. This name originally referred to late manifestations of the disease, but has been usurped as a label for clinical syndromes that may or may not be associated with previous Lyme borrelia exposure (47).
The reasons for persistent symptoms in some patients after treatment for Lyme disease are not known. Theories include residual damage to tissue, slow resolution of the inflammatory state, and/or a form of cytokine-induced sickness behavior due to previously high levels of circulating cytokines. Systemic inflammatory cytokines exert adverse effects on neurobehavioral function in other conditions, independent of a CNS infection (8). Nonspecific symptoms such as fatigue and pain are common in the general population as well. Because no test can prove absence of Lyme borrelia infection in humans, animal models have been used to examine antibiotic efficacy (reviewed in (48, 49)). Most investigations have had methodological concerns, including suboptimal antibiotic dosing and introduction of infection in ways that did not model the inoculum size and route by which humans usually become infected (i.e., from the bite of a single Ixodes species tick). Even so, antibiotic failures defined by the presence of cultivable spirochetes are rare and usually occur in the setting of immunodeficiency or inadequate antibiotic dosing. A recent study in mice employed two–photon intravital microscopy to examine the fate of B. burgdorferi after antibiotics (50). Treatment rapidly eliminated viable spirochetes but B. burgdorferi inflammatory products (antigens and DNA) could be detected for extended periods adjacent to cartilage and in certain tissues such as the entheses. Viable spirochetes were not found, although xenodiagnosis performed by feeding uninfected ticks on the mice occasionally detected borrelia DNA. These results provide insight into the significance of borrelia DNA that can occasionally be detected in treated animals when spirochetes cannot be demonstrated by culture and in those instances in humans in which borrelia DNA may be detected after antibiotics for Lyme disease (41, 49, 51).
Four placebo-controlled trials of extended antibiotic therapy for PTLDS have been conducted (52). These showed either no durable benefit (n=3) or a benefit in fatigue only (n=1) with an unacceptable adverse event rate from parenteral therapy. Persistence of Lyme borrelia could not be demonstrated by culture or PCR. These trials form the basis of the recommendation to consider symptomatic treatment of PTLDS following guidelines for chronic fatigue syndrome or fibromyalgia.
Conclusion
Lyme disease is a relatively common tick-transmitted infection that is associated with a characteristic pattern of cutaneous, nervous system, cardiac and musculoskeletal manifestations. EM is the only sign for which a diagnosis can be considered on clinical grounds alone and in its absence, serologic tests should be used to support a diagnosis of Lyme disease. All of the objective clinical manifestations of the disorder occur as a consequence of the host inflammatory response to the pathogen. Our understanding of the pathogenetic events that account for inflammation and specific disease manifestations caused by Lyme borrelia are continuing to evolve, particularly in the area of Lyme arthritis pathogenesis and the persistence of symptoms after treatment.
Acknowledgments
Supported by NIH R01AI AI085798 and the Harold J. Jockers Professorship
Disclosures: Dr. Wormser reports receiving research grants from Immunetics, Inc., BioRad, DiaSorin, Inc., and bioMérieux SA. He owns equity in Abbott; has been an expert witness in malpractice cases involving Lyme disease; is an unpaid board member of the American Lyme Disease Foundation; has been an expert witness regarding Lyme disease in a disciplinary action for the Missouri Board of Registration for the Healing Arts; and has been a consultant to Baxter for Lyme vaccine development.
References
- 1.Kurtenbach K, Hanincova K, Tsao JI, Margos G, Fish D, Ogden NH. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nat Rev Microbiol. 2006;4(9):660–9. doi: 10.1038/nrmicro1475. [DOI] [PubMed] [Google Scholar]
- 2.Steere AC, Malawista SE, Snydman DR, Shope RE, Andiman WA, Ross MR, et al. Lyme arthritis: an epidemic of oligoarticular arthritis in children and adults in three connecticut communities. Arthritis Rheum. 1977;20(1):7–17. doi: 10.1002/art.1780200102. [DOI] [PubMed] [Google Scholar]
- 3.Wormser GP, Brisson D, Liveris D, Hanincova K, Sandigursky S, Nowakowski J, et al. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J Infect Dis. 2008;198(9):1358–64. doi: 10.1086/592279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanincova K, Mukherjee P, Ogden NH, Margos G, Wormser GP, Reed KD, et al. Multilocus sequence typing of Borrelia burgdorferi suggests existence of lineages with differential pathogenic properties in humans. PLoS One. 2013;8(9):e73066. doi: 10.1371/journal.pone.0073066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bockenstedt LK. Lyme disease. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O’Dell JR, editors. Kelley’s Textbook of Rheumatology. 9. Philadelphia: Elsevier Saunders; 2013. pp. 1815–28. [Google Scholar]
- 6.Shapiro ED. Clinical practice. Lyme disease. N Engl J Med. 2014;370(18):1724–31. doi: 10.1056/NEJMcp1314325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steere AC, Schoen RT, Taylor E. The clinical evolution of Lyme arthritis. Ann Intern Med. 1987;107(5):725–31. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- 8.Halperin JJ. Nervous system Lyme disease. Handb Clin Neurol. 2014;121:1473–83. doi: 10.1016/B978-0-7020-4088-7.00099-7. [DOI] [PubMed] [Google Scholar]
- 9.CDC. Three sudden cardiac deaths associated with Lyme carditis – United States, November 2012 – July 2013. 2013 Dec 13; 2013 [cited 2013 December 13, 2013 Available from: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6249a1.htm?s_cid=mm6249a1_w. [PMC free article] [PubMed]
- 10.Smith BG, Cruz AI, Jr, Milewski MD, Shapiro ED. Lyme disease and the orthopaedic implications of Lyme arthritis. J Am Acad Orthop Surg. 2011;19(2):91–100. doi: 10.5435/00124635-201102000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daikh BE, Emerson FE, Smith RP, Lucas FL, McCarthy CA. Lyme arthritis: a comparison of presentation, synovial fluid analysis, and treatment course in children and adults. Arthritis Care Res (Hoboken) 2013;65(12):1986–90. doi: 10.1002/acr.22086. [DOI] [PubMed] [Google Scholar]
- 12.Steere AC, Klitz W, Drouin EE, Falk BA, Kwok WW, Nepom GT, et al. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J Exp Med. 2006;203(4):961–71. doi: 10.1084/jem.20052471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strle K, Shin JJ, Glickstein LJ, Steere AC. Association of a Toll-like receptor 1 polymorphism with heightened Th1 inflammatory responses and antibiotic-refractory Lyme arthritis. Arthritis Rheum. 2012;64(5):1497–507. doi: 10.1002/art.34383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones KL, McHugh GA, Glickstein LJ, Steere AC. Analysis of Borrelia burgdorferi genotypes in patients with Lyme arthritis: High frequency of ribosomal RNA intergenic spacer type 1 strains in antibiotic-refractory arthritis. Arthritis Rheum. 2009;60(7):2174–82. doi: 10.1002/art.24812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012;10(2):87–99. doi: 10.1038/nrmicro2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell TM, Johnson BJ. Lyme disease spirochaetes possess an aggrecan-binding protease with aggrecanase activity. Mol Microbiol. 2013;90(2):228–40. doi: 10.1111/mmi.12276. [DOI] [PubMed] [Google Scholar]
- 17.Coburn J, Leong J, Chaconas G. Illuminating the roles of the Borrelia burgdorferi adhesins. Trends Microbiol. 2013;21(8):372–9. doi: 10.1016/j.tim.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Taeye SW, Kreuk L, van Dam AP, Hovius JW, Schuijt TJ. Complement evasion by Borrelia burgdorferi: it takes three to tango. Trends Parasitol. 2013;29(3):119–28. doi: 10.1016/j.pt.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Norris SJ. Antigenic variation with a twist–the Borrelia story. Mol Microbiol. 2006;60(6):1319–22. doi: 10.1111/j.1365-2958.2006.05204.x. [DOI] [PubMed] [Google Scholar]
- 20.Weis JJ, Bockenstedt LK. Host Response. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 413–42. [Google Scholar]
- 21.Duray PH. Histopathology of clinical phases of human Lyme disease. Rheum Dis Clin North Am. 1989;15(4):691–710. [PubMed] [Google Scholar]
- 22.Chung Y, Zhang N, Wooten RM. Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS One. 2013;8(12):e84980. doi: 10.1371/journal.pone.0084980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rupprecht TA, Koedel U, Fingerle V, Pfister HW. The pathogenesis of Lyme neuroborreliosis: from infection to inflammation. Mol Med. 2008;14(3–4):205–12. doi: 10.2119/2007-00091.Rupprecht. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strle K, Stupica D, Drouin EE, Steere AC, Strle F. Elevated levels of IL-23 in a subset of patients with post-Lyme disease symptoms following erythema migrans. Clin Infect Dis. 2014;58(3):372–80. doi: 10.1093/cid/cit735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montgomery RR, Lusitani D, de Boisfleury Chevance A, Malawista SE. Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J Infect Dis. 2002;185(12):1773–9. doi: 10.1086/340826. [DOI] [PubMed] [Google Scholar]
- 26.Steere AC, Glickstein L. Elucidation of Lyme arthritis. Nat Rev Immunol. 2004;4(2):143–52. doi: 10.1038/nri1267. [DOI] [PubMed] [Google Scholar]
- 27.Shen S, Shin JJ, Strle K, McHugh G, Li X, Glickstein LJ, et al. Treg cell numbers and function in patients with antibiotic-refractory or antibiotic-responsive Lyme arthritis. Arthritis Rheum. 2010;62(7):2127–37. doi: 10.1002/art.27468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henningsson AJ, Tjernberg I, Malmvall BE, Forsberg P, Ernerudh J. Indications of Th1 and Th17 responses in cerebrospinal fluid from patients with Lyme neuroborreliosis: a large retrospective study. J Neuroinflammation. 2011;8:36. doi: 10.1186/1742-2094-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malawista SE. Resolution of Lyme arthritis, acute or prolonged: a new look. Inflammation. 2000;24(6):493–504. doi: 10.1023/a:1007079705485. [DOI] [PubMed] [Google Scholar]
- 30.Ecklund K, Vargas S, Zurakowski D, Sundel RP. MRI features of Lyme arthritis in children. AJR Am J Roentgenol. 2005;184(6):1904–9. doi: 10.2214/ajr.184.6.01841904. [DOI] [PubMed] [Google Scholar]
- 31.Wormser GP, Nadelman RB, Schwartz I. The amber theory of Lyme arthritis: initial description and clinical implications. Clin Rheumatol. 2012;31(6):989–94. doi: 10.1007/s10067-012-1964-x. [DOI] [PubMed] [Google Scholar]
- 32.Vudattu NK, Strle K, Steere AC, Drouin EE. Dysregulation of CD4+CD25(high) T cells in the synovial fluid of patients with antibiotic-refractory Lyme arthritis. Arthritis Rheum. 2013;65(6):1643–53. doi: 10.1002/art.37910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katchar K, Drouin EE, Steere AC. Natural killer cells and natural killer T cells in Lyme arthritis. Arthritis Res Ther. 2013;15(6):R183. doi: 10.1186/ar4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drouin EE, Seward RJ, Strle K, McHugh G, Katchar K, Londono D, et al. A novel human autoantigen, endothelial cell growth factor, is a target of T and B cell responses in patients with Lyme disease. Arthritis Rheum. 2013;65(1):186–96. doi: 10.1002/art.37732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steere AC, Angelis SM. Therapy for Lyme arthritis: strategies for the treatment of antibiotic-refractory arthritis. Arthritis Rheum. 2006;54(10):3079–86. doi: 10.1002/art.22131. [DOI] [PubMed] [Google Scholar]
- 36.Nimmrich S, Becker I, Horneff G. Intraarticular corticosteroids in refractory childhood Lyme arthritis. Rheumatol Int. 2014 doi: 10.1007/s00296-013-2923-9. [DOI] [PubMed] [Google Scholar]
- 37.Wormser GP, Dattwyler RJ, Shapiro ED, Halperin JJ, Steere AC, Klempner MS, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089–134. doi: 10.1086/508667. [DOI] [PubMed] [Google Scholar]
- 38.CDC. Southern tick-associated rash illness. 2011 Oct 21; [cited November 17, 2013]; Available from: http://www.cdc.gov/stari/
- 39.Aguero-Rosenfeld ME, Wang G, Schwartz I, Wormser GP. Diagnosis of Lyme borreliosis. Clin Microbiol Rev. 2005;18(3):484–509. doi: 10.1128/CMR.18.3.484-509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson C, Hojvat S, Johnson B, Petersen J, Schriefer M, Beard B, et al. Concerns regarding a new culture method for Borrelia burgdorferi not approved for the diagnosis of Lyme disease. Morbidity and Mortality Weekly Report (MMWR) 2014;63(15):333. [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, McHugh GA, Damle N, Sikand VK, Glickstein L, Steere AC. Burden and viability of Borrelia burgdorferi in skin and joints of patients with erythema migrans or Lyme arthritis. Arthritis Rheum. 2011;63(8):2238–47. doi: 10.1002/art.30384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avery RA, Frank G, Eppes SC. Diagnostic utility of Borrelia burgdorferi cerebrospinal fluid polymerase chain reaction in children with Lyme meningitis. Pediatr Infect Dis J. 2005;24(8):705–8. doi: 10.1097/01.inf.0000172903.14077.4c. [DOI] [PubMed] [Google Scholar]
- 43.Eshoo MW, Crowder CC, Rebman AW, Rounds MA, Matthews HE, Picuri JM, et al. Direct molecular detection and genotyping of Borrelia burgdorferi from whole blood of patients with early Lyme disease. PLoS One. 2012;7(5):e36825. doi: 10.1371/journal.pone.0036825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wormser GP, Schriefer M, Aguero-Rosenfeld ME, Levin A, Steere AC, Nadelman RB, et al. Single-tier testing with the C6 peptide ELISA kit compared with two-tier testing for Lyme disease. Diagn Microbiol Infect Dis. 2013;75(1):9–15. doi: 10.1016/j.diagmicrobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yazdany J, Schmajuk G, Robbins M, Daikh D, Beall A, Yelin E, et al. Choosing wisely: the American College of Rheumatology’s Top 5 list of things physicians and patients should question. Arthritis Care Res (Hoboken) 2013;65(3):329–39. doi: 10.1002/acr.21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nadelman RB, Hanincova K, Mukherjee P, Liveris D, Nowakowski J, McKenna D, et al. Differentiation of reinfection from relapse in recurrent Lyme disease. N Engl J Med. 2012;367(20):1883–90. doi: 10.1056/NEJMoa1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feder HM, Jr, Johnson BJ, O’Connell S, Shapiro ED, Steere AC, Wormser GP, et al. A critical appraisal of “chronic Lyme disease”. N Engl J Med. 2007;357(14):1422–30. doi: 10.1056/NEJMra072023. [DOI] [PubMed] [Google Scholar]
- 48.Wormser GP, Schwartz I. Antibiotic treatment of animals infected with Borrelia burgdorferi. Clin Microbiol Rev. 2009;22(3):387–95. doi: 10.1128/CMR.00004-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bockenstedt LK, Radolf JD. Editorial commentary: xenodiagnosis for posttreatment lyme disease syndrome: resolving the conundrum or adding to it? Clin Infect Dis. 2014;58(7):946–8. doi: 10.1093/cid/cit942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bockenstedt LK, Gonzalez DG, Haberman AM, Belperron AA. Spirochete antigens persist near cartilage after murine Lyme borreliosis therapy. J Clin Invest. 2012;122(7):2652–60. doi: 10.1172/JCI58813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marques A, Telford SR, 3rd, Turk SP, Chung E, Williams C, Dardick K, et al. Xenodiagnosis to detect Borrelia burgdorferi infection: a first-in-human study. Clin Infect Dis. 2014;58(7):937–45. doi: 10.1093/cid/cit939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klempner MS, Baker PJ, Shapiro ED, Marques A, Dattwyler RJ, Halperin JJ, et al. Treatment trials for post-Lyme disease symptoms revisited. Am J Med. 2013;126(8):665–9. doi: 10.1016/j.amjmed.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]