

Abstract
Chemokine CXCL8 and its receptor CXCR1 are key mediators in combating infection and have also been implicated in the pathophysiology of various diseases including chronic obstructive pulmonary disease (COPD) and cancer. CXCL8 exists as monomers and dimers but monomer alone binds CXCR1 with high affinity. CXCL8 function involves binding two distinct CXCR1 sites – the N-terminal domain (Site-I) and the extracellular/transmembrane domain (Site-II). Therefore, higher monomer affinity could be due to stronger binding at Site-I or Site-II or both. We have now characterized the binding of a human CXCR1 N-terminal domain peptide (hCXCR1Ndp) to WT CXCL8 under conditions where it exists as both monomers and dimers. We show that the WT monomer binds the CXCR1 N-domain with much higher affinity and that binding is coupled to dimer dissociation. We also characterized the binding of two CXCL8 monomer variants and a trapped dimer to two different hCXCR1Ndp constructs, and observe that the monomer binds with ∼10- to 100-fold higher affinity than the dimer. Our studies also show that the binding constants of monomer and dimer to the receptor peptides, and the dimer dissociation constant, can vary significantly as a function of pH and buffer, and so the ability to observe WT monomer peaks is critically dependent on NMR experimental conditions. We conclude that the monomer is the high affinity CXCR1 agonist, that Site-I interactions play a dominant role in determining monomer vs. dimer affinity, and that the dimer plays an indirect role in regulating monomer function.
Keywords: human CXCR1 N-domain, CXCL8, solution NMR, monomer-dimer equilibrium, affinity
Introduction
Humans express ∼50 chemokines that play diverse and fundamental roles from trafficking immune cells and organogenesis to combating infection.1–4 Not surprisingly, a dysregulation in chemokine function has been implicated in the pathophysiology of various autoimmune and inflammatory diseases and cancer.3,5 Most chemokines exist as monomers and dimers, and mediate their function by activating G protein-coupled receptors (GPCR).4,6 Chemokine CXCL8 (also known as interleukin-8, IL-8) has a rich history, is one of the best-characterized members of the family, and mediates its function by activating two receptors, CXCR1 and CXCR2.7–9 The CXCL8 structure was the first for any chemokine, and revealed it to be a dimer.10,11 Subsequent structural and functional studies using trapped CXCL8 monomer and dimer variants have shown that the monomer binds with higher affinity and is a more potent activator of CXCR1.12–15
On the basis of structure-function studies, a two-step two-site model has been proposed for CXCL8 binding and receptor activation.16–19 According to this model, the first step involves interactions between CXCL8 N-loop residues and CXCR1 N-terminal residues (defined as Site-I) and the second step involves interactions between CXCL8 N-terminal residues and CXCR1 extracellular/transmembrane residues (defined as Site-II) (Fig. 1). Therefore, differences in binding affinities and activities between the monomer and dimer could be due to differences in Site-I or Site-II interactions or both.
Figure 1.
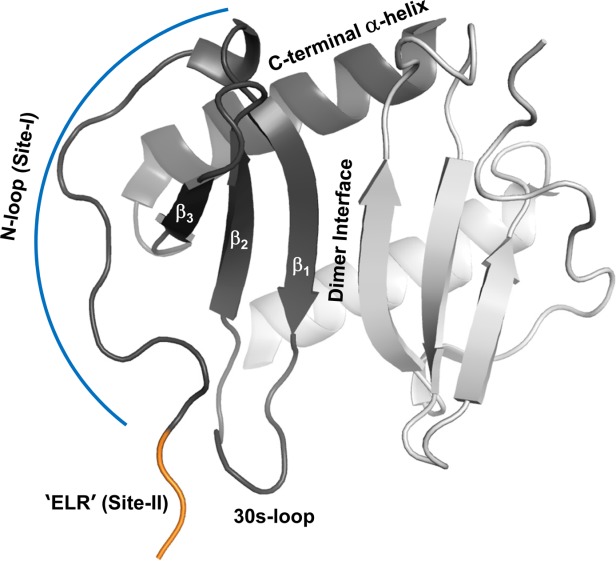
A schematic of CXCL8 dimer. The different secondary structural regions and the receptor binding sites are highlighted in one of the monomers.
Measuring the binding affinities of WT monomer or dimer alone to Site-I or Site-II of the intact receptor is challenging at many levels, and is beyond the capabilities of currently available methodologies. Binding to intact receptors is generally determined by competitive binding using radiolabeled ligands, and can provide the binding constant of only the monomer, because the monomer binds with higher affinity. It is also not possible to selectively bind to Site-I or Site-II unless binding to one of the sites is abolished by mutagenesis. However, a divide-and-conquer approach to characterizing Site-I interactions is possible by studying binding to isolated receptor N-domain peptides; this has been done using fluorescence, nuclear magnetic resonance (NMR), and isothermal titration calorimetry (ITC) techniques.19–29 In particular, a number of studies have characterized the binding of receptor peptides to the WT dimer and various monomer constructs using solution NMR spectroscopy.19,23–28 However, there is a lack of consensus among the various NMR studies regarding the binding affinities of the monomer and dimer, whether the monomer binds with higher affinity or the same affinity as the dimer, and whether binding and dimer dissociation are coupled. The differences among these studies could be due to differences in experimental conditions such as the choice of the buffer, CXCR1 peptide (human vs. rabbit and peptide length), and/or the monomer construct.
We have now overcome the limitations of the previous studies by characterizing the binding of a human CXCR1 N-domain peptide (hCXCR1Ndp) to WT CXCL8 under conditions where it exists as both monomers and dimers. We were able to simultaneously track the binding of both the WT monomer and WT dimer from the same titration experiment, and show that the WT monomer binds hCXCR1Ndp with much higher affinity and that binding is coupled to dimer dissociation. We also characterized the binding of WT CXCL8 and of two designed monomers and a trapped dimer to different hCXCR1Ndps under different buffer conditions. Our data consistently show that the monomer binds with much higher affinity, and that the actual binding affinities for both monomer and dimer can vary significantly depending on the receptor peptide construct and buffer conditions. Our studies also show that the NMR peaks corresponding to the minor WT monomer population can be easily missed unless care is taken to optimize various experimental conditions such as protein concentration and buffer, and that using a monomer construct that does not capture native monomer activity can lead to incorrect conclusions.
Results and Discussion
A summary of the previous NMR studies of CXCR1 receptor peptide binding to WT CXCL8 dimer is shown in Table I. Three of the studies using the human peptide reported binding affinities between 70 and 550 µM and did not report dimer dissociation of the chemokine,24,26,27 whereas our studies using the rabbit peptide indicated that binding and chemokine dimer dissociation were coupled.21,23 Therefore, the question arises whether dimer dissociation is unique to the rabbit peptide, or if the studies using human peptide were not optimal for detecting the monomer peaks. Besides the receptor peptides, these studies also vary with respect to the use of different buffers, different starting protein concentrations, and different final protein–peptide molar ratios resulting in different final bound populations. We now show that human receptor peptide binding is also coupled to dimer dissociation, and that the ability to detect monomer peaks is critically dependent on experimental variables and not due to differences between human and rabbit sequences.
Table I.
Summary of CXCL8 Binding to CXCR1 N-Terminal Domain Peptides Reported in Literature
| CXCL8 variant | CXCR1 | Technique | Buffer conditions | [CXCL8], µM | Kd (µM) | Reference |
|---|---|---|---|---|---|---|
| WT dimer | h40mera | NMR | 50 mM Pi, pH 6.7 | 750 | 170 | Clubb et al., 199424 |
| WT dimer | h38mer | NMR | 20 mM HEPES, pH 5.5 | 100 | 70 | Park et al., 26,44 |
| WT dimer | h21mer | NMR | 50 mM Pi, 150 mM NaCl, pH 6.5 | ≥200 | 550 | Kendrick et al., 27 |
| WT dimerb | r24mer | NMR | 50 mM acetate, pH 6.0 | 90c | n.d. | Ravindran et al., 200923 |
| R26C dimer | r24mer | NMR | 50 mM acetate, pH 6.0 | 93c | 360 | Ravindran et al., 200923 |
| 1–66 monomer | h40mer | NMR | 50 mM acetate, pH 5.5 | 130–200 | <10 | Barter and Stone, 25 |
| 1–66 monomer | h40mer | ITC | 50 mM MOPS, pH 7.0 | 9.6 | Barter and Stone, 25 | |
| L25Y/V27R monomer | h21mer | NMR | 50 mM Pi, 150 mM NaCl, pH 6.5 | ≥200 | 460 | Kendrick et al., 27 |
| 1–66 monomer | r24mer | NMR | 50 mM acetate, pH 6.0 | 115c | 12 | Ravindran et al., 200923 |
| L25NMe monomer | r34mera | ITC | 50 mM HEPES, 50 mM NaCl, pH 8.0 | 6.0 | Fernando et al., 21 | |
| 1–66 monomer | r34mera | ITC | 50 mM Pi, 50 mM NaCl, pH 8.0 | 8.7 | Fernando et al., 22 | |
| 1–66 monomer | r34mera | ITC | 50 mM HEPES, 50 mM NaCl, pH 8.0 | 8.6 | Fernando et al., 22 | |
| 1–66 monomer | r34mera | ITC | 50 mM Tris, 50 mM NaCl, pH 8.0 | 7.5 | Fernando et al., 22 |
| CXCR1 constructs | ||||||
|---|---|---|---|---|---|---|
| h40mera | MSNITDPQMWDFDDLNFTGMPPADEDYSPSMLETETLNKY | |||||
| h38merd | MSNITDPQMWDFDDLNFTGMPPADEDYSPSMLETETLN | |||||
| h29mer | MSNITDPQMWDFDDLNFTGMPPADEDYSP | |||||
| h21mer | MWDFDDLNFTGMPPADEDYSP | |||||
| r24mer | LWTWFEDEFANATGMPPVEKDYSP | |||||
| r34mera | LWTWFEDEFANATGMPPVEKDYSPSLVVTQTLNK | |||||
| CXCL8 constructs | |
|---|---|
| WT | SAKELRCQCIKTYSKPFHPKFIKELRVIESGPHCANTEIIVKLSDGRELCLDPKENWVQRVVEKFLKRAENS |
| R26Ce | SAKELRCQCIKTYSKPFHPKFIKELCVIESGPHCANTEIIVKLSDGRELCLDPKENWVQRVVEKFLKRAENS |
| L25NMee | SAKELRCQCIKTYSKPFHPKFIKELRVIESGPHCANTEIIVKLSDGRELCLDPKENWVQRVVEKFLKRAENS |
| V27P/E29Pe | SAKELRCQCIKTYSKPFHPKFIKELRPIPSGPHCANTEIIVKLSDGRELCLDPKENWVQRVVEKFLKRAENS |
| L25Y/V27Re | SAKELRCQCIKTYSKPFHPKFIKEYRRIESGPHCANTEIIVKLSDGRELCLDPKENWVQRVVEKFLKRAENS |
| 1–66 | SAKELRCQCIKTYSKPFHPKFIKELRVIESGPHCANTEIIVKLSDGRELCLDPKENWVQRVVEKFL |
h, human; r, rabbit; Pi, phosphate.
native cysteine mutated to serine
dimer dissociation coupled to r24mer binding
personal communication (Ravindran, A)
extra GS on the N-terminus and hexa His-tag on the C-terminus
R26C – Arg26 is substituted with a Cys (in red); L25NMe - Leu25 (in red italic) backbone amide is substituted with a methyl group; V27P/E29P – Val27 and Glu29 are substituted with a Pro (in red); L25Y/V27R – Leu25 and Val27 are substituted with Tyr and Arg, respectively (in red).
NMR experiments are routinely acquired at as high a concentration as possible due to the inherent insensitivity of NMR spectroscopy. Further, if solubility and availability are not an issue, there is no reason not to use high concentrations for characterizing simple bimolecular interactions with the exception that excess titration of the receptor peptide could be limited by its solubility. However, characterizing binding to two species such as the dimer and monomer from the same titration experiment is less straightforward. The ability to detect both the dimer and monomer population in the NMR spectrum is crucially dependent on multiple variables including, among other things, protein characteristics, concentration, protein : peptide ratio, and the four equilibrium constants (between the monomer and dimer, KM-D; monomer and receptor peptide, KM-Ndp; dimer and receptor peptide, KD-Ndp; and between the dimer-bound receptor peptide and monomer-bound receptor peptide, KD.Ndp-M.Ndp) (Fig. 2).
Figure 2.
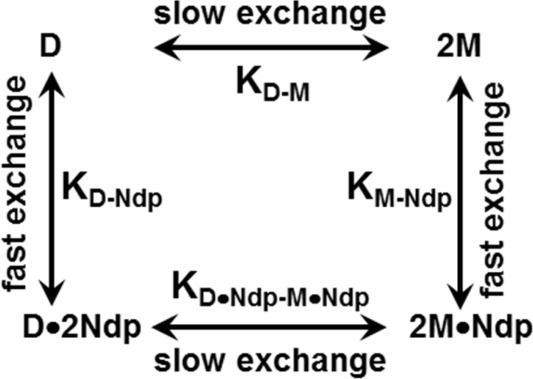
The linkage scheme showing the various equilibria of CXCL8 monomer and dimer binding to the CXCR1 N-domain.
Under NMR conditions, the peaks corresponding to the CXCL8 monomer and dimer are in slow exchange33; therefore, in principle, distinct peaks corresponding to the monomer and dimer can be observed. However, the peaks corresponding to the monomer/dimer and receptor peptide-bound monomer/dimer are in fast exchange; therefore, only one peak (weight averaged of the free and bound species) is observed (Figs. 2 and 3).23 As all four equilibria are coupled, a stronger monomer peak at the end of the titration would indicate that the monomer binds the receptor peptide more strongly, promoting dissociation of the free dimer and the receptor peptide-bound dimer.
Figure 3.
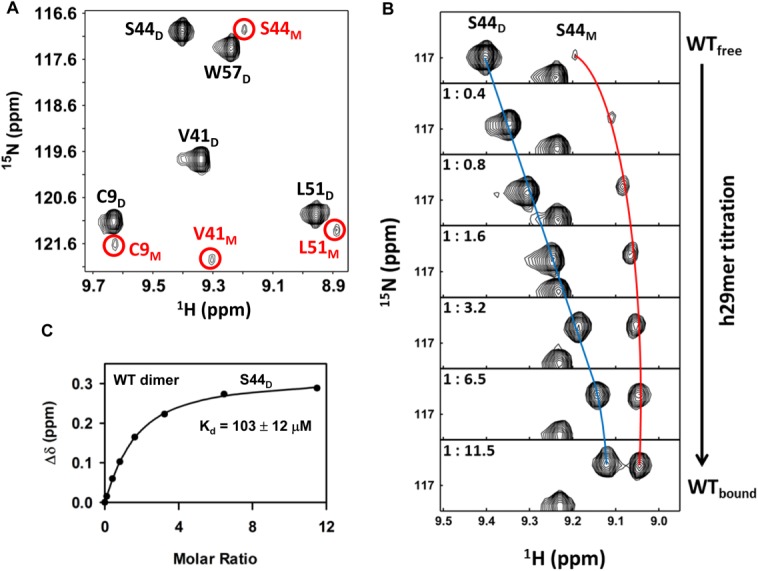
Binding of h29mer to WT CXCL8. A: A selected region of the 1H-15N HSQC spectrum showing dimer and monomer peaks (circled and labeled in red) of a 90 µM WT CXCL8 sample in 50 mM sodium phosphate buffer at pH 7.0. B: A selected region of the 1H-15N HSQC spectrum showing binding-induced chemical shift changes for the S44D (dimer) and S44M (monomer) resonances. The individual panels correspond to different points (molar ratios highlighted in the panels) over the course of the titration. The blue and red lines trace the movement of the dimer and monomer peaks. C: The apparent dissociation constant (Kd) curves obtained by fitting the binding-induced chemical shift changes for S44D resonances. Average Kd from a subset of 10–12 residues is indicated.
The KM-D of CXCL8 is sensitive to pH and buffer conditions and could vary by as much as ∼100-fold, with values ranging from ∼0.1 to 18 µM having been reported in the literature.30–34 Therefore, detecting monomer peaks is dependent on being able to exploit weaker dimer affinities as a function of pH and/or buffer, ideal protein concentrations (the lower the better), and being able to titrate excess receptor peptide to saturate the bound protein population. We were able to obtain a good quality HSQC spectrum of 100 µM WT CXCL8 using our 600 and 800 MHz NMR instruments equipped with cryoprobes in <30 min. Therefore, we measured HSQC spectra of ∼100 µM WT CXCL8 in 50 mM sodium phosphate buffer as a function of pH, and observed distinct monomer peaks at pH 7.0 and above, indicating that dimer association is weaker at higher pH (data not shown).
We first characterized the binding of human CXCR1 N-domain 29mer peptide (h29mer) to WT CXCL8 in 50 mM phosphate, pH 7.0. The HSQC spectrum of WT CXCL8 at pH 7.0 showing prominent dimer peaks and weaker but distinct monomer peaks is shown in Figure 3(A). On successive addition of the h29mer, we observed not only binding-induced chemical shift changes for both the monomer and dimer but also the relative intensity of the monomer becoming stronger [ Fig. 3(B)]. The increase in the monomer-h29mer population indicates that the WT monomer binds the receptor peptide with higher affinity than does the WT dimer. We calculate KD-Ndp as 103 µM, and that 87% of CXCL8 exists in the bound form at the end of the titration [ Fig. 3(C)]. Considering that the dimer dissociates during the course of the titration, we calculated the upper limit of KD-Ndp as 140 µM (assuming 50% of the protein exists as a dimer at the end of the titration), indicating dimer dissociation results in at most a 2-fold error in the measured binding constants. We also carried out titration experiments in 50 mM sodium phosphate buffer at pH 6.0 (Table II). Dimer formation is stronger at pH 6.0 than at pH 7.0, as evidenced by the fact that, at pH 6.0, there was no evidence of the monomer peak in the free spectrum, and the dimer affinity was much stronger for the h29mer (17 µM). We were still able to observe the bound monomer peaks but only at the last titration point [ Fig. 4(A)]. The relative population of the bound monomer was lower at pH 6.0 than at pH 7.0 [ Fig. 4(B)]. We could not measure the binding affinity of the WT monomer due to uncertainties in the monomer concentration, especially during the early time points that determine the binding isotherm. However, binding studies using the designed monomer provide the KM-Ndp as <5 µM at both pH 6 and 7 (see below), indicating the monomer binds with much higher affinity compared with the dimer (Table II).
Table II.
Summary of NMR Titration Experiments of hCXCR1Ndp Binding to CXCL8 Variants
| Complex | Buffer conditions | Temp | CXCL8 conc. Start/end (μM)c | Final Peptide Conc. (μM) | Molar ratio | FB (%) | Kd (μM)d |
|---|---|---|---|---|---|---|---|
| WT + h29mera,b | 50 mM Pi, pH 7.0 | 30°C | 90/62 | 711 | 1:11.5 | 87 ± 2 | 103 ± 12 |
| WT + h29mera,b | 50 mM Pi, pH 6.0 | 30°C | 100/80 | 850 | 1:10.5 | 98 ± 1 | 17 ± 4 |
| WT + h21mera,b | 50 mM Pi, 150 NaCl, pH 6.5 | 25°C | 125/20 | 2660 | 1:134 | 79 ± 3 | 700 ± 90 |
| R26C + h29mer | 50 mM Pi, pH 7.0 | 30°C | 150/129 | 1029 | 1:11.2 | 85 ± 3 | 172 ± 24 |
| 1–66 monomer + h29mer | 50 mM Pi, pH 6.0 | 30°C | 200/135 | 1135 | 1:8.4 | >99 | <5 |
| V27P/E29P monomer + h29mer | 50 mM Pi, pH 7.0 | 30°C | 100/65 | 777 | 1:11.9 | >99 | <5 |
| V27P/E29P monomer + h21mer | 50 mM Pi, pH 7.0 | 30°C | 100/72 | 461 | 1:6.4 | 90 ± 3 | 43 ± 10 |
| V27P/E29P monomer + h21mer | 50 mM Pi, 150NaCl, pH 6.5 | 25°C | 100/69 | 945 | 1:13.8 | 88 ± 2 | 118 ± 14 |
| L25Y/V27R monomer + h21mer | 50 mM Pi, pH 7.0 | 30°C | 100/63 | 506 | 1:8 | 77 ± 5 | 135 ± 25 |
| L25Y/V27R monomer + h21mer | 50 mM Pi, 150 NaCl, pH 6.5 | 25°C | 150/62 | 1176 | 1:19 | 72 ± 2 | 432 ± 25 |
WT dimer dissociation was coupled to receptor peptide binding
Kd calculated for the dimer.
Concentrations are reported in monomer units.
Kd values are averages calculated from a subset of 8–15 residues; Pi, phosphate.
Figure 4.
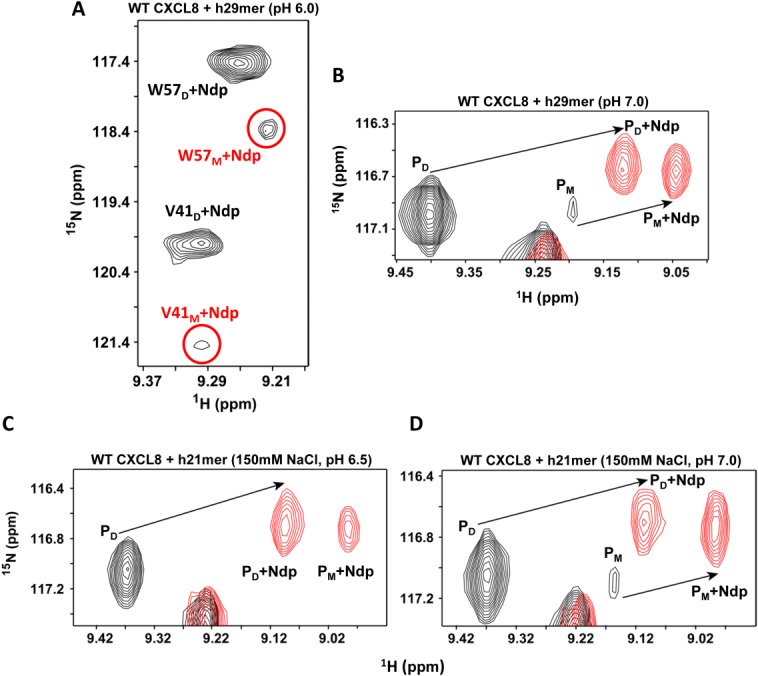
Binding of hCXCR1Ndp and dimer dissociation are coupled. A: A selected region of the 1H-15N HSQC spectrum showing strong dimer and weak monomer peaks (circled and labeled in red) of the last titration point (molar ratio 1 : 10.5) of WT CXCL8 (80 µM) bound to h29mer in 50 mM sodium phosphate buffer pH 6.0. B: A selected region of the 1H-15N HSQC (of the Ser44 resonance) shows binding-induced dimer dissociation for h29mer in 50 mM sodium phosphate buffer pH 7.0; C: for h21mer in 50 mM sodium phosphate, 150 mM NaCl, pH 6.5; and D: for h21mer in 50 mM sodium phosphate, 150 mM NaCl, pH 7.0. In panel C, the protein concentration at the end of the titration was 20 µM. The peaks in black represent the spectra of the free WT CXCL8 and in red of the hCXCR1Ndp-bound form. PD and PM are the free dimer and monomer, while PD + Ndp and PM +Ndp are the bound form respectively. The free monomer peak is not observed at pH 6.5. Note the increase in intensity of the bound monomer peak and the decrease in intensity of the bound dimer peak, indicating a shift in the equilibrium to the monomer-bound form.
We finally characterized the binding of the human CXCR1 N-domain 21mer peptide (h21mer) to WT CXCL8 using the buffer conditions of Kendrik et al.27 These authors recently characterized the binding of a h21mer to WT CXCL8 in 50 mM sodium phosphate, 150 mM NaCl, pH 6.5 buffer, and reported very weak binding (Kd ∼550 µM) and further reported that binding did not lead to dimer dissociation (Table I). We also observed that the binding was weak (Kd ∼700 µM) but nevertheless could observe dimer dissociation [ Fig. 4(C)]. We could detect the monomer peaks because of the large protein:peptide ratio and the low CXCL8 concentration (20 µM) at the end of the titration. The chemical shifts of the new monomer peaks were closer to the shift observed for the monomer peak at pH 7.0 in the h29mer binding data [ Fig. 4(B,C)], indicating that the new peak arises predominantly from the bound monomer. The binding affinity of the V27P/E29P monomer (KM-Ndp 118 µM) under the same buffer conditions provides further evidence that the monomer binds with higher affinity compared to the dimer. We also collected HSQC spectra of free CXCL8 and in presence of excess h21mer at the same low concentration of 20 µM sample at pH 7.0 containing 150 mM NaCl [ Fig. 4(D)]. The data show that the relative intensity of the monomer compared to dimer is weaker in the free form but stronger in the presence of receptor peptide indicating favorable binding.
These data together emphasize how differences in protein concentration, long acquisition times (that determine the signal-to-noise [s/n] ratio), and buffer conditions can be optimally exploited for detecting the minor monomer population. These studies also indicate that the binding affinities of both monomer and dimer can vary substantially as a function of buffer conditions, but the CXCL8 monomer always binds with higher affinity under all buffer conditions.
We also determined the binding affinity of a trapped CXCL8 dimer, generated by introducing a disulfide bond about the two-fold (R26C) symmetry across the dimer interface. We have shown previously that the structure of the trapped dimer is essentially identical to the WT dimer.14 We observe that the chemical shift perturbation profile of the trapped dimer is similar to that of the WT dimer (Fig. 5), and that the measured binding affinity is also similar to that of the WT dimer (Table II) providing further evidence that the binding affinity of the dimer is much weaker than that of the monomer.
Figure 5.
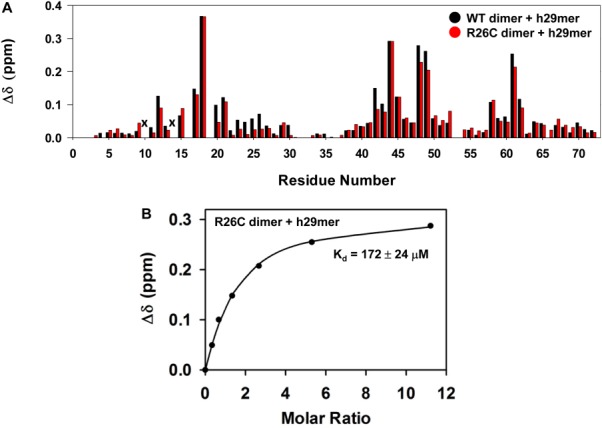
Binding of WT dimer vs. R26C dimer to h29mer. A: Histogram plot showing h29mer binding-induced chemical shift changes (Δδ) for the WT dimer (black) and R26C trapped dimer (red). I10 and S14 are broadened out (indicated by x). B: The dissociation constant (Kd) curves obtained by fitting the binding-induced chemical shift changes for S44 resonance are shown for the R26C dimer. Average Kd from a subset of 10–12 residues is indicated.
Binding affinities of the monomer
Our studies as described above have shown that NMR detection of the WT monomer is straightforward, but measuring its binding affinity is challenging due to uncertainties in the monomer concentration. An alternate approach would be to characterize a designed CXCL8 monomer variant—an ideal monomer construct must retain the structural and functional characteristics of the WT monomer and should require the least number of substitutions/modifications to achieve this goal. We have used two different monomer constructs in this study—the 1–66 deletion mutant that is missing the last 6 residues, and the V27P/E29P mutant. The 1−66 deletion mutant (referred to as the 1−66 monomer) was designed on the basis that the dimer structure revealed that residues 67−72 were involved in packing interactions with residues of the other monomer. The 1−66 monomer shows WT monomer-like receptor activity, and we and others have used this monomer as a surrogate for the WT monomer in various structural and functional studies.22,23,25,28,34 More recently, we designed the V27P/E29P mutant on the basis that disrupting backbone H-bonding interactions across the dimer interface should result in a monomer. We have shown that the V27P/E29P mutant is indeed a monomer (referred to as the V27P/E29P monomer) and that it has WT monomer-like activity.35
The binding affinities of both the 1–66 and V27P/E29P monomers for the h29mer from our NMR titrations were determined to be <5 µM [Table II, Fig. 6(B)]. A previous NMR study of binding of the 1–66 monomer to a h40mer also reported the KM-Ndp as between 1 and 10 µM.25 Accurate determination of the binding constant from NMR titrations must satisfy the requirement that the starting CXCL8 concentration is in the order of ∼0.5 × Kd and no more than 5 × Kd36; we have used CXCL8 concentrations (∼100 to 200 µM) that are much higher. Nevertheless, these observations together provide compelling evidence that the monomer, compared with dimer, binds the CXCR1 N-terminal domain with much higher affinity.
Figure 6.
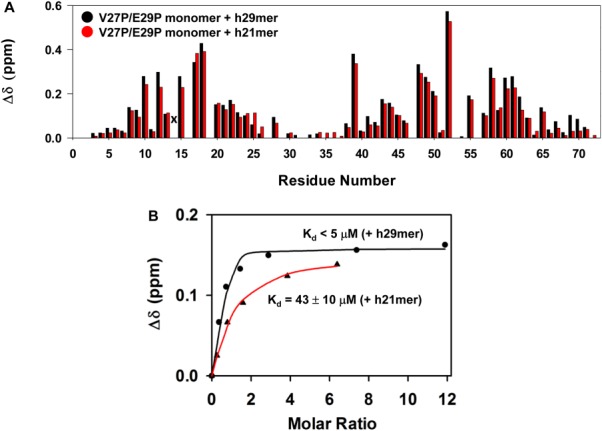
Binding of CXCL8 monomer to h29mer and h21mer. A: A histogram plot showing h29mer (black) versus h21mer (red) binding-induced chemical shift changes in the V27P/E29P monomer. Residue S14 is broadened out (indicated by x), and residues 16, 19, 32, and 53 are prolines. B: A representative plot for measuring the dissociation constant (Kd) by fitting binding-induced chemical shift changes for S44 resonance is shown. The average Kd from a subset of 10–12 residues is indicated.
We also characterized the binding of the h21mer to the V27P/E29P monomer on the basis that previous studies had shown that a 21mer is sufficient for CXCL8 binding.37 The chemical shift perturbation profiles for both h21mer and h29mer were essentially identical except that the binding affinity of the h21mer (KM-Ndp 43 µM) was lower (Fig. 6, Table II). These observations confirm that a 21mer sequence contains all of the residues that mediate binding as any binding interactions of the additional eight residues of the h29mer would have resulted in an altered CSP profile compared with the 21mer. Considering the CSP profile and the maximum CSP are essentially the same for the two peptides, we propose that the increased affinity of the h29mer is most likely due to reduced entropic penalty on binding of the longer 29mer compared with the 21mer though a more favorable enthalpy of binding also cannot be ruled out.
Recently, Kendrik et al. reported using NMR that the binding of the h21mer to the L25Y/V27R monomer was much weaker (440 µM).27 In addition to the weaker binding of the h21mer, this lower affinity could also be due to the use of a buffer containing 150 mM NaCl and/or due to the intrinsic lower affinity of the L25Y/V27R monomer. Therefore, we characterized the binding of the h21mer to the V27P/E29P and L25Y/V27R monomers in 50 mM sodium phosphate, 150 mM NaCl, pH 6.5 buffer, and to the L25Y/V27R monomer in 50 mM sodium phosphate pH 7.0 buffer (Supporting Information, Figs. S1 and S2). The chemical shift perturbation profiles between the buffer systems were essentially identical, but the binding affinity in the buffer containing 150 mM NaCl was lower (Fig. 7, Table II). Further, the binding affinity of the L25Y/V27R monomer was weaker compared with the V27P/E29P monomer, indicating that the L25Y/V27R monomer is intrinsically less active compared to other monomer constructs (Table II). These studies provide compelling evidence of how differences in receptor peptide sequence, monomer construct, and buffer conditions together can result in >100-fold differences in binding affinity.
Figure 7.
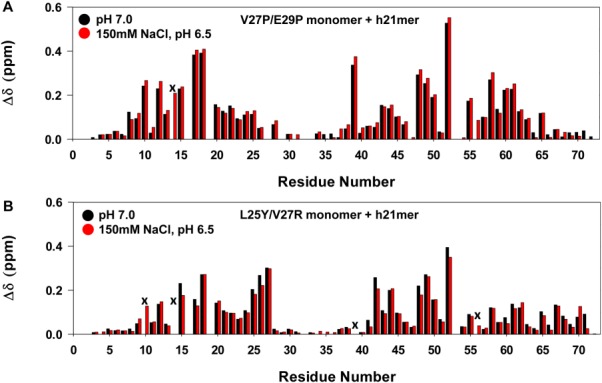
Binding of monomer variants to h21mer at different pH and buffer conditions. A: Histogram plot showing h21mer binding-induced chemical shift changes in the V27P/E29P monomer at pH 7.0 (black) and 150 mM NaCl, pH 6.5 (red). S14 is broadened out (indicated by x). B: Histogram plot showing h21mer binding-induced chemical shift changes in L25Y/V27R monomer at pH7.0 (black) and 150 mM NaCl, pH6.5 (red). Data for residues I10 and N56 at pH 7, and S14 and I39 at both pHs are not shown due to peak broadening (indicated by x).
The L25Y/V27R mutant was first described by Daly et al. and was generated by this group in their effort to understand the molecular basis of CXCL8 and CXCL4 in myeloid progenitor proliferation.38 These authors and subsequently William et al. characterized the activity of the L25Y/V27R mutant for neutrophils (that express both CXCR1 and CXCR2).38,39 Daly et al. also measured the binding affinity of this mutant for CXCR2 using a transfected CHO cell line. The mutant was observed to be less active in many of the assays, and most importantly, the mutant has not been exclusively characterized for CXCR1 function. Single and double Tyr or Phe substitutions for Leu25 and Val27 have also been shown to result in reduced binding affinities.40,41 NMR spectrum of L25Y/V27R shows that it is folded and has native-like structure (Supporting Information, Fig. S1), indicating that differences between the monomer affinities are not as much as due to differences in structures but due to differences in conformational dynamics. We have previously observed for a number of CXCL8 mutants that there is no correlation between structural similarity at the HSQC level and functional activity.28
Comparison between rabbit and human CXCR1 peptides
Previous studies had shown that human CXCL8 binds rabbit CXCR1 with affinity and specificity like human CXCR1.42,43 The human and rabbit receptor N-terminal domains show high sequence similarity, and early studies had also shown that both human and rabbit receptor peptides bind CXCL8 with similar affinities.43 We had previously characterized the binding of rabbit CXCR1 N-domain 24mer (r24mer) and 34mer (r34mer) peptides to the WT and the 1–66 monomer using NMR, ITC, and fluorescence methods.19–23,28 We observe that binding is coupled to CXCL8 dimer dissociation, and that the 1–66 monomer binds both peptides with ∼10 µM affinity (Table I). Our current data using two human CXCR1 N-domain h21mer and h29mer peptides confirm our earlier observations using rabbit r24mer and r34mer peptides that the monomer is the high-affinity CXCR1 ligand and that binding promotes dimer dissociation.
Structural basis of the differential binding of the monomer and dimer
Previous and our current studies show that dimer interface residues are not involved in binding and also that only the monomer of the dimer is involved in binding to the receptor N-terminal domain (Fig. 1). The structure of a trapped monomer designed by introducing a methyl group substitution of a backbone amide proton of the dimer interface residue Leu25 is known, and is observed to be similar to the monomer of the dimer12; however, amide exchange and backbone dynamics measurements have shown that the monomer is conformationally more flexible.12,13,35 The CXCR1 structure and solution studies of the peptides have shown that the N-terminal domain lacks structure and is natively unfolded, but adopts a definite structure on binding.26,44–46
Comparison of the chemical shift perturbation profiles between the WT dimer and the V27P/E29P monomer also show that the extent of perturbation is lower for the dimer; furthermore, residues such as D52, E55, I39, V58 are highly perturbed in the monomer but less in the dimer (Fig. 8). The structures reveal that these residues cannot be involved in direct interactions, indicating that chemical shift changes must be due to indirect interactions. Therefore, it is possible that the conformational flexibility of the monomer allows it bind more efficiently, which is not possible for the more structured dimer. High-resolution structures and knowledge of the dynamics of both monomer and dimer in the CXCR1 peptide-bound form are essential to gain deeper insights into the molecular basis of the differences in the binding interactions.
Figure 8.
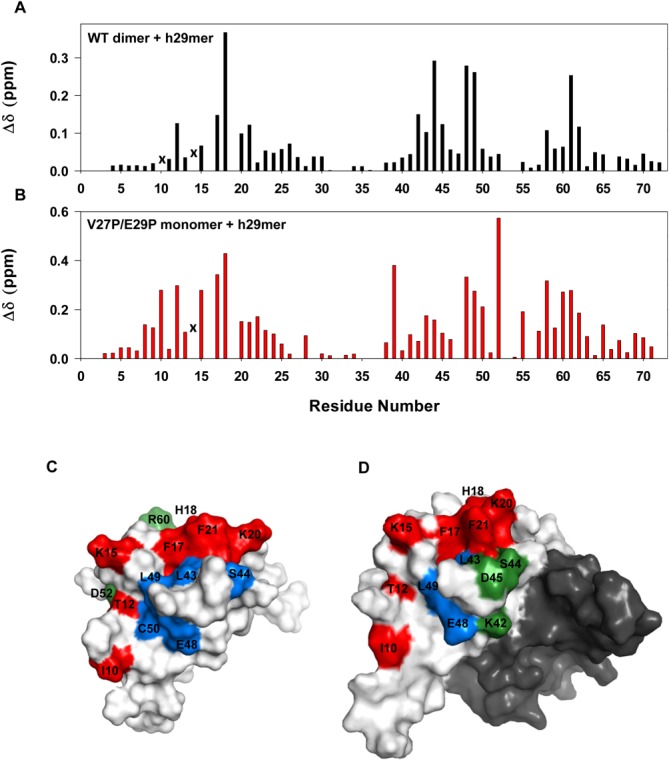
Binding of WT dimer and V27P/E29P monomer to h29mer. Histogram plots showing h29mer binding-induced chemical shift changes in WT dimer (A) and V27P/E29P monomer (B). Data for residues I10 in dimer and S14 in monomer and dimer are not shown as they are broadened out (indicated by x). C, D: Molecular surface plots of the monomer and dimer. In the case of dimer, binding residues are highlighted in one monomer and the other monomer is shaded in dark gray for clarity. The perturbed N-loop residues are painted in red, residues that are differentially perturbed in monomer versus dimer in green, and all others in blue. Residues I39, E55, V58, V61, V62, and F65 are buried and so not visible in the surface plots.
Binding at Site-II is mediated by the CXCL8 N-terminal “Glu-Leu-Arg” residues.16 The structures of both the CXCL8 monomer and dimer reveal that these residues are unstructured and so it is very likely that the binding affinities of both the monomer and dimer for Site-II are similar and that the differences in Site-I interactions determine the binding affinities to the intact receptor.
Functional relevance of monomer and dimer affinities
CXCL8 mediates function by activating two receptors, CXCR1 and CXCR2. Considering that the CXCL8 monomer and dimer show similar CXCR2 affinities, selective activation of CXCR1 by the CXCL8 monomer over the dimer could have important implications for their in vivo function.15 The receptor sequences reveal large differences in the N-terminal residues, indicating that the Site-I interactions dictate differences between the monomer and dimer and between CXCR1 and CXCR2 selectivity and affinity. Indeed, our current studies provide compelling evidence that Site-I interactions play an important role in determining high-affinity CXCR1 binding of the monomer. These studies are also relevant for designing peptide decoys, because inhibitors that disrupt Site-I interactions could function as drugs in a clinical setting.47 Finally, our studies also emphasize how various experimental parameters such as protein concentration, buffer conditions, binding constants of the various equilibria, protein–peptide ratio, NMR instrumentation and acquisition time, protein and peptide solubility together must be optimal to be able to detect the peaks corresponding to the minor monomer population.
Materials and Methods
Cloning, expression, and purification
The WT CXCL8, R26C obligate dimer, V27P/E29P monomer, (1–66) monomer, L25Y/V27R monomer, and the human CXCR1 29mer peptide (h29mer) were recombinantly expressed in Escherichia coli strain BL21(DE3) and purified as discussed earlier.20 The clone for the L25Y/V27R mutant was generated on the WT CXCL8 background by performing iterative cycles of mutagenesis using the Stratagene QuikChange site-directed mutagenesis protocol.48 The DNA sequence encoding the h29mer peptide (MSNITDPQMWDFDDLNFTGMPPADEDYSP) was custom-synthesized (Genscript), amplified by PCR, and inserted into the pET32 Xa/LIC vector using a ligation-independent cloning method. The human CXCR1 21mer (h21mer) peptide was prepared using solid-phase peptide synthesis (MWDFDDLNFTGMPPADEDYSP) (Aapptec (KY, USA). The 15N-labeled proteins were produced by growing cells in minimal medium containing 15NH4Cl as the sole nitrogen source. The purity and molecular weight of the proteins and peptides were confirmed using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS)
Nuclear magnetic resonance (NMR) spectroscopy
The starting protein concentrations for the HSQC titrations experiments were ∼100–200 µM. The titration experiments were performed at two different conditions, 50 mM sodium phosphate pH 7.0, 30°C, and 50 mM sodium phosphate, 150 mM NaCl pH 6.5, 25°C. The 15N-labeled CXCL8 samples were prepared in the respective buffers containing 1 mM sodium azide, 1 mM sodium 2,2-dimethyl-2-silapentane sulfonate (DSS), and 10% 2H2O (v/v). Stock solutions (∼2 mM) of the CXCR1 N-domain peptides in the same buffer were used for the titration experiments. NMR HSQC titration experiments were acquired using a Bruker Avance III 800 MHz (equipped with a TXI cryoprobe) or 600 MHz (equipped with a QCI cryoprobe) spectrometers. The spectra were processed using NMRPipe,49 and analyzed using NMRView,50 or Bruker Topspin 3.2 software.
Binding interactions were monitored by adding aliquots of CXCR1 peptide to the 15N-labeled proteins essentially until no change in chemical shifts were observed in the 1H-15N HSQC spectra. Experimental details such as buffer, starting and final CXCL8 and peptide concentrations, final peptide : protein molar ratio, and bound population are provided in Table II. The apparent binding constants (Kd) were determined by fitting the chemical shift changes as a function of peptide : protein molar ratios as described previously.23 The observed chemical shift change was calculated using the equation, Δδ = &(δH2 + (0.2δN)2), where δH and δN are the 1H and 15N chemical shift changes, respectively. The calculated Kd is the average obtained from fitting the titration curves of 8-15 well resolved amide resonances.
Acknowledgments
The authors thank Dan Nguyen for help with protein expression and purification, and Dr. Tianzhi Wang (UTMB) for help with the NMR instrumentation.
Glossary
- HSQC
heteronuclear single quantum coherence
- NMR
nuclear magnetic resonance
- Ndp
N-terminal domain peptide
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A. Chemokines and chemokine receptors: an overview. Front Biosci. 2009;14:540–551. doi: 10.2741/3261. [DOI] [PubMed] [Google Scholar]
- 2.Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–716. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 4.Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res. 2011;317:590–601. doi: 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem J. 2008;409:635–649. doi: 10.1042/BJ20071493. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol. 2002;42:469–499. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- 7.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 8.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 9.Stillie R, Farooq SM, Gordon JR, Stadnyk AW. The functional significance behind expressing two IL-8 receptor types on PMN. J Leukoc Biol. 2009;86:529–543. doi: 10.1189/jlb.0208125. [DOI] [PubMed] [Google Scholar]
- 10.Clore GM, Appella E, Yamada M, Matsushima K, Gronenborn AM. Three-dimensional structure of interleukin 8 in solution. Biochemistry. 1990;29:1689–1696. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 11.Baldwin ET, Weber IT, St Charles R, Xuan JC, Appella E, Yamada M, Matsushima K, Edwards BF, Clore GM, Gronenborn AM, Wlodawer A. Crystal structure of interleukin 8: symbiosis of NMR and crystallography. Proc Natl Acad Sci USA. 1991;88:502–506. doi: 10.1073/pnas.88.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajarathnam K, Clark-Lewis I, Sykes BD. 1H NMR solution structure of an active monomeric interleukin-8. Biochemistry. 1995;34:12983–12990. doi: 10.1021/bi00040a008. [DOI] [PubMed] [Google Scholar]
- 13.Rajarathnam K, Sykes BD, Kay CM, Dewald B, Geiser T, Baggiolini M, Clark-Lewis I. Neutrophil activation by monomeric interleukin-8. Science. 1994;264:90–92. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 14.Rajarathnam K, Prado GN, Fernando H, Clark-Lewis I, Navarro J. Probing receptor binding activity of interleukin-8 dimer using a disulfide trap. Biochemistry. 2006;45:7882–7888. doi: 10.1021/bi0605944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nasser MW, Raghuwanshi SK, Grant DJ, Jala VR, Rajarathnam K, Richardson RM. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J Immunol. 2009;183:3425–3432. doi: 10.4049/jimmunol.0900305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajagopalan L, Rajarathnam K. Structural basis of chemokine receptor function—a model for binding affinity and ligand selectivity. Biosci Rep. 2006;26:325–339. doi: 10.1007/s10540-006-9025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wells TN, Power CA, Lusti-Narasimhan M, Hoogewerf AJ, Cooke RM, Chung CW, Peitsch MC, Proudfoot AE. Selectivity and antagonism of chemokine receptors. J Leukoc Biol. 1996;59:53–60. doi: 10.1002/jlb.59.1.53. [DOI] [PubMed] [Google Scholar]
- 18.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joseph PR, Sarmiento JM, Mishra AK, Das ST, Garofalo RP, Navarro J, Rajarathnam K. Probing the role of CXC motif in chemokine CXCL8 for high affinity binding and activation of CXCR1 and CXCR2 receptors. J Biol Chem. 2010;285:29262–29269. doi: 10.1074/jbc.M110.146555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajagopalan L, Rajarathnam K. Ligand selectivity and affinity of chemokine receptor CXCR1. Role of N-terminal domain. J Biol Chem. 2004;279:30000–30008. doi: 10.1074/jbc.M313883200. [DOI] [PubMed] [Google Scholar]
- 21.Fernando H, Chin C, Rösgen J, Rajarathnam K. Dimer dissociation is essential for interleukin-8 (IL-8) binding to CXCR1 receptor. J Biol Chem. 2004;279:36175–36178. doi: 10.1074/jbc.C400283200. [DOI] [PubMed] [Google Scholar]
- 22.Fernando H, Nagle GT, Rajarathnam K. Thermodynamic characterization of interleukin-8 monomer binding to CXCR1 receptor N-terminal domain. FEBS J. 2007;274:241–251. doi: 10.1111/j.1742-4658.2006.05579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ravindran A, Joseph PR, Rajarathnam K. Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain: role of coupled interactions and dynamics. Biochemistry. 2009;48:8795–8805. doi: 10.1021/bi901194p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clubb RT, Omichinski JG, Clore GM, Gronenborn AM. Mapping the binding surface of interleukin-8 complexed with an N-terminal fragment of the type 1 human interleukin-8 receptor. FEBS Lett. 1994;338:93–97. doi: 10.1016/0014-5793(94)80123-1. [DOI] [PubMed] [Google Scholar]
- 25.Barter EF, Stone MJ. Synergistic interactions between chemokine receptor elements in recognition of interleukin-8 by soluble receptor mimics. Biochemistry. 2012;51:1322–1331. doi: 10.1021/bi201615y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park SH, Casagrande F, Cho L, Albrecht L, Opella SJ. Interactions of interleukin-8 with the human chemokine receptor CXCR1 in phospholipid bilayers by NMR spectroscopy. J Mol Biol. 2011;414:194–203. doi: 10.1016/j.jmb.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kendrick AA, Holliday MJ, Isern NG, Zhang F, Camilloni C, Huynh C, Vendruscolo M, Armstrong G, Eisenmesser EZ. The dynamics of interleukin-8 and its interaction with human CXC receptor I peptide. Protein Sci. 2014;23:464–480. doi: 10.1002/pro.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joseph PR, Sawant KV, Isley A, Pedroza M, Garofalo RP, Richardson RM, Rajarathnam K. Dynamic conformational switching in the chemokine ligand is essential for G Protein coupled-receptor activation. Biochem J. 2013;456:241–51. doi: 10.1042/BJ20130148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Girrbach M, Meliciani I, Waterkotte B, Berthold S, Oster A, Brurein F, Strunk T, Wadhwani P, Berensmeier S, Wenzel W, Schmitz K. A fluorescence polarization assay for the experimental validation of an in silico model of the chemokine CXCL8 binding to receptor-derived peptides. Phys Chem Chem Phys. 2014;16:8036–8043. doi: 10.1039/c3cp53850h. [DOI] [PubMed] [Google Scholar]
- 30.Burrows SD, Doyle ML, Murphy KP, Franklin SG, White JR, Brooks I, McNulty DE, Scott MO, Knutson JR, Porter D, Young PR, Hensley P. Determination of the monomer-dimer equilibrium of interleukin-8 reveals it is a monomer at physiological concentrations. Biochemistry. 1994;33:12741–12745. doi: 10.1021/bi00209a002. [DOI] [PubMed] [Google Scholar]
- 31.Paolini JF, Willard D, Consler T, Luther M, Krangel MS. The chemokines IL-8, monocyte chemoattractant protein-1, and I-309 are monomers at physiologically relevant concentrations. J Immunol. 1994;153:2704–2717. (Erratum in 1996;156(8):following 3088) [PubMed] [Google Scholar]
- 32.Rajarathnam K, Kay CM, Clark-Lewis I, Sykes BD. Characterization of quaternary structure of interleukin-8 and functional implications. Methods Enzymol. 1997;287:89–105. doi: 10.1016/s0076-6879(97)87009-7. [DOI] [PubMed] [Google Scholar]
- 33.Lowman HB, Fairbrother WJ, Slagle PH, Kabakoff R, Liu J, Shire S, Hebert CA. Monomeric variants of IL-8: effects of side chain substitutions and solution conditions upon dimer formation. Protein Sci. 1997;6:598–608. doi: 10.1002/pro.5560060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin H, Hayes GL, Darbha NS, Meyer E, LiWang PJ. Investigation of CC and CXC chemokine quaternary state mutants. Biochem Biophys Res Commun. 2005;338:987–999. doi: 10.1016/j.bbrc.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 35.Joseph PR, Poluri KM, Gangavarapu P, Rajagopalan L, Raghuwanshi S, Richardson RM, Garofalo RP, Rajarathnam K. Proline substitution of dimer interface β-strand residues as a strategy for the design of functional monomeric proteins. Biophys J. 2013;105:1491–1501. doi: 10.1016/j.bpj.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Attwood MR, Borkakoti N, Bottomley GA, Conway EA, Cowan I, Fallowfield AG, Handa BK, Jones PS, Keech E, Kirtland SJ, Williams G, Wilson FX. Identification and characterisation of an inhibitor of interleukin-8: a receptor based approach. Bioorganic Med Chem Lett. 1996;6:1869–1874. [Google Scholar]
- 38.Daly TJ, LaRosa GJ, Dolich S, Maione TE, Cooper S, Broxmeyer HE. High activity suppression of myeloid progenitor proliferation by chimeric mutants of interleukin 8 and platelet factor 4. J Biol Chem. 1995;270:23282–23292. doi: 10.1074/jbc.270.40.23282. [DOI] [PubMed] [Google Scholar]
- 39.Williams MA, Cave CM, Quaid G, Robinson C, Daly TJ, Witt D, Lentsch AB, Solomkin JS. Interleukin 8 dimerization as a mechanism for regulation of neutrophil adherence-dependent oxidant production. Shock. 2005;23:371–376. doi: 10.1097/01.shk.0000158140.58335.54. [DOI] [PubMed] [Google Scholar]
- 40.Lusti-Narasimhan M, Power CA, Allet B, Alouani S, Bacon KB, Mermod JJ, Proudfoot AE, Wells TN. Mutation of Leu25 and Val27 introduces CC chemokine activity into interleukin-8. J Biol Chem. 1995;270:2716–2721. doi: 10.1074/jbc.270.6.2716. [DOI] [PubMed] [Google Scholar]
- 41.Horcher M, Rot A, Aschauer H, Besemer J. IL-8 derivatives with a reduced potential to form homodimers are fully active in vitro and in vivo. Cytokine. 1998;10:1–12. doi: 10.1006/cyto.1997.0251. [DOI] [PubMed] [Google Scholar]
- 42.LaRosa GJ, Thomas KM, Kaufmann ME, Mark R, White M, Taylor L, Gray G, Witt D, Navarro J. Amino terminus of the interleukin-8 receptor is a major determinant of receptor subtype specificity. J Biol Chem. 1992;267:25402–25406. [PubMed] [Google Scholar]
- 43.Gayle RB, Sleath PR, Srinivason S, Birks CW, Weerawarna KS, Cerretti DP, Kozlosky CJ, Nelson N, Vanden Bos T, Beckmann MP. Importance of the amino terminus of the interleukin-8 receptor in ligand interactions. J Biol Chem. 1993;268:7283–7289. [PubMed] [Google Scholar]
- 44.Park SH, Casagrande F, Das BB, Albrecht L, Chu M, Opella SJ. Local and global dynamics of the G protein-coupled receptor CXCR1. Biochemistry. 2011;50:2371–2380. doi: 10.1021/bi101568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skelton NJ, Quan C, Reilly D, Lowman H. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure. 1999;7:157–168. doi: 10.1016/S0969-2126(99)80022-7. [DOI] [PubMed] [Google Scholar]
- 46.Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, Opella SJ. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szpakowska M, Fievez V, Arumugan K, van Nuland N, Schmit JC, Chevigné A. Function, diversity and therapeutic potential of the N-terminal domain of human chemokine receptors. Biochem Pharmacol. 2012;84:1366–1380. doi: 10.1016/j.bcp.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Johnson BA, Blevins RA. NMR view: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information