

Abstract
The conformational energy landscape of a protein determines populations of all possible conformations of the protein and also determines the kinetics of the conversion between the conformations. Interaction with ligands influences the conformational energy landscapes of proteins and shifts populations of proteins in different conformational states. To investigate the effect of ligand binding on partial unfolding of a protein, we use Escherichia coli dihydrofolate reductase (DHFR) and its functional ligand NADP+ as a model system. We previously identified a partially unfolded form of DHFR that is populated under native conditions. In this report, we determined the free energy for partial unfolding of DHFR at varying concentrations of NADP+ and found that NADP+ binds to the partially unfolded form as well as the native form. DHFR unfolds partially without releasing the ligand, though the binding affinity for NADP+ is diminished upon partial unfolding. Based on known crystallographic structures of NADP+-bound DHFR and the model of the partially unfolded protein we previously determined, we propose that the adenosine-binding domain of DHFR remains folded in the partially unfolded form and interacts with the adenosine moiety of NADP+. Our result demonstrates that ligand binding may affect the conformational free energy of not only native forms but also high-energy non-native forms.
Keywords: DHFR, NADP+, partial unfolding, proteolysis, ligand binding
Introduction
Proteins assume various conformations even under native conditions.1 The experimentally observed structures are ensemble-averaged structures and dominated by the most populated compactly folded forms (the native forms). Though much less in population than the native forms, various non-native conformations, including partially or even globally unfolded forms, also exist in equilibrium with the native forms.2–4 The conformational energy landscape of a protein determines the relative populations of all the possible conformations and the kinetics of conversion between the conformations.5 Protein folding can be described as a process in which a protein finds the global minimum by traversing through multiple local energy minima along one or more low-energy paths on its conformational energy landscape.6–8
The environment and chemical components of the system have critical influences on the conformational energy landscapes of proteins. Proteins denature at an elevated temperature or at a high concentration of a chemical denaturant because the native forms are no longer at global minima in their conformational energy landscapes.9 Ligand binding is also coupled with shifts in the population of each conformation on the conformational energy landscape of proteins, which is strongly relevant to protein functions, such as enzyme catalysis, allostery, and signal transduction.10–12 In most cases, we observe the consequence of ligand binding only on the structure of the native form, for example, the transition from an apo-form to a ligand-bound form. However, ligand binding may alter the relative energies of multiple non-native conformations of a protein and reshape the conformational energy landscape of the protein.10 Interactions between ligands and non-native conformations have significant implications in protein folding. Proteins frequently form partially unfolded intermediates along their folding routes on the conformational energy landscapes.13 Alteration of the energy of the folding intermediates by interactions with ligands would affect the rate and the efficiency of folding.14 When multiple routes are available for folding, interactions with ligands may dictate which route is most favorable.15 Designing a ligand that can modulate folding of a protein through interactions with folding intermediates is a potential way to improve folding efficiency of a target protein. Ligands with this property could also be used as drugs to facilitate folding of proteins with disease-causing mutations. Therefore, the complete picture of the effect of ligand binding on the conformational energy landscape requires elucidation of the effect of ligand binding on these non-native conformations.
In this study, we investigate the effect of NADP+ on partial unfolding of Escherichia coli dihydrofolate reductase (DHFR; EC 1.5.1.3). We have recently demonstrated the presence of a high-energy partially unfolded form of DHFR that is transiently populated under native conditions.16 Using native-state proteolysis, we determined the free energy required for this partial unfolding (4.9 kcal/mol). Also, analyzing the effect of mutations, we determined the structure of the partially unfolded form. Our results suggested that two loops near the active site of the protein (F-G loop and Met20 loop; Fig. 1) are mostly unfolded, and the hydrophobic cluster covered by these loops is disrupted and exposed to solvent. Still, the adenosine-binding domain of DHFR remains mostly folded in the partially unfolded form. Here we examine the effect of NADP+ on the energetics of partial unfolding in DHFR using native-state proteolysis. For its catalysis, DHFR interacts with two substrates (NADPH and dihydrofolate) and two products (NADP+ and tetrahydrofolate). It is well known that DHFR experiences a series of conformational changes in its catalytic cycle according to the bound ligands.17,18 With an understanding of the structure of the partially unfolded form and the energetics of partial unfolding, we now probe the effect of ligand binding on a high-energy non-native conformation of DHFR. As the ligand, we chose NADP+, which is one of the products from the catalytic reaction. Moreover, due to the relatively weak binding affinity of NADP+, we can investigate partial unfolding of DHFR under conditions ranging from non-saturating to saturating concentrations of NADP+. As well as on wild-type DHFR, we also investigate the effect of NADP+ on the partial unfolding of I61A, I91A, and L110V DHFR to gain insight on the effect of NADP+ on the structure of the partially unfolded DHFR.
Figure 1.
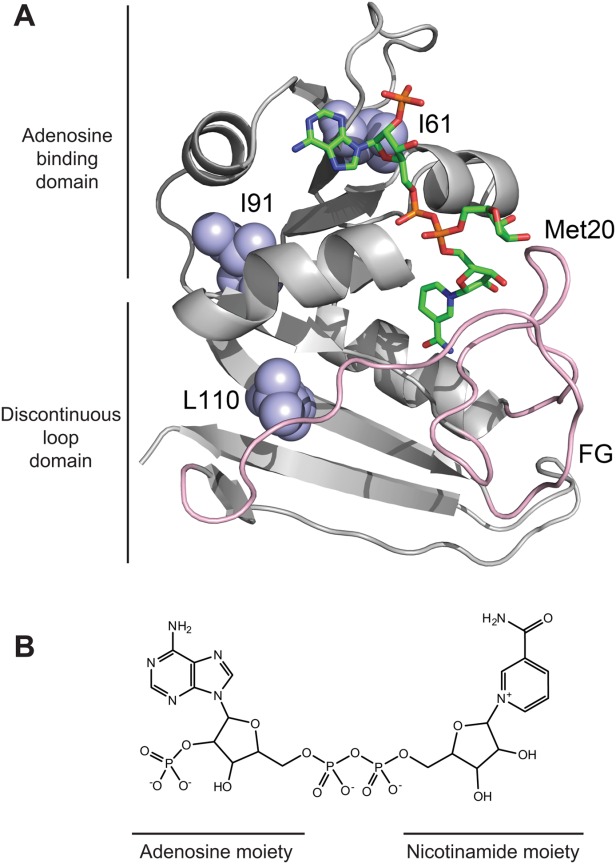
The structure of NADP+-bound DHFR and NADP+. (A) The backbone of DHFR (PDB: 1RX9) is represented as a ribbon with NADP+ as sticks. The sugar group of the nicotinamide moiety is observed in two different conformations in the crystallographic structure. The nicotinamide group is not observable in the conformation pointing to bulk water. Ile61, Ile91, and Leu110 are shown in light blue spheres. The F-G and Met20 loops are colored pink. The image was created with PyMOL. (B) The chemical structure of NADP+ is shown. The adenosine and nicotinamide moieties are indicated accordingly.
Results
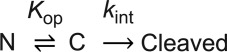
Kinetic model for native-state proteolysis with a ligand
We have previously investigated the partially unfolded form of DHFR by native-state proteolysis.16 From native-state proteolysis, we determine the equilibrium constant for partial unfolding by comparing the proteolysis rate of a protein by a non-specific protease with that of an unstructured peptide substrate by the same protease. The typical kinetic model for native-state proteolysis is:In Scheme 1, Kop is the equilibrium constant between the native form (N) and the cleavable form (C), and kint is the rate constant for the proteolysis of the cleavable form. When the proteolysis step is rate-limiting (EX2-like condition), the apparent rate constant for overall proteolysis (kp) is expressed as:
Scheme 1.
•••
| (1) |
By determining kp experimentally and approximating kint with the rate constant for proteolysis of a peptide substrate, one can calculate Kop by Eq. (1) and also the free energy for unfolding to the cleavable form (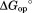 ) from Kop. By applying this approach to DHFR, we found that proteolysis of DHFR by thermolysin occurs through a partially unfolded form, which is similar to a known folding intermediate (IHF) of the protein.16
) from Kop. By applying this approach to DHFR, we found that proteolysis of DHFR by thermolysin occurs through a partially unfolded form, which is similar to a known folding intermediate (IHF) of the protein.16
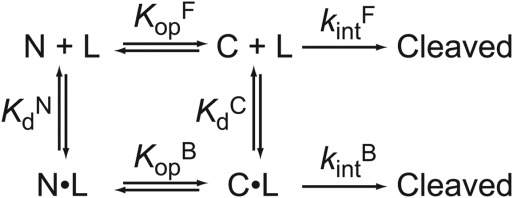
To determine the effect of ligand binding on the partial unfolding in DHFR, we modified the kinetic model. Assuming that both the native and partially unfolded form of DHFR can bind a ligand, we used the following kinetic model with four states:
The four states are the free native form (N), the free partially-unfolded form (C), the bound native form (N·L), and the bound partially-unfolded form (C·L). and
and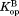 are the equilibrium constants for partial unfolding in the free form and the bound form, respectively.
are the equilibrium constants for partial unfolding in the free form and the bound form, respectively.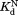 and
and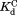 are the dissociation equilibrium constants for the native form and the partially unfolded form, respectively.
are the dissociation equilibrium constants for the native form and the partially unfolded form, respectively.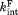 and
and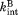 are the rate constant for the proteolysis of the free partially-unfolded form (C) and the bound partially-unfolded form (C·L), respectively. An analogous model has been used to assess ligand binding to a partially unfolded form by native-state hydrogen exchange.19
are the rate constant for the proteolysis of the free partially-unfolded form (C) and the bound partially-unfolded form (C·L), respectively. An analogous model has been used to assess ligand binding to a partially unfolded form by native-state hydrogen exchange.19
When the proteolysis step is rate-limiting in Scheme 2, the four states can be assumed to be in equilibrium. Also, we approximate both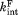 and
and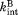 with kint determined with a peptide substrate. Because thermolysin is a non-specific protease and partial unfolding in DHFR is relatively large in scale, kint is not likely to change significantly upon binding to a ligand. Under this condition, the apparent rate constant for overall proteolysis (kp) is determined by the product of kint and the fraction of the protein in the cleavable forms (C and C·L). Therefore, the apparent rate constant for overall proteolysis (kp) is expressed as a function of [L]:
with kint determined with a peptide substrate. Because thermolysin is a non-specific protease and partial unfolding in DHFR is relatively large in scale, kint is not likely to change significantly upon binding to a ligand. Under this condition, the apparent rate constant for overall proteolysis (kp) is determined by the product of kint and the fraction of the protein in the cleavable forms (C and C·L). Therefore, the apparent rate constant for overall proteolysis (kp) is expressed as a function of [L]:
Scheme 2.
•••
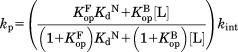 |
(2) |
Because KopF << 1 and KopB << 1, Eq. (2) is simplified to:
| (3) |
We define the apparent free energy required for partial unfolding at a given ligand concentration (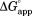 ) as
) as
| (4) |
By combining Eq. (3) and Eq. (4), we obtain
| (5) |
At a very low ligand concentration,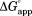 would converge to
would converge to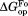 (= –RT ln
(= –RT ln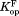 ), the free energy for partial unfolding in the free form. At a very high ligand concentration,
), the free energy for partial unfolding in the free form. At a very high ligand concentration,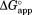 would converge at
would converge at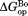 (= –RTln
(= –RTln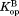 ), the free energy for partial unfolding in the bound form. By setting
), the free energy for partial unfolding in the bound form. By setting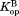 to 0, Eq. (5) is recast to explain the effect of ligand concentration on
to 0, Eq. (5) is recast to explain the effect of ligand concentration on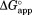 for a model in which proteolysis occurs only through the free partially-unfolded form (C):
for a model in which proteolysis occurs only through the free partially-unfolded form (C):
| (6) |
Different from Eq. (5), ΔG°app in this case increases infinitely as the ligand concentration increases.
Native-state proteolysis in the presence of NADP+
As the ligand for this study, we chose NADP+, one of the products of DHFR. During its catalytic cycle, DHFR does not exist in a binary complex with NADP+ but with NADPH. Still, we used NADP+ instead of NADPH because the binding affinity of NADP+ (Kd = 24 μM)20 is more suitable than that of NADPH (Kd = 0.33 μM)20 for native-state proteolysis. To know the free ligand concentration ([L]) accurately, we need a ligand concentration significantly greater than the protein concentration. In the case of NADPH, the minimum concentration of a ligand necessary for our assay (∼10 μM) is much greater than its Kd. In the case of NADP+, we can use ligand concentrations both below and above the Kd value, and therefore the use of Eq. ((6)) is valid.
In the presence of NADP+, we investigated partial unfolding of I61A, I91A, and L110V DHFR as well as wild-type DHFR. We chose the three DHFR variants to probe the structure of the potential NADP+-bound partially unfolded form. According to our previous study, Ile61 and Ile91 maintain a large portion of their native contacts in the partially unfolded form while Leu110 loses most of its native contacts in the partially unfolded form.16 Also, the three residues do not contact NADP+ directly in the native structure (Fig. 1). We determined the rate of proteolysis of DHFR by thermolysin at varying concentrations of NADP+ (0.010–2.0 mM) (Supporting Information Fig. S1). We calculated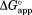 from kp for each NADP+ concentration using Eq. (4). When
from kp for each NADP+ concentration using Eq. (4). When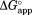 is plotted against NADP+ concentration,
is plotted against NADP+ concentration,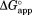 increases as NADP+ concentration increases (Fig. 2). This increase in
increases as NADP+ concentration increases (Fig. 2). This increase in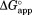 indicates that NADP+ suppresses partial unfolding of DHFR. As our kinetic model predicts,
indicates that NADP+ suppresses partial unfolding of DHFR. As our kinetic model predicts,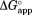 values approach a plateau at high concentrations of NADP+.
values approach a plateau at high concentrations of NADP+.
Figure 2.
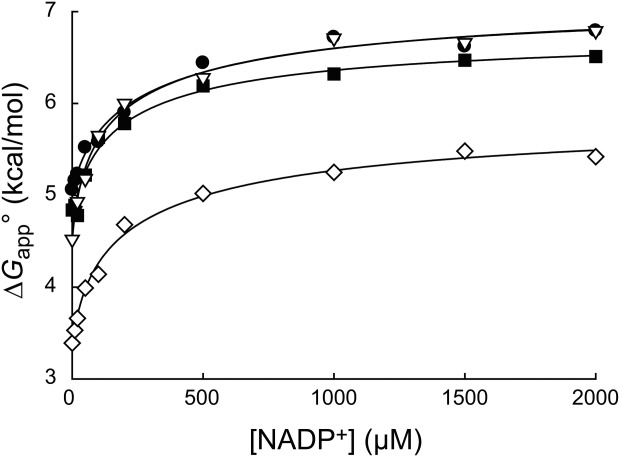
Effect of NADP+ binding on partial unfolding. The apparent free energy for partial unfolding (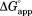 ) of wild-type (•), I61A (▽), I91A (▪), and L110V (⋄) DHFR is plotted against the concentration of NADP+. The curves were fit to Eq. ((6)). The standard errors of
) of wild-type (•), I61A (▽), I91A (▪), and L110V (⋄) DHFR is plotted against the concentration of NADP+. The curves were fit to Eq. ((6)). The standard errors of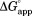 are smaller than the size of the symbols.
are smaller than the size of the symbols.
The curve for each variant was fit to Eq. ((6)) to determine the values of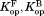 , and
, and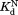 (Table I). The
(Table I). The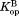 values for all four curves clearly have non-zero values (P < 0.001), which indicate that DHFR can access the partially unfolded form without releasing the ligand. In other words, NADP+ can bind to the partially unfolded form of DHFR. As observed in the trend in Figure 2, the
values for all four curves clearly have non-zero values (P < 0.001), which indicate that DHFR can access the partially unfolded form without releasing the ligand. In other words, NADP+ can bind to the partially unfolded form of DHFR. As observed in the trend in Figure 2, the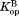 values are much smaller than the
values are much smaller than the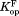 values. Binding of NADP+ suppresses the partial unfolding of DHFR.
values. Binding of NADP+ suppresses the partial unfolding of DHFR.
Table I.
Equilibrium Constants for Partial Unfolding and NADP+ Dissociation
| DHFR |
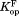 (×10−6)a (×10−6)a
|
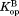 (×10−6)a (×10−6)a
|
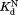 (µM)a (µM)a
|
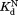 ITC (µM)b ITC (µM)b
|
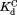 (µM)c (µM)c
|
|---|---|---|---|---|---|
| Wild type | 190 ± 20 | 5.5 ± 1.5 | 47 ± 11 | 34 ± 6 | 1600 ± 600 |
| I61A | 470 ± 50 | 5.9 ± 1.1 | 17 ± 3 | 24 ± 2 | 1400 ± 400 |
| I91A | 330 ± 50 | 11 ± 3 | 33 ± 9 | 60 ± 7 | 1000 ± 400 |
| L110V | 3400 ± 300 | 49 ± 11 | 26 ± 5 | 52 ± 4 | 1800 ± 600 |
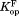 ,
,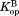 , and
, and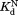 were determined by fitting to Eq. ((6)).
were determined by fitting to Eq. ((6)).
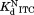 was determined by isothermal titration calorimetry.
was determined by isothermal titration calorimetry.
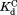 was calculated from the relationship of
was calculated from the relationship of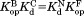 .
.
The standard errors of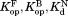 , and
, and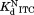 are from curve-fitting. The standard errors of
are from curve-fitting. The standard errors of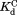 are from error propagation.
are from error propagation.
The similar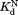 values of the DHFR variants to that of wild-type DHFR (Table I) show that the mutations do not affect binding to NADP+. To confirm the validity of our kinetic model, we determined
values of the DHFR variants to that of wild-type DHFR (Table I) show that the mutations do not affect binding to NADP+. To confirm the validity of our kinetic model, we determined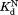 independently by isothermal titration calorimetry (Table I). For wild-type and I61A DHFR, the
independently by isothermal titration calorimetry (Table I). For wild-type and I61A DHFR, the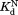 values determined by isothermal titration calorimetry agree well with those determined by proteolysis kinetics within the error. For I91A and L110V, the
values determined by isothermal titration calorimetry agree well with those determined by proteolysis kinetics within the error. For I91A and L110V, the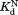 values determined by isothermal titration calorimetry are about 2–3 times higher than those determined by proteolysis kinetics. Still, the
values determined by isothermal titration calorimetry are about 2–3 times higher than those determined by proteolysis kinetics. Still, the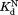 values within a similar range confirm that the kinetic model is valid.
values within a similar range confirm that the kinetic model is valid.
The thermodynamic cycle in Scheme 1 also allows us to determine the dissociation constant for the NADP+-bound partially unfolded form (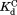 ) from the three other equilibrium constants using the relationship of
) from the three other equilibrium constants using the relationship of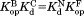 . For the four DHFRs,
. For the four DHFRs,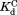 ranges from 0.8 to 1.7 mM (Table I). The similar
ranges from 0.8 to 1.7 mM (Table I). The similar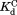 values for wild-type DHFR and the variants indicate that the mutations do not interfere with the binding of NADP+ to the partially unfolded form. The
values for wild-type DHFR and the variants indicate that the mutations do not interfere with the binding of NADP+ to the partially unfolded form. The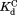 values much greater than
values much greater than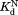 also show that partial unfolding diminishes the binding affinity to NADP+.
also show that partial unfolding diminishes the binding affinity to NADP+.
We calculated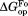 and
and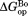 from
from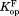 and
and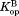 , respectively (Table II). The
, respectively (Table II). The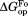 values are close to 5 kcal/mol for wild-type, I61A, and I91A DHFR and 3.4 kcal/mol for L110V DHFR.
values are close to 5 kcal/mol for wild-type, I61A, and I91A DHFR and 3.4 kcal/mol for L110V DHFR.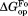 matches very well to the
matches very well to the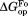 value that we previously determined by native-state proteolysis of the free form of each variant (Table II).16 The
value that we previously determined by native-state proteolysis of the free form of each variant (Table II).16 The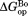 values are close to 7 kcal/mol for wild-type, I61A, and I91A DHFR and 6.0 kcal/mol for L110V DHFR. The effects of mutations on
values are close to 7 kcal/mol for wild-type, I61A, and I91A DHFR and 6.0 kcal/mol for L110V DHFR. The effects of mutations on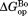 (
(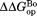 ) are quite similar to those on
) are quite similar to those on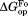 (
(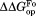 ). At least near the mutated residues, the structure of the bound partially-unfolded form is similar to that of the free partially-unfolded form. The difference between
). At least near the mutated residues, the structure of the bound partially-unfolded form is similar to that of the free partially-unfolded form. The difference between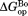 and
and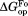 (
(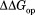 ) shows the degree of the protection against partial unfolding by NADP+. For wild-type DHFR, partial unfolding in the NADP+-bound form requires 2.2 kcal/mol more of free energy than partial unfolding in the free form.
) shows the degree of the protection against partial unfolding by NADP+. For wild-type DHFR, partial unfolding in the NADP+-bound form requires 2.2 kcal/mol more of free energy than partial unfolding in the free form.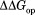 is similar for the three mutant DHFRs as well.
is similar for the three mutant DHFRs as well.
Table II.
Free Energies for Partial Unfolding in the Free and Bound Form of DHFR
| DHFR |
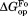 (kcal/mol)a (kcal/mol)a
|
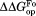 (kcal/mol)b (kcal/mol)b
|
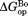 (kcal/mol)a (kcal/mol)a
|
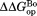 (kcal/mol)c (kcal/mol)c
|
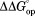 (kcal/mol)d (kcal/mol)d
|
|---|---|---|---|---|---|
| Wild Type | 5.06 ± 0.06 | – | 7.1 ± 0.2 | – | 2.0 ± 0.2 |
| I61A | 4.52 ± 0.06 | −0.54 ± 0.08 | 7.1 ± 0.1 | 0.0 ± 0.2 | 2.6 ± 0.1 |
| I91A | 4.73 ± 0.09 | −0.33 ± 0.11 | 6.7 ± 0.2 | −0.4 ± 0.3 | 2.0 ± 0.2 |
| L110V | 3.35 ± 0.05 | −1.71 ± 0.08 | 5.9 ± 0.1 | −1.2 ± 0.2 | 2.6 ± 0.1 |
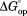 values were calculated with the equilibrium constants in Table1.
values were calculated with the equilibrium constants in Table1.
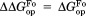 (mutant) −
(mutant) − 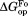 (wild type).
(wild type).
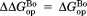 (mutant) −
(mutant) − 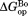 (wild type).
(wild type).
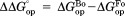 .
.
The standard errors are from error propagation.
To demonstrate visually the effect of NADP+ binding on partial unfolding, we present our results in energy diagrams (Fig. 3). The diagrams show the relative free energy of the native and the partially unfolded forms in the presence of 10 mM NADP+ as well as the relative free energies of each form in free DHFR. We only measured proteolysis of DHFR in the presence of up to 2.0 mM NADP+. However, to show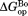 in the diagram, we calculated the free energies at 10 mM NADP+ with which
in the diagram, we calculated the free energies at 10 mM NADP+ with which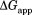 would converge at
would converge at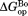 [Eq. ((6)) and Fig. 2]. As shown in the diagram, the partially unfolded form is stabilized by NADP+, though not as significantly as the native form is. Additional increases in the ligand concentration will not increase the energy difference between the native and the partially unfolded forms, because both forms are saturated with NADP+, and the energy of both forms decrease to the same extent with the increase in the concentration of NADP+. For comparison, the energy diagram at 2.0 mM NADP+, with which the partially unfolded forms are not fully saturated, is also shown in Supporting Information Figure S2.
[Eq. ((6)) and Fig. 2]. As shown in the diagram, the partially unfolded form is stabilized by NADP+, though not as significantly as the native form is. Additional increases in the ligand concentration will not increase the energy difference between the native and the partially unfolded forms, because both forms are saturated with NADP+, and the energy of both forms decrease to the same extent with the increase in the concentration of NADP+. For comparison, the energy diagram at 2.0 mM NADP+, with which the partially unfolded forms are not fully saturated, is also shown in Supporting Information Figure S2.
Figure 3.
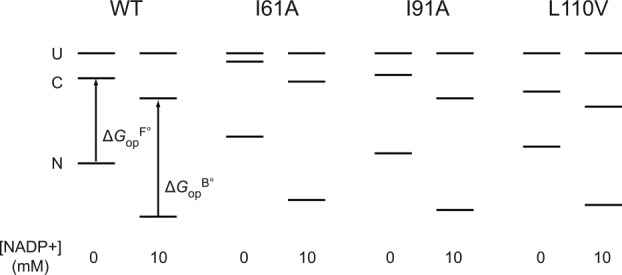
Consequences of NADP+ binding on free energy. The diagram is scaled according to the relative free energy of the native (N), partially unfolded (C), and globally unfolded (U) forms in the presence and absence of 10 mM NADP+. The global stabilities of wild-type DHFR and the variants are from our previous study.16 We estimated the effect of 10 mM NADP+ on the stability of the native form of DHFR with the NADP+ concentration and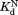 using the relationship of
using the relationship of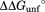 = RTln(1+[L]/Kd),41 where R is the gas constant and T is the temperature in Kelvin. We determined the relative free energy of the free and bound partially unfolded forms by adding
= RTln(1+[L]/Kd),41 where R is the gas constant and T is the temperature in Kelvin. We determined the relative free energy of the free and bound partially unfolded forms by adding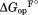 and
and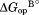 to the relative free energy of the free and bound native forms, respectively. Here we assume that proteolysis occurs mostly through bound partially-unfolded forms at 10 mM NADP+.
to the relative free energy of the free and bound native forms, respectively. Here we assume that proteolysis occurs mostly through bound partially-unfolded forms at 10 mM NADP+.
Discussion
We have studied partial unfolding of DHFR by native-state proteolysis in the presence of NADP+ to understand the effects of this ligand on the protein's conformational energy landscape, specifically interaction with a partially unfolded form. NADP+ slows proteolysis but does not slow it infinitely. As the kinetic model in Scheme 2 predicts, proteolysis occurs through the NADP+-bound partially-unfolded form at high concentrations of the ligand. Proteolysis through the NADP+-bound form is slower than through the free form because NADP+ binds to the native form more tightly than to the cleavable form (Tables I and II).
Structure of NADP+-bound partially-unfolded form
Based on our knowledge of the cleavable form, we make some inferences about the structural origin of its lower affinity for NADP+. In the previous investigation, we found that the cluster of Leu8, Leu110, and Leu112 loses most of its native contacts in the partially unfolded form.16 We infer that the F-G and Met20 loops that these residues contact (Fig. 1) are unfolded in the partially unfolded form. These loops close over the nicotinamide moiety of NADP+ in the native structure (Fig. 1). The lower affinity of NADP+ to the partially unfolded form likely results from unfolding of the binding site for the nicotinamide moiety. Still, NADP+ binds to the partially unfolded form through the interaction between the adenosine moiety of NADP+ and the adenosine-binding domain, which remains folded in the partially unfolded form.
Crystallographic structures of NADPH- or NADP+-bound DHFR also reveal that the binding of the adenosine and nicotinamide moieties can be uncoupled.21 The crystallographic structure of NADPH-bound DHFR showed only 75% occupancy of the nicotinamide moiety in the binding pocket.21 In the case of NADP+, occupancy of the nicotinamide moiety is even lower so that the electron density of the moiety is almost undetectable. The structure shown in Figure 1 also shows two different conformations of the nicotinamide moiety. In one conformation, the nicotinamide moiety is properly bound to the binding pocket. In the other conformation, however, the nicotinamide moiety points to the bulk water, and only the ribose group is visible. Figure 4 shows schematic representations of the two different conformations of NADP+. These structural observations suggest that the adenosine moiety contributes much more greatly to the binding affinity of NADP+ to DHFR than the nicotinamide moiety. Our finding that NADP+ still binds to the partially unfolded form is consistent with the structural studies. The unfolding of the FG and Met20 loops in the partially unfolded form diminishes, but does not abolish, the binding of NADP+. The association of the adenosine moiety of NADP+ with the native-like adenosine-binding domain and the plausible interaction with the diphosphate group seem primarily responsible for the binding of NADP+ to the partially unfolded form [ Fig. 4(C,D)].
Figure 4.
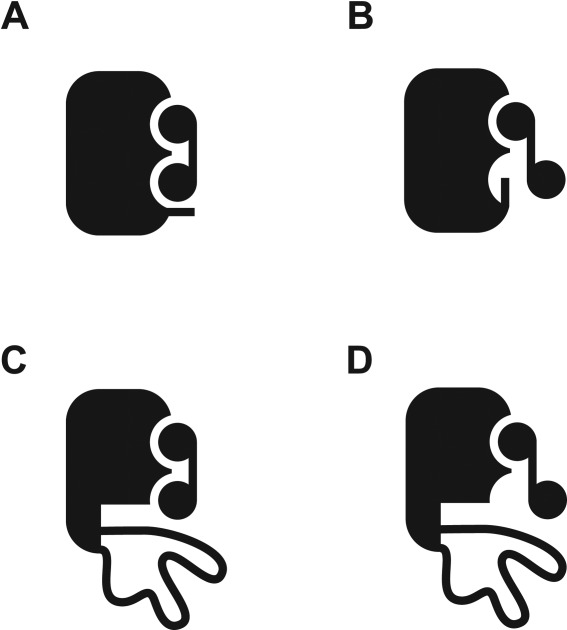
Binding of NADP+ to the native and the partially unfolded DHFR. The modes of interactions between NADP+ and DHFR in the native and the partially unfolded forms are shown in schematic presentations. (A) NADP+ bound to the native DHFR in the closed conformation. (B) NADP+ bound to the native DHFR in the occluded conformation. The nicotinamide moiety is not in the binding site. (C) NADP+ bound to the partially unfolded DHFR with the nicotinamide moiety in the conformation analogous to that in panel A. (D) NADP+ bound to the partially unfolded DHFR with the nicotinamide moiety in the conformation analogous to that in panel B.
The uncoupling of binding of the adenosine and nicotinamide moieties of NADP+ is relevant to the catalytic mechanism of DHFR.21,22 DHFR utilizes NADPH to reduce 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate. After NADPH is oxidized, and prior to release of NADP+ from DHFR, the active site is occluded by the Met20 loop and the nicotinamide moiety is no longer bound; this transient form associates with only the adenosine moiety [ Fig. 4(B)].22 After release of NADP+, the adenosine moiety of NADPH binds DHFR, while the nicotinamide site still remains occluded. It is not until 5,6,7,8-tetrahydrofolate is released that the Met20 loop opens, allowing the nicotinamide of NADPH to bind the active site.
A computational study showed that the NADP+ binding site is energetically independent of the region that we found to be unfolded in the partially unfolded form. Pan et al. investigated the connection of NADP+ binding and folding of DHFR with the COREX algorithm.23 This model assesses an ensemble of conformational states in which a unique set of residues are unfolded, and the population of each state is calculated according to the energetic penalty of unfolding. Using this model, they determined the probability of each residue to be folded if the entire NADP+ binding site is folded. The NADP+ binding site has few correlations with residues in the β strands F (containing L110), G, and H. The F-G loop even shows negative correlation with the NADP+ binding site; the loop is less likely to be unfolded when the NADP+ binding site is unfolded. The absence of positive correlation between the NADP+ binding site and the region unfolded in the cleavable form is consistent with our observation that DHFR can still unfold partially with bound NADP+.
Ligand binding by partially unfolded forms
Ligand binding by partially unfolded forms has also been observed in other proteins. The effects of partial unfolding on ligand binding span a broad spectrum. A protein may maintain its full binding affinity or lose its affinity completely to a ligand upon partial unfolding. Partial unfolding of E. coli maltose binding protein is not affected by the presence of its ligand.24 Proteolysis of the protein by thermolysin occurs through unfolding of two C-terminal helices, and the rate of proteolysis is identical whether maltose is present or not. When hydrogen exchange of cytochrome c was monitored in the presence of antibodies, protection factors of a group of residues in cytochrome c are shown to increase by the binding of E3 and C3 antibodies.25 Because the change in protection factors is not even across the structure of cytochrome c, the antibodies also likely stabilize a partially unfolded form of cytochrome c. Free energies for opening of free and bound SH3 domain in the presence of a peptide ligand were investigated using the same model that we use in this study.19 Some residues of SH3 do exchange through a bound form, but these residues do not map to a common element of the structure. This pattern of exchange in SH3 is thought to result from the promotion of local fluctuations relative to global unfolding upon binding, as opposed to binding of a partially unfolded form. The complex of S protein with S peptide (ribonuclease S) did not show any evidence of unfolding in the bound form.26 It was found that exchange occurs only through the dissociated complex, the extended S peptide and a partially unfolded S protein. Whether a protein can unfold partially without releasing its ligand must be dependent on the structural cooperativity between the unfolding site and the ligand binding site. In a highly cooperative system, partial unfolding would be strongly coupled with release of the ligand, and the bound partially unfolded form would be extremely unfavorable and not detectable. In a less cooperative system, a protein would unfold partially with a ligand bound, though with reduced binding affinity. The uncoupling in the binding of the adenosine moiety and the nicotinamide moiety of NADP+ to DHFR results in somewhat low cooperativity between the partial unfolding and binding of NADP+ in DHFR, which allows us to detect the bound partially unfolded form under native conditions.
Our findings support the growing evidence that small molecules can interact with non-native forms of proteins during folding. Cofactors and ligands have been found to interact with proteins before translation is complete,27 and metal cofactors can interact with intermediate forms and even unfolded forms.28 Also, some inhibitors stabilize transition states of protein folding,29,30 and an enzyme was shown to acquire partial catalytic activity in a folding intermediate form.31 We previously discovered that ATP binds to a folding intermediate of glyceraldehyde-3-phosphate dehydrogenase but does not bind to the native form.14 Binding with non-native forms may facilitate folding by protecting folding intermediates against misfolding or degradation or by enhancing folding rates.
Frieden's group has conducted refolding experiments of DHFR in the presence of 100 µM NADP+.32 However, they did not observe any influence of NADP+ on the amplitude of the fluorescence during the formation of I1 or the following intermediates I2 or I3, and concluded that NADP+ only binds the native form. We measured a Kd value of 1.4 mM for binding of NADP+ to the partially unfolded form, which we believe is equivalent to I1. Our result is consistent with what Frieden's group observed because only 7% of the partially unfolded form or I1 would be bound to NADP+ in the presence of 100 µM NADP+.
The ligand binding that we observed may be relevant to DHFR in vivo. The cellular concentrations of NADP+ and NADPH in E. coli are estimated to be 2 µM and 300 µM, respectively.33 Assuming similar binding of NADPH to the partially unfolded form as NADP+ (Kd ∼ 1.4 mM), we estimate ∼20% of the kinetic intermediate bound to NADPH in E. coli. The binding affinity of NADPH to the native DHFR is ∼70-fold greater than that of NADP+.20 Though the binding site for the nicotinamide moiety is largely unfolded, it is still possible that NADPH also binds to the partially unfolded form better than NADP+ and associates with the folding intermediate to a greater extent than we estimate here.
Here we show that NADP+ not only binds to the native form but also a partially unfolded form of DHFR. Although binding to the partially unfolded form is weak, NADP+ or NADPH may provide DHFR with a mild stabilization or protection during folding. Our result contributes to a body of evidence that demonstrates a ligand can affect the entire conformational energy landscape of a protein, not just the stability of the native form.
Materials and Methods
Materials
We expressed DHFR in E. coli BL21(DE3)pLysS cells grown to OD600 of 0.6 and induced with 500 µM isopropyl-β-D-thiogalactopyranoside (IPTG). We purified DHFR by DEAE Sepharose Fast Flow (GE Healthcare Life Sciences; Piscataway, NJ) anion exchange chromatography and Superdex 200 (GE Healthcare Life Sciences; Piscataway, NJ) size exclusion chromatography. The production of I61A, I91A, and L110V DHFR has been reported previously.16 All the DHFR proteins we used in this study have C85A/C152S mutations.34 For simplicity, we refer to C85A/C152S DHFR as wild-type DHFR. We dissolved lyophilized thermolysin (Type X; Sigma-Aldrich; St. Louis, MO) in 2.5M NaCl, 10 mM CaCl2.35 Concentrations of all proteins were determined by absorbance at 280 nm using extinction coefficients determined according to their amino acid composition.36 Less than 10% of purified DHFR was bound to the cofactor, NADPH, as determined by NADPH titration. NADP+ monosodium salt (Sigma-Aldrich; St. Louis, MO) was dissolved in 20 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, and the pH was adjusted to 8.0 with NaOH. The concentration of NADP+ was determined by absorbance at 260 nm according to an extinction coefficient of 18,000M−1 cm−1.37
Proteolysis
We initiated proteolysis of DHFR by adding thermolysin to DHFR in buffer to achieve final conditions of 0.10 mg/mL DHFR, 20 mM Tris-HCl buffer (pH 8.0), 100 mM NaCl, 10 mM CaCl2, 80 µg/mL Thermolysin, and varying concentrations of NADP+ (0.010–2.0 mM). At the desired time, 15-μL aliquots were quenched with 5 μL of 50 mM EDTA. Quenched samples were separated by SDS-PAGE. Gels were stained with SYPRO Red Protein Gel Stain (Life Technologies; Grand Island, NY), and fluorescent images were taken with a Typhoon scanner (GE Healthcare Life Sciences; Piscataway, NJ). Intact protein gel bands were quantified from images with ImageJ software. The apparent rate constants of proteolysis (kobs) were determined by fitting the change in band intensity over time to a first-order rate equation in OriginPro 8.5.1 (OriginLab; Northampton, MA). We have recently reported that, due to product inhibition, proteolysis of intact proteins may deviate from first-order kinetics, and kobs may be underestimated.38 In DHFR, however, we have shown that ΔGop° is still well consistent with ΔGunf°,16 which confirms that ΔGop° is determined reliably by native-state proteolysis.
We calculated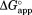 from kobs and kint according to Eq. (4). We estimated kint using the kcat/Km value for cleavage of a generic thermolysin substrate, 2-aminobenzoyl-Ala-Gly-Leu-Ala-4-nitrobenzylamide.39 We confirmed that NADP+ does not inhibit thermolysin up to 2.0 mM NADP+. We determined
from kobs and kint according to Eq. (4). We estimated kint using the kcat/Km value for cleavage of a generic thermolysin substrate, 2-aminobenzoyl-Ala-Gly-Leu-Ala-4-nitrobenzylamide.39 We confirmed that NADP+ does not inhibit thermolysin up to 2.0 mM NADP+. We determined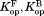 , and
, and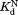 by fitting the plot of
by fitting the plot of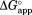 versus [NADP+] in Figure 2 to Eq. ((6)).
versus [NADP+] in Figure 2 to Eq. ((6)).
In our analysis with Eq. ((6)), we used the total NADP+ concentration instead of free NADP+ concentration. When the NADP+ concentration is as low as 10 μM, the free NADP+ concentration changes up to ∼10% from the release of bound NADP+ during proteolysis. This change in the free NADP+ concentration is negligible when the NADP+ concentration is greater than 100 μM. For simplicity, we did not count in this change in the NADP+ concentration in our analysis of proteolysis kinetics. When we used the initial free NADP+ concentration instead of the total NADP+ concentration in Eq. ((6)), the change in the fitting parameters was negligible.
Isothermal titration calorimetry
Isothermal titration calorimetry was performed using a MicroCal iTC200 (GE Healthcare Life Sciences; Piscataway, NJ) calorimeter. We prepared NADP+ and DHFRs in 20 mM Tris–HCl buffer (pH 8.0) containing 100 mM NaCl, and 10 mM CaCl2. 1.5 mM NADP+ was titrated in 21 injections into 58 µM (L110V) or 76 µM (wild-type, I61A, and I91A) DHFR with 3-min intervals. Thermograms were automatically integrated with NITPIC40 and fit with SEDPHAT, assuming a single NADP+ binding site per DHFR molecule.
Glossary
- DHFR
C85A/C152S E. coli dihydrofolate reductase
- EDTA
ethylenediaminetetraacetic acid
- IPTG
isopropyl-β-D-thiogalactopyranoside
- NADP+
β-nicotinamide adenine dinucleotide phosphate
- PAGE
polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Hilser VJ, Garcia-Moreno EB, Oas TG, Kapp G, Whitten ST. A statistical thermodynamic model of the protein ensemble. Chem Rev. 2006;106:1545–1558. doi: 10.1021/cr040423+. [DOI] [PubMed] [Google Scholar]
- 2.Chamberlain AK, Marqusee S. Touring the landscapes: partially folded proteins examined by hydrogen exchange. Structure. 1997;5:859–863. doi: 10.1016/s0969-2126(97)00240-2. [DOI] [PubMed] [Google Scholar]
- 3.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNase H. Nature Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 5.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 6.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nature Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 7.Bai Y. Protein folding pathways studied by pulsed- and native-state hydrogen exchange. Chem Rev. 2006;106:1757–1768. doi: 10.1021/cr040432i. [DOI] [PubMed] [Google Scholar]
- 8.Englander SW. Protein folding intermediates and pathways studied by hydrogen exchange. Annu Rev Biophys Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 9.Dill KA, Shortle D. Denatured states of proteins. Annu Rev Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 10.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 11.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nature Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim PS, Baldwin RL. Intermediates in the folding reactions of small proteins. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 14.Liu PF, Park C. Selective stabilization of a partially unfolded protein by a metabolite. J Mol Biol. 2012;422:403–413. doi: 10.1016/j.jmb.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 15.Krantz BA, Sosnick TR. Engineered metal binding sites map the heterogeneous folding landscape of a coiled coil. Nature Struct Biol. 2001;8:1042–1047. doi: 10.1038/nsb723. [DOI] [PubMed] [Google Scholar]
- 16.Kasper JR, Liu PF, Park C. Structure of a partially unfolded form of Escherichia coli dihydrofolate reductase provides insight into its folding pathway. Prot Sci. 2014 doi: 10.1002/pro.2555. doi: 10.1002/pro.2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bystroff C, Kraut J. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry. 1991;30:2227–2239. doi: 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- 18.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 19.Wildes D, Marqusee S. Hydrogen exchange and ligand binding: ligand-dependent and ligand-independent protection in the Src SH3 domain. Prot Sci. 2005;14:81–88. doi: 10.1110/ps.04990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 21.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 22.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 23.Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang Y, Park C. Mapping transient partial unfolding by protein engineering and native-state proteolysis. J Mol Biol. 2009;393:543–556. doi: 10.1016/j.jmb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Mayne L, Paterson Y, Cerasoli D, Englander S. Effect of antibody binding on protein motions studied by hydrogen-exchange labeling and two-dimensional NMR. Biochemistry. 1992;31:10678–10685. doi: 10.1021/bi00159a006. [DOI] [PubMed] [Google Scholar]
- 26.Chakshusmathi G, Ratnaparkhi G, Madhu P, Varadarajan R. Native-state hydrogen-exchange studies of a fragment complex can provide structural information about the isolated fragments. Proc Natl Acad Sci USA. 1999;96:7899–7904. doi: 10.1073/pnas.96.14.7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fedorov AN, Baldwin TO. Cotranslational protein folding. J Biol Chem. 1997;272:32715–32718. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- 28.Wittung-Stafshede P. Role of cofactors in protein folding. Acc Chem Res. 2002;35:201–208. doi: 10.1021/ar010106e. [DOI] [PubMed] [Google Scholar]
- 29.Sancho J, Meiering EM, Fersht AR. Mapping transition states of protein unfolding by protein engineering of ligand-binding sites. J Mol Biol. 1991;221:1007–1014. doi: 10.1016/0022-2836(91)80188-z. [DOI] [PubMed] [Google Scholar]
- 30.Adler E, Wolfenden R. Inhibitor binding in the transition state for unfolding of adenosine deaminase. Bioorg Chem. 1994;22:216–225. [Google Scholar]
- 31.Bemporad F, Gsponer J, Hopearuoho HI, Plakoutsi G, Stati G, Stefani M, Taddei N, Vendruscolo M, Chiti F. Biological function in a non-native partially folded state of a protein. EMBO J. 2008;27:1525–1535. doi: 10.1038/emboj.2008.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frieden C. Refolding of Escherichia coli dihydrofolate reductase: sequential formation of substrate binding sites. Proc Natl Acad Sci USA. 1990;87:4413–4416. doi: 10.1073/pnas.87.12.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nature Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwakura M, Jones BE, Luo J, Matthews CR. A strategy for testing the suitability of cysteine replacements in dihydrofolate reductase from Escherichia coli. J Biochem. 1995;117:480–488. doi: 10.1093/oxfordjournals.jbchem.a124733. [DOI] [PubMed] [Google Scholar]
- 35.Inouye K, Kuzuya K, Tonomura B. Sodium chloride enhances markedly the thermal stability of thermolysin as well as its catalytic activity. Biochim Biophys Acta. 1998;1388:209–214. doi: 10.1016/s0167-4838(98)00189-7. [DOI] [PubMed] [Google Scholar]
- 36.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption-coefficient of a protein. Prot Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merck & Co. The Merck index. NJ: Merck: Whitehouse Station; 2006. [Google Scholar]
- 38.Kasper JR, Andrews EC, Park C. Product inhibition in native-state proteolysis. PLoS ONE. 2014;9:e111416. doi: 10.1371/journal.pone.0111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morihara K, Tsuzuki H. Thermolysin: kinetic study with oligopeptides. Eur J Biochem. 1970;15:374–380. doi: 10.1111/j.1432-1033.1970.tb01018.x. [DOI] [PubMed] [Google Scholar]
- 40.Keller S, Vargas C, Zhao H, Piszczek G, Brautigam C, Schuck P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal Chem. 2012;84:5066–5073. doi: 10.1021/ac3007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pace CN, McGrath T. Substrate stabilization of lysozyme to thermal and guanidine hydrochloride denaturation. J Biol Chem. 1980;255:3862–3865. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information