

Abstract
PII protein is one of the largest families of signal transduction proteins in archaea, bacteria, and plants, controlling key processes of nitrogen assimilation. An intriguing characteristic for many PII proteins is that the three ligand binding sites exhibit anticooperative allosteric regulation. In this work, PII protein from Synechococcus elongatus, a model for cyanobacteria and plant PII proteins, is utilized to reveal the anticooperative mechanism upon binding of 2-oxoglutarate (2-OG). To this end, a method is proposed to define the binding pocket size by identifying residues that contribute greatly to the binding of 2-OG. It is found that the anticooperativity is realized through population shift of the binding pocket size in an asymmetric manner. Furthermore, a new algorithm based on the dynamic correlation analysis is developed and utilized to discover residues that mediate the anticooperative process with high probability. It is surprising to find that the T-loop, which is believed to be responsible for mediating the binding of PII with its target proteins, also takes part in the intersubunit signal transduction process. Experimental results of PII variants further confirmed the influence of T-loop on the anticooperative regulation, especially on binding of the third 2-OG. These discoveries extend our understanding of the PII T-loop from being essential in versatile binding of target protein to signal-mediating in the anticooperative allosteric regulation. Proteins 2014; 82:1048–1059.
Keywords: PII protein, allosteric regulation, signal transduction, population shift, dynamic correlation analysis
INTRODUCTION
The PII protein, as one of the most widely distributed families of signal transduction proteins, has key functions in terms of regulation of central metabolic processes by sensing signals from the carbon, nitrogen and energy status within the cells.1,2 It is able to regulate the activity of not only key metabolic and regulatory enzymes, but also transcription factors and even transport proteins.3–5 The control of PII protein on nitrogen-storage metabolism is due to the binding of PII protein toward N-acetyl-l-glutamate kinase (NAGK) in cyanobacteria and plants which controls the biosynthesis of arginine.6–8 The binding of PII is a response to the cellular concentration of central metabolites ATP, ADP, and 2-oxoglutarate (2-OG), which reflect the cellular energy, carbon and nitrogen abundance.2,5,9 These metabolites bind to PII in an interdependent manner, thereby transmitting metabolic information into structural states.2,10 Another intriguing characteristic is that the three ATP binding sites and the three 2-OG binding sites each exhibit negative cooperativity, which allows PII to sense a wide range of metabolite concentrations.10–13
As revealed from numerous crystal structures, PII proteins show a highly conserved three-dimensional structure. It is composed of homotrimers with each subunit of 12 to 13 kDa and two ferredoxin-like fold-containing cores (βαβ-βαβ) (Fig. 1).2,14 There is a long and flexible T-loop extending outward from each subunit and it is believed that the flexibility of these T-loops is essential to mediate the versatile functions of PII protein.2 Furthermore, each subunit comprises two small loops (B and C loop) in the intersubunit clefts, which take part in the binding of ATP or ADP.15–17 In the presence of Mg-ATP, up to three 2-OG molecules can bind per trimer with the exception that Arabidopsis thaliana PII, which binds 2-OG also in the presence of ADP.18,19 From the crystal structure of the Synechococcus elongatus PII protein in complex with ATP, Mg2+, and 2-OG, the site of 2-OG was located in the vicinity between the subunit clefts and the base of the T loop.11 More importantly, besides PII trimers with all the three sites fully occupied, structures with one or two 2-OG molecules bound per PII trimer were also obtained, which make it possible to reveal the basis for the anticooperative allosteric regulation upon binding of 2-OG.
Figure 1.
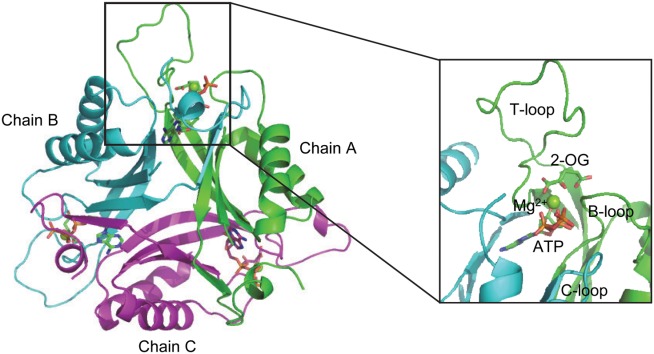
Overall structure of PII protein with ligand in the binding pocket. Chain A, B and C are shown in cartoon and colored in green, cyan and purple, respectively. ATP and 2-OG are shown in sticks; Mg2+ in sphere.
In this work, the PII protein from S. elongatus is utilized to reveal the anticooperative allosteric mechanism from the point of view of population shift. Population shift or re-distribution of protein conformational states has been proved to be a powerful concept for rationalizing binding mechanisms and allosteric regulations.20–23 It emphasizes that the process of ligand binding merely shifts the population of conformational states in a dynamic ensemble of the protein. Recent experiments also show that conformational states in a pre-existing equilibrium can influence protein function.24–26 To this end, we first identified residues that contribute greatly to the binding of 2-OG and based on this, we then proposed a method to define the binding pocket size. The anticooperative mechanism can be well revealed by considering the population shift of the binding pocket size. Furthermore, a new algorithm is developed based on dynamic correlation analysis and used to identify residues that mediate the allosteric regulation upon the binding of 2-OG with high probability. Predictions concerning “unexpected” roles of residues on the PII T-loop for signal mediation in the anticooperative process are experimentally verified.
MATERIALS AND METHODS
Structures and systems preparation
Crystal structures of PII protein with different number of bound 2-OG (PDB code: 2XZW) were employed. Residues missed in the original PDB file were added using Modeller 9v227 and the enumeration algorithm28 was used to increase the accuracy of the calculated structure. Three PII protein structures with different states of ligand binding were constructed: PII protein with 2-OG only in the binding pocket of chain A (PIIOG1); PII protein with 2-OG in both chain A and B (PIIOG2); PII protein with 2-OG in both chain A, B and C (PIIOG3). For each case, ATP and Mg2+ exist in each of the subunits.
Each structure was first neutralized by adding sodium counter ions randomly with the xLeap module of Amber 10.029 and then solvated in a rectangular box of TIP3P water molecules30 with a solute-wall distance of 12 Å. The solvated systems were energy-minimized prior to the molecular dynamics simulations. Each system was minimized by six consecutive rounds with each round of 1500 steps. Harmonic constraints were applied to all non hydrogen atoms with the strength of 500, 400, 300, 200, 100, and 0 kcal mol−2 at each round. After that, the system was slowly heated from 0 K to 300 K in 120 ps. Then the systems were conducted in a NVT ensemble for 50 ps followed by a 100 ps simulation in a NPT ensemble.
Molecular dynamics simulations
The molecular dynamics simulations were performed with the Sander module Amber 10.0 using the force field developed by Cornell et al.31 with a periodic boundary condition in the NPT ensemble at 300 K. Langevin dynamics was used with the collision frequency of 1.0 ps−1 and constant pressure of 1 atm. The Shake algorithm32 was applied to fix all covalent bonds. The Particle Mesh Ewald (PME) method33 was used to treat long-range electrostatic interactions. A residue-based cutoff of 10 Å was applied to the noncovalent interactions. No constraint was applied during molecular dynamics simulations which lasted 1.0 ns for each system to get an equilibrium state. A time step of 2.0 fs was used and the coordinates of the simulated complexes were saved every 1.0 ps.
Binding free energy calculations
The MM-GBSA approach,34 which is implemented in the Amber program, was applied to compute the binding free energy (ΔGbinding) of ligand. The ΔGbinding was determined according to the following equations:
| 1 |
| 2 |
| 3 |
where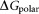 is the free energy for the polar interaction;
is the free energy for the polar interaction;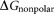 is for the nonpolar interaction;
is for the nonpolar interaction;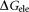 is the electrostatic interaction free energy;
is the electrostatic interaction free energy;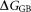 is the electrostatic solvation free energy;
is the electrostatic solvation free energy;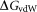 is the van der Waals interaction energy,
is the van der Waals interaction energy,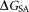 is the nonpolar solvation free energy. Default parameters were used to calculate the overall binding free energy and its decomposition at residue level.
is the nonpolar solvation free energy. Default parameters were used to calculate the overall binding free energy and its decomposition at residue level.
Hydrogen-bond interactions analysis
Since the molecular dynamics simulations include hydrogen atoms, it is possible to make an analysis of hydrogen-bond properties. The hydrogen-bond interactions, which were performed with the Ptraj module of Amber 10.0, are assumed to be present if the participating heavy atoms are 3.5 Å apart and the angle formed between the heavy atoms and donated hydrogen is larger than 120°.
Dynamic correlation analysis
Dynamic correlation analysis was employed to identify residues that mediate the signal transduction during the anticooperative process. This approach is based on the correlation coefficient between each pair of residues obtained from the trajectory of molecular dynamics simulations. The correlation coefficients are calculated according to the following equation:
| 4 |
where C(Rs,Rt) is the correlation coefficient between residues s and t. Rs and Rt are the coordinates of residues s and t; <Rs>N and <Rt>N are the average coordinates of residues s and t calculated based on the whole trajectory and N is the total number of snapshots recorded in the trajectory during the molecular dynamics simulations.
The residue m that mediates the signal transduction between residues s and t with the largest probability is identified according to the following equation:
| 5 |
where C(Rs,Ri,Rt) is the correlation coefficient between residues s and t mediated by residue i; C(Rs,Ri) is the correlation coefficient between residues s and i; C(Ri,Rt) is the correlation coefficient between residues i and t; | | indicates absolute value; N is the total residue number of the protein. The residue holding the maximal value of C(Rs,Ri,Rt) is considered to mediate the signal transduction between residues s and t with the largest probability (C(Rs,Rm,Rt)). After the first round, residues mediate the signal transduction between residues s and m with the largest probability is to be discovered and the same for residues m and t. The procedure is continued until no more residues could be found.
Overexpression and purification of PII variants
Site-directed mutagenesis of glnB and cloning into the Strep tag fusion vector pASK-IBA3 (IBA GmbH, Göttingen, Germany) was performed and described previously.35 The vectors were transformed into Escherichia coli strain RB906036 and purified using affinity chromatography as described earlier.7
ITC measurements
ITC experiments were performed using a VP-ITC microcalorimeter (MicroCal) in buffer containing 10 mM Na2HPO4, 1.8 mM KH2PO4, 25 mM NaCl, 10 mM KCl, 1 mM MgCl2 and 1 mM ATP (pH 7.5); 2 mM of the titrant 2-OG were dissolved in the same buffer. After an initial 2 µL injection 6 µL were injected 45 times into the 1.4285 mL cell with stirring at 155 rpm at a temperature of 25°C. The PII concentration in the cell was 33 µM (trimer concentration). The binding isotherms were calculated from received data and fitted to a three-site binding model with the MicroCal ORIGIN software (Northampton, MA) as indicated.
RESULTS
Definition of 2-OG binding pocket size
Structures of PII trimers with all three sites fully occupied and with one or two bound molecules of 2-OG not only shed first light on structural basis of anticooperativity but help revealing the anticooperative mechanism from perspectives of dynamics. The population shift of binding pocket size employed here to demonstrate the allosteric effects upon 2-OG binding, is defined by identifying residues that contribute greatly to the binding of 2-OG through decomposition of binding free energy followed by analysis of hydrogen-bond interactions.
It is reported that 2-OG is bound to a Mg2+ ion, which is coordinated by three phosphates of ATP, and by ionic interactions with the highly conserved residues Gln39 and Lys58 together with the B- and T-loop backbone interactions.11 Table 1 lists the overall binding free energy for structures with different ligand binding states. The slight differences among their overall binding free energy mainly result from the polar interactions
mainly result from the polar interactions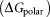 , namely the electrostatic interaction energy
, namely the electrostatic interaction energy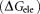 and the electrostatic solvation free energy
and the electrostatic solvation free energy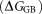 . When examining the energy contribution of each residue [Fig. 2(A)], it can be seen that Arg9, sequence Phe36-Gly41 of the PII T-loop, Lys58 and Gly87 are the main contributors to the energy differences upon 2-OG binding. Among these residues, Arg9 makes contributions to the binding of the first and third 2-OG although it does not make large contributions when the second 2-OG binds to the binding pocket. Gln39 influences the ligand binding most in the sequence Phe36-Gly41. It is evident that both Lys58 and Gly87 make large and positive contributions to the ligand binding. Therefore, Arg9, Gln39, Lys58, and Gly87 were finally chosen as the residue-level indicators to describe the size of the binding pocket.
. When examining the energy contribution of each residue [Fig. 2(A)], it can be seen that Arg9, sequence Phe36-Gly41 of the PII T-loop, Lys58 and Gly87 are the main contributors to the energy differences upon 2-OG binding. Among these residues, Arg9 makes contributions to the binding of the first and third 2-OG although it does not make large contributions when the second 2-OG binds to the binding pocket. Gln39 influences the ligand binding most in the sequence Phe36-Gly41. It is evident that both Lys58 and Gly87 make large and positive contributions to the ligand binding. Therefore, Arg9, Gln39, Lys58, and Gly87 were finally chosen as the residue-level indicators to describe the size of the binding pocket.
Table I.
Binding Free Energies Computed by the MM-GBSA Method upon Binding of the First, Second, and Third 2-OG Toward the Binding Pocket of Chain A, B, and C of PII Protein (Unit: kcal mol−1)
| Energy terms | PIIOG1 | PIIOG2 | PIIOG3 |
|---|---|---|---|
| ΔGele | −295.3 | −344.0 | −236.4 |
| ΔGvdw | −2.6 | −2.7 | −4.2 |
| ΔGMMa | −297.9 | −346.7 | −240.6 |
| ΔGnp | −2.7 | −2.7 | −2.7 |
| ΔGGB | 226.4 | 277.4 | 181.8 |
| ΔGsolvb | 223.7 | 274.7 | 179.0 |
| ΔGbindingc | −74.1 | −72.0 | −61.6 |
ΔGMM = ΔGele + ΔGvdw.
ΔGsolv = ΔGnp + ΔGGB.
ΔGbinding= ΔGMM + ΔGsolv.
Figure 2.
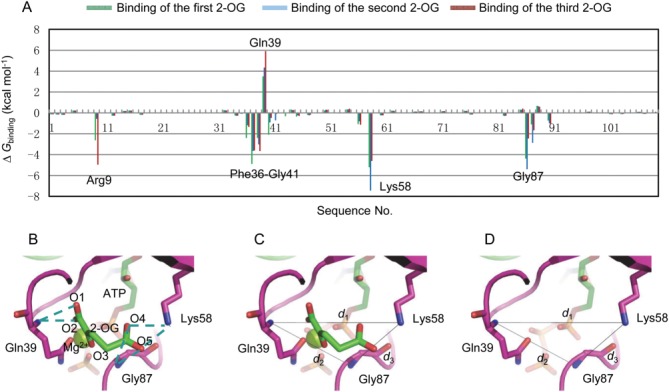
Definition of the binding pocket size. (A) Binding free energy decomposed at residue level for PII protein. (B) Hydrogen bonds interactions (dashed lines) between Gln39, Lys58, Gly87, and 2-OG. Definition of binding pocket size by d1, d2 and d3 with (C) or without (D) the binding of 2-OG. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Since hydrogen-bond interactions always play an important role for ligand binding, hydrogen-bond interactions analysis is employed to identify atoms that can be used to describe the size of the binding pocket at an atomic level. The analysis was conducted between the residues Arg9, Gln39, Lys58, Gly87, and 2-OG. As shown in Figure 2(B), hydrogen-bonds are formed between the main chain N atom of Gln39 and O1 (O2) of 2-OG, the side chain N atom of Lys58 and O4 (O5) of 2-OG. There are also hydrogen-bonds between the main chain N atom of Gly87 and the O4 (O5) of 2-OG. However, no hydrogen-bond is formed between Arg9 and 2-OG due to their long distance although Arg9 also makes contributions to the binding of 2-OG because of its positively charged side chain. Thus, the main chain N atom of Gln39, the side chain N atom of Lys58 and the main chain N atom of Gly87 are selected as the atom-level indicators to describe the binding pocket size. Then three distance parameters d1 (the distance between the main chain N atom of Gln39 and the side chain N atom of Lys58), d2 (the distance between the main chain N atom of Gln39 and the main chain N atom of Gly87) and d3 (the distance between the side chain N atom of Lys58 and the main chain N atom of Gly87) are defined to reflect the size of the binding pocket with or without the binding of 2-OG [Fig. 2(C,D)].
Population shift of binding pocket size upon 2-OG binding
With the definition of the binding pocket size, it is possible to quantitatively investigate the population shift of the binding pockets in response to the binding of 2-OG. In order to investigate the anticooperativity in the context of distribution shift from unbound to bound PII, molecular dynamics simulations of unbound PII trimer (PDB code: 1QY7) were carried out and the distributions of the binding pocket size (d1, d2, d3) were calculated (Fig. 3). It is found that although the distances measured from the crystal structure show that they are the same among the monomers (purple dots in Fig. 3), the simulation results show some difference. For instance, d1 is 10.07 Å and 10.05 Å for chain A and chain B, whereas it is 9.65 Å for chain C. d2 of chain A (9.85 Å) is somewhat larger than that of chain B (8.10Å) and chain C (8.17Å). There are also difference for d3 among chain A, chain B and chain C.
Figure 3.
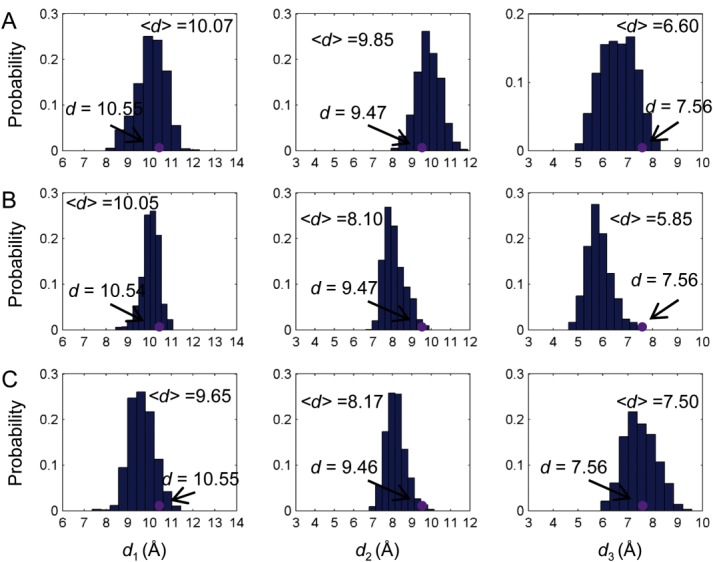
Distribution of the binding pocket size without ligand binding. Distance distributions of d1, d2 and d3 for chain A (A), chain B (B), and chain C (C). Purple dots are distances measured based on the crystal structures. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Upon binding of the first 2-OG towards chain A of PII trimer, the shift of the binding pocket size of chain B and C is determined and compared with that of chain A. As shown in Figures 3(B) and 4(A), the pocket size of chain B becomes larger as indicated by the distance parameters defined above. In particular, distance d1 increases from 10.05 Å to 11.47 Å although d2 and d3 show little changes. By contrast, the binding pocket of chain C becomes smaller upon the binding of the first 2-OG. This can be seen from the distance d2, which decreases from 8.17 Å to 6.29 Å and d3 which deceases from 7.50 Å to 5.62 Å [Figs. 3(C) and 4(B)]. There are only slight changes in terms of d1.
Figure 4.
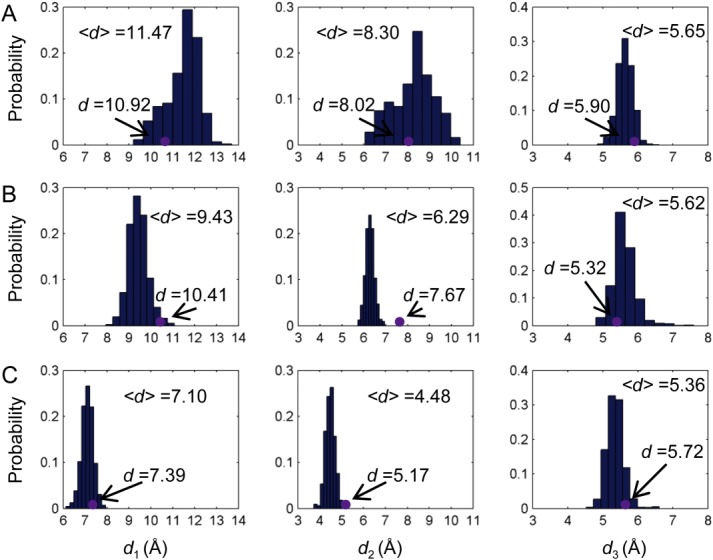
Distribution of the binding pocket size upon ligand binding. Distance distributions of d1, d2, and d3 for chain B (A) and chain C (B) when the first 2-OG binds towards the binding pocket of chain A as well as distance distributions for chain C (C) when the second 2-OG binds toward the binding pocket of chain B. Purple dots are distances measured based on the crystal structures. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
When the second 2-OG binds to chain B, the binding pocket size of chain C shrinks further because of the decrease of d1 from 9.43 Å to 7.10 Å and d2 from 6.29 Å to 4.48 Å [Fig. 4(C,D)]. Again the distance change of d3 is very small, only from 5.62 Å to 5.72 Å. The small distance change of d3 upon the binding of either the first or the second 2-OG may be due to the fact that Lys58 and Gly87 are located in more stable structure areas than Gln39, which resides in the flexible T-loop. The large distance shifts of d1 and d2 in response to the ligand binding illustrate the role of PII T-loop in the anticooperative process as well as providing further proof of its role in the ligand binding process.
By defining the binding pocket size with residues that make great contributions to the ligand binding, the anticooperative mechanism of PII protein in response to 2-OG can be well explained as a population shift of the binding pocket size (Fig. 5). Shifts of the binding pocket to become either bigger (in the case of chain B upon the binding of the first 2-OG) or smaller (in the case of chain C upon the binding of the first or the second 2-OG) will definitely decrease the binding opportunities of 2-OG and result in the negative cooperativity of PII trimer. Results from the ITC measurements suggest that the binding of 2-OG to the first binding site is driven by both enthalpy and entropy (ΔH < 0, TΔS > 0) with the enthalpy having the bigger impact. The situation is similar for the binding to the second binding site, but here the entropy has the bigger impact. The binding of 2-OG to the third binding site, on the other hand, is clearly driven by the enthalpy (ΔH < 0, TΔS < 0). The thermodynamic considerations fit well with the binding model illustrated by the change of the binding pocket size. They imply that occupation of the first and second site lead to a more disordered conformation because of the change of the binding pocket size, whereas occupation of the third site gives a more ordered state because all three sites are now occupied with 2-OG. That is why this reaction is exclusively enthalpy driven.
Figure 5.
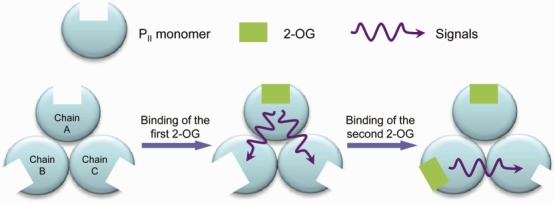
Sketch of the change of binding pocket size upon the binding of 2-OG. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Residues mediating the anticooperative signal transduction
Dynamic correlation analysis [Eq. 4] was first used to obtain the correlation coefficients between each pair of residues when the first 2-OG binds toward the binding pocket of chain A [Fig. 6(A)] and the second 2-OG toward chain B [Fig. 6(B)]. The number of residues correlated with high correlation coefficient (>0.5) provides a quantitative parameter about the signal-mediating function of each residue [Fig. 6(C,D)]. Then, residues that mediate the signal transduction process with high probability were identified using the algorithm derived from the dynamic correlation analysis with residues identified to illustrate the binding pocket size as Rs or Rt in Eq. (5).
Figure 6.
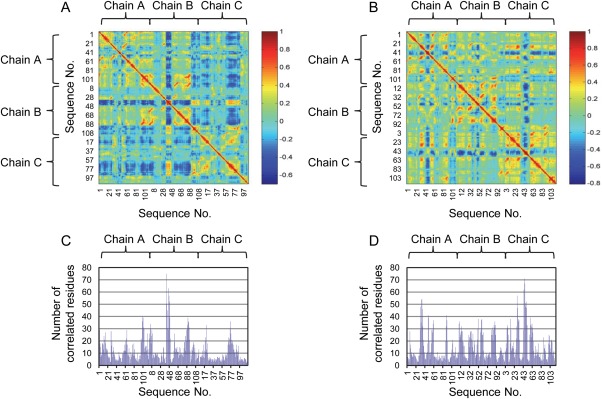
Dynamic correlation analysis of PII protein upon binding of 2-OG. Correlation coefficients for each pair of residues upon the binding of the first (A) and the second (B) 2-OG in the case of wild-type PII protein. Number of correlated residues with the correlation coefficient value over 0.5 upon the binding of the first (C) and the second (D) 2-OG. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
By comparing Figure 6(A,C), we found that the sequence 41 to 50 of chain B shows the largest numbers of correlated residues upon the binding of the first 2-OG. Besides, the sequence 81 to 94 of chain B and the sequence 70 to 81 of chain C also show large numbers of correlated residues. Particularly, 21 residues are primarily involved in the signal transduction process from the binding pocket of chain A to that of chain B, of which 7 belong to chain A and 14 belong to chain B [Table 2 and Fig. 7(A)]. In the case of signaling process from chain A to C, 13 residues contribute to the process, wherein 8 of them belong to chain A and 5 of them belong to chain C [Table 2 and Fig. 7(B)]. Among these residues, Gln39, Lys40, Ser49, Lys58, Leu59, Gly87, and Asp88 are involved in the signal transduction from chain A to both chain B and C. In the case when the second 2-OG binds towards the binding pocket of chain B [Fig. 6(B,D)], the sequence 43 to 51 of chain C shows the largest numbers of correlated residues. Besides, the sequence 29 to 40 of chain A also shows large numbers of correlated residues. In particular, 14 residues contribute largely to the signal transduction process from chain B to C, of which 6 belong to chain B and 8 belong to chain C [Table 2 and Fig. 7(C)].
Table II.
Major Residues Mediating the Signal Transduction from the Binding Site of Chain A to that of Chain B and Chain C upon Binding of the First 2-OG as well as from Chain B to Chain C upon Binding of the Second 2-OG
| Residue no. | Chain A to B | Chain A to C | Chain B to C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | A | B | C | A | B | C | |
| Phe36a,b | √ | ||||||||
| Gly37a,b | √ | ||||||||
| Arg38a,b | √ | √ | |||||||
| Gln39a,b | √ | √ | √ | √ | √ | √ | |||
| Lys40 | √ | √ | |||||||
| Gly41a,b | √ | ||||||||
| Gln42a,b | √ | ||||||||
| Thr43 | √ | ||||||||
| Glu44a | √ | ||||||||
| Tyr46a,b | √ | ||||||||
| Arg47a,b | √ | ||||||||
| Gly48a,b | √ | √ | |||||||
| Ser49a,b | √ | √ | √ | √ | |||||
| Leu56a | √ | ||||||||
| Lys58a,b | √ | √ | √ | √ | √ | √ | |||
| Leu59a,b | √ | √ | |||||||
| Ile86 | √ | √ | |||||||
| Gly87a,b | √ | √ | √ | √ | √ | √ | |||
| Asp88a,b | √ | √ | √ | ||||||
| Lys90 | √ | ||||||||
| Ile91 | √ | ||||||||
| Phe92 | √ | ||||||||
| Arg103 a | √ | ||||||||
The symbol √ indicates to which chain the residue belongs to in each case.
Residues identified within the time period of 1.0 and 1.5 ns.
Residues identified within the time period of 1.0, 1.5, and 2.0 ns.
Figure 7.
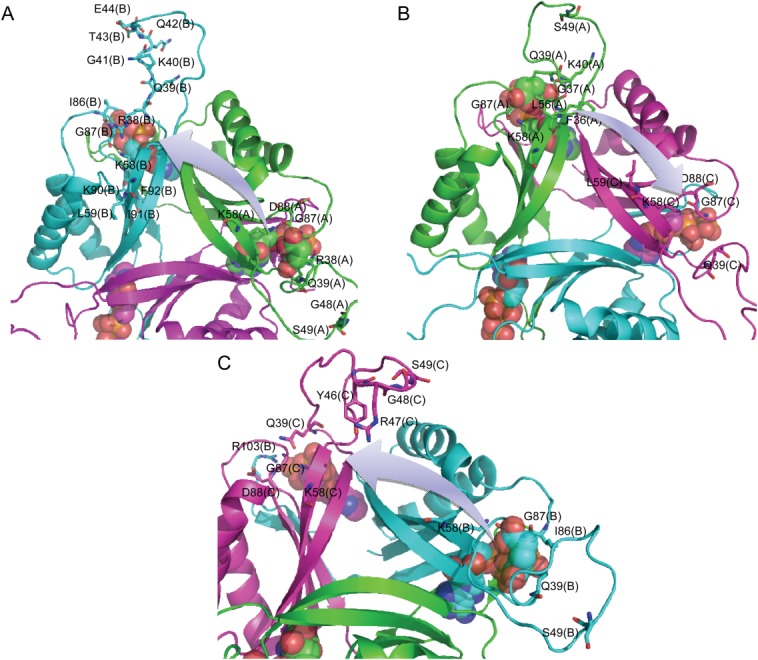
Major residues mediating the signal transduction from the binding site of chain A to that of chain B (A) and chain C (B) upon binding of the first 2-OG as well as from chain B to chain C (C) upon binding of the second 2-OG. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
It is not surprising to find that Gln39, Lys58, and Gly87 are involved in each case of ligand binding state due to the fact that they comprise the binding pocket. Besides these three residues, Ser49 and Asp88 are also involved in not only the signaling process from the binding pocket of chain A to B and C upon the binding of first 2-OG but also from chain B to C upon the binding of second 2-OG. Especially, it is interesting to find that Ser49, which is located at the T-loop of PII protein and is an important site to regulate the binding of S. elongatus PII toward its target protein through phosphorylation7,37 also plays a key role in the signal transduction process. In addition, residues Lys40 and Leu59 take part in the signaling process from the binding pocket of chain A to B and C upon the binding of first 2-OG. Residues Gly48 and Ile86 take part in the signaling process from the binding pocket of chain A to B upon the binding of first 2-OG and chain B to C upon the binding of second 2-OG.
Since allosteric proteins undergo structural fluctuations over a wide range of timescales (from femtoseconds to milliseconds or longer), residues mediating signal transduction may differ during different periods of time. The signal-mediating residues identified in this work are based on correlation matrix obtained within the first nanosecond upon binding of ligand. To test whether similar signal-mediating residues can be indentified from correlation matrix obtained from a longer simulation, molecular dynamics simulations were carried out for another nanosecond. Then signal-mediating residues were identified within the time period of 1.5 and 2.0 separately and compared with the results from the first nanosecond (Table 2). As expected, most of the residues keep their function in the signal transduction process. Among the 23 residues identified within the time period of 1.00 ns, 17 are identified within the time period of 1.0 and 1.50 ns. Fourteen are identified within the time period of 1.00, 1.50, and 2.0 ns. The identification of PII T-loop residues, especially Ser49 and its neighboring residues, indicates their significant roles in the signaling process during the alloteric regulation.
Anticooperativity of PII variants by 2-OG
Considering the specificity of Ser49 in the function of PII protein, two variants PII-Ser49Gly and PII-Ser49Asp were constructed and their anticooperativity was measured by isothermal titration calorimetry (ITC) approach. The former is constructed to investigate the role of the side chain in the signal transduction process and the later is used to mimic the phosphorylation modification of Ser49. As shown in Figure 8, the anticooperativity of PII-Ser49Gly is decreased by approx. 19 % upon the binding of the second 2-OG (Kd2/Kd1 from 3.2 to 2.6). The anticooperativity is weakened by approximately 31% upon the binding of the third 2-OG (Kd3/Kd1 from 22.1 to 15.3). In the case where Ser49 is replaced by aspartate acid, the anticooperativity (Kd2/Kd1) is slightly changed from 3.2 to 3.5 upon the binding of the second 2-OG. However, the anticooperativity is strengthened by 19.9% upon the binding of the third 2-OG (Kd3/Kd1 from 22.1 to 26.5) which means PII-Ser49Asp is able to sense a wider range of metabolite concentrations than the wild type if it is expressed in the organism. Moreover, the influence of site mutation on the binding of the third 2-OG is stronger than the second 2-OG as expected by the dynamic correlation analysis.
Figure 8.
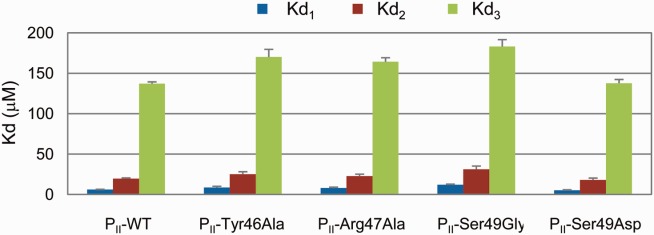
Anticooperativity of PII variants by 2-OG measured using ITC method. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In order to further confirm the role of T-loop in the anticooperative allosteric regulation of PII, two neighboring sites (Tyr46 and Arg47) that are also located in the mid of the T-loop were chosen and tested. Since these two sites hold large and even positive charged (in the case of Arg47) side chain, both of them were mutated to alanine to decrease the contributions of their side chains and the anticooperativity of the variants were measured using the ITC method. From Figure 8, it can be seen that both variants show the same influence on the anticooperativity upon the binding of the second 2-OG (Kd2/Kd1 from 3.2 to 2.9 and 2.8, respectively). However, Tyr46Ala shows a little stronger influence than Arg47Ala on the anticooperativity upon the binding of the third 2-OG. Tyr46Ala decreases the anticooperativity (Kd3/Kd1) from 22.1 to 19.6 while the anticooperativity is decreases from 22.1 to 20.5 in the case of Arg47Ala. Compared with Ser49, Tyr46 and Arg47 show less influence on the anticooperativity of PII. This is also expected by the dynamic correlation analysis. From these experimental results it can be seen that although the T-loops are long and extend outward from each subunit, they indeed take part in the anticooperative allosteric regulation of PII upon binding of 2-OG. Modifications of residues located at the T-loop can influence the anticooperativity positively or negatively.
DISCUSSION
The concept of protein allostery went back to the MWC model (also known as the concerted model or symmetry model) put forth by Monod et al. and the KNF model (also known as the sequential or “induced fit” model) described by Koshland et al.38,39 Both sought to account for allostery based on gross properties of the transition between two well-defined end states. The two models differ most in their assumptions about subunit interaction and the preexistence of both states. The concerted model of allostery postulates that subunits are connected in such a way that a conformational change in one subunit is necessarily conferred to all other subunits. Thus, all subunits must exist in the same conformation. The sequential model of allosteric regulation assumes that ligand molecules bind via an induced fit protocol, which converts a subunit from one state to another. However, it does not propagate the conformational change to adjacent subunits. Instead, ligand-binding at one subunit only slightly alters the structure of other subunits so that their binding sites are more receptive or rejective to the ligand. In the case of PII protein, it is known that 2-OG binds in a sequential manner and the binding of 2-OG at one subunit decreases the binding of 2-OG at the other subunits.11 Thus, it seems that the KNF model is more suitable to describe the anticooperativity of PII by 2-OG. Nevertheless, the underlying mechanism behind the sequential binding of 2-OG is still unclear. Thus it is necessary to get a deeper understanding of the anticooperative allosteric regulation of PII protein in order to reveal the underlying mechanism. To this end, new models and approaches are needed to shed more light into the anticooperativity of PII by 2-OG.
Considering the fact that the function of a protein is ultimately governed by its dynamics in most cases, proteins are inherently dynamical molecules that undergo structural fluctuations over a wide range of timescales.40,41 More recent thermodynamic models of allostery has expanded the understanding and definition of allostery by emphasizing that rather than only in two conformational states, proteins exist in ensembles. Thus, allosteric regulation is considered to be accompanied by population shift in conformational ensembles.42–44 In this work, the concept of binding pocket size is proposed to describe the conformational ensembles. From the re-distribution of the distances that define the binding pocket size, the population shifts proceeding upon binding of 2-OG are well characterized. These discoveries shed now light into the anticooperative allosteric regulation of PII protein by first confirming that the binding of 2-OG at one subunit changes the population of the binding pocket site of the other subunits, which subsequently influence the binding of 2-OG at the other subunits. Furthermore, this influence is conducted in an asymmetric manner due to the different population shifts of the second and the third binding pocket size upon binding of the first 2-OG.
To uncover signal transduction processes within allosteric proteins, several approached have been developed so far. A co-evolutionary approach, which is based on sequence information of the protein family, was employed to identify amino acid interactions in each protein family.45,46 Another novel approach to reveal intramolecular signal transduction networks was proposed based on a new protein dynamics model of energy dissipation.47,48 Since correlations in the atomic displacements (dynamic correlation analysis based on fluctuation dynamics) indicate collective motion and are therefore of potential relevance to protein function, correlated motions have been detected in molecular dynamics simulations and used to make deductions concerning dynamical aspects of protein function.49–54 In a recent study, molecular dynamics simulation was coupled with co-evolutionary analysis to identify a key residue-residue interaction network which is responsible for the allosteric regulation and signal transduction within allosteric enzymes.55,56 In this work, a new approach derived from dynamic correlation analysis is utilized to reveal how the signal is transferred from the binding site of one subunit to the other subunits upon binding of ligand. This approach highlighted amino acid residues mediating the signaling process with high probability. It was surprising to find the important role of PII T-loop in the signal transduction process since the long T-loop extends outward from each subunit and is believed to be responsible for mediating the binding of PII with its target proteins.35,57 In particular, it is found that Ser49 also takes part in the allosteric process. The particular impact of this site relies on the fact that it is an important site to regulate the binding of PII towards its target protein N-acetyl-l-glutamate kinase through phopshorylation.7 Experimental results further confirmed the role of T-loop in influencing the anticooperativity of PII, especially on binding of the third 2-OG. These resuls not only indicate that the population shift of the binding pocket size is highly correlated to the ligand binding through protein dynamics but also illustrate the versatile function of PII T-loop in the anticooperative allosteric regulation. From the perspective of application, it is possible to alter the sensitivity of PII protein to a much wider range of metabolite concentrations by modifying the residues discovered through the new dynamic correlation analysis approach.
CONCLUSIONS
The dynamics of the binding pocket is well characterized by defining its size with residues that contribute greatly to the binding of 2-OG. The anticooperative mechanism of PII protein upon the binding of 2-OG can be well explained by population shift of the binding pocket size. Residues mediating the anticooperative regulation with high probability are discovered by using the new algorithm developed based on dynamic correlation analysis. It is found that Ser49, which is located in the flexible T-loop of PII, plays an important role in the allosteric signaling, as illustrated by experimental results of the anticooperativity of the variants PII-Ser49Gly and PII-Ser49Asp. Overall, the results presented here extend our understanding of the PII T-loop from being essential in versatile binding of target protein to signal-mediating in the anticooperative allosteric regulation.
Acknowledgments
The work of CWM and APZ was partially supported by “Die Landesexzellenzinitiative Hamburg” through the project SynBio.
REFERENCES
- 1.Chellamuthu VR, Alva V, Forchhammer K. From cyanobacteria to plants: conservation of PII functions during plastid evolution. Planta. 2013;237:451–462. doi: 10.1007/s00425-012-1801-0. [DOI] [PubMed] [Google Scholar]
- 2.Forchhammer K. P(II) signal transducers: novel functional and structural insights. Trends Microbiol. 2008;16:65–72. doi: 10.1016/j.tim.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Espinosa J, Forchhammer K, Burillo S, Contreras A. Interaction network in cyanobacterial nitrogen regulation: PipX, a protein that interacts in a 2-oxoglutarate dependent manner with PII and NtcA. Mol Microbiol. 2006;61:457–469. doi: 10.1111/j.1365-2958.2006.05231.x. [DOI] [PubMed] [Google Scholar]
- 4.Feria Bourrellier AB, Valot B, Guillot A, Ambard-Bretteville F, Vidal J, Hodges M. Chloroplast acetyl-CoA carboxylase activity is 2-oxoglutarate-regulated by interaction of PII with the biotin carboxyl carrier subunit. Proc Natl Acad Sci USA. 2010;107:502–507. doi: 10.1073/pnas.0910097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leigh JA, Dodsworth JA. Nitrogen regulation in bacteria and archaea. Annu Rev Microbiol. 2007;61:349–377. doi: 10.1146/annurev.micro.61.080706.093409. [DOI] [PubMed] [Google Scholar]
- 6.Bossa C, Anselmi M, Roccatano D, Amadei A, Vallone B, Brunori M, Nolam AD. Extended molecular dynamics simulation of the carbon monoxide migration in sperm whale myoglobin. Biophys J. 2004;86:3855–3862. doi: 10.1529/biophysj.103.037432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinrich A, Maheswaran M, Ruppert U, Forchhammer K. The Synechococcus elongatus PII signal transduction protein controls arginine synthesis by complex formation with N-acetyl-l-glutamate kinase. Mol Microbiol. 2004;52:1303–1314. doi: 10.1111/j.1365-2958.2004.04058.x. [DOI] [PubMed] [Google Scholar]
- 8.Maheswaran M, Urbanke C, Forchhammer K. Complex formation and catalytic activation by the PII signaling protein of N-acetyl-L-glutamate kinase from Synechococcus elongatus strain PCC 7942. J Biol Chem. 2004;279:55202–55210. doi: 10.1074/jbc.M410971200. [DOI] [PubMed] [Google Scholar]
- 9.Sant'Anna FH, Trentini DB, de Souto Weber S, Cecagno R, da Silva SC, Schrank IS. The PII superfamily revised: a novel group and evolutionary insights. J Mol Evol. 2009;68:322–336. doi: 10.1007/s00239-009-9209-6. [DOI] [PubMed] [Google Scholar]
- 10.Jiang P, Ninfa AJ. Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry. 2007;46:12979–12996. doi: 10.1021/bi701062t. [DOI] [PubMed] [Google Scholar]
- 11.Fokina O, Chellamuthu VR, Forchhammer K, Zeth K. Mechanism of 2-oxoglutarate signaling by the Synechococcus elongatus PII signal transduction protein. Proc Natl Acad Sci USA. 2010;107:19760–19765. doi: 10.1073/pnas.1007653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forchhammer K, Hedler A. Phosphoprotein PII from cyanobacteria–analysis of functional conservation with the PII signal-transduction protein from Escherichia coli. Eur J Biochem. 1997;244:869–875. doi: 10.1111/j.1432-1033.1997.00869.x. [DOI] [PubMed] [Google Scholar]
- 13.Jiang P, Ninfa AJ. Alpha-ketoglutarate controls the ability of the Escherichia coli PII signal transduction protein to regulate the activities of NRII (NtrB) but does not control the binding of PII to NRII. Biochemistry. 2009;48:11514–11521. doi: 10.1021/bi901158h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helfmann S, Lu W, Litz C, Andrade SL. Cooperative binding of MgATP and MgADP in the trimeric PII protein GlnK2 from Archaeoglobus fulgidus. J Mol Biol. 2010;402:165–177. doi: 10.1016/j.jmb.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 15.Sakai H, Wang H, Takemoto-Hori C, Kaminishi T, Yamaguchi H, Kamewari Y, Terada T, Kuramitsu S, Shirouzu M, Yokoyama S. Crystal structures of the signal transducing protein GlnK from Thermus thermophilus HB8. J Struct Biol. 2005;149:99–110. doi: 10.1016/j.jsb.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Cheah E, Carr PD, van Heeswijk WC, Westerhoff HV, Vasudevan SG, Ollis DL. GlnK, a PII-homologue: structure reveals ATP binding site and indicates how the T-loops may be involved in molecular recognition. J Mol Biol. 1998;282:149–165. doi: 10.1006/jmbi.1998.1979. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Carr PD, Clancy P, Garcia-Dominguez M, Forchhammer K, Florencio F, Vasudevan SG, Tandeau de Marsac N, Ollis DL. The structures of the PII proteins from the cyanobacteria Synechococcus sp. PCC 7942 and Synechocystis sp. PCC 6803. Acta Crystallogr Sect D Biol Crystallogr. 2003;59:2183–2190. doi: 10.1107/s0907444903019589. [DOI] [PubMed] [Google Scholar]
- 18.Ninfa AJ, Jiang P. PII signal transduction proteins: sensors of alpha-ketoglutarate that regulate nitrogen metabolism. Curr Opin Microbiol. 2005;8:168–173. doi: 10.1016/j.mib.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Smith CS, Weljie AM, Moorhead GB. Molecular properties of the putative nitrogen sensor PII from Arabidopsis thaliana. Plant J. 2003;33:353–360. doi: 10.1046/j.1365-313x.2003.01634.x. [DOI] [PubMed] [Google Scholar]
- 20.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat Rev Drug Discov. 2002;1:198–210. [Google Scholar]
- 21.Freire E. The propagation of binding interactions to remote sites in proteins: analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc Natl Acad Sci USA. 1999;96:10118–10122. doi: 10.1073/pnas.96.18.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kern D, Zuiderweg ERP. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malmendal A, Evenäs J, Forsén S, Akke M. Structural dynamics in the C-terminal domain of calmodulin at low calcium levels. J Mol Biol. 1999;293:883–899. doi: 10.1006/jmbi.1999.3188. [DOI] [PubMed] [Google Scholar]
- 25.Martinez KL, Gohon Y, Corringer PJ, Tribet C, Mérola F, Changeux JP, Popot JL. Allosteric transitions of Torpedo acetylcholine receptor in lipids, detergent and amphipols: molecular interactions vs. Physical constraints. FEBS Lett. 2002;528:251–256. doi: 10.1016/s0014-5793(02)03306-9. [DOI] [PubMed] [Google Scholar]
- 26.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 27.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 28.Fiser A, Do RKG, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods R. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorgensen WL. Revised TIPS for simulations of liquid water and aqueous solutions. J Chem Phys. 1982;77:4156–4163. [Google Scholar]
- 31.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 32.Jayaram B, Sprous D, Beveridge DL. Solvation free energy of biomolecules: parameters for a modified generalized born model consistent with the AMBER force field. J Phys Chem ser B. 1998;102:9571–9576. [Google Scholar]
- 33.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of Cartesian equations of motion of a system with constraints-molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 34.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 35.Llácer JL, Contreras A, Forchhammer K, Marco-Marín C, Gil-Ortiz F, Maldonado R, Fita I, Rubio V. The crystal structure of the complex of PII and acetylglutamate kinase reveals how PII controls the storage of nitrogen as arginine. Proc Natl Acad Sci USA. 2007;104:17644–17649. doi: 10.1073/pnas.0705987104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bueno R, Pahel G, Magasanik B. Role of glnB and glnD gene products in regulation of the glnALG operon of Escherichia coli. J Bacteriol. 1985;164:816–822. doi: 10.1128/jb.164.2.816-822.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forchhammer K. Global carbon/nitrogen control by PII signal transduction in cyanobacteria: from signals to targets. FEMS Microbiol Rev. 2004;28:319–333. doi: 10.1016/j.femsre.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 39.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 40.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 41.Olsson MH, Parson WW, Warshel A. Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis. Chem Rev. 2006;106:1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 42.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 43.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luque I, Leavitt SA, Freire E. The linkage between protein folding and functional cooperativity: two sides of the same coin? Annu Rev Biophys Biomol Struct. 2002;31:235–256. doi: 10.1146/annurev.biophys.31.082901.134215. [DOI] [PubMed] [Google Scholar]
- 45.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 46.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication inproteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 47.Ma CW, Xiu ZL, Zeng AP. A new concept to reveal protein dynamics based on energy dissipation. PLoS One. 2011;6:e26453. doi: 10.1371/journal.pone.0026453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma CW, Xiu ZL, Zeng AP. Discovery of intramolecular signal transduction network based on a new protein dynamics model of energy dissipation. PLoS One. 2012;7:e31529. doi: 10.1371/journal.pone.0031529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burillo S, Luque I, Fuentes I, Contreras A. Interactions between the nitrogen signal transduction protein PII and N-acetyl glutamate kinase in organisms that perform oxygenic photosynthesis. J Bacteriol. 2004;186:3346–3354. doi: 10.1128/JB.186.11.3346-3354.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daniel RM, Dunn RV, Finney JL, Smith JC. The role of dynamics in enzyme activity. Annu Rev Biophys Biomol Struct. 2003;32:69–92. doi: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- 51.Garcia AE, Hummer G. Conformational dynamics of cytochrome c: correlation to hydrogen exchange. Proteins. 1999;36:175–191. [PubMed] [Google Scholar]
- 52.Showalter SA, Hall KB. A functional role for correlated motion in the N-terminal RNA-binding domain of human U1A protein. J Mol Biol. 2002;322:533–542. doi: 10.1016/s0022-2836(02)00804-5. [DOI] [PubMed] [Google Scholar]
- 53.Keskin O, Durell SR, Bahar I, Jernigan RL, Covell DG. Relating molecular flexibility to function: a case study of tubulin. Biophys J. 2002;83:663–680. doi: 10.1016/S0006-3495(02)75199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ertekin A, Nussinov R, Haliloglu T. Association of putative concave protein-binding sites with the fluctuation behavior of residues. Protein Sci. 2006;15:2265–2277. doi: 10.1110/ps.051815006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Z, Rappert S, Sun JB, Zeng AP. Integrating molecular dynamics and co-evolutionary analysis for target prediction and engineering of allosteric regulation of aspartokinases for amino acids production. J Biotechnol. 2011;154:248–254. doi: 10.1016/j.jbiotec.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 56.Chen Z, Meyer WQ, Rappert S, Sun JB, Zeng AP. Coevolutionary analysis enables rational deregulation of allosteric enzyme inhibition in Corynebacterium glutamicum for lysine production. Appl Environ Microbiol. 2011;77:4352–4360. doi: 10.1128/AEM.02912-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeth K, Fokina O, Forchhammer K. An engineered PII protein variant that senses a novel ligand: atomic resolution structure of the complex with citrate. Acta Crystallogr D: Biol Crystallogr. 2012;68:901–908. doi: 10.1107/S0907444912016447. [DOI] [PubMed] [Google Scholar]