

Abstract
HP1 proteins are transcriptional regulators that, like histones, are targets for post-translational modifications defining an HP1-mediated subcode. HP1γ has multiple phosphorylation sites, including serine 83 (S83) that marks it to sites of active transcription. In a guinea pig model for Shigella enterocolitis, we observed that the defective type III secretion mxiD Shigella flexneri strain caused more HP1γ phosphorylation in the colon than the wild-type strain. Shigella interferes with HP1 phosphorylation by injecting the phospholyase OspF. This effector interacts with HP1γ and alters its phosphorylation at S83 by inactivating ERK and consequently MSK1, a downstream kinase. MSK1 that here arises as a novel HP1γ kinase, phosphorylates HP1γ at S83 in the context of an MSK1-HP1γ complex, and thereby favors its accumulation on its target genes. Genome-wide transcriptome analysis reveals that this mechanism is linked to up-regulation of proliferative gene and fine-tuning of immune gene expression. Thus, in addition to histones, bacteria control host transcription by modulating the activity of HP1 proteins, with potential implications in transcriptional reprogramming at the mucosal barrier.
Keywords: CBX3, colon, Heterochromatin protein 1, MSK1, Shigella
See also: H Ashida & C Sasakawa (November 2014)
Introduction
The sensing of stress signals and their transduction into appropriate response is crucial for the adaptation and survival of any organism. The evolutionary conserved mitogen-activated protein kinase (MAPK) pathways transduce a large variety of external signals, leading to a wide range of cellular responses, one of which is the innate immune response to infection. The three parallel MAPKs cascades, the JNK, p38, and ERK cascades, contribute to subsequent inflammatory and innate immune responses by regulating the expression of numerous effector genes at transcriptional or post-transcriptional levels (Yang et al, 2003). Therefore, to counteract host immunity, pathogenic bacteria have evolved virulence strategies that intercept the MAPK signaling, mostly by injecting virulence effector proteins through their type III secretion system (T3SS) into the host cell (Shan et al, 2007). In an earlier study, we showed the Shigella bacterial species, a causal agent of bacillary dysentery in humans, delivered the T3SS virulence effector OspF in host epithelial cells, to directly inactivate both ERK and p38 MAPK signaling in the nucleus of infected cells (Arbibe et al, 2007). OspF inactivates MAPKs through a phosphothreonine lyase activity (Li et al, 2007) and therefore belongs to a small family of bacterial effectors harboring phosphothreonine lyase activity like SpvC from Salmonella and HopAI1 from Pseudomonas syringae (Zhang et al, 2007; Mazurkiewicz et al, 2008). This phosphothreonine lyase activity irreversibly inactivates the dual-phosphorylated host MAPKs (pT-X-pY) through beta elimination of the phosphate group, converting the phosphothreonine residue required for MAPK activity into a dehydrobutyrine (Dhb). The newly formed Dhb residue is irreversibly dephosphorylated because of the missing OH group (for review, Brennan & Barford, 2009). We previously showed that bacterially injected OspF principally located to the nucleus of infected cells to inactivate MAPK nuclear signaling, thereby causing a transcriptional repression of a limited number of host immune genes (Arbibe et al, 2007). From microarray analysis, we identified a narrow set of OspF target genes encompassing the immediate-early genes and some NF-κB responsive genes, and by chromatin immunoprecipitation, we showed that OspF precisely blocks nuclear MAPK induced histone H3 phosphorylation at the promoter of its target immunes genes. Therefore, OspF acts as an epigenetic regulator reprogramming the host transcriptional response by irreversibly inactivating nuclear MAPKs, which results in an immunosuppressive environment favorable for the bacterium life at the mucosal surface.
Regulation of transcription involves post-translational histone modifications controlling the recruitment of epigenetic regulators, among which are the Heterochromatin Protein 1 family members (HP1α, HP1β and HP1γ in humans) that specifically recognize and bind histone H3 methylated at lysine 9 through their “histone reader domain” called the chromodomain. Although initial studies revealed a key role for HP1 proteins in heterochromatin formation and gene silencing, recent progress has shed light on a positive role for HP1 in euchromatic gene expression. As a matter of fact, the HP1γ isoform plays an unexpected role in active transcription on a large range of promoters (for review, Kwon & Workman, 2011). For example, on the HIV1 LTR and the survivin promoter, HP1γ is most abundant after activation of transcription at a time when HP1α and HP1β are evicted (Smallwood et al, 2007; Mateescu et al, 2008). Consistent with this, HP1γ concentrates in the coding region of active genes and interacts with the elongating RNA polymerase II (RNAPII) and the pre-mRNA (Vakoc et al, 2005; Lomberk et al, 2006). At these intragenic positions, HP1γ may stabilize ongoing transcription and/or participate in RNA maturation. Genome-wide analyses implicate HP1γ in both alternative splicing and co-transcriptional RNA processing, presumably by favoring the recruitment of the splicing machinery (Saint-André et al, 2011; Ameyar-Zazoua et al, 2012; Smallwood et al, 2012). Overall, these studies suggest that HP1 proteins are transcriptional regulators, their functional impact on gene expression depending on their association with various partner proteins. The search for additional mechanisms regulating the functional outcome of HP1 protein on transcription points out the implication of HP1 post-translational modifications. Notably, phosphorylation of HP1γ at serine 83 (S83) located within the hinge of the protein defines a subpopulation exclusively located to euchromatin, targeted to the site of transcriptional elongation, and with abrogated HP1γ silencing activity in a transcriptional reporter assay (Lomberk et al, 2006). This infers the existence of a second layer of transcriptional regulation on top of the histone code, earlier referred to as a “HP1 subcode”, governed by the dynamic of nuclear kinases phosphorylating HP1γ and with a fundamental impact on its bioactivity.
Interestingly, a phosphoproteome analysis performed on macrophages stimulated by LPS in vitro showed that TLR4 activation had a strong impact on the cellular phosphorylation state, with sub-data analysis revealing multiple phosphorylation sites on HP1γ, including S83 (Weintz et al, 2010). Nevertheless, the regulation of HP1γ by HP1 kinases remains weakly characterized in living cells and, upon innate immune challenge, the modulation of HP1γ phosphorylation state poorly investigated.
In this report, we provide the seminal observation that the bacterial pathogen Shigella flexneri modulates HP1γ phosphorylation in the colon. Notably, colonic infection with the proinflammatory non-invasive mxiD Shigella mutant that does not assemble the T3SS needle and therefore does not secrete T3SS effectors, dramatically up-regulated HP1γ phosphorylation, while a weaker induction was observed in response to the wild-type (WT) invasive Shigella strain. Our in vitro approach identified the T3SS virulence effector OspF as a modulator of HP1γ phosphorylation. We showed that OspF directly interacted with HP1γ and inactivated the ERK-downstream kinase MSK1 that we identified as a major HP1 kinase. A transcriptome analysis of HP1γ null cell lines re-complemented or not with HP1γ revealed that many genes known to be under the transcriptional control of OspF during Shigella infection are dependent on HP1γ for their regulation. Stimulation of the cells with an activator of the MAPK pathway further showed that HP1γ seems to function as a moderator of the amplitude of the innate immune response, while also promoting specificity in the signaling, properties that makes it a very likely target for bacterial takeover. Finally, an S83A mutation in HP1γ confirmed that phosphorylation at this position is important for the normal function of the protein, but is insufficient to abolish its role in the innate immune response.
Results
Shigella modulates HP1γ phosphorylation state in vivo
To investigate in vivo the impact of bacterial challenge on HP1γ phosphorylation, we used a guinea pig model of Shigellosis in which bacterial infection induces a severe and acute rectocolitis, reproducing human bacillary dysentery (Shim et al, 2007). Guinea pigs were administered intrarectally either with PBS (control group), the S. flexneri 5a (WT) strain, or the non-invasive mxiD that does not assemble the T3SS needle and therefore does not secrete effectors. Eight hours post-infection, the animals were sacrificed. Both bacterial challenges induced a potent inflammatory infiltrate composed of PMN in the submucosa and laminar propria, or at proximity of the bacterial infiltrate, providing evidence for the activation of the immune response (Supplementary Fig S1). To follow HP1γ in the colon, the tissues were double stained with monoclonal anti-HP1γ or polyclonal anti-phospho S83 HP1γ (HP1γS83p) antibodies, and with DAPI to visualize DNA, then examined by fluorescent confocal microscopy. While both anti-HP1γ antibodies displayed a nuclear signal, the anti-HP1γS83p staining showed a unique punctuate pattern co-localizing with DAPI-light euchromatic regions, in agreement with the strictly euchromatic localization of HP1γS83p (Supplementary Fig S2). HP1γ expression was detected in the lamina propria and in the epithelial cells, while the most differentiated enterocytes in the upper part of villi were devoid of HP1γ staining (Fig1A). Phosphorylation at HP1γS83 was weak in the control groups (PBS), but increased strongly upon bacterial challenge with the non-invasive mxiD strain, the most intense signals being observed at the lamina propria, and the epithelial layer (Fig1A). The WT Shigella strain also induced the HP1γS83p signal, albeit weaker in intensity, as shown by the quantification of the HP1γS83p/HP1γ total ratio, with signals being mostly located at the lamina propria (Fig1A and B). Thus, we conclude that bacterial challenge promoted HP1γ phosphorylation in the colon, this effect being substantially alleviated upon invasive Shigella challenge.
Figure 1. HP1γ immunostaining in the distal colon of guinea pigs following intra-rectal challenge with Shigella flexneri strains.

- Samples of the distal colon were taken 7 h after infection with the T3SS defective mxiD Shigella pGFP(e–h), WT Shigella pGFP (i–l) or PBS treated as control (a–d) and co-stained with anti-HP1γ (a, e, and i) or anti-HP1γS83p (b, f, and j) antibodies and merged with DAPI (d, h, and l). GFP allowed for visualization of the bacterium (c, g, and k). The star indicated the submucosa, and the arrow showed the terminally differentiated columnar absorptive enterocytes devoid of HP1γ.
- Fluorescence intensity ratios between HP1γS83p and total HP1γ. HP1γS83p staining was quantified on HP1γ-positive nuclei in each field. The intensity of fluorescence for each channel of interest was measured, and the ratio of fluorescence intensity between HP1γS83p and total HP1γ signals was calculated as described in Materials and Methods. Statistical analysis was performed as described in Materials and Methods.
Shigella targets HP1γ phosphorylation at S83 through the injection of the phosphothreonine lyase OspF
We further develop an in vitro approach to identify bacterial mechanisms modulating the HP1γS83p signal in cells. Initially, we observed that activation of protein kinase C upstream of the MAPK pathway with a phorbol ester (PMA) resulted in increase phosphorylation at HP1γS83p (Fig2A, lanes 1–5). The inducibility of this phosphorylation event was substantially altered when cells were pre-infected with a wild-type (WT) Shigella flexneri strain (Fig2A, lanes 6–10). Furthermore, PMA-induced HP1γS83p was impaired by the MEK inhibitor U0126, indicating that the ERK pathway drives formation of HP1γS83p in this experimental setting (Fig2B, compare lanes 6–7). OspF is a virulence effector irreversibly inactivating MAPK though a phosphothreonine lyase activity (Li et al, 2007). To examine the impact of this bacterial effector on HP1γS83p level, we infected HeLa cells with either a WT or an ospF-deficient Shigella strain (ospF strain). Immunoblot analysis showed that the ospF mutant strain induced formation of HP1γS83p signal as efficiently as did a PMA treatment, while the Shigella WT strain had no effect (Fig2C, compare lanes 2–5 to 6–9). Overall, these data indicated that OspF blocks formation of HP1γS83p, while conversely, MAPK activators promotes this phosphorylation event. We have previously shown that OspF localizes into the nucleus of infected cells (Arbibe et al, 2007). Therefore, we hypothesized that OspF interacts with HP1γ to block a nuclear MAPK signaling cascade controlling HP1γ phosphorylation. To explore this, we immunoprecipitated endogenous HP1γ from nuclear extracts of HeLa cells transiently expressing the virulence effector. This resulted in co-immunoprecipitation of OspF (Fig2D). GST pull-down experiments indicated that this interaction was direct and dependent of the C-terminal HP1 chromoshadow domain (CSD) (Supplementary Fig S3). The functional impact of this interaction on HP1γ phosphorylation was illustrated by an extensive dephosphorylation at HP1γS83 when HP1γ was co-immunoprecipitated with OspF (Fig2E, compare lanes 1 to lanes 2). Overall, these results show that OspF directly interacted with HP1γ causing its dephosphorylation in Shigella-infected cells.
Figure 2. The virulence effector OspF blocks formation of HP1γS83p in Shigella-infected cells.

- Shigella inhibits PMA-induced formation of HP1γS83p. HeLa cells were infected with the Shigella flexneri invasive strain (WT) and stimulated or not by PMA at the indicated times. Western blots were performed with anti-HP1γS83p, anti-HP1γ, and anti-actin antibodies.
- Pharmacological inhibition of the MAPK/MSK1 pathway blocks formation of HP1γS83p. HeLa cells were pretreated for 1 h with the MEK1 inhibitor U0126 or MSK1 inhibitor H89 and stimulated by PMA. Western blots were performed with anti-HP1γS83p, anti-HP1γ, anti-H3K9me2S10p, or anti-H3 antibodies.
- HeLa cells were infected with the Shigella flexneri invasive (WT) or the ospF-deficient (ospF) strains at the indicated time or stimulated by PMA (60 min). Western blots were performed with anti-HP1γS83p, anti-HP1γ, and anti-actin antibodies.
- Immunoprecipitation of endogenous HP1γ from HeLa cells overexpressing OspF. HeLa cells were transiently transfected with the indicated plasmids. Cellular extract were immunoprecipitated with mouse IgG as a negative control or HP1γ antibodies. Western blots were performed with OspF and HP1γ antibodies. The arrow indicates the OspF signal.
- Dephosphorylation at S83 in HP1γ immunoprecipitates containing OspF. HeLa cells were transiently transfected with the indicated plasmids and cellular extract immunoprecipitated with HP1γ antibody in the absence or the presence of DNase I. Western blots were performed with anti-HP1γS83p, anti-HP1γ, anti-OspF, or anti-ERK antibodies.
Source data are available online for this figure.
OspF is chromatin-bound and modulates HP1γ association to its target genes
HP1γS83 phosphorylation was reported to target HP1γ to sites of transcriptional elongation (Lomberk et al, 2006). We therefore investigated the impact of OspF on the recruitment of HP1γ to the chromatin of its target genes. We first characterized OspF chromatin binding properties. We noticed that treating the nuclear extracts with DNase I that favors recovery of the nucleosome-associated fraction of HP1γ resulted in more efficient co-immunoprecipitation of OspF, suggesting that OspF bound chromatin (Fig1E, compare lane 2–3). To further characterize OspF chromatin association, we forced expression of OspF in HeLa cells and used chromatin immunoprecipitation (ChIP) assays with an anti-OspF antibody to show that OspF WT was bound to chromatin at the promoter of the target gene IL8 upon stimulation of the cells with TNF (Fig3A). In contrast, OspF was not detected at the promoters of the NFKBIA, or CD44 genes not regulated by the bacterial effector (Fig3A and Supplementary Fig S4). Therefore, OspF was recruited at the genes it specifically regulated. Next, we looked at the impact of OspF on HP1γ chromatin association at those genes. This was best followed using non-cross-linked HeLa chromatin in “native ChIP” assays. In these assays, infection with WT Shigella, although it resulted in approx. Tenfold transcriptional activation of the IL8 promoter (Supplementary Fig S4), caused either no change or a moderate decrease in the levels of HP1γ recruitment. Only infection with the mutant ospF strain that does not interfere with HP1γ phosphorylation induced accumulation of this protein on the IL8 promoter and coding regions. This is consistent with a requirement for HP1γS83 phosphorylation for reaching association of HP1γ with active genes. Importantly, the WT Shigella strain had no impact on HP1γ accumulation at a series of non-OspF target genes including NFKB1, NFKBIA, or CD44 (Fig3B). Overall, these results showed that OspF bound to selected chromatin sites and, in consistence with its ability to dephosphorylate HP1γ, interfered with normal association of this protein with its target genes.
Figure 3. OspF is chromatin-bound and modulates HP1γ chromatin association at its target immune genes.
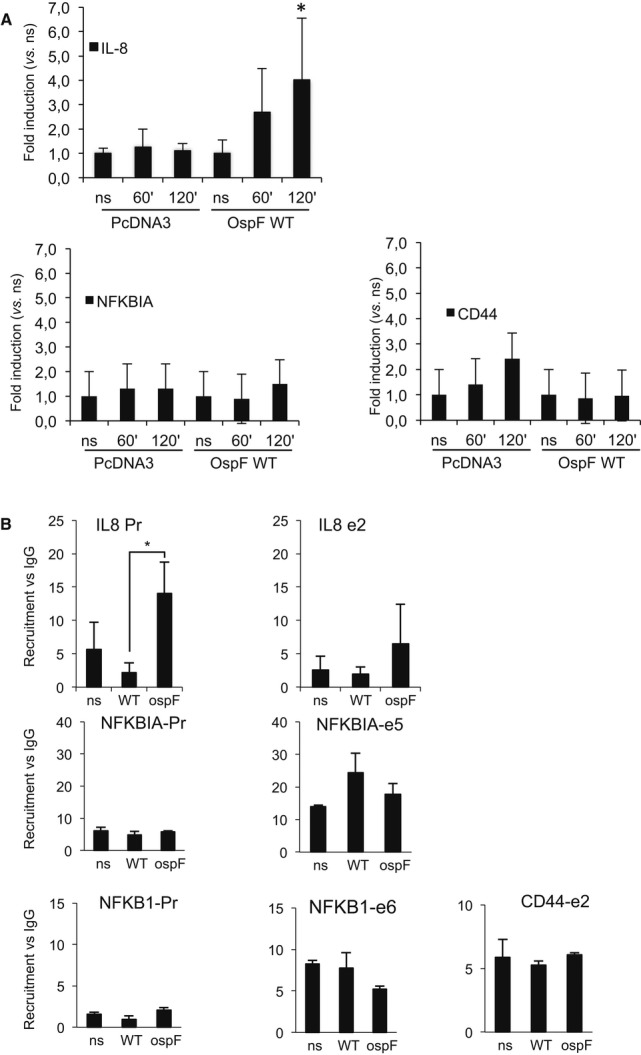
- HeLa cells were transfected with the indicated plasmids and stimulated by TNF at the indicated times. Chromatin immunoprecipitation experiments (ChIP) were performed using anti-OspF antibodies. Enrichment in chromatin was quantified by qPCR using indicated primers at the IL8, CD44, and NFKBIA genes. Enrichment values are the mean of three independent experiments. *P < 0.05 when compared to the non-stimulated cells (ns).
- OspF modulates HP1γ chromatin association. HeLa cells were infected with the Shigella flexneri invasive strain (WT) or the OspF-deficient strain (ospF). ChiPs were performed using anti-HP1γ antibodies or mouse IgG as control. Enrichment in chromatin was quantified by qPCR using indicated primers at the IL8, NFKB1, and NFKBIA genes (Pr, proximal promoter; e2, exon 2). Enrichment values are the mean of three independent experiments. Significance of the differences was estimated using Student's t-test with a threshold at 5%.
MSK1 is a new HP1 kinase regulating HP1γ phosphorylation state in Shigella-infected cells
We next sought to identify the MAPK-activated kinase responsible for HP1γS83 phosphorylation. A possible candidate was MSK1 known to phosphorylate histone H3 at serine 10 (H3S10). This kinase is immediately down-stream of ERK and its activation by phosphorylation at serine 360 (P-Ser360 MSK1) was, as expected, blocked in cells infected by a WT Shigella strain and restored when the ospF mutant strain was used instead (Fig4A). Interestingly, treatment of HeLa cells with the MSK1-specific inhibitor H89 blocked phosphorylation at both H3S10 and HP1γS83 upon activation of the MAPK pathway with PMA (Fig2B, compare lanes 3–4). Furthermore, in MSK1/2 double null mouse embryonic fibroblasts (MEFs), levels of HP1γS83p were markedly reduced when compared to WT MEFs (Fig4B, lanes 9–10). In these WT MEFs, infection with the WT Shigella strain resulted in decreased levels HP1γS83p, while this phosphorylation was induced when using the ospF mutant strain, consistent with our data collected with HeLa cells (Fig4B, compare lanes 1–3, and 4–5, and densitometric analysis). By contrast, in the MSK1/2 double null MEFs, neither infection with the WT nor the ospF mutant Shigella strain had an impact on accumulation of the HP1γS83p signal (Fig4B, compare lanes 6 to 7–10), while the ability of the ospF mutant strain to induce ERK phosphorylation remained intact (Fig4B, compare lanes 7–8 to lanes 9–10). In vitro kinase assays revealed that MSK1 directly phosphorylated both HP1α and HP1γ but not HP1β (Fig4C). Mutation to alanine of HP1γS83 and neighboring serines (S85, S87, S89, and S92) further showed that under these conditions, HP1γS83, a residue conserved in HP1α and HP1γ isoforms but not HP1β, was the predominant residue modified by MSK1 thus confirming the specificity of the reaction (Fig4D, compare lane 4 to lanes 5–8). Importantly, OspF did not inhibit the ability for MSK1 to induce HP1γ phosphorylation in an in vitro kinase assay (Supplementary Fig S5). Overall, these data indicated that the MSK kinases play a major role in the modulation of the HP1γS83p signal in Shigella-infected cells and that OspF targeting of this nuclear MAPK signaling pathway was the main mechanism by which this bacterial effector altered HP1γ phosphorylation state.
Figure 4. The MSK1 kinase drives HP1γS83p formation in Shigella-infected cells.

A OspF leads to the accumulation of a dephosphorylated and inactive form of MSK1 into Shigella-infected cells. HeLa cells were infected with the Shigella flexneri invasive (WT) or the ospF-deficient (ospF) strains at the indicated times or PMA (60 min). Western blots were performed with anti-MSK1S360p or MSK1 antibodies.
B The MSK1 kinase modulates the HP1γS83p signal in Shigella-infected cells. Primary MEFs WT and MSK1/2 double null (MEF MSK1/2−/−) were infected with the Shigella flexneri invasive strain (WT) or the OspF-deficient strain (ospF) at the indicated times. Western blots were performed with anti-HP1γS83p, anti-HP1γ, anti-phospho-ERK, and anti-actin antibodies. Lower panel: Densitometric quantification of the signal obtained by Western blot. For each time point, the results are expressed as the level of HP1γS83p signal related to the total amount of HP1γ.
C, D Kinase assays showing that MSK1 directly phosphorylates HP1γ at the S83 residue. Purified active MSK1 was incubated with histones, GST or GST-HP1α, GST-HP1β, GST-HP1γ or the indicated GST-HP1γ mutant fusion proteins. Kinase assay was performed at 30°C during 1 h in the presence of [γ-32P]-ATP followed by autoradiography.
Source data are available online for this figure.
These observations prompted us to probe for an interaction between MSK1 and HP1γ. Co-immunoprecipitation experiments with either anti-P-Ser360 MSK1 or anti-HP1γ antibodies in HeLa cell extracts showed that HP1γ, ERK, and activated MSK1 form a tri-complex enriched upon PMA stimulation (Fig5A). Use of an anti-HP1γS83p antibody strongly improved detection of ERK in the co-immunoprecipitate, suggesting that in the tri-complex, HP1γ is phosphorylated (Fig5B, compare lane 3–6). Consistent with this, confocal microscopy showed a high coincidence of immunostaining between HP1γS83p and the active form of MSK1 (P-Ser360 MSK1) (mean overlap coefficient of 0.83, the mean value obtained with an irrelevant protein such as p53 being < 0.1, Fig5C and Supplementary Fig S6A). Also, a co-localization was detectable upon bacterial infection between MSK1 and HP1γS83p when the HP1γS83p signal was triggered by the ospF mutant strain (mean overlap coefficient of at 0.84, Fig5D and Supplementary Fig S6B). Pull-down experiments finally showed that GST-MSK1 directly interacted with full-length recombinant HP1α and HP1γ proteins, in a CSD-dependent manner (Supplementary Fig S7). Overall, these data identified MSK1 as a novel kinase phosphorylating HP1γ at S83 in the context of a molecular tri-complex between the phosphorylated pool of HP1γ and the ERK/MSK1 kinases.
Figure 5. MSK1 forms a molecular complex with the phosphorylated pool of HP1γ in cells.

- PMA stimulation leads to the formation of molecular tri-complex between the HP1γ and the ERK/MSK1 kinases. HeLa cells were stimulation by PMA (60 min), and cellular extracts were immunoprecipitated with mouse IgG as a negative control or anti-MSK1S360p or anti-HP1γ antibodies. Western blots were performed with anti-MSK1, anti-ERK, and anti-HP1γ antibodies.
- The phosphorylated pool of HP1γ pulls down the ERK kinase. Cellular extracts from HeLa cells were immunoprecipitated with mouse IgG as a negative control or anti-HP1γS83p, or anti-HP1γ antibodies. Western blots were performed with anti-ERK and anti-HP1γS83p antibodies.
- Confocal microscopy showing high coincidence of immunostaining between HP1γS83p and active MSK1. Immunofluorescence was performed with anti-HP1γS83p (red) and anti-MSK1S360p (green) antibodies.
- Confocal microscopy showing the immunostaining between HP1γS83p (red) and MSK1 upon infection with the WT and ospF mutant strains (green).
Source data are available online for this figure.
HP1γ promotes MSK1 chromatin association and regulates MAPK-dependent immune gene expression
The identification of a MSK1-HP1γ molecular complex led us to hypothesize that HP1γ stabilizes MSK1 to the chromatin template. To investigate this, we isolated nuclear-soluble and chromatin-bound fractions from either WT or HP1γ null MEFs, as previously described (Zhao et al, 1998). PMA stimulation of the cells resulted in rapid accumulation of a pool of MSK1 in the chromatin-bound fraction with a peak at 30 min, and this phenomenon was strongly intensified in the presence of HP1γ (Fig6A, lanes 9–16). At the same time, a pool of HP1γ phosphorylated at S83 also appeared in the chromatin-bound fraction, consistent with this compartment being highly transcriptionally active (Xu et al, 1986). A depletion experiment with siRNAs finally confirmed that recruitment of MSK1 to the IL8 promoter in HeLa cells was also dependent on HP1γ (Fig6B and Supplementary Fig S8). Altogether, these results indicated that HP1γ was required for optimal recruitment of MSK1 to its target genes and thereby played a not previously described role in assisting MAPK signal transduction.
Figure 6. HP1γ bridges MSK1 at the chromatin and regulates the expression of specific genes.
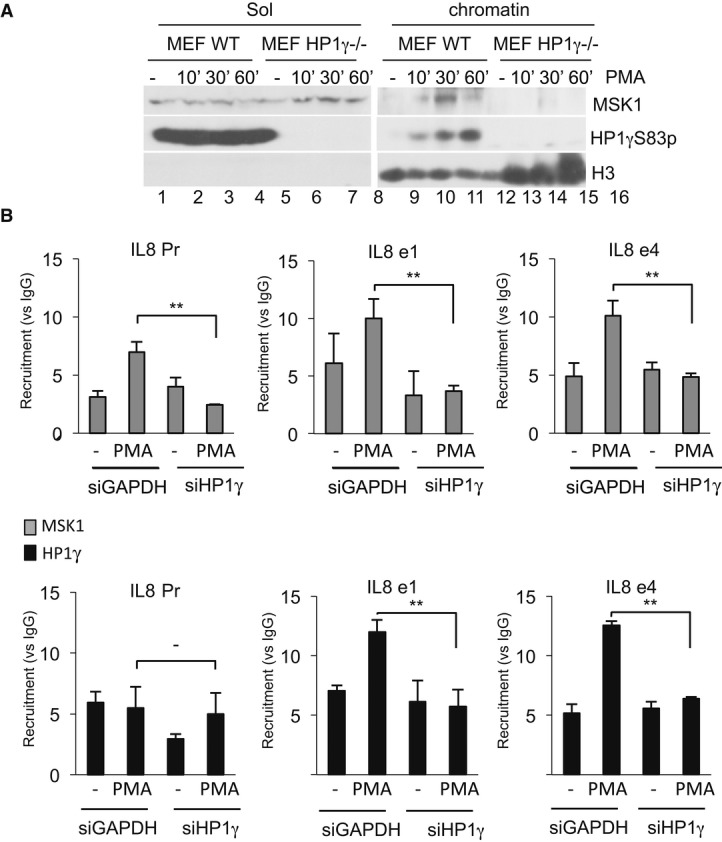
- WT and HP1γ null mouse fibroblasts were stimulated by PMA at the indicated times. Nuclear extract were processed as indicated in Materials and Methods. Western blots were performed with anti-MSK1, anti-HP1γS83p, and anti-histone H3 antibodies.
- HeLa cells were transfected with siHP1γ or, as a control, siGAPDH and stimulated by PMA (40 min). ChIPs were performed using anti-HP1γ or anti-MSK1 antibodies, or unrelated mouse IgGs as a control. Results are expressed as fold induction versus IgG for each point (**P < 0.01). Enrichment values are the mean of two independent experiments. Significance of the differences was estimated using Student's t-test with a threshold at 5%.
Source data are available online for this figure.
To acquire a more global insight in the role of HP1γ in signal transduction, we established mouse embryonic fibroblast (MEF)-derived HP1γ null cell lines stably re-complemented with either wild-type HP1γ, or a HP1γS83A mutant not phosphorylatable at S83 (Supplementary Fig S9A). These cells were then used to examine the effect HP1γ/PMA stimulation on genome-wide transcription by next generation sequencing. Approximately 2,000 transcripts were up- or down-regulated 1.5-fold or more when re-complemented with wild-type HP1γ (Supplementary Fig S9B and C). As anticipated, many of the down-regulated transcripts were encoded by repeated DNA sequences known to be silenced by HP1 proteins. Transcripts encoded by the interferon-inducible IFI44, IFIT3, and OAS1b genes previously described as HP1γ targets were also repressed by re-expression of HP1γ (Lavigne et al, 2009) (Supplementary Fig S9D). These observations that were confirmed by RT-PCR validated our cellular system as suitable for the study of the impact of HP1γ on transcription.
A large population of the transcripts regulated by HP1γ WT was also regulated by HP1γS83A, although approximately 500 transcripts were affected only by HP1 WT, and 1,761 additional transcripts were affected only by HP1γS83A, confirming that the phospho-mutant was affected in its activity (Supplementary Fig S9B and C). Upon stimulation of the cells with PMA, 973 transcripts were affected only in the absence of WT HP1γ (Fig7A). As a large fraction of these transcripts were encoded by DNA repeats, this observation illustrates the importance of HP1 proteins in keeping vestigial viral sequences in check. Inversely, mapping of annotated genes to KEGG pathways interestingly showed that several pathways remained unaffected by PMA stimulation in the absence of HP1γ (Fig7B). For example, only in the presence of HP1γ did we observe up-regulation of a significant number of genes within the DNA Replication KEGG pathway. A similar observation was made for the Pyrimidine Metabolism or Cell Cycle KEGG pathways. In addition, when pathways were affected by PMA in the absence of HP1γ, re-introduction of HP1γ seemed to oppose this effect. This was in particular seen for MAP Kinase Signaling and Base Excision Repair KEGG pathways. Finally, we noted that HP1γ had a very coherent effect within each significantly affected pathway, causing either solely activation or solely repression (with the exception of the p53 signaling pathway). Together, these observations suggested that HP1γ broaden the impact of PMA stimulation while simultaneously fine-tuning its specificity and attenuating the amplitude of its effects. We noted also that HP1 WT and HP1γS83A had similar effects on most pathways, suggesting that the S83A mutation only partially altered the ability of HP1γ to respond to PMA.
Figure 7. HP1γ affects the transcriptional impact of PMA.

- Venn diagram showing the number of transcripts regulated by PMA stimulation in HP1γ null (blue), HP1γ WT (red), and HP1γS83A (green) MEF-derived cell lines (≥ 1.5-fold change in expression, ≥ 20 reads, P < 0.05).
- KEGG functional categories significantly enriched (P < 0.05) for genes differentially expressed in the indicated conditions, depicted according to their P-value [−log10(P-value)].
- Fold induction by PMA of a subset of OspF target genes (≥ 2-fold change in expression in HP1γ null) in each of the indicated cell lines. Data and P-values are provided in Supplementary Table S1.
To particularly explore the effect of HP1γ on OspF targets, we selected 64 genes previously characterized as activated by S. flexneri infection only in the absence of OspF, having unambiguous mouse homologs, and being transcriptionally affected in the MEF HP1γ null cells upon re-complementation with either HP1γ or HP1γS83A. A majority of these genes were unaffected by the reintroduction of HP1γ at basal levels (Supplementary Table S1, green lane). In contrast, all these genes except 3 were transcriptionally activated by the PMA treatment, illustrating the similarities between PMA stimulation and ospF infection, and suggesting that many MAPK targets are conserved in the MEF-derived cells (Supplementary Table S1). In agreement with the conclusions from the KEGG pathway analysis, HP1γ opposed the transcriptional activation of a large majority of the 64 genes upon stimulation of the cells by PMA (compare red and blue bars Fig7C). A similar effect was observed with HP1γS83A (compare red and green bars Fig7C). These effects were validated by RT-qPCR for a series of genes (Supplementary Table S2). This series of validation experiments also allowed us to further verify on a subset of these genes that there was a similarity between the response of the cells to PMA and that observed upon S. flexneri infection (Supplementary Table S2).
Discussion
In this study, we provide the first evidence that bacteria such as the enteropathogen Shigella flexneri control the epigenetic of their host by altering the activity of a chromatin reader, the HP1 protein. In an in vivo model of rectocolitis, we showed that the non-invasive—albeit proinflammatory—mxiD Shigella mutant promotes pronounced HP1γ phosphorylation in the colon when compared to wild-type Shigella. A phosphoproteome analysis of Toll-like receptor-activated macrophages reported that LPS caused major dynamic changes in the cell phosphorylation state, with sub-data analysis indicating multiple phosphorylation sites on HP1γ, including S83 (Weintz et al, 2010). In the context of our in vivo model of colonic infection, one can reasonably conceived that bacterial challenge might directly—through LPS release—or indirectly initiated proinflammatory signaling cascade(s), leading to increase HP1γ phosphorylation at multiple residues, including the S83 residue monitored in our study. As such, phosphorylation at S83 (S93 in mice) can be envisaged as a biomarker reflecting the activation of pro-inflammatory pathways targeting HP1γ at multiple phosphorylation sites. The modulation of this biological event by invasive Shigella was somehow consistent with the known ability of Shigella to develop strategies dampening inflammation for its own survival. From our in vitro analysis, we identified the T3SS virulence effector OspF, a phosphothreonine lyase targeting nuclear MAPK signaling, as the main cause leading to HP1γ dephosphorylation. Our data further indicated that OspF exhibited gene-specific chromatin-bound properties and directly interacted with HP1γ, thereby dephosphorylating and reducing HP1γ association to the chromatin template. HP1 proteins are histone code readers that recognize and specifically bind methylated histone H3 through their conserved chromodomain. HP1 was first discovered in Drosophila as a dominant suppressor of position-effect variegation and a major component of heterochromatin that represses gene activity (Eissenberg et al, 1990). However, these proteins are extensively modified in a manner analogous to histones, by phosphorylation, acetylation, methylation, formylation, sumoylation, and others (LeRoy et al, 2009), and experimental evidence indicates that post-translational modifications like phosphorylation and sumoylation at the central region of the protein (the hinge regions), which is rich in serine and lysine residues, modulate their sub-nuclear location and bioactivity. For example, SUMOylated lysines in HP1α were shown to target this HP1 to pericentric heterochromatin. Inversely, HP1γ phosphorylation at S83 defined a strictly euchromatic subpopulation, associated with the elongated form of the RNAPII and with an abrogated silencing activity in a transcriptional reporter assay (Lomberk et al, 2006; Maison et al, 2011). Various consensus sites for protein kinases such as casein kinases, PKA and PKC, have been identified and mutation of the corresponding phospho-residues were shown to reduce HP1 silencing activities in Drosophila (Zhao et al, 2001). Overall, these studies suggested that multiple kinase pathways might phosphorylate and therefore modulate the bioactivity of this epigenetic regulator. However, the regulation of HP1γ by HP1 kinases remained poorly characterized in living cells and virtually unexplored in the context of host–pathogen interaction. Our work identified MSK1 as an HP1 kinase, playing a major role in the modulation of the HP1γS83p signal in Shigella-infected cells. MSK1 is a nuclear kinase phosphorylating transcription factors such as CREB, ATF1, and the p65 subunit of NF-κB as well as components of the nucleosomal response like histone H3 and HMG-14 (Wiggin et al, 2002; Vermeulen et al, 2003). Consequently, in response to mitogen or pro-inflammatory stimuli, MSK1 positively regulates the expression of early responsive genes and a subset of immune NF-κB genes such as IL6. On the other hand, MSK1/2 invalidation demonstrated that MSK1/2 is important for the expression of anti-inflammatory molecules such as the MAPK phosphatase DUSP1 (MKP-1) and the cytokine IL-10, thereby limiting the extend of the inflammatory response in various mice models of acute and chronic inflammation (Ananieva et al, 2008; Bertelsen et al, 2011). Therefore, MSK1 transcriptionally regulates the expression of various classes of immune genes, which somehow reflects its ability to interact with and phosphorylate various transcriptional regulators. Our work showed that active MSK1 formed a tight nuclear complex with the epigenetic regulator HP1γ that aimed at stabilizing MSK1 on the chromatin template of its immune target genes. Such ability for HP1 to “bring” a kinase to the chromatin template is reminiscent of the S. pombe HP1 homologue Swi6 that recruits the protein kinase Hsk1-Dfp1 to positively regulate replications origins at pericentromeric region (Bailis et al, 2003). Therefore, our work extends the function of HP1γ as a chromatin integrator for nuclear kinases in higher eukaryotes.
A direct functional consequence of the MSK1/HP1γ molecular interplay was the induction of HP1γ phosphorylation at S83 that was impeded by the virulence effector OspF. Once injected by the T3SS into host cells, OspF bound chromatin at its target genes, providing a mechanisms sustaining gene selectivity, while the molecular identity of the anchoring system remains elusive. Our study identified HP1γ as a new OspF interactant and showed that OspF dephosphorylated HP1γ chiefly by inactivating the MAPK/MSK1 pathway. Consistent with the requirement for HP1γS83 phosphorylation to reach association of HP1γ to sites of transcription elongation, OspF uncoupled recruitment of HP1γ from transcriptional activation. Thus, nuclear MAPK inactivation by OspF has two consequences at the chromatin level, one previously demonstrated consisting in decreasing the content of histone marks permissive for transcription (reduction in the content of phospho-acetylated histone H3) (Arbibe et al, 2007), the second being to impair HP1γ signaling at OspF target gene (Fig8, model A/B). We interrogated the impact of HP1γ and its phosphorylation on gene regulation by RNA seq, using HP1γ null cell lines rescued with HP1γ either WT or with a S83A mutation. This showed that, as a “versatile” transcriptional regulator, HP1γ is operating either as a repressor or a transcriptional activator. However, the RNAseq data also indicated that within a functional group, all genes affected by HP1γ were regulated in a same way (all up, or all down) illustrating the very coherent impact of this protein. Furthermore, HP1γ re-complementation resulted in a significant enrichment in targets related to regulation of cell cycle and proliferation, purine metabolism, DNA replication and repair, these pathways being not statistically enriched in the PMA-stimulated HP1γ null cells. Previous reports have detected high levels of HP1γ protein associated with enhanced cell proliferation and oncogenesis in colon, breast, and cervical cancers (Takanashi et al, 2009; Abe et al, 2011; Slezak et al, 2013). Also, HP1γ S83 was reported enriched at the mitotic spindle, suggesting an additional role in proper mitotic cell division (Grzenda et al, 2013). Our in vivo analysis indicated that terminally differentiated columnar absorptive enterocytes were devoid of HP1γ, while the expression was detectable in the basal level of the crypt that contained the proliferative cell compartment. Overall, these observations convey the idea that HP1γ is an important factor for cell division, with a potential impact on crypt regeneration, especially in the context of immune stimuli, which triggers HP1γ phosphorylation in the colon.
Figure 8. Model: OspF targets the nuclear MAPK/HP1γ signaling pathway and reveals the function of the MSK1/HP1γ cross talk.

- In unstimulated cells (ns), HP1 is brought to the promoter by histone H3K9 methylation and acts as a transcriptional repressor.
- Shigella flexneri infection results in activation of the MAPK/MSK1 pathway, but this activation is rapidly dampened by OspF that dephosphorylates ERK. The combined effect of these two events is a moderate transcriptional activation of IL8. This causes HP1γ to leave the promoter, while inactivation of ERK prevents it from getting phosphorylated and join the transcription machinery. Consequently, accumulation of HP1γ at the IL8 gene drops.
- Upon ospF Shigella infection or treatment with PMA, the MAPK nuclear pathway is fully triggered, levels of histone H3 marks permissive for transcription are increased (H3S10), and the transcription machinery is recruited. Activation of the MAPK nuclear pathway induces HP1γ phosphorylation and allows for its recruitment to IL8 and other similarly regulated immune genes.
- In MEF KO cells, the loss of HP1γ eliminates an important checkpoint in the control of immune gene activation, leading to a deregulated possibly excessive gene expression in response to MAPK activation.
We further looked at the impact of HP1γ re-complementation on immune gene expression, with a particular focus on the OspF target genes that we identified in an earlier study (Arbibe et al, 2007). As a global picture, we found that both HP1γ WT and HP1γS83A protein exerted similarly effect on gene expression, notably by reducing gene inducibility in response to PMA stimulation. Clearly, in the context of PMA or bacterial stimulations, a single phospho-mutation was not sufficient to abrogate the biological impact of HP1γ, this suggesting that multiple phosphorylation sites modified upon cell activation control the biological activity of the protein. In this context, an extensive proteomic analysis is required to elucidate the additional phospho-residues or modifications involved in the control of HP1 bioactivity and to determine how these new modifications are targeted upon Shigella infection.
Our model predicts that HP1γ recruitment is required for fine-tuning of immune gene expression upon cell activation (Fig8, Model C). HP1γ was reported to decrease the RNAPII elongation rate at the CD44 gene to facilitate inclusion of alternative exons (Saint-André et al, 2011), and it is tempting to speculate that HP1γ slowing down RNAPII might alter gene promoter activity by affecting the speed with which the polymerase moves away from the promoter and makes room for the binding of the next polymerase. Importantly, our model implies that loss of HP1γ eliminates an important checkpoint in the control of immune gene activation, leading to the super-induction of the gene (Fig8, Model D).
In conclusion, our work showed that bacteria such as the enteropathogen, Shigella flexneri, in addition to their effect on histone modifications, controls the epigenetic of their host by altering the activity of chromatin readers like HP1. The development of in vivo models targeting HP1γ will be essential for understanding the transcriptional impact of this epigenetic regulator in the context of infectious diseases. Nevertheless, our study illustrates the ability for some bacterial effector to modulate chromatin-borne information, providing invaluable tools to integrate these events in the field of innate immunity. As such, the modulation of HP1γ phosphorylation upon bacterial challenge in the colon suggested functional links with mucosal defense or repair. Thus, the development of in vivo approaches targeting HP1γ and its phosphorylation in the intestine is the next challenge to be taken up to solve these important physiological issues.
Materials and Methods
Cell culture and bacterial strains
Mouse embryonic fibroblasts (MEFs) from HP1γ null mice were provided by Dr. Florence Cammas (IRCM, Montpellier, France). Stable recomplementations of the MEF HP1γ null mice were obtained by retroviral transduction according to the method of Nakatani and Ogryzko (Nakatani & Ogryzko, 2003), using the retroviral pOZ-N backbone vector. The human HP1γ cDNA was subcloned between XhoI and NotI restriction sites of the pOZ-N vector and the S83A mutant obtained by the QuickChange Site-Directed Mutagenesis kit (Agilent Technologies). MEF MSK1/2 null mice were provided by Dr. Simon Arthur (School of Life Sciences, University of Dundee, UK). MEF and HeLa cells were cultured in DMEM medium supplemented with 10% fetal calf serum.
Bacterial strains and cellular infection
The wild-type Shigella flexneri serotype 5a strain (WT), the ospF-deficient (ospF), and the T3SS defective mxiD Shigella mutant strains have been previously described (Allaoui et al, 1993; Arbibe et al, 2007). Cells were infected with the Shigella strains at a multiplicity of infection (MOI) of 35. Infection was initiated by synchronization step for 10 min. After incubation for 15 min at 37°C, gentamycin was added in the medium to 50 μg/ml to kill extracellular bacteria.
Cytokines, inhibitors, and antibodies
Recombinant human TNF-α, phorbol 12-myristate 13-acetate (PMA), SB203580, and PD98059 were purchased from Alexis Corporation (Lausen, Switzerland); U0126 was supplied by Promega Biotec (Madison, WI), and H89 obtained from Calbiochem-Novabiochem International (San Diego, CA). The following antibodies were used: HP1γ antibody (1G6, Euromedex) and anti-ERK antibody (4696, Cell Signaling) for immunoblot, anti-HP1γS83p antibody (ab45270), anti-phospho Ser360-MSK1 (ab81294) from Abcam, and p53 antibody (DO1, Santa Cruz). For immunoprecipitation and ChIP analysis, HP1γ antibody (42s2, Upstate) and MSK1 antibody from Bethyl (A302-747A) were used and the quality control of the antibodies used for ChIP is provided in Supplementary Fig S10A. Quality control of the HP1γ antibodies was also checked by the loss of signal detection upon knockdown of the corresponding protein in the immunoprecipitate and input (Supplementary Fig S10B). The specificity of the polyclonal OspF antibody has been previously described (Arbibe et al, 2007).
Plasmids and in vitro interaction experiments
pRK5myc-OspF encoding Myc-tagged OspF and its catalytically inactive form (H104L) have been previously described (Arbibe et al, 2007). GST-HP1 fusions proteins were constructed in pGEX3X and HA-tagged HP1 fusions in pET41 plasmids. HA-tagged HP1α ΔDim is a truncation mutant lacking the last helix of the chromoshadow domain (HP1α 1–157). GST-HP1γ mutants were obtained using the QuickChange Site-Directed Mutagenesis kit (Stratagene). pGEX4T2-OspF encoding GST-OspF was previously described (Arbibe et al, 2007). Purified active MSK1 (millipore) was coupled to glutathione–sepharose beads. Pull-down experiments were performed in ELB buffer (50 mM HEPES pH 7, 250 mM NaCl, 1 mM EDTA, 0.1% NP-40, 1× Complete protease inhibitor cocktail from Roche). Bound proteins were eluted in 100 mM Tris pH 8, 20 mM glutathione, resolved by SDS-PAGE and detected by Western blotting.
siRNA transfections
siRNAs targeting HP1γ were described earlier (Mateescu et al, 2008). The siRNAs targeting GAPDH were designed by Dharmacon. All siRNAs were synthesized as ON-TARGETplus grade from Dharmacon. siRNAs were transfected with Lipofectamine RNAiMAX (Invitrogen) for 72 h according to the manufacturer's instructions.
In vitro kinase assay
GST, GST-HP1γ WT, and GST-HP1γ mutants were produced and purified according to the manufacturer's instructions (GE Healthcare Life Sciences). A 5 μg aliquot of the purified proteins was incubated at 30°C for 20 min in kinase buffer (50 mM Tris pH 7.4, 10 mM MgCl2, 1 mm DTT), supplemented with 100 μM ATP ([γ-32P] ATP) and 100 ng of purified PKA or purified active MSK1 (Millipore), Phosphorylated GST or fusion proteins were subjected to SDS–PAGE and phosphate incorporation analyzed using PhosphorImager technology.
In vitro OspF assay
For in vitro competition assays between OspF and MSK1, 120 ng of GST-HP1γ was phosphorylated by 200 ng of active MSK1 (14-548; Millipore) for 1 h at 30°C in the presence of 0, 1, or 2 μg of GST-OspF in 30 μl MSK1 reaction buffer as described by the manufacturer, followed by immunoblotting. Control reactions were performed in MSK1 reaction buffer on 150 ng of phosphorylated GST-ERK1 (14-439; Millipore).
Analysis of mRNA expression
Total RNA was extracted using commercially available kits (Qiagen or Macherey-Nagel) with DNase treatment. Reverse transcription was carried out with SuperScript III (Invitrogen) and random hexanucleotides for 1 h at 50°C on 1 μg RNA, quantified with a NanoDrop (Thermo Scientific). Real-time qPCR was carried out on a Stratagene Mx3005p with Brilliant II SYBR Green kits (Stratagene) according to the manufacturer's instructions. Primer sequences are described in Supplementary Table S3.
NativeChIP
Non-cross-linked chromatin was isolated as described previously (Saint-André et al, 2011), digested with TURBO DNase and sonicated (three cycles of 10 s on a Diagenode Bioruptor). For nativeChIP, the chromatin was incubated for 3 h at 4°C with 1 μg of indicated antibodies (or non-immune IgG as negative control). Saturated magnetic beads coupled to protein A or G or to anti-rabbit or anti-mouse antibodies (Dynabeads) were used to recover the complexes. After 2 h of incubation, beads were washed extensively and nucleic acids were purified. DNA bound to the immunoprecipitated proteins was quantified by qPCR. Primer sequences are described in Supplementary Table S3.
Immunofluorescence staining
The EasyLink Cy5 Conjugation kit was used to conjugate the polyclonal anti-phospho Ser360-MSK1 with Cy5 (Abcam, ab102877). Immunofluorescence analysis on HeLa cells was performed after 20 min of fixation with paraformaldehyde and 10 min of permeabilization with 0.5% Triton X-100 in PBS. Cells were first labeled with the polyclonal anti-HP1γS83p (ab45270) and in a second step with the Cy5 phospho MSK1 conjugated antibody. The goat anti-human MSK1 antibody was purchased from R&D Systems (AF2518). The images were acquired and analyzed by confocal microscopy. The analysis is based on the evaluation of color components of the selected pair of channels (red, green) in a selected region of interest (the nucleus). The background was corrected, and a coefficient was produced by the software to estimate the degree of co-localization. The approximate estimation of co-localization was presented in a two-dimensional scatter gram.
Intrarectal S. flexneri inoculation and colon immunostainings
Young guinea pigs (Hartley, < 150 g) were infected intrarectally with PBS as control, or 1010 CFU exponentially grown Shigella flexneri pGFP or mxiD Shigella pGFP as described (Shim et al, 2007). In total, 15 guinea pigs were assigned randomly to three groups (each group 5 animals) and experiments were performed on two independent occasions. Infection occurred during 7 h before animals were sacrificed and the distal colon collected. Following 60 min PFA 4% fixation, infected guinea pig colon samples were washed in PBS, incubated at 4°C in PBS containing 12% sucrose overnight, then in PBS with 18% sucrose overnight, and frozen in optimum cutting temperature (OCT) formulation (Sakura) on dry ice. Seven-micrometer sections were obtained using a cryostat CM-3050 (Leica). Sections were permeabilized with 0.1% Triton X-100 and 1% saponine in PBS for 15 min, and washed three times in PBS. Samples were fixed with 3% BSA and 0.1% Triton X-100 in PBS for 1 h, then washed three times in PBS. Primary antibodies used were mouse monoclonal to HP1γ (1:500, ab56978, Abcam), rabbit polyclonal to phosphoserine 83 HP1γ (1:500, ab45270, Abcam), diluted in 0.1% Triton X-100 and 1% BSA in PBS, and incubated for 90 min. Sections were washed three times in PBS, and secondary antibodies were used: anti-mouse cy5 (1:1,000; GE Healthcare), anti-rabbit cy3 (1:1,000; Jackson ImmunoResearch), diluted in 0.1% Triton X-100 and 1% BSA in PBS, and incubated for 1 h. DAPI (1:2,000; Life Technologies) was also added to stain nuclei and anti-myeloperoxidase (MPO) antibody to detect polymorphonuclear neutrophils. Fluorescently labeled tissues were observed using a laser-scanning confocal microscope (Leica SP5). Image analysis was performed using ImageJ software. Regions of interest (ROIs) were drawn manually around HP1γ positive nuclei on each field. Three to 6 fields were analyzed per sample, and 25–37 ROIs were drawn per field (same number of ROI per field per condition). The intensity of fluorescence for each channel of interest was measured, and the ratio of fluorescence intensity between phosphoserine 83 HP1γ and total HP1γ was calculated. Unpaired t-test with Welch correction was used to analyze the ratio of fluorescence intensity between phosphoserine 83 HP1γ and total HP1γ. Values of P < 0.05 were considered statistically significant.
RNA sequencing
MEF-derived cells either HP1γ null or re-complemented with either HP1γ WT or HP1γS83A were either not stimulated or exposed to PMA for 1 h. Total RNA was depleted from ribosomal RNA then sequenced by Next Generation sequencing with a 50+ bp standard. Sequencing was performed by the IGBMC Microarray and Sequencing platform, member of the France Genomique program. RNA Seq data analysis was performed by GenoSplice (http://www.genosplice.com). First, reads were aligned onto the mouse genome (mm9) using STAR v.2.3.0, with an exon–exon junction database built using annotations from version 2013_2 of FAST-DB (see http://www.easana.com). For each gene present in FAST DB v2013_2, reads aligning on constitutive regions (that are not prone to alternative splicing) were counted. Based on these read counts, normalization and differential gene expression were performed using DESeq (v1.12.0 on R v3.0.0). EASANA was used for visualization of results (http://www.easana.com). The RNA seq data have been submitted to the GEO database, and the accession number is GSE60283.
Acknowledgments
We thank Dr. Simon Arthur for providing us the MEF MSK1/2 null mice (School of Life Sciences, University of Dundee, UK). We also thank Dr. Eric Batsché for discussion and technical support. The work was supported by grant from the «Agence National de la Recherche» (ANR-MIME). Habiba Harouz received a fellowship from the «fondation pour la recherche médicale» (FRM).
Author contributions
All the authors contributed to the discussion of this work. HH and CR designed, performed, and analyzed the experiments. BMM and BM performed the in vivo experiments and immunolabeling. CM performed an in-depth analysis of the RNA seq data. LA conceived the project, performed experiments, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Abe K, Naruse C, Kato T, Nishiuchi T, Saitou M, Asano M. Loss of heterochromatin protein 1 gamma reduces the number of primordial germ cells via impaired cell cycle progression in mice. Biol Reprod. 2011;85:1013–1024. doi: 10.1095/biolreprod.111.091512. [DOI] [PubMed] [Google Scholar]
- Allaoui A, Sansonetti PJ, Parsot C. MxiD, an outer membrane protein necessary for the secretion of the Shigella flexneri lpa invasins. Mol Microbiol. 1993;7:59–68. doi: 10.1111/j.1365-2958.1993.tb01097.x. [DOI] [PubMed] [Google Scholar]
- Ameyar-Zazoua M, Rachez C, Souidi M, Robin P, Fritsch L, Young R, Morozova N, Fenouil R, Descostes N, Andrau JC, Mathieu J, Hamiche A, Ait-Si-Ali S, Muchardt C, Batsché E, Harel-Bellan A. Argonaute proteins couple chromatin silencing to alternative splicing. Nat Struct Mol Biol. 2012;19:998–1004. doi: 10.1038/nsmb.2373. [DOI] [PubMed] [Google Scholar]
- Ananieva O, Darragh J, Johansen C, Carr JM, McIlrath J, Park JM, Wingate A, Monk CE, Toth R, Santos SG, Iversen L, Arthur JS. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol. 2008;9:1028–1036. doi: 10.1038/ni.1644. [DOI] [PubMed] [Google Scholar]
- Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol. 2007;8:47–56. doi: 10.1038/ni1423. [DOI] [PubMed] [Google Scholar]
- Bailis JM, Bernard P, Antonelli R, Allshire RC, Forsburg SL. Hsk1-Dfp1 is required for heterochromatin-mediated cohesion at centromeres. Nat Cell Biol. 2003;5:1111–1116. doi: 10.1038/ncb1069. [DOI] [PubMed] [Google Scholar]
- Bertelsen T, Iversen L, Riis JL, Arthur JSC, Bibby BM, Kragballe K, Johansen C. The role of mitogen- and stress-activated protein kinase 1 and 2 in chronic skin inflammation in mice. Exp Dermatol. 2011;20:140–145. doi: 10.1111/j.1600-0625.2010.01153.x. [DOI] [PubMed] [Google Scholar]
- Brennan DF, Barford D. Eliminylation: a post-translational modification catalyzed by phosphothreonine lyases. Trends Biochem Sci. 2009;34:108–114. doi: 10.1016/j.tibs.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, James TC, Foster-Hartnett DM, Hartnett T, Ngan V, Elgin SC. Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc Natl Acad Sci USA. 1990;87:9923–9927. doi: 10.1073/pnas.87.24.9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzenda A, Leonard P, Seo S, Mathison AJ, Urrutia G, Calvo E, Iovanna J, Urrutia R, Lomberk G. Functional impact of Aurora A-mediated phosphorylation of HP1γ at serine 83 during cell cycle progression. Epigenetics Chromatin. 2013;6:21. doi: 10.1186/1756-8935-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Workman JL. The changing faces of HP1: from heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. BioEssays. 2011;33:280–289. doi: 10.1002/bies.201000138. [DOI] [PubMed] [Google Scholar]
- Lavigne M, Eskeland R, Azebi S, Saint-André V, Jang SM, Batsche E, Fan H-Y, Kingston RE, Imhof A, Muchardt C. Interaction of HP1 and Brg1/Brm with the globular domain of histone H3 is required for HP1-mediated repression. PLoS Genet. 2009;5:e1000769. doi: 10.1371/journal.pgen.1000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Weston JT, Zee BM, Young NL, Plazas-Mayorca MD, Garcia BA. Heterochromatin protein 1 is extensively decorated with histone code-like post-translational modifications. Mol Cell Proteomics. 2009;8:2432–2442. doi: 10.1074/mcp.M900160-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou J-M, Shao F. The phosphothreonine lyase activity of a bacterial type III effector family. Science. 2007;315:1000–1003. doi: 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
- Lomberk G, Bensi D, Fernandez-Zapico ME, Urrutia R. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat Cell Biol. 2006;8:407–415. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- Maison C, Bailly D, Roche D, Montes de Oca R, Probst AV, Vassias I, Dingli F, Lombard B, Loew D, Quivy JP, Almouzni G. SUMOylation promotes de novo targeting of HP1α to pericentric heterochromatin. Nat Genet. 2011;43:220–227. doi: 10.1038/ng.765. [DOI] [PubMed] [Google Scholar]
- Mateescu B, Bourachot B, Rachez C, Ogryzko V, Muchardt C. Regulation of an inducible promoter by an HP1beta-HP1gamma switch. EMBO Rep. 2008;9:267–272. doi: 10.1038/embor.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurkiewicz P, Thomas J, Thompson JA, Liu M, Arbibe L, Sansonetti P, Holden DW. SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol Microbiol. 2008;67:1371–1383. doi: 10.1111/j.1365-2958.2008.06134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Ogryzko V. Immunoaffinity purification of mammalian protein complexes. Meth Enzymol. 2003;370:430–444. doi: 10.1016/S0076-6879(03)70037-8. [DOI] [PubMed] [Google Scholar]
- Saint-André V, Batsche E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol. 2011;18:337–344. doi: 10.1038/nsmb.1995. [DOI] [PubMed] [Google Scholar]
- Shan L, He P, Sheen J. Intercepting host MAPK signaling cascades by bacterial type III effectors. Cell Host Microbe. 2007;1:167–174. doi: 10.1016/j.chom.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Shim D-H, Suzuki T, Chang S-Y, Park S-M, Sansonetti PJ, Sasakawa C, Kweon M-N. New animal model of shigellosis in the Guinea pig: its usefulness for protective efficacy studies. J Immunol. 2007;178:2476–2482. doi: 10.4049/jimmunol.178.4.2476. [DOI] [PubMed] [Google Scholar]
- Slezak J, Truong M, Huang W, Jarrard D. HP1γ expression is elevated in prostate cancer and is superior to Gleason score as a predictor of biochemical recurrence after radical prostatectomy. BMC Cancer. 2013;13:148. doi: 10.1186/1471-2407-13-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood A, Estève P-O, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21:1169–1178. doi: 10.1101/gad.1536807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood A, Hon GC, Jin F, Henry RE, Espinosa JM, Ren B. CBX3 regulates efficient RNA processing genome-wide. Genome Res. 2012;22:1426–1436. doi: 10.1101/gr.124818.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanashi M, Oikawa K, Fujita K, Kudo M, Kinoshita M, Kuroda M. Heterochromatin protein 1gamma epigenetically regulates cell differentiation and exhibits potential as a therapeutic target for various types of cancers. Am J Pathol. 2009;174:309–316. doi: 10.2353/ajpath.2009.080148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintz G, Olsen JV, Frühauf K, Niedzielska M, Amit I, Jantsch J, Mages J, Frech C, Dölken L, Mann M, Lang R. The phosphoproteome of toll-like receptor-activated macrophages. Mol Syst Biol. 2010;6:371. doi: 10.1038/msb.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JSC. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Barnard MB, Rose SM, Cockerill PN, Huang SY, Garrard WT. Transcription termination and chromatin structure of the active immunoglobulin kappa gene locus. J Biol Chem. 1986;261:3838–3845. [PubMed] [Google Scholar]
- Yang S-H, Sharrocks AD, Whitmarsh AJ. Transcriptional regulation by the MAP kinase signaling cascades. Gene. 2003;320:3–21. doi: 10.1016/s0378-1119(03)00816-3. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shao F, Li Y, Cui H, Chen L, Li H, Zou Y, Long C, Lan L, Chai J, Chen S, Tang X, Zhou JM. A Pseudomonas syringae effector inactivates MAPKs to suppress PAMP-induced immunity in plants. Cell Host Microbe. 2007;1:175–185. doi: 10.1016/j.chom.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, Crabtree GR. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 1998;95:625–636. doi: 10.1016/s0092-8674(00)81633-5. [DOI] [PubMed] [Google Scholar]
- Zhao T, Heyduk T, Eissenberg JC. Phosphorylation site mutations in heterochromatin protein 1 (HP1) reduce or eliminate silencing activity. J Biol Chem. 2001;276:9512–9518. doi: 10.1074/jbc.M010098200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.