

Abstract
The current convergence of molecular and pharmacological data provides unprecedented opportunities to gain insights into the relationships between the two types of data. Multiple forms of large scale molecular data, including but not limited to gene and microRNA transcript expression, DNA somatic and germline variations from Next-Generation DNA and RNA Sequencing, and DNA copy number from array comparative genomic hybridization are all potentially informative when one attempts to recognize the panoply of potentially influential events both for cancer progression and therapeutic outcome. Concurrently, there has also been a substantial expansion of the pharmacological data being accrued in a systematic fashion. For cancer cell lines, the National Cancer Institute cell line panel (NCI-60), the Cancer Cell Line Encyclopedia (CCLE), and the collaborative Genomics of Drug Sensitivity in Cancer (GDSC) databases all provide subsets of these forms of data. For the patient derived data, The Cancer Genome Atlas (TCGA) provides analogous forms of genomic information along with treatment histories. Integration of these data in turn relies on the fields of statistics and statistical learning. Multiple algorithmic approaches may be chosen from, depending on the data being considered, and the nature of the question being asked. Combining these algorithms with prior biological knowledge, the results of molecular biological studies, and the consideration of genes as pathways or functional groups provides both the challenge and the potential of the field. The ultimate goal is to provide a paradigm shift in the way that drugs are selected to provide a more targeted and efficacious outcome for the patient.
Keywords: mutations, genetic variants, drug resistance, biomarkers, pharmacogenomics
“Omic” data as a driver for pharmacological decision-making
Progress in scientific understanding often goes hand-in-hand with advances in technology. This applies to pharmacology as well, as the advent of multiple platforms providing “omic” forms of data has created the opportunity for a systems-based understanding for both cancer progression and pharmacological response. These platforms include (but are not limited to) those that provide data on i) gene transcript expression, ii) microRNA expression, iii) DNA copy number, iv) DNA methylation, v) DNA sequencing, and vi) RNA sequencing. Perhaps chief among these currently is DNA and RNA sequencing, driven largely by the combination of cheaper sequencing and the biological importance of genomic information. Sequencing results are now being used both for molecular diagnosis, and the identification of disease-specific mutations (Choi et al. 2012; Doherty and Bamshad 2012; Dyment et al. 2012). In the cancer context, they have been used for the identification of cancer-susceptibility and drug-sensitivity associated genes (Banerji et al. 2012; Cromer et al. 2012; Johnston et al. 2012; Koo et al. 2012). Both germline and somatic variants are informative in this context (Gillis et al. 2013; Hertz 2013). The presence of variants in non-cancerous genomes (such as the 1000 Genomes and ESP5400)1,2, as well as databases such as the National Center for Biotechnology Information (NCBI) database of Single Nucleotide Polymorphisms (dbSNP)3, and the Catalogue Of Somatic Mutations in Cancer (COSMIC)4 may be determined, placing those variants in either a non-cancerous (1000 Genomes, ESP5400, dbSNP) or cancerous (COSMIC) context. Ultimately, the goal will be the use of sequencing for precision oncology (Abaan et al. 2013; Cronin and Ross 2011).
The large-scale cell line panels
Although cancer cell lines lack the complexities of clinical cancer tissues (Weinstein 2012), they do present rich sources of data that has led to improved understanding of cancer physiology and pharmacological response. We review here three important initiatives that have been undertaken in recent years that approach cell lines in a more systematic fashion, and have generated information that may be applied in a systems biological fashion to cancer therapeutics.
The Cancer Cell Line Encyclopedia (CCLE) is a collaborative effort of the BROAD Institute and the Novartis Institutes for Biomedical Research5. It includes 1,046 cell lines and has molecular data for the i) mRNA expression, ii) DNA copy number, iii) single nucleotide polymorphisms (from SNP array) and iv) mutational status of ~1,600 genes selected to remove likely germline events. It also includes pharmacological profiles for 24 anticancer drugs across approximately half of the cell lines (507 cell lines) (Barretina et al. 2012).
The Genomics of Drug Sensitivity in Cancer (GDSC) project is a collaborative effort of the Wellcome Trust Sanger Institute and the Massachusetts General Hospital Cancer Center that combines genomics data from the Cancer Genome Project (CGP) and drug activity data6. It includes 778 cell lines and has molecular data for i) mRNA expression, ii) point mutations for 64 genes by sequencing, iii) gene amplifications and deletions, iv) microsatellite instability, and v) frequently occurring DNA rearrangements (Garnett et al. 2012). Pharmacological profiles for 138 anticancer drugs versus 715 cell lines have also been done (Haibe-Kains et al. 2013).
The cell lines of the US National Cancer Institute (the NCI-60) provided the first of the cell line panels, developed in the mid 80’s under the leadership of the Developmental Therapeutics Program (DTP) (Paull et al. 1989) (Shoemaker 2006). It includes 60 cell lines, and its molecular data include: i) karyotypic analysis with multiple parameters of genomic instability (Roschke et al. 2003), ii) DNA copy number (Bussey et al. 2006; Reinhold et al. 2010; Reinhold et al. 2014), iii) single nucleotide polymorphisms (Ruan et al. 2012), iv) whole exome sequencing identification of 140,171 variants (germline and somatic) (Abaan et al. 2013), v) gene transcript expression (done using five platforms) (Liu et al. 2010; Reinhold et al. 2012), vi) microRNA expression (Blower PE et al. 2007; Liu et al. 2010), vii) global proteomic analysis (Moghaddas Gholami et al. 2013), and viii) metabolite profiling (Jain et al. 2012). Additional, more narrow profiles for the NCI-60 include; i) DNA fingerprinting (Lorenzi et al. 2009), ii) putative tumor stem cell marker identification (Stuelten et al. 2010), iii) genotyping of HLA class I and II antigens (Adams et al. 2005), iv) nuclear receptor and ABC transporter quantitative RT-PCR expression profiling (Holbeck et al. 2010a; Szakacs et al. 2004), and v) reverse-phase protein lysate microarrays (Nishizuka et al. 2003). To date, over 100,000 compounds, including ~50,000 natural products are available for analysis (Holbeck et al. 2010b). Of these, there are currently 50,163 publicly available at the DTP web site, 49,512 non-public, and 20,002 compounds, including 114 FDA-approved and 55 clinical trial drugs available through CellMiner (Reinhold et al. 2012; Shankavaram et al. 2009)7,8. It is by far the largest publicly accessible cell line panel compound and drug database worldwide (Weinstein 2012). Additional phenotypic assays include: i) ionizing radiation response (Amundson et al. 2008), ii) homologous recombination and base excision repair phenotype identification (Stults et al. 2011), iii) apoptotic sensitivity and CD95 Type I or II status (Algeciras-Schimnich et al. 2003), iv) S-1 phase arrest induced by DNA damage (Garner and Eastman 2011), and v) rhodamine efflux as a measure of multidrug resistance (Lee et al. 1994).
Each of these datasets has its assets and liabilities. The CCLE and GDSC offer large numbers of cell lines, thus providing more extensive coverage in terms of disease tissue of origin, with 25, 16, and 9 for CCLE, GDSC, and the NCI-60, respectively. A longer list of specific disease histology’s is available for CCLE and GDS as well. The increased numbers of cell lines for CCLE and GDS also provide increased ability to identify less common or disease-specific mutations. The NCI-60 offers a uniquely large number of “omic” molecular data profiles, a number of more narrow molecular profiles, and the additional forms of phenotypic data described above. Perhaps most importantly, the NCI-60 has the largest compound library. Its natural products and compounds include many with activity profiles that are unlike anything currently in the clinic, an underappreciated asset for the field (Holbeck et al. 2010b). Each of these three cell line panels is undergoing augmentation.
Cell line databases overlap and consistency
There are 1324 cell lines in total for the CCLE, GDSC, and NCI-60 combined, with various amounts of overlap for the different data types. The overlap comparisons for the i) cell lines, ii) common drugs, iii) number of cell lines with drug data, iv) number of cell lines with transcript expression data, and v) number of cell lines with DNA variant data are summarized in Table 1. Such overlaps enable data cross-validation and comparison, and potentially extrapolation between panels.
Table 1.
Overlapping data between the CCLE, GDS, and NCI-60
| Common cell lines
|
Common drugs
|
Cell lines with drug data
|
Cell lines with expression dataa
|
Cell lines with variant datab
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCLE | GDSC | NCI-60 | CCLE | GDSC | NCI-60c | CCLE | GDSC | NCI-60 | CCLE | GDSC | NCI-60 | CCLE | GDSC | NCI-60 | |
| CCLE | 1046 | 503 | 44 | 24 | 15 | 10 | 507 | 302 | 44 | 1036 | 471 | 44 | 995 | 478 | 44 |
| GDSC | 778 | 59 | 138 | 44 | 715 | 56 | 725 | 56 | 714 | 55 | |||||
| NCI-60 | 60 | 20,007 | 60 | 60 | 60 | ||||||||||
Transcript expression.
DNA variant data from sequencing.
Based on the drugs in CellMiner version 1.5.
Prior work comparing the transcript expression profiles of the CCLE and GDSC (CGP) found the data to be well correlated with a median correlation of 0.85 (Haibe-Kains et al. 2013). In addition, comparisons of mutations in the CCLE and GDSC found robust relationship by two-sided Wilcoxon rank-sum test (p value of 1×10−16) (Haibe-Kains et al. 2013). However, the result for the pharmacological data is more problematic. For the CCLE and GDSC, comparison of 15 overlapping drugs yielded only two drugs with good correlations (r = 0.61 and 0.53) (Haibe-Kains et al. 2013). For the NCI-60, ten drugs also have data in the CCLE database. Of these, two (20%) have significant correlations. Of the 44 drugs that have available data for both the NCI-60 and the GDSC, 18 (41%) show significant correlations (by Spearman’s correlation, p<0.01 for both comparisons).
Multiple factors are likely to contribute to these inconsistencies. Assay types and time point are surely contributors, as CCLE measures ATP levels at 72–84 hours, GDSC measures nucleic acids for adherent, or oxidation-reduction levels for suspended cells at 72 hours, and the DTP measures total protein at 48 hours (Barretina et al. 2012) (Garnett et al. 2012) (Rubinstein et al. 1990). Additional factors that have been raised include range of drug concentrations tested, choice of an estimator for summarizing the drug dose response curve, gene-expression profiles, and computational approach (Haibe-Kains et al. 2013; Weinstein and Lorenzi 2013). This has brought into question the identification of genomic predictors using these pharmacological data (Haibe-Kains et al. 2013). However, as all information is not obtained from a single assay or specific time, these datasets should most likely be viewed as being complementary, once reproducibility (within assay type) and proper concentration values (for each drug) are obtained.
Large-scale clinical data, the TCGA
The Cancer Genome Atlas (TCGA)9 is a collaborative effort between the NCI and the National Human Genome Research Institute (NHGRI). It applies analytical genomic approaches to clinical tissue samples based on principles similar to those utilized in the cell line panels. It currently includes data from ~9,939 patients from 33 cancer types. It has molecular data for: i) DNA copy number, ii) SNP genotyping, iii) DNA methylation, iv) exon sequencing of at least 1,200 genes, v) gene transcript expression, and vi) microRNA profiling. In addition, TCGA provides patient therapy information. However, interpreting the therapeutic data is complex because patients receive drug combinations, which are not uniform for all patients.
The integration of statistical learning, biology, and pharmacology
The patterns that exist between molecular and pharmacological parameters may be interrogated in either direction, that is to say starting with drug activity and considering what genomic molecular events might be involved, or starting with the genomic characteristics and considering their potential influence on pharmacology. The selection of which analytical method to use to identify biologically-related events is affected by what is currently known, and the particular questions of interest. It remains a challenging endeavor, especially because the factors that affect drug activity may be multifactorial.
Compound activity and genomic profiling data over well-characterized cell line panels allows computational prediction of molecular drug response determinants. The computational techniques exist in a continuum of complexity (Figure 1). Simple analyses begin with correlation-based filters that prioritize genomic features whose cell line profiles are significantly correlated with the corresponding compound activity profile (by Pearson’s or Spearman’s correlation). This methodology has the ability to recognize robustly correlated parameters. Examples of results of this type of analysis, are: i) ABCB1 expression and doxorubicin activity, ii) the activities of DNA-damaging drugs and SLFN11 expression, and iii) BRAF V600E mutational status and vemurafenib activity (Abaan et al. 2013; Gmeiner et al. 2010; Reinhold et al. 2012; Zoppoli et al. 2012). It also may be used to identify broad trends in pharmacological response (Liu et al. 2010). It is at the core of the COMPARE algorithm pioneered by the NCI-DTP (Paull et al. 1989), and it has been further developed and applied in multiple studies (Covell 2008; Liu et al. 2010; Reinhold et al. 2012; Ross et al. 2000; Shankavaram et al. 2009; Weinstein et al. 1997). Statistical tests, such as t-tests, can also be applied to rank molecular events that differ between sensitive and resistant lines for further evaluation (Covell 2012) (Kwei et al. 2012). The obtained gene sets can be evaluated for enrichment of genes associated with particular biological pathways and processes, using tools such as GoMiner10, and Gene Set Enrichment Analysis (GSEA) 11 (Zeeberg et al. 2003). Notably, these analyses based on fundamental statistical tests can be ‘reversed’ in a sense by beginning with a binary gene expression or other phenotypic profile, and screening for compounds with significant differences in activity between the two specified classes.
Figure 1.
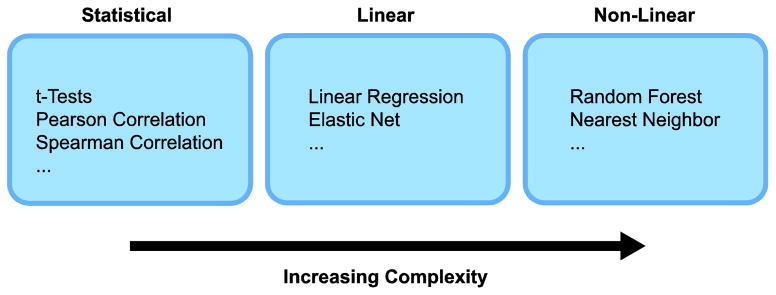
Bio statistical methods used for the determination of biomarkers for drug response. Complexity increases from left to right.
While these methods emphasize discovery of individual correlated and potentially causal and predictive features, more complex approaches attempt to identify limited sets of interacting response markers. Biologically, this makes sense if one presumes that there are potentially many molecular events that might affect pharmacological response that act in a cumulative fashion. Following this rationale, both the CCLE and GDSC projects have applied the elastic net algorithm (Zou and Hastie 2005), a linear regression-based method, to infer multivariate drug response predictors combining diverse molecular data types (Garnett et al. 2012). The elastic net attempts to reconstruct a drug activity profile as a weighted sum of a limited number of genomic features, and includes feature weights and signs to indicate the relative strength and direction of influence. More complex, non-linear methodologies (i.e. random forests and nearest neighbor methods) have also been used to predict drug response using proteomic profiles of the NCI-60 (Ma et al. 2006). A potential shortcoming of many non-linear techniques is the limited interpretability of their results, with the response often not being explicitly modeled as a simple function of the predictive features.
An overarching concern with these mathematically formulated methods is their ability to recognize the biologically meaningful features from the large number of inputs. An aid in this area is the application of sound statistical learning principles, such as the strict separation of data used to develop and evaluate predictive models (Baggerly and Coombes 2009; Coombes et al. 2007; Papillon-Cavanagh et al. 2013). Other ways to achieve greater biological specificity have considered combining binary genomic features using Boolean set operations, with union, intersection and difference operations acting as proxies of molecular redundancy, synergy, and resistance, respectively (Masica and Karchin 2013).
A promising new direction for enhancing all these techniques is to leverage prior biological knowledge, such as molecular interactions derived from biological pathways using literature-curated resources, or computationally inferred gene regulatory networks (Croft et al. 2011; Kohn 2001; Kohn et al. 2006; Margolin et al. 2006). Promising results have been achieved by mapping gene expression values to metabolic networks to predict drug targets for known anticancer drugs in the NCI-60 (Folger et al. 2011). Extensions to the elastic net have also been proposed to prioritize predictor sets that are associated in a network of established molecular interactions (Li and Li 2008). A limitation of these methodologies comes from the incomplete state of biological knowledge, such as the ability to find comprehensive and high quality interaction networks. Projects such as Pathway Commons are designed to aggregate disparate interaction databases and should help these methods advance (Cerami et al. 2011).
Observations with translational implication
From the cell line panels, several potentially important associations have been identified between cancer-specific genomic alterations and pharmacological responses. From the CCLE panel: i) MEK inhibitor efficacy was associated with AHR expression in NRAS mutant cell lines, ii) sensitivity to topoisomerase inhibitors was predicted by SLFN11 expression, and iii) IGF1R inhibitor response was correlated with plasma cell lineage (Barretina et al. 2012). From the Massachusetts General Hospital Cancer Center: i) activating mutations in EGFR and amplifications of EGFR and HER2 were shown to affect the EGFR inhibitor erlotinib, ii) amplifications in MET were shown to affect the MET inhibitor PHA665752, and iii) the V600E BRAF mutation was shown to affect the BRAF inhibitor AZ628 (McDermott et al. 2007). From the GDSC, EWS-FLI1 translocation containing cells were associated with sensitivity to PARP inhibitors (Garnett et al. 2012). The association of EGFR mutations with the response of erlotinib provides a clear example of translation from cell lines to clinic: erlotinib-refractory cell lines and patient tumors both specifically have wild type EGFR (McDermott and Settleman 2009). From the NCI-60, i) the activity of MDM2-TP53 interaction inhibitor nutlin was shown to be dependent upon TP53 mutational status, ii) BRAF V600E mutational status was associated with activity of vemurafenib and MEK inhibitors, iii) a multifactorial combination of ERBB1 and 2 expression and RAS-RAF-PTEN mutational status was associated with the activity of erlotinib, and iv) ATAD5 mutational status was associated with the DNA-damaging bleomycin, zorbamycin, and peplomycin activities (Abaan et al. 2013). Specific examples of working back and forth between the molecular and compound data are found in two recent publications. In the first, drugs activities were used to identify an important novel molecular factor in pharmacology, SLFN11 expression (Gmeiner et al. 2010; Zoppoli et al. 2012). In the second, important molecular events (RUNX1 and CBFB expression) in core binding factor leukemia were used to identify candidate lead compounds. Ro5-3335, a drug developed for other purposes (treatment of HIV (Cupelli and Hsu 1995)) was identified informatically, and subsequently these genes were shown to have a causal relationship to the identified compound (Cunningham et al. 2012).
From the TCGA, multiple informative results are being reported. Gene mutations involved in squamous cell cancer of the head and neck, such as FAT1, MLL2, TGFRBR2, HLA-A, and NFE212 have been identified (Mountzios et al. 2014), providing potential targets for pharmacological intervention. The association between connexin 43 and temozolomide resistance found in cell lines has potential clinical relevance, given the elevated levels of CX43 observed in glioblastoma tumor samples (Munoz et al. 2014). Used in conjunction with CCLE data, numerous candidate metabolic vulnerabilities were identified, many of which can be targeted with FDA-approved drugs (Aksoy et al. 2014).
Future perspectives
There has been, and continues to be debate about the role of the large-scale cell line and cancer “omic” databases and how they mesh with traditional one gene at a time molecular biological approaches. It has been argued that progress in molecular biology is being jeopardized by the shift in funding to the large-scale “omics” projects (Weinberg 2010). While what proportion of the funding should go to either type of research may be debated, and traditional molecular biology certainly has in the past and will in the future continue to make important contributions, to pretend that a patient’s cancer or pharmacological response can be viewed or understood as a one gene event based on isogenic systems is likely to be depending on the exception rather than the rule. An additional ongoing tension between these disciplines is the time frames over which they occur. While a single large-scale analysis typically generates multiple potentially important hypotheses, often times with broad implications, traditional molecular biology proceeds at a slow pace and tends to be narrow in focus. However, the reality is that each of the disciplines needs and is enhanced by the other.
Even if one accepts the potential of the “omics” approaches, there are multiple challenges remaining to bring this potential to fruition. Technically, considerations such as cell line selection and number, compound selection and number, clinical sample selection and handling, and assay types all are key. Furthermore, the attention to experimental detail, quality control, and reproducibility exceed that required for less systematic studies.
Biologically, it is likely that the field will need to adopt a more pathway inclusive view of molecular events. Even in the robustly correlated BRAF V600E vemurafenib example, (Garnett et al. 2012) (Abaan et al. 2013), patients tend to respond well for a three to six month period of time and then relapse. Of course in patients, every combination of pathway modification occurs, including those that affect BRAF influence. The logical outcome is that when making treatment decisions, the specific complexity of the patient’s disease needs to be considered. An example from the NCI-60 data of this type of complexity is that although the LOXIMVI melanoma contains the V600E alteration in BRAF, it has reduced sensitivity to the BRAF inhibitor vemurafenib as compared to the other cell lines with that mutation. Variations that occur in known pharmacologically important genes, such as drug targets, and those involved in apoptosis and survival, DNA damage response, drug efflux, and metabolic vulnerabilities will also require extra scrutiny.
Pharmacologically, the previously discussed result inconsistencies between the CCLE, GDSC, and the NCI-60 place an interpretive burden on the field. Further study to clarify these inconsistencies is indicated, with some level of cross-institutional collaboration or cooperation desirable. Certainly having each institution doing more of the same drugs at identical concentrations is a sensible starting point. However, it remains likely that different assay types done at different time points will give continue to give real and different results, and these may have some specificity for the individual drugs. The goal here for all should be to make these differences informative and complimentary, not confusing and uninterruptable. Algorithmically, there are multiple approaches to choose from, with the choice being influenced by the data being considered and nature of the comparison being made. For dominant factor considerations, correlation will work well, however, the statistical learning approaches will likely dominate as the numbers of contributing factors being considered and size and numbers of the databases increase. Statistics can provide an assessment of significance to aid in the interpretation of results.
Unfortunately, it is likely that identification of those molecular events that affect pharmacology based on the above-described approaches will be generally inadequate. Some form of integration of biological knowledge into the algorithmic approaches, such as those described previously will likely be requisite (Croft et al. 2011; Folger et al. 2011; Li and Li 2008; Margolin et al. 2006). This biological knowledge may take the form of pathways, functional grouping of genes that work together, genes known to influential in pharmacology, or results from some large scale assay (example, the kinase inhibitor screen (McDermott and Settleman 2009; McDermott et al. 2007)).
While the obstacles are numerous and daunting, the need to better understand the determinants of response and resistance to both the currently approved, as well as an increasing number of drugs in preclinical development provides an impetus to continued research and development. We look forward to a paradigm-shift in the diagnosis and selection of pharmacological treatments based on genomic and proteomic technologies.
Supplementary Material
Figure 2.

A schematic representation of the various databases that might be used to move towards providing hypothesis for selection of pharmacological agents. Acronyms: CCLE: Cancer Cell Line Encyclopedia; GDSC: Genomics of Drug Sensitivity in Cancer; NCI-60: the sixty cell lines of the US National Cancer Institute; TCGA: The Cancer Genome Atlas.
Acknowledgments
Our studies are supported by the Center for Cancer Research, Intramural Program of the National Cancer Institute (Z01 BC 006150). This work was also supported in part by a MSKCC Genome Data Analysis Center grant (U24 CA143840) awarded as part of the National Cancer Institute (NCI)/National Human Genome Research Institute (NHGRI).
Footnotes
No conflicts of interest to report.
References
- Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, Pineda M, Gindin Y, Jiang Y, Reinhold WC, Holbeck SL, Simon RM, Doroshow JH, Pommier Y, Meltzer PS. The Exomes of the NCI-60 Panel: A Genomic Resource for Cancer Biology and Systems Pharmacology. Cancer Research. 2013;73:4372–82. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S, Robbins FM, Chen D, Wagage D, Holbeck SL, Morse HC, 3rd, Stroncek D, Marincola FM. HLA class I and II genotype of the NCI-60 cell lines. J Transl Med. 2005;3:11. doi: 10.1186/1479-5876-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy BA, Demir E, Babur O, Wang W, Jing X, Schultz N, Sander C. Prediction of individualized therapeutic vulnerabilities in cancer from genomic profiles. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algeciras-Schimnich A, Pietras EM, Barnhart BC, Legembre P, Vijayan S, Holbeck SL, Peter ME. Two CD95 tumor classes with different sensitivities to antitumor drugs. Proc Natl Acad Sci U S A. 2003;100:11445–50. doi: 10.1073/pnas.2034995100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundson SA, Do KT, Vinikoor LC, Lee RA, Koch-Paiz CA, Ahn J, Reimers M, Chen Y, Scudiero DA, Weinstein JN, Trent JM, Bittner ML, Meltzer PS, Fornace AJ., Jr Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 2008;68:415–24. doi: 10.1158/0008-5472.CAN-07-2120. [DOI] [PubMed] [Google Scholar]
- Baggerly KA, Coombes KR. Deriving Chemosensitivity from Cell Lines: Forensic Bioinformatics and Reproducible Research in High-Throughput Biology. Annals of Applied Statistics. 2009;3:1309–1334. doi: 10.1214/09-Aoas291. [DOI] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–9. doi: 10.1038/nature11154. nature11154 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower PE, Verducci JS, Lin SZJ, Chung JH, Dai Z, Liu CG, Reinhold W, Lorenzi PL, Kaldjian EP, Croce CM, Weinstein JN, WS MicroRNA expression profiles for the NCI-60 cancer cell panel. Mol Cancer Ther. 2007 May;6(5):1483–91. doi: 10.1158/1535-7163.MCT-07-0009. [DOI] [PubMed] [Google Scholar]
- Bussey KJ, Chin K, Lababidi S, Reimers M, Reinhold WC, Kuo WL, Gwadry F, Ajay, Kouros-Mehr H, Fridlyand J, Jain A, Collins C, Nishizuka S, Tonon G, Roschke A, Gehlhaus K, Kirsch I, Scudiero DA, Gray JW, Weinstein JN. Integrating data on DNA copy number with gene expression levels and drug sensitivities in the NCI-60 cell line panel. Mol Cancer Ther. 2006;5:853–67. doi: 10.1158/1535-7163.MCT-05-0155. 5/4/853 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Schultz N, Bader GD, Sander C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–90. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, Choi KG, Jung SC, Kim HS, Hyun YS, Nakhro K, Lee HJ, Woo HM, Chung KW. Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth Disease. Hum Mutat. 2012 doi: 10.1002/humu.22143. [DOI] [PubMed] [Google Scholar]
- Coombes KR, Wang J, Baggerly KA. Microarrays: retracing steps. Nature Medicine. 2007;13:1276–1277. doi: 10.1038/Nm1107-1276b. [DOI] [PubMed] [Google Scholar]
- Covell DG. Connecting chemosensitivity, gene expression and disease. Trends Pharmacol Sci. 2008;29:1–5. doi: 10.1016/j.tips.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Covell DG. Integrating constitutive gene expression and chemoactivity: mining the NCI60 anticancer screen. PLoS One. 2012;7:e44631. doi: 10.1371/journal.pone.0044631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D’Eustachio P, Stein L. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–7. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, Carling T. Identification of Somatic Mutations in Parathyroid Tumors Using Whole-Exome Sequencing. J Clin Endocrinol Metab. 2012 doi: 10.1210/jc.2012-1743. jc.2012-1743 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin M, Ross JS. Comprehensive next-generation cancer genome sequencing in the era of targeted therapy and personalized oncology. Biomark Med. 2011;5:293–305. doi: 10.2217/bmm.11.37. [DOI] [PubMed] [Google Scholar]
- Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VR, Dehdashti SJ, Reinhold WC, Alemu L, Zhao L, Yeh JR, Sood R, Pommier Y, Austin CP, Jeang KT, Zheng W, Liu P. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proc Natl Acad Sci U S A. 2012;109:14592–7. doi: 10.1073/pnas.1200037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupelli LA, Hsu MC. The human immunodeficiency virus type 1 Tat antagonist, Ro 5-3335, predominantly inhibits transcription initiation from the viral promoter. J Virol. 1995;69:2640–3. doi: 10.1128/jvi.69.4.2640-2643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty D, Bamshad MJ. Exome sequencing to find rare variants causing neurologic diseases. Neurology. 2012 doi: 10.1212/WNL.0b013e3182617170. WNL.0b013e3182617170 [pii] [DOI] [PubMed] [Google Scholar]
- Dyment DA, Cader MZ, Chao MJ, Lincoln MR, Morrison KM, Disanto G, Morahan JM, De Luca GC, Sadovnick AD, Lepage P, Montpetit A, Ebers GC, Ramagopalan SV. Exome sequencing identifies a novel, multiple sclerosis susceptibility variant in the TYK2 gene. Neurology. 2012 doi: 10.1212/WNL.0b013e3182616fc4. WNL.0b013e3182616fc4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner KM, Eastman A. Variations in Mre11/Rad50/Nbs1 status and DNA damage-induced S-phase arrest in the cell lines of the NCI60 panel. BMC Cancer. 2011;11(206):1–13. doi: 10.1186/1471-2407-11-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis N, Patel J, Innocenti F. Clinical Implementation of Germ Line Cancer Pharmacogenetic Variants During the Next-Generation Sequencing Era. Clin Pharmacol Ther. 2013 doi: 10.1038/clpt.2013.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmeiner WH, Reinhold WC, Pommier Y. Genome-wide mRNA and microRNA profiling of the NCI 60 cell-line screen and comparison of FdUMP[10] with fluorouracil, floxuridine, and topoisomerase 1 poisons. Mol Cancer Ther. 2010;9:3105–14. doi: 10.1158/1535-7163.MCT-10-0674. 9/12/3105 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJ, Quackenbush J. Inconsistency in large pharmacogenomic studies. Nature. 2013;504:389–93. doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz DL. Germline pharmacogenetics of paclitaxel for cancer treatment. Pharmacogenomics. 2013;14:1065–84. doi: 10.2217/pgs.13.90. [DOI] [PubMed] [Google Scholar]
- Holbeck S, Chang J, Best AM, Bookout AL, Mangelsdorf DJ, Martinez ED. Expression profiling of nuclear receptors in the NCI60 cancer cell panel reveals receptor-drug and receptor-gene interactions. Mol Endocrinol. 2010a;24:1287–96. doi: 10.1210/me.2010-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol Cancer Ther. 2010b;9:1451–60. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–4. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, Mullikin JC, Biesecker LG. Secondary Variants in Individuals Undergoing Exome Sequencing: Screening of 572 Individuals Identifies High-Penetrance Mutations in Cancer-Susceptibility Genes. Am J Hum Genet. 2012 doi: 10.1016/j.ajhg.2012.05.021. S0002-9297(12)00304-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn KW. Molecular interaction maps as information organizers and simulation guides. Chaos. 2001;11:84–97. doi: 10.1063/1.1338126. [DOI] [PubMed] [Google Scholar]
- Kohn KW, Aladjem MI, Kim S, Weinstein JN, Pommier Y. Depicting combinatorial complexity with the molecular interaction map notation. Mol Syst Biol. 2006;2:51. doi: 10.1038/msb4100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, Quek R, Loong S, Yeoh KW, Yap SP, Lee KA, Lim LC, Tan D, Goh C, Cutcutache I, Yu W, Ng CC, Rajasegaran V, Heng HL, Gan A, Ong CK, Rozen S, Tan P, Teh BT, Lim ST. Janus Kinase 3-Activating Mutations Identified in Natural Killer/T-cell Lymphoma. Cancer Discov. 2012 doi: 10.1158/2159-8290.CD-12-0028. 2159–8290.CD-12-0028 [pii] [DOI] [PubMed] [Google Scholar]
- Kwei KA, Baker JB, Pelham RJ. Modulators of sensitivity and resistance to inhibition of PI3K identified in a pharmacogenomic screen of the NCI-60 human tumor cell line collection. PLoS One. 2012;7:e46518. doi: 10.1371/journal.pone.0046518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol Pharmacol. 1994;46:627–38. [PubMed] [Google Scholar]
- Li C, Li H. Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics. 2008;24:1175–82. doi: 10.1093/bioinformatics/btn081. [DOI] [PubMed] [Google Scholar]
- Liu H, D’Andrade Petula, Fulmer-Smentek Stephanie, Lorenzi Philip, Kohn Kurt W, Weinstein John N, Pommier Yves G, Reinhold WC. MCT. mRNA and microRNA expression profiles integrated with drug sensitivities of the NCI-60 human cancer cell lines. 2010;9(5):1080–1091. doi: 10.1158/1535-7163.MCT-09-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi P, Reinhold W, Varma S, Hutchinson A, Pommier Y, Chanock S, Weinstein J. DNA fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther. 2009;8:713–24. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Ding Z, Qian Y, Shi X, Castranova V, Harner EJ, Guo L. Predicting cancer drug response by proteomic profiling. Clin Cancer Res. 2006;12:4583–9. doi: 10.1158/1078-0432.CCR-06-0290. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masica DL, Karchin R. Collections of Simultaneously Altered Genes as Biomarkers of Cancer Cell Drug Response. Cancer Res. 2013;73:1699–1708. doi: 10.1158/0008-5472.Can-12-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott U, Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol. 2009;27:5650–9. doi: 10.1200/JCO.2009.22.9054. [DOI] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, Rothenberg SM, Supko JG, Sordella R, Ulkus LE, Iafrate AJ, Maheswaran S, Njauw CN, Tsao H, Drew L, Hanke JH, Ma XJ, Erlander MG, Gray NS, Haber DA, Settleman J. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007;104:19936–41. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddas Gholami A, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M, Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4:609–20. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Mountzios G, Rampias T, Psyrri A. The mutational spectrum of squamous-cell carcinoma of the head and neck: Targetable genetic events and clinical impact. Ann Oncol. 2014 doi: 10.1093/annonc/mdu143. [DOI] [PubMed] [Google Scholar]
- Munoz JL, Rodriguez-Cruz V, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014;5:e1145. doi: 10.1038/cddis.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka S, Charboneau L, Young L, Major S, Reinhold W, Waltham M, Kouros-Mehr H, Bussey K, Lee J, Espina V, Munson P, Petricoin IE, Liotta L, Weinstein J. Proteomic profiling of the NCI60 cancer cell lines using new high-density ‘reverse-phase’ lysate microarrays. Proc Natl Acad Sci U S A. 2003;100:14229–34. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon-Cavanagh S, De Jay N, Hachem N, Olsen C, Bontempi G, Aerts HJ, Quackenbush J, Haibe-Kains B. Comparison and validation of genomic predictors for anticancer drug sensitivity. J Am Med Inform Assoc. 2013;20:597–602. doi: 10.1136/amiajnl-2012-001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull K, Shoemaker R, Hodes L, Monks A, Scudiero D, Rubinstein L, Plowman J, Boyd M. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- Reinhold WC, Mergny JL, Liu H, Ryan M, Pfister TD, Kinders R, Parchment R, Doroshow J, Weinstein JN, Pommier Y. Exon array analyses across the NCI-60 reveal potential regulation of TOP1 by transcription pausing at guanosine quartets in the first intron. Cancer Res. 2010;70:2191–203. doi: 10.1158/0008-5472.CAN-09-3528. 0008-5472.CAN-09-3528 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, Pommier Y. CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Varma S, Sousa F, Sunshine M, Abaan OD, Davis SR, Reinhold SW, Kohn KW, Morris J, Doroshow J, Pommier Y. NCI-60 whole exome sequencing and pharmacological CellMiner analyses. PLOS ONE. 2014 doi: 10.1371/journal.pone.0101670. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschke A, Tonon G, Gehlhaus K, McTyre N, Bussey K, Lababidi S, Scudiero D, Weinstein J, Kirsch I. Karyotypic Complexity of the NCI-60 Drug-Screening Panel. Cancer Research. 2003;63:8634–47. [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–35. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- Ruan X, Kocher JP, Pommier Y, Liu H, Reinhold WC. Mass Homozygotes Accumulation in the NCI-60 Cancer Cell Lines As Compared to HapMap Trios, and Relation to Fragile Site Location. PLoS One. 2012;7:e31628. doi: 10.1371/journal.pone.0031628. PONE-D-11-09941 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–8. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- Shankavaram UT, Varma S, Kane D, Sunshine M, Chary KK, Reinhold WC, Pommier Y, Weinstein JN. CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics. 2009;10:277. doi: 10.1186/1471-2164-10-277. 1471-2164-10-277 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Stuelten CH, Mertins SD, Busch JI, Gowens M, Scudiero DA, Burkett MW, Hite KM, Alley M, Hollingshead M, Shoemaker RH, Niederhuber JE. Complex display of putative tumor stem cell markers in the NCI60 tumor cell line panel. Stem Cells. 2010;28:649–60. doi: 10.1002/stem.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Shelton BJ, Pierce AJ. Recombination phenotypes of the NCI-60 collection of human cancer cells. BMC Mol Biol. 2011;12:23. doi: 10.1186/1471-2199-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakacs G, Annereau J, Lababidi S, Shankavaram U, Arciello A, Bussey K, Reinhold W, Guo Y, Kruh G, Reimers M, Weinstein J, Gottesman M. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. 2004 Aug;6(2):129–37. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Weinberg R. Point: Hypotheses first. Nature. 2010;464:678. doi: 10.1038/464678a. [DOI] [PubMed] [Google Scholar]
- Weinstein JN. Drug discovery: Cell lines battle cancer. Nature. 2012;483:544–5. doi: 10.1038/483544a. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Lorenzi PL. Cancer: Discrepancies in drug sensitivity. Nature. 2013;504:381–3. doi: 10.1038/nature12839. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–9. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- Zeeberg B, Feng W, Wang G, Wang M, Fojo A, Sunshine M, Narasimhan S, Kane D, Reinhold W, Lababidi S, Bussey K, Riss J, Barrett J, Weinstein J. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4(4):R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, Doroshow JH, Pommier Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A. 2012;109:15030–5. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2005;67:301–320. doi: 10.1111/J.1467-9868.2005.00503.X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.