

Abstract
Xenopus tropicalis has been developed as a model organism for developmental biology, providing a system offering both modern genetics and classical embryology. Recently, the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated (CRISPR/Cas) system for genome modification has provided an additional tool for Xenopus researchers to achieve simple and efficient targeted mutagenesis. Here, we provide insights into experimental design and procedures permitting successful application of this technique to Xenopus researchers, and offer a general strategy for performing loss-of-function assays in F0 and subsequently F1 embryos.
Keywords: CRISPR, Disease model, sgRNA design, Amphibian mutagenesis, Targeted mutagenesis, Loss-of-function, Off-target effects, Genome engineering
1 Introduction
Xenopus has long been a favored model organism for developmental and cell biology due to its unique combination of advantageous features including: the ability to acquire large numbers of eggs (or oocytes) and embryos; the availability of relatively simple techniques for microinjection of mRNA, DNA, protein, or antisense morpholino oligonucleotides for gain-of-function or loss-of-function (LOF) experiments; ease of transgenesis allowing modern molecular developmental and biochemical studies; and its suitability for classical explant/transplantation embryology. Although many studies use Xenopus laevis, an allotetraploid species with a long generation time, for embryological and cell biological studies, recently Xenopus tropicalis has emerged as a new model organism (Harland & Grainger, 2011). Forward and reverse genetic approaches have identified developmental mutants and their causative genes (Abu-Daya, Khokha, & Zimmerman, 2012). However, the number of characterized mutants to date is small. Mutational screens, including directed approaches such as Targeting Induced Local Lesions In Genomes (e.g., Fish et al., 2014) are laborious, and new approaches to efficiently edit the genome are urgently needed. Recent technological advances have allowed researchers to readily perform targeted gene editing in many organisms. Two major methods that have been used are zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), both of which have been successfully applied in Xenopus (Ishibashi, Cliffe, & Amaya, 2012; Lei et al., 2012; Nakajima, Nakai, Okada, & Yaoita, 2013; Nakajima, Nakajima, Takase, & Yaoita, 2012; Nakajima & Yaoita, 2013; Suzuki et al., 2013; Young et al., 2011). Most recently, Type II CRISPR/Cas (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated) technology has been developed for genome modification. This system was first identified as part of the naturally occurring bacterial adaptive defense mechanism (Fineran & Dy, 2014; Hsu, Lander, & Zhang, 2014; Terns & Terns, 2014), and now has been successfully applied in numerous organisms (Sander & Joung, 2014) including X. tropicalis to effect targeted genome modification (Blitz, Biesinger, Xie, & Cho, 2013; Guo et al., 2014; Nakayama et al., 2013), providing an additional tool for Xenopus researchers to achieve simple and efficient targeted mutagenesis. The application of newly available genome engineering tools in the X. tropicalis system, with its sequenced diploid genome, high degree of synteny with the human genome, and conservation of key developmental processes, will make this an outstanding model organism for studying human genetic disease and developmental pathologies.
Here, we present a general protocol for CRISPR/Cas9-mediated targeted mutations in X. tropicalis including a strategy for performing LOF experiments in F0 mutagenized animals and subsequently F1 animals.
2 Principle
CRISPR/Cas9 creates genome modifications using a common biological mechanism across taxa, and is described briefly here. The Type II CRISPR/Cas system uses Cas9 (an RNA-guided DNA endonuclease) for genome editing. In bacteria, Cas9 cleaves target DNA by forming a complex with two small RNAs, a CRISPR RNA (crRNA) that has complementary sequence to the target DNA and the trans-activating CRISPR RNA (tracrRNA) that base pairs with the crRNA. For efficient cleavage, the target DNA must be followed by a sequence called the protospacer (a complementary sequence targeted by a specific crRNA) adjacent motif (PAM; Fig. 17.1), which varies among bacterial species. Streptococcus pyogenes Cas9 is most widely used for genome editing in eukaryotic systems, and its PAM sequence is NGG (where N can be any nucleotide), although NAG can also function at lower efficiency (Anders, Niewoehner, Duerst, & Jinek 2014; Terns &Terns, 2014) . For genome editing applications, a portion of the crRNA has been fused to the tracrRNA to create a cassette for production of synthetic (or single) guide RNAs (sgRNAs) (Hwang et al., 2013; Mali, Yang, et al., 2013). A target sequence (~ 20 bp) is added to this cassette to create the final sgRNA, which then directs Cas9 to specific sites in the genome for cleavage. In Xenopus, sgRNAs are coinjected together with either Cas9 mRNA or protein into fertilized eggs or early embryos (Blitz et al., 2013; Guo et al., 2014; Nakayama et al., 2013) (Fig. 17.2). Following Cas9-mediated scission of the target site, double-strand breaks are often imperfectly repaired by nonhomologous end-joining (NHEJ), which frequently leads to insertion and deletion mutations (indels).
Figure 17.1.
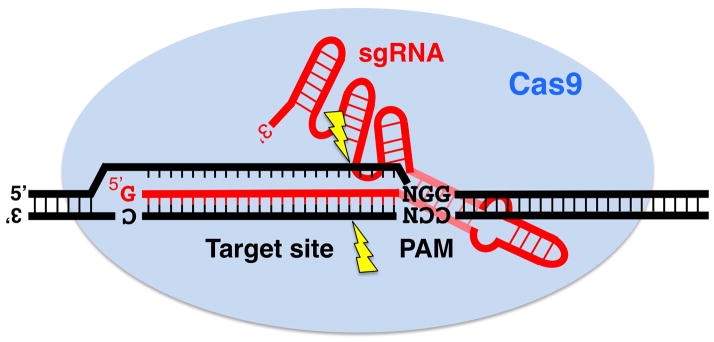
Schematic representation of CRISPR/Cas9-mediated target cleavage. Cas9 protein (oval) and sgRNA together recognize the target sequence. The PAM, NGG, while recognized by Cas9, does not base pair with the sgRNA. Cleavage (indicated by lightning bolts) occurs within the target to create a double-stranded break, which is then repaired by an error-prone NHEJ mechanism.
Figure 17.2.

Strategy of CRISPR/Cas9-mediated mutagenesis in Xenopus tropicalis. Schematic representation of targeted mutagenesis of the tyrosinase (tyr) gene, as an example of a generalized workflow to create mutants. Homozygous mutants at the tyr locus display oculocutaneous albinism. Cas9 mRNA or protein is coinjected with a tyr-specific sgRNA into one-cell stage fertilized eggs (top, left). F0 embryos are mosaics: they are comprised of populations of cells containing different mutant alleles and wild-type tissue. In the case of tyr targeting, biallelic loss of gene function results in deficient melanin synthesis, which is observed as a loss of pigmentation in the eyes and skin. Mosaic F0 animals can be intercrossed to produce nonmosaic (compound heterozygous or homozygous) mutants (tyr−/−), as well as carriers (tyr+/−), and homozygous wild-type F1 progeny.
The resultant embryos are mosaic; cells bearing combinations of mutant and wild-type (unsuccessfully targeted or repaired perfectly) alleles are present in various proportions. The relative amounts of mutant to wild type are likely a function of both the timing of mutagenesis and efficacy of individual sgRNAs to direct Cas9 to target sites. Mutant phenotypes may be scorable in F0 mosaic animals. Intercrosses between F0 adults can be performed to create nonmosaic F1s that are expected to be primarily compound heterozygotes (containing two different mutant allelic variants). Alternatively, in the F2 generation, homozygous mutants can be created that carry a single allelic variant.
3 Protocol
3.1 Background knowledge and experimental equipment
In this chapter, the authors assume that readers have sufficient experimental knowledge, basic molecular biology skills, and experience handling Xenopus, including general embryo manipulation and microinjection technique (Sive, Grainger, & Harland, 2010). All necessary equipment is commonly available in laboratories already equipped for Xenopus embryo research and no special equipment is required for the CRISPR-mediated mutagenesis.
3.2 sgRNA design
The first step in designing a CRISPR/Cas mutagenesis strategy is to identify possible target sites in the gene of interest. A target-adjacent PAM sequence is an absolute requirement for efficient mutagenesis; it is important to note that the PAM sequence is not included in the sgRNA itself. A further constraint on the target sequence is due to the use of in vitro transcription from promoters such as T7, T3, or SP6 to produce sgRNAs, which function optimally with an initiator guanine (G) residue (Fig. 17.3A). If one uses a plasmid template for making sgRNA by in vitro transcription as originally described (e.g., Hwang et al., 2013), the target must start with GG due to cohesive end requirements of the cloning strategy. Alternatively, the template can be prepared using a PCR-based method (e.g., Nakayama et al., 2013), which is easier and faster, and allows more flexibility for target sequence selection because it requires only a single 5′ G (+ 1). The typical length of target sequence included in the design of an sgRNA (i.e., the genomic target sequence before the PAM) is 20 bp, but a recent report (Fu, Sander, Reyon, Cascio, & Joung, 2014) suggests that shorter (i.e., as short as 17 bp) targets function as efficiently as 20 bp and have fewer potential off-target sites. In summary, one needs to find G(N)16–19 target sequence (total length is 17–20 bp), followed by NGG in the genome. Any effects of decreasing the target length have not been rigorously tested in Xenopus, and therefore, it remains unknown whether shorter target sequences will work as efficiently in this system.
Figure 17.3.

PCR-based template synthesis for sgRNA in vitro transcription. (A) Two long, partially overlapping oligonucleotides are annealed and serve as primers for fill-in reactions by a thermostable polymerase. The 5′-oligo (primer) contains the T7 (or T3 or SP6) promoter; the transcription start site (G) corresponds to the first base of the target region (17–20 bp), followed by a portion of the sgRNA backbone. This region is complementary to the 3′-end of the 3′-oligo (primer) that contains the remainder of the sgRNA backbone sequence required for proper RNA folding. (B) Agarose gel (2%) electrophoresis of 2 μl of a PCR reaction (+) compared to no PCR (−) confirms successful sgRNA template synthesis. M, 100-bp DNA ladder. (C) Agarose gel (2%) electrophoresis of ~ 200 ng sgRNA following in vitro transcription, using the DNA template shown in (B). Example of heat-denatured (+) and nondenatured (−) sgRNA is shown. The RNA size marker lane (M) shown here was loaded on the same gel and was digitally spliced to place it adjacent to the sample lanes in this image.
Potential CRISPR/Cas9 target sequences in the genome can be identified by manually locating PAM sequences within the region of interest. Finding a guanine nucleotide 17–20 bp upstream of a PAM would identify a potential CRISPR target. Recent publications, however, suggest that target sites do not necessarily need to start with G. Any sequence followed by a PAM in the genome may be targeted by simply adding an extra G or GG not encoded in the genome sequence (Ansai & Kinoshita, 2014 and references therein). This approach may be a workable strategy when no other suitable option can be found. Once the target site is chosen, the next step is to determine whether the selected target sequence has similarity to other regions that may lead to off-target mutagenesis. This can be done bioinformatically using the raw genome sequence database (e.g., Blitz et al., 2013), or using a Web-based homology search engine (http://gggenome.dbcls.jp/en/) (Nakayama et al., 2013).
A number of other online tools are now available to search the X. tropicalis genome to identify optimal target and putative off-target sites. These include CHOPCHOP (https://chopchop.rc.fas.harvard.edu/index.php), CRISPR/Cas9 target predictor (http://crispr.cos.uni-heidelberg.de), CRISPRdirect (http://crispr.dbcls.jp/), E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html), GT-Scan (http://gt-scan.braembl.org.au/gt-scan/), and Cas-OFFinder (http://www.rgenome.net/cas-offinder/). Each tool offers a variety of input and output options, and depending on the purpose and knowledge of users different tools may be preferable. Notable features offered by many of the tools include: options for PCR primer design to assay CRISPR efficiency in target regions and to check for off-target mutagenesis (along with the inclusion of genomic location information for putative off-target regions); the ability to vary target length and 5′ nucleotide identity; the option to search for alternative PAM sequences; and the ability to target 5′ or 3′ regions of ORFs for the purpose of introducing N- or C-terminal protein modifications by homologous recombination, among others.
3.2.1 Considerations in target site choice
The success of a gene targeting strategy will be influenced by location of the mutation within the gene. While repair of double-strand breaks by NHEJ results in indels, most are short deletions. By chance, approximately two-thirds of indels will result in premature termination of protein translation and hence more 5′ target sites might be predicted to yield stronger effects. The remaining indels may or may not result in loss of gene function depending on their effects on protein structure. Therefore, choosing target sites within sequence encoding folded domains may be more advantageous for generating LOF mutations because even in-frame mutations may disrupt proper domain folding, resulting in loss of protein activity or stability. Folded domains are often recognizable as highly conserved sequences across species, or protein families, and protein fold search tools such as Pfam (http://pfam.xfam.org/search) can be used to assist in identification of these regions.
Genes encoding multiple protein isoforms need careful examination to ensure that site choice results in proper targeting of all possible isoforms. However, there may be cases where it is desirable to target specific isoforms while maintaining the function of others. A one-size-fits-all strategy is impossible, and therefore application of these ideas will need to be tailored to each gene's unique characteristics. Since the efficiency of cleavage at different target sites is variable and unpredictable, multiple independent target sites should be explored to identify the best sgRNA (see Section 4.1 for further discussion).
3.3 sgRNA template construction
Two sources of template can be used for the in vitro synthesis of sgRNAs. One is plasmid-based and requires subcloning (e.g., Guo et al., 2014; Hwang et al., 2013) and the other uses a PCR-based strategy for making linear DNA templates, shown schematically in Fig. 17.3.
3.3.1 Template assembly by PCR: Primers
The 5′ primer is unique to each target site and has the form
5′-TAATACGACTCACTATA G(N)16–19 GTTTTAGAGCTAGAAATAGCAAG-3′,
where G(N)16–19 is the target sequence of interest, designed as described above, and double underlined is the T7 promoter. T3 or SP6 promoters could be substituted, for example, SP6 would work better for GA(N)15–18.
The 3′ primer is common to all sgRNA templates and is as follows:
5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3′
Both primers above encode the original backbone published in Hwang et al. (2013). To date, we have tested another backbone, sgRNA(F + E), which has a modified hairpin reported to have stronger activity (Chen et al., 2013). However, we have not observed significant enhancement of mutagenesis activity using the sgRNA(F + E) backbone in Xenopus (our unpublished observations).
3.3.2 Template assembly by PCR: Assembly conditions
High fidelity polymerase (e.g., Platinum Pfx DNA polymerase, Invitrogen) is used to generate PCR-based templates. Assembly reactions are performed in a final volume of 100 μl as follows:
| PCR assembly reaction | |
|---|---|
| 10 × Buffer | 10 μl |
| 25 mM dNTP mixture | 1.2 μl |
| 50 mM Mg2SO4 | 2 μl |
| 5′ Primer (100 pmol/μl) | 2 μl |
| 3′ Primer (100 pmol/μl) | 2 μl |
| DNA polymerase | 1 μl |
| H2O (DNAse/RNAse-free) | To 100 μl |
Cycling conditions are
94 °C for 5 min.
10 cycles (up to 20 cycles) of 94 °C for 20 s, 58 °C for 20 s, 68 °C for 15 s.
68 °C for 5 min.
A typical result is shown in Fig. 17.3B. After confirming that a single product has been synthesized, templates are column-purified using, for example, QIAquick® PCR Purification Kit (Qiagen) or DNA Clean & Concentrator™-5 (Zymo Research) and DNA is eluted with 30–50 μl of RNase-free water. The concentration should be between 30 and 80 ng/μl or more, but, if lower, PCR may need to be repeated to increase yield.
3.3.3 In vitro transcription of sgRNA
We use the MEGAscript® T7 Transcription Kit (Life Technologies) following the manufacturer's recommended reaction mixture using up to 8 μl of template (0.25–0.6 μg if PCR-generated and 1 μg if using linearized plasmid) in 20 μl final volume. Incubations are 4 h to overnight at 37 °C to maximize yield, followed by DNAse digestion. Subsequent purification of sgRNA can be performed using either the LiCl precipitation (which is usually not recommended for small RNAs, but has been successful for sgRNAs) or by the phenol–chloroform extraction/NH4OAc precipitation method. sgRNA is dissolved in RNase-free water (5–30 μl depending on pellet size). A minimal RNA concentration of 100 ng/μl is desired to create “cocktails” for microinjection (see Section 3.4.1) and quality is further assessed by gel electrophoresis (see Fig. 17.3C). Secondary structure of sgRNAs can result in multiple bands that are readily resolved by denaturation before electrophoresis. Heat-denaturation can be performed by incubating at 60 °C for 2 min in 60–80% formamide followed by quick cooling on ice for 2 min. The yield of sgRNA is variable and template dependent. Amounts as high as 100 μg of sgRNA have been achieved from a single reaction. However, if yield is unacceptably low, longer incubation times may improve the outcome. If this approach fails, adding an extra 5′ sequence (5′-GCAGC-3′) to the end of the 5′-oligo (Michinori Toriyama, personal communication and our observations, see note added in proof 1) has been shown to enhance T7 polymerase reactions (Baklanov, Golikova, & Malygin, 1996). Other alternatives are to use the plasmid-based strategy for template construction (see Section 3.3), or choose an alternative target site.
Note added in proof 1: We now recommend the use of an improved 5′ primer which ensures higher efficiency of T7 polymerase-mediated transcription under the same experimental conditions as described: 5′-GCAGCTAATACGACTCACTATAG(N)16-19GTTTTAGAGCTAGAAATA-3′.
3.4 Procedure for microinjection
Optimal amounts of sgRNA and Cas9 used in embryo microinjections will vary depending both on the target site and experimental goals. For example, F0 analyses will require relatively high doses to observe LOF phenotypes in a high proportion of animals. However, there are scenarios where F0 LOF is either not possible (e.g., F0 knockouts cannot be achieved for maternal RNAs) or insufficient (e.g., phenotypic variability between mosaic LOF animals may not provide consistent results), and creation of lines carrying mutant alleles might be preferable. High doses of sgRNA and Cas9 that cause lethal phenotypes or are otherwise sterile would preclude the possibility of creating lines. Therefore, moderate doses may be needed to create fertile adults for germline transmission. In short, empirical determination of optimal doses will be required.
3.4.1 Doses of sgRNA and Cas9
To achieve mutations at a high efficiency, sgRNA doses ranging from 50 to 500 pg appear to be sufficient in many cases (Blitz et al., 2013; Guo et al., 2014; Nakayama et al., 2013). We recommend testing a range from 50 to 200 pg as increased toxicity can be observed at high doses (Guo et al., 2014).
Cas9 can be microinjected as either mRNA or protein. Three Cas9 plasmid templates have been successfully used in X. tropicalis (Blitz et al., 2013; Guo et al., 2014; Nakayama et al., 2013). Since native S. pyogenes Cas9 mRNA has weak activity in Xenopus (Nakayama et al., 2013), successful Cas9 constructs have incorporated two key modifications: a codon optimization more compatible with vertebrate systems and fusion to nuclear localization signals (NLSs). We recommend using appropriate mMessage mMachine Kits (Life Technologies) and following standard conditions to make capped Cas9 mRNAs. These should be quality-tested by A260 quantification and gel electrophoresis.
Table 17.1 summarizes effective mRNA doses that can be used as a starting point for experimental design. Depending on the source, Cas9 mRNAs are generally effective in the dose range from ~ 300 pg to 3–4 ng levels and researchers will need to empirically determine optimal doses. Although increasing toxicity has been reported with moderate doses of Cas9 prepared from pCS2 + 3xFLAG-NLS-SpCas9-NLS (Guo et al., 2014), this mRNA showed no apparent toxicity at doses up to 2 ng/embryo in our experiments (our unpublished observations). Recently, a recombinant Cas9 protein (containing an NLS) has become available (PNA Bio, Inc.). We have successfully targeted the tyrosinase (tyr) gene using this protein with no toxic effects on embryogenesis. Cas9 protein is reconstituted by dissolving 50 μg Cas9 protein in 40 μl of nuclease-free water, creating a stock at 1.25 μg/μl in 20 mM HEPES (pH 7.5), 150 mM NaCl, and 1% sucrose. This Cas9 solution is stored in small aliquots at − 80 °C and diluted in nuclease-free water to create “cocktails” for embryo injections. Cas9 protein is equally efficient in creating mutations at the tyr locus as coinjected Cas9 mRNAs transcribed from available plasmids (Fig. 17.4A and B). We find that 900–1200 ng of Cas9 protein coinjected with 200 pg of tyr sgRNA efficiently produces albino tadpoles (whereas lower doses of Cas9 protein under this regimen are less effective), at a mutation frequency comparable to 2 ng of Cas9 mRNA. An additional benefit of the availability of Cas9 protein is the opportunity to test efficiency of newly designed sgRNAs in vitro prior to microinjection (see Section 3.4.2).
Table 17.1.
Cas9 mRNA and sgRNA dosing for CRISPR/Cas9 mutagenesis in X. tropicalis
| Plasmid/protein | Cas9 dose range (per embryo) | sgRNA dose range (per embryo) | References |
|---|---|---|---|
| Cas9 (w/NLS) protein | 900–1200 pg protein | 50–200 pg | This study |
| pXT7-Cas9 | 0.55–3.2 ng mRNA (up to 6 nga) | 25–200 pg (200 pg eacha) | Nakayama et al. (2013) |
| pCasX | 3–4 ng mRNA | 150 pg | Blitz et al. (2013) |
| pCS2 + 3xFLAG-NLS-SpCas9-NLS | 200–500 pg mRNA | 1–2000 pg | Guo et al. (2014) |
| Our recommendations for RNAs | 300–2000 pg mRNA | 50–200 pg | See text for details |
Cas9 mRNA and sgRNA doses for simultaneous targeting of two different sites.
Figure 17.4.
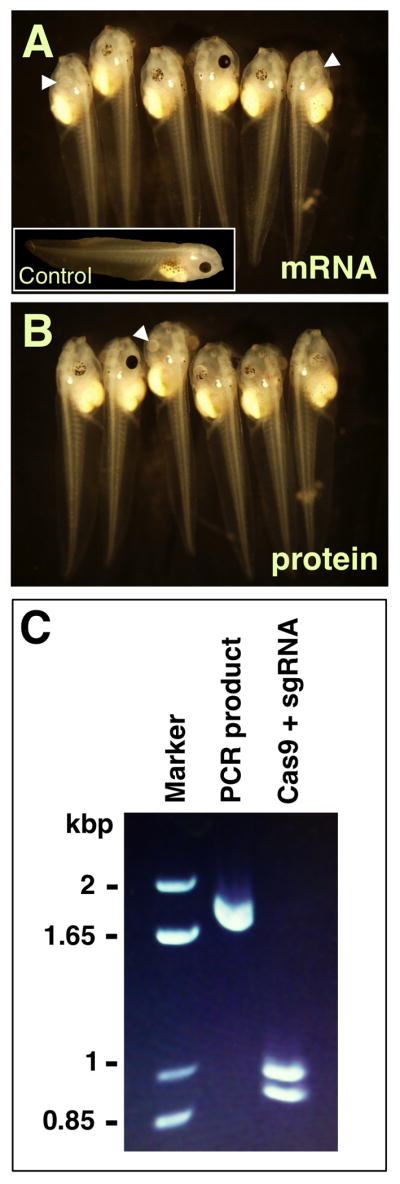
Cas9 protein-mediated mutagenesis and in vitro cleavage assays. sgRNA targeting the tyr gene was coinjected with either Cas9 mRNA (A) or protein (B) and representative embryos are shown. Inset is an uninjected control embryo. White arrowheads mark embryos displaying complete albinism. (C) The result of an in vitro Cas9 cleavage assay performed using a PCR product containing an asymmetrically located tyr target site.
3.4.2 Sidebar: Cas9 protein in vitro cleavage assays
An advantage of using Cas9 protein is that one can assay for sgRNA function using an in vitro cleavage assay prior to testing it in vivo. It seems reasonable to presume that sgRNAs that fail to efficiently direct Cas9 cleavage in vitro would probably also underperform in vivo. While success in vitro may not forecast success in vivo, we anticipate that this assay can serve to help researchers eliminate poor sgRNA candidates.
To assess a sgRNA's function in vitro, we have tested the tyr sgRNA on a DNA fragment amplified from the X. tropicalis genome (Fig. 17.4C). In vitro assays are performed in a 10 μl final volume as follows:
| Cas9 in vitro cleavage assay reaction | |
|---|---|
| 10 × NEB buffer 3 | 1 μl |
| 10 × NEB BSA (diluted from 100 ×) | 1 μl |
| Target DNA (PCR amplicon) | ~ 250 ng |
| Cas9 protein | 0.5–1 ng |
| sgRNA | 250 pg |
| H2O (DNAse/RNAse-free) | To 10 μl |
The reaction is incubated at 37 °C for 1 h. We recommend using the manufacturer's protocol for RNAse digestion and inactivation of the reaction; samples are then analyzed by gel electrophoresis (Fig. 17.4C).
3.4.3 Procedure for embryo microinjection
Many factors are critical for successful Cas9-mediated mutagenesis in X. tropicalis. In vitro fertilizations are preferable to natural matings because they produce synchronous populations of embryos. It is recommended that injections begin shortly after fertilization to maximize the number/extent of animals displaying the strongest phenotypes. We begin de-jellying 10 min postfertilization (mpf; fertilization is defined here as the time of flooding of the eggs with medium after sperm addition; Ogino, McConnell, & Grainger, 2006). We strive to complete de-jellying within 5–10 min and after washing the one-cell embryos, injections can begin approximately 20 mpf. In some batches of embryos, single-site injections produced the strongest phenotypes during the earliest 10 min of injection (30 mpf) and incrementally declined thereafter (our unpublished data). Injections at later time points may have improved outcomes if multiple injection sites are used, but this needs further investigation. Injection regimens can be applied to later stage embryos, which may be useful for achieving a “poor man's” tissue-specific knockout—by taking advantage of the fate map to target specific blastomeres for injection. Detailed procedures for in vitro fertilization, microinjection, and culture of X. tropicalis embryos are published in Ogino et al. (2006). We routinely microinject sgRNA/Cas9 cocktails in a volume of 1–4 nl per embryo, with the injection site located in the animal hemisphere. Some researchers prefer to coinject a lineage tracer as a component of their cocktails to permit elimination of embryos that were not properly injected. Inclusion of 5 ng fluorescent dextran/nl (Life Technologies, fluorescein (D-1845) or rhodamine dextrans (D-1818)) into sgRNA/Cas9 cocktails has no deleterious effects on mutagenesis.
3.5 Assessment of mutagenesis: Genotyping
A number of methods have been employed for detecting induced indels; examples include the T7 endonuclease I (Kim, Lee, Kim, Cho, & Kim, 2009) and Surveyor assays (Guschin et al., 2010). These detect mismatches occurring between wild-type and mutant DNAs created after denaturation and annealing of mixtures of PCR amplicons from target regions. Another method, the high resolution melting assay, detects differences in melting temperature between wild-type and mutant amplicons (Wittwer, Reed, Gundry, Vandersteen, & Pryor, 2003). Here, we describe the DSP (direct sequencing of PCR amplicons) assay (Nakayama et al., 2013): a rapid, initial screening assay for targeting efficiency in which target regions are PCR amplified from single mosaic F0 embryos and sequenced directly to detect the presence of indels within the population of amplicons.
3.5.1 Embryo lysis and PCR
Individual embryos are transferred to 0.2-ml PCR tubes containing 100 μl of lysis buffer (50 mM Tris [pH 8.8], 1 mM EDTA, 0.5% Tween 20) containing freshly added proteinase K at a final concentration of 200 μg/ml. Embryos are incubated at 56 °C for 2 h to overnight, followed by incubation at 95 °C for 10 min to inactivate proteinase K. Lysates are centrifuged for 10 min at 4 °C and 1 μl aliquots are used directly in 25 μl PCR reactions. Individual embryos from blastula stages to approximately stage 40 (Nieuwkoop & Faber, 1967) lysed in this manner generally yield sufficient DNA for many PCR reactions. Similarly prepared samples from later stages typically require 10- to 20-fold dilution prior to use in PCR. The PCR primers ideally should be designed to amplify 300–400 bp genomic regions containing the target sequence. Successful PCR should be confirmed by gel electrophoresis, followed by column-purification of the PCR reaction. Amplicons are then subjected to Sanger sequencing using either primer used for the PCR.
3.5.2 Evaluation of sequencing results and subsequent identification of specific indels
If indels have occurred, the PCR product will contain a mixture of heterogeneous fragments with different mutations. The profile will show sequence heterogeneity in peaks in the mutated region (Fig. 17.5A). Subcloning PCR products from the population and sequencing of individual clones confirms the results of the DSP assay (Fig. 17.5B) that successful mutagenesis has occurred. The advantages of the DSP assay include its ease of use and the ability to visualize that the genomic region where sequence perturbations are observed corresponds to the intended target site. We have detected mutations by DSP assay in embryos with rates of mutagenesis of ~ 25% or higher, but generally cannot detect rates at lower levels (our unpublished observations), demonstrating the moderate sensitivity of the assay. Notably, a strength of this assay may be that it is not overly sensitive: since F0 mosaic animals with low rates of germline transmission necessitate laborious screening of F1s, it may be a useful standard to only raise F0 animals for subsequent mating that have mutagenesis levels detectable by DSP assay. This standard would improve one's chances of achieving adult F0 animals with germline transmission rates of at least one in four, which is not too burdensome for genotypic screening of F1s.
Figure 17.5.

DSP assay. (A) Representative results of DSP assays. Single embryos (top, uninjected; bottom, injected with indicated RNAs) were lysed and the targeted genomic region was PCR amplified; amplicons were then directly sequenced. Perturbation of peaks (double-headed arrow) on the 3′ side of the PAM region (note that in this specific case, the target is on the antisense strand) seen in the sample of the injected embryo suggests indel events occurred between the target sequence (lightly shaded area where multiple peaks overlap) and the PAM region (darkly shaded). (B) The PCR amplicon from the injected embryo was recloned and sequenced to show the profile of individual mutations found in mosaic individuals (bottom sequence alignments). Dashes (-) indicate gaps. The numbers in parentheses indicate the frequency of each mutation pattern seen in the total number of sequenced clones, suggesting that a mutation frequency of 36% (i.e., 4/11) can be detected as a positive targeting event by the DSP assay. The figure is adapted and modified from Nakayama et al. (2013).
4 Discussion
We have described basic and essential tips for CRISPR-mediated mutagenesis in X. tropicalis. Here, we briefly discuss additional aspects of CRISPR/Cas that may apply to Xenopus.
4.1 Multiple targeting strategy: Avoiding off-target problems and simpler genotyping of F1 animals
A problem with all targeted nuclease mutagenesis strategies (ZFNs, TALENs, and CRISPR/Cas9) is the possibility of creating mutations at unintended sites located elsewhere in the genome. These off-target sites bear sequence similarity to the intended target site and therefore sequence similarity-based searches are one means of limiting off-target mutagenesis. Studies on CRISPR/Cas9 performed both in vitro and in bacteria have shown that a perfect match between sgRNA and the DNA target in an 8- to 12-base region of the target sequence proximal to the PAM, known as the “seed sequence,” is crucial for Cas9-mediated cleavage (see Fineran & Dy, 2014; Hsu et al., 2014; Sander & Joung, 2014 and references therein). Mismatches are tolerated in the target nucleotides that are distal to the seed sequence, which could lead to off-target cleavage. As discussed in Section 3.2, a number of Web-based search tools have been developed that predict both target and off-target sites for sgRNAs in X. tropicalis. When choosing between potential sites to target, one should avoid sequences with off-target mismatches that are largely located distal to the seed sequence. Also, one should keep in mind that sequences followed by the NAG PAM can also be off-target sites, therefore off-target analysis should include both NGG and NAG PAMs.
One possible avenue for reducing off-target mutagenesis is the use of paired nickases (Mali, Aach, et al., 2013; Ran et al., 2013). The Cas9 D10A mutant is incapable of creating double-strand breaks, but instead nicks one strand at the target site. Using a pair of sgRNAs with target sites of appropriate orientation and spacing, in conjunction with Cas9 D10A, permits mutagenesis at the paired site without creating double-strand breaks at sgRNA single (off-target) sites across the genome. Another approach would be to use FokI fusions to dCas9, a doubly mutated Cas9 with no nuclease activity (Guilinger, Thompson, & Liu, 2014; Tsai et al., 2014).
This approach similarly involves choosing two nearby target sites to direct Cas9–FokI to the genome, which only functions as a dimer to cleave DNA. Both strategies are limited by the ability to find two nearby target sequences with appropriate directionality and spacing. Neither approach has been reported in Xenopus (see note added in proof 2).
Off-target mutagenesis may not be a major concern for LOF assays in Xenopus. Studies by Blitz et al. (2013) and Guo et al. (2014) found no evidence for off-target mutagenesis in X. tropicalis, similar to studies in mouse embryos (Wang et al., 2013; Yang et al., 2013), but unlike reports using cultured cells (Cradick, Fine, Antico, & Bao, 2013; Fu et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013). This suggests that in whole organisms off-target cleavage may be negligible.
A strategy we recommend when performing F0 analyses to ensure that a phenotype is due to mutagenesis of the targeted gene is to replicate the phenotype using multiple independent sgRNAs targeting the same gene (Nakayama et al., 2013). It is unlikely that multiple sgRNAs would result in mutation of the same off-target gene. An alternative would be to rescue the phenotype by expressing a wild-type mRNA (Guo et al., 2014; Nakayama et al., 2013). These approaches provide strong supporting evidence for an on-target effect and refute the notion that a phenotype is due to off-target mutagenesis.
If the study of F0 mosaic animals is not desirable, then various breeding strategies for creating non-mosaic animals may be employed. Ideally, one would construct a mutant that contains the same lesion at both alleles; however, since multiple generations are needed to achieve this, a time consuming process, analysis of phenotypes may be significantly delayed. Matings between F0 mosaic animals will produce non-mosaic F1s that are (primarily) compound heterozygotes (Fig. 17.2). Because F0 mutagenesis results in a variety of indels in the same target site, F1 offspring will likely be difficult to genotype. These problems can be overcome using two nonoverlapping sgRNA target sites within a gene. A mating between F0 animals mutated at different target sites will produce F1 offspring that can more readily be genotyped. Thus, compound heterozygotes mutated at nonoverlapping target sites permit more rapid phenotypic analyses.
Note added in proof 2: We have confirmed that the nickase version of Cas9 (D10A) can mediate efficient mutagenesis using paired sgRNAs in Xenopus.
4.2 Further applications of CRISPR-mediated mutagenesis in Xenopus
As with morpholino antisense oligo-mediated knockdowns, it may be possible to study the effects of multigene knockouts using CRISPR/Cas. Coinjection of multiple sgRNAs, each targeting a different site or gene, will likely permit such analyses. This is especially useful in situations where redundancy, possibly caused by gene duplications, makes phenotypic assays of gene function problematic.
The use of multiple sgRNAs also permits examining the role of cis-regulatory elements. Target sites flanking an element can be used to delete the intervening sequence as has been shown by promoter deletion by Nakayama et al. (2013). This approach in principle can be used to create larger gene deletions though the frequency of successful mutagenesis by this strategy needs to be determined.
Another application is homology-directed recombination (HDR)-dependent gene targeting, i.e., introduction of point mutations, or knocking in larger genetic elements such as sequence tags or transgenes into any site in the genome (see Sander & Joung, 2014 and references therein). This is a future challenge for Xenopus researchers.
The CRISPR-mediated mutagenesis strategy outlined herein may provide a means for LOF analysis in the F0 animal. However, the variable mosaicism of F0 embryos may ultimately interfere with some analyses and therefore improvements to the efficiency of CRISPR/Cas in Xenopus would be valuable. Major advantages of the Xenopus system include the ease of manipulating oocytes and sperm and these techniques may offer new avenues to reduce mosaicism. The host-transfer method (Olson, Hulstrand, & Houston, 2012) allows for in vitro culturing and manipulation of oocytes, which are then implanted back into the body cavity of a host female for proper ovulation that is necessary for successful fertilization. Oocytes may be injected with Cas9 protein and sgRNA(s) leading to a mutant gamete with only one gene copy. Fertilization of manipulated eggs with wild-type sperm would result in nonmosaic heterozygotes. Sperm can be similarly manipulated. It may be feasible to incubate decondensed sperm nuclei (Hirsch et al., 2002; Kroll & Amaya, 1996) with Cas9 protein–sgRNA cocktails to create mutations, and to inject these into unfertilized eggs to create nonmosaic heterozygotes. In both cases, further technology development is desirable to achieve HDR. The host-transfer method may be especially useful in this regard because oocytes have naturally high homologous recombination activity (Carroll, Wright, Wolff, Grzesiuk, & Maryon, 1986).
Acknowledgments
This work was supported in part by NIH grants EY022954, EY018000, OD010997, and by the Sharon Stewart Aniridia Trust to R. M. G. Funding was also provided by NSF and NIH grants 1147270 and HD073179, respectively, to K. W. Y. C., and NIH grant HD080684 to I. L. B. Fellowship funding that contributed to this work was awarded by the UVA Society of Fellows to M. B. F. Some preliminary experiments were performed during the 2013 and 2014 Cold Spring Harbor Laboratory Xenopus Courses, which the authors would like to acknowledge. The authors also thank Amy Sater, Jamina Oomen-Hajagos, Jerry Thomsen, Michinori Toriyama, Doug Houston, and Rob Steele for sharing unpublished information, and past and current members in Grainger Lab, especially Marilyn Fisher and Cristina D'Ancona, for help in experiments and discussion. We also thank Yonglong Chen for pCS2 + 3xFLAG-NLS-SpCas9-NLS and Jianzhong Jeff Xi for pXT7-Cas9. Finally, the authors would also like to gratefully acknowledge the support of Xenbase and the National Xenopus Resource and their staffs.
References
- Abu-Daya A, Khokha MK, Zimmerman LB. The Hitchhiker's guide to Xenopus genetics. Genesis. 2012;50:164–175. doi: 10.1002/dvg.22007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansai S, Kinoshita M. Targeted mutagenesis using CRISPR/Cas system in medaka. Biology Open. 2014;3:362–371. doi: 10.1242/bio.20148177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baklanov MM, Golikova LN, Malygin EG. Effect on DNA transcription of nucleotide sequences upstream to T7 promoter. Nucleic Acids Research. 1996;24:3659–3660. doi: 10.1093/nar/24.18.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz IL, Biesinger J, Xie X, Cho KWY. Biallelic genome modification in F0 Xenopus tropicalis embryos using the CRISPR/Cas system. Genesis. 2013;51:827–834. doi: 10.1002/dvg.22719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D, Wright SH, Wolff RK, Grzesiuk E, Maryon EB. Efficient homologous recombination of linear DNA substrates after injection into Xenopus laevis oocytes. Molecular and Cellular Biology. 1986;6:2053–2061. doi: 10.1128/mcb.6.6.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Research. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Dy RL. Gene regulation by engineered CRISPR-Cas systems. Current Opinion in Microbiology. 2014;18:83–89. doi: 10.1016/j.mib.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Fish MB, Nakayama T, Fisher M, Hirsch N, Cox A, Reeder R, et al. Xenopus mutant reveals necessity of rax for specifying the eye field which otherwise forms tissue with telencephalic and diencephalic character. Developmental Biology. 2014;395:317–330. doi: 10.1016/j.ydbio.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High- frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR- Cas nuclease specificity using truncated guide RNAs. Nature Biotechnology. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to Fok1 nuclease improves the specificity of genome modification. Nature Biotechnology. 2014;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang T, Hu Z, Zhang Y, Shi Z, Wang Q, et al. Efficient RNA/ Cas9-mediated genome editing in Xenopus tropicalis. Development. 2014;141:707–714. doi: 10.1242/dev.099853. [DOI] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods in Molecular Biology. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- Harland RM, Grainger RM. Xenopus research: Metamorphosed by genetics and genomics. Trends in Genetics. 2011;27:507–515. doi: 10.1016/j.tig.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch N, Zimmerman LB, Gray J, Chae J, Curran KL, Fisher M, et al. Xenopus tropicalis transgenic lines and their use in the study of embryonic induction. Developmental Dynamics. 2002;225:522–535. doi: 10.1002/dvdy.10188. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR- Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Cliffe R, Amaya E. Highly efficient bi-allelic mutation rates using TALENs in Xenopus tropicalis. Biology Open. 2012;1:1273–1276. doi: 10.1242/bio.20123228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll KL, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- Lei Y, Guo X, Liu Y, Cao Y, Deng Y, Chen X, et al. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs) Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17484–17489. doi: 10.1073/pnas.1215421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA- guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Nakai Y, Okada M, Yaoita Y. Targeted gene disruption in the Xenopus tropicalis genome using designed TALE nucleases. Zoological Science. 2013;30:455–460. doi: 10.2108/zsj.30.455. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Nakajima T, Takase M, Yaoita Y. Generation of albino Xenopus tropicalis using zinc-finger nucleases. Development, Growth & Differentiation. 2012;54:777–784. doi: 10.1111/dgd.12006. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Yaoita Y. Comparison of TALEN scaffolds in Xenopus tropicalis. Biology Open. 2013;2:1364–1370. doi: 10.1242/bio.20136676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Fish MB, Fisher M, Oomen-Hajagos J, Thomsen GH, Grainger RM. Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis. 2013;51:835–843. doi: 10.1002/dvg.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin) Amsterdam: North-Holland; 1967. [Google Scholar]
- Ogino H, McConnell WB, Grainger RM. High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nature Protocols. 2006;1:1703–1710. doi: 10.1038/nprot.2006.208. [DOI] [PubMed] [Google Scholar]
- Olson DJ, Hulstrand AM, Houston DW. Maternal mRNA knock-down studies: Antisense experiments using the host-transfer technique in Xenopus laevis and Xenopus tropicalis. Methods in Molecular Biology. 2012;917:167–182. doi: 10.1007/978-1-61779-992-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High- throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nature Biotechnology. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Early development of Xenopus laevis: A laboratory manual. Cold Spring Harbor Press; 2010. [Google Scholar]
- Suzuki KT, Isoyama Y, Kashiwagi K, Sakuma T, Ochiai H, Sakamoto N, et al. High efficiency TALENs enable F0 functional analysis by targeted gene disruption in Xenopus laevis embryos. Biology Open. 2013;2:448–452. doi: 10.1242/bio.20133855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns RM, Terns MP. CRISPR-based technologies: Prokaryotic defense weapons repurposed. Trends in Genetics. 2014;30:111–118. doi: 10.1016/j.tig.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature Biotechnology. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. High-resolution genotyping by amplicon melting analysis using LCGreen. Clinical Chemistry. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JJ, Cherone JM, Doyon Y, Ankoudinova I, Faraji FM, Lee AH, et al. Efficient targeted gene disruption in the soma and germ line of the frog Xenopus tropicalis using engineered zinc-finger nucleases. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7052–7057. doi: 10.1073/pnas.1102030108. [DOI] [PMC free article] [PubMed] [Google Scholar]