

Abstract
Background & Aims
A fenestrated phenotype is characteristic of liver sinusoidal endothelial cells (LSECs), but liver sinusoids become defenestrated during fibrosis and other liver diseases. Thrombospondin-1 (TSP1) is a matrix glycoprotein with pro-fibrotic effects, and the CD47-binding fragment of TSP1 also has anti-angiogenic effects in endothelial cells. We hypothesized that the CD47-binding fragment of TSP1 could induce defenestration in LSECs through the Rho-Rho kinase (ROCK)-myosin pathway.
Methods
Freshly isolated rat LSECs were treated with TSP1 or CD47-binding peptides of TSP1. LSEC fenestration was assessed with scanning electron microscopy, and myosin phosphorylation was assessed with immuno-fluorescence.
Results
Treating LSECs with TSP1 caused a dose-dependent loss of fenestrae, and this effect could not be blocked by SB-431542, the TGF-β1 receptor inhibitor. A CD47-binding fragment of TSP1, p4N1, was able to induce defenestration, and a CD47-blocking antibody, B6H12, was able to suppress p4N1-induced defenestration. The p4N1 fragment also caused contraction of fenestra size, correlated with an increase in myosin activation. Pretreatment with Y-237642 (a ROCK inhibitor) prevented p4N1-induced myosin activation and fenestrae decrease. Simvastatin has also been shown to antagonize Rho-ROCK signalling, and we found that simvastatin pretreatment protected LSECs from p4N1-induced myosin activation and defenestration.
Conclusions
We conclude that CD47 signals through the Rho-ROCK-myosin pathway to induce defenestration in LSECs. In addition, our results show that simvastatin and Y-237642 have a beneficial impact on fenestration in vitro, providing an additional explanation for the efficacy of these compounds for regression of liver fibrosis.
Keywords: capillarization, discontinuous endothelium, mechanical signal transduction, myosin light chain
Thrombospondin 1(TSP1) is a matricellular protein secreted during tissue damage and is an essential component of the inflammatory response (1–3). In the liver, TSP1 is secreted mainly by the activated hepatic stellate cells and to a lesser extent by the Kupffer cells and endothelial cells (4). One of the well-studied functions of TSP1 in liver includes the activation of latent transforming growth factor-β1 (LTGF-β1) by binding and dissociating the latency associated peptide bound to the active TGF-β1 dimer (5). After liver injury, TSP1-mediated TGF-β1 activation contributes to the high bioavailability and fibrogenic activity of TGF-β1 (6).
Thrombospondin 1 is also a potent inhibitor of angiogenesis (7). The C-terminal domain of TSP1 binds the cell surface receptors CD36 (glycoprotein IV) and CD47 (Integrin Associated Protein, IAP, or Rh-related antigen), and both receptors have anti-angiogenic effects. The ligation of CD47 has been shown to activate Rho and Rho kinase (ROCK), causing myosin contractility with decreased migration (8). TSP1 binding to CD47 causes multiple parallel effects that oppose vasodilation by inhibiting: VEGFR2 activation (9), NO-stimulated activation of sGC (10) and adenylate cyclase activation (11). Although there are multiple studies showing TSP1 to have a pro-fibrotic role in liver and other organs, and many studies showing TSP1 to have anti-angiogenic and hypertensive roles in endothelial cells, still there is little known about fibrotic roles of TSP1 in endothelial cells. Liver sinusoidal endothelial cells (LSECs) are an intriguing system in which to address this question because altered cell morphology and contractile effects could affect their filtration functions.
Liver sinusoidal endothelial cells form a porous barrier between the sinusoidal blood and the space of Disse. LSEC have small (100–200 nm) openings called fenestrae, which lack basement membrane or diaphragm. Fenestrae allow free transfer of substrates between blood and hepatocytes and loss of fenestrae (defenestration) hinders the normal exchange function carried out by the sinusoids (12). Defenestration is a hallmark of LSEC capillarization, which is a pathological de-differentiation process that leads to formation of an organized basement (13,14). Capillarization precedes liver fibrosis (15) and has been linked with other diseases including alcohol-induced fibrosis (16), steatohepatitis (17) and atherosclerosis (18). Liver toxins (e.g. ethanol, Dimethylnitrosoamine etc.) have been found to decrease fenestration. Targeted gene-delivery of hepatocyte growth factor to the activated hepatic stellate cells in DMN-induced fibrotic rats was shown to regress liver fibrosis together with re-fenestration of hepatic sinusoids (19). Agents that disrupt actomyosin filaments (e.g. Cytochalasin B, Latrunculin A etc.) caused an increase in LSEC fenestration (12), and blocking the calcium-MLCK (myosin light chain kinase) pathway decreased fenestration (20,21), suggesting that myosin motor activity, in conjunction with actin filaments, can decrease fenestration.
In the light of previous work indicating LSEC fenestration to be regulated by actomyosin, we asked whether TSP1 alters LSECs fenestration, and whether this effect is mediated by cytoskeletal signalling. We found that addition of TSP1 to freshly isolated LSECs caused a dose-dependent decrease in fenestration, independent of TGF-β1 signalling. The CD47-binding fragment of TSP1 (p4N1) was sufficient to induce defenestration, and a CD47-blocking antibody prevented this effect. To test whether the Rho/ROCK/myosin pathway is involved, we inhibited ROCK signalling and found LSEC protected from p4N1-induced defenestration. p4N1 alone could induce myosin phosphorylation, but it had no effect on myosin in the presence of ROCK inhibitor. Finally, we found that simvastatin, a statin drug with the side effect of suppressing Rho/ROCK (22), was effective at protecting LSEC from p4N1-induced defenestration. Our results indicate a role for the C-terminal domain of TSP1 in inducing LSEC defenestration via cytoskeletal signalling. Furthermore, statin drugs and therapeutic antibodies against CD47 may provide protection against this effect.
Materials and methods
Ethics statement
Cell isolation procedures were performed on male Wistar rats, with initial weights of 250–300 gm, housed in the NUS animal holding unit (AHU). Rats were handled in strict accordance with good animal practices to ameliorate suffering, and all animal work was approved by the Institutional Animal Care and Use Committee (IACUC approval 014/09), National University of Singapore, Singapore.
Endothelial cell isolation
The method of LSEC isolation has been described earlier (23). The liver cells were harvested by a modified two-step in situ collagenase perfusion and were centrifuged at 50g for 2 min to separate the hepatocytes. The supernatant, containing a mixture of non-parenchymal cells, was layered on top of a two-step Percoll (Sigma Aldrich, St. Louise, MO, USA) gradient (25–50%). Endothelial cell isolation from the supernatant was further carried out by density-gradient centrifugation for 20 min at 900g. The intermediate zone between the two density layers was enriched in LSECs, which were plated on 12 mm coverslips (Electron Microscopy Sciences, Hatfield, PA, USA) coated with collagen Type I (Thermo Fisher Scientific, Waltham, MA, USA) at a density of 2.5e5 cells/cm2. For seeding the cells, supplemented M131 medium (M131 medium + microvascular growth supplement; Gibco/Invitrogen, Carlsbad, CA, USA) was used. After 2 h of attachment, the medium was changed to basal M131 medium [M131 medium+2% foetal bovine serum (HyClone, Logan, UT, USA)]. The purity of the cultures was >95% as determined by acetylated-LDL (Invitrogen, Carlsbad, USA) uptake and also by examining the cells under SEM where <5% of the cells were devoid of fenestrae.
Experiments with LSECs
For all experiments, freshly isolated LSECs were seeded on collagen-coated dishes for 2 h in supplemented M131 medium, after which the medium was changed to basal M131 medium with addition of peptides, inhibitors or small molecules. To show the long-term, general effect of TSP1 on LSECs, cells in Fig. 1 were treated for 12 h with different doses (0 ng/ml, 10 ng/ml and 100 ng/ml) of purified human platelet TSP1 (Millipore, Billerica, MA, USA) in basal M131 medium and were fixed for SEM preparation. For all subsequent experiments (Fig. 7), 1 h treatments were used, as 1 h exposure to TSP1 was sufficient to induce significant levels of defenestration (Fig. 2A). For experiments involving the inhibitor SB-431542 (R&D systems, Minneapolis, MN, USA) and TGF-β1 (R&D systems), cells were pretreated with 20 μM (24) of inhibitor for 1 h before addition of 100 ng/ml of TSP1 or 5 ng/ml of TGF-β1 [which has a very short half-life (25)] for 1 h, followed by fixation for SEM analysis. The CD47-binding peptides and the mutated peptides (Anaspec, Fremont, CA, USA) of TSP1 (Fig. 3A) were used at a concentration of 1 μM (26) in basal M131 media. For experiments with other compounds, LSECs were first incubated with 10 μM of Y-237642 (Sigma Alderich, St. Louis, MO, USA), or 1 μg/ml of B6H12 (BD Pharmigen, San Diego, CA, USA), or 1 μM (27) of simvastatin (Sigma Alderich, MO, USA), in basal M131 medium for 1 h followed by addition of 1 μM p4N1 or p4G1. The samples were fixed after 1 h and further processed for SEM.
Fig 1.
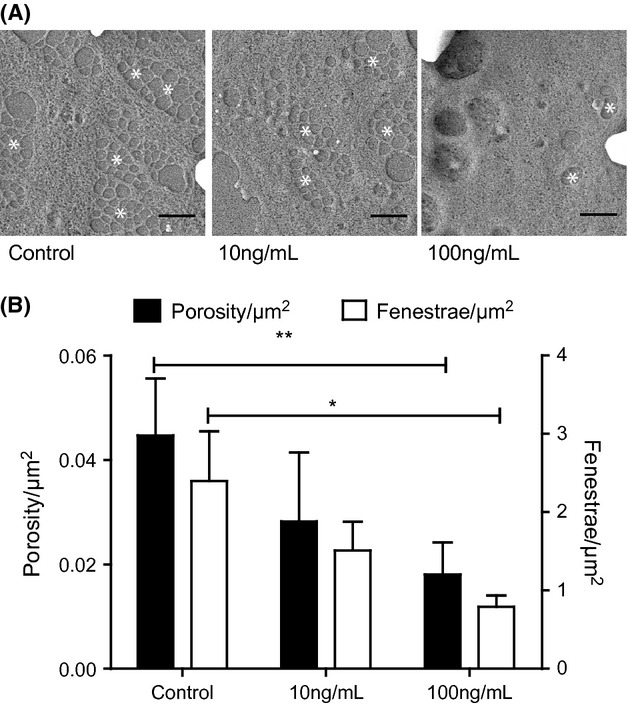
Thrombospondin 1 (TSP1) induced LSEC defenestration. (A) SEM images of LSEC after treatment with different doses (0 ng/ml, 10 ng/ml and 100 ng/ml) of TSP1 for 12 h. Scale bars represent 1 μm. White asterisks (*) indicate fenestrae or clusters of fenestrae (sieve plates). (B) Porosity/μm2 (black bars, represent fenestrae area) and Fenestrae number/μm2 (white bars, represent the absolute number of fenestrae) observed in LSEC after treatment with different doses of TSP1 (* represents P < 0.05 with Student's t-test).
Fig 7.

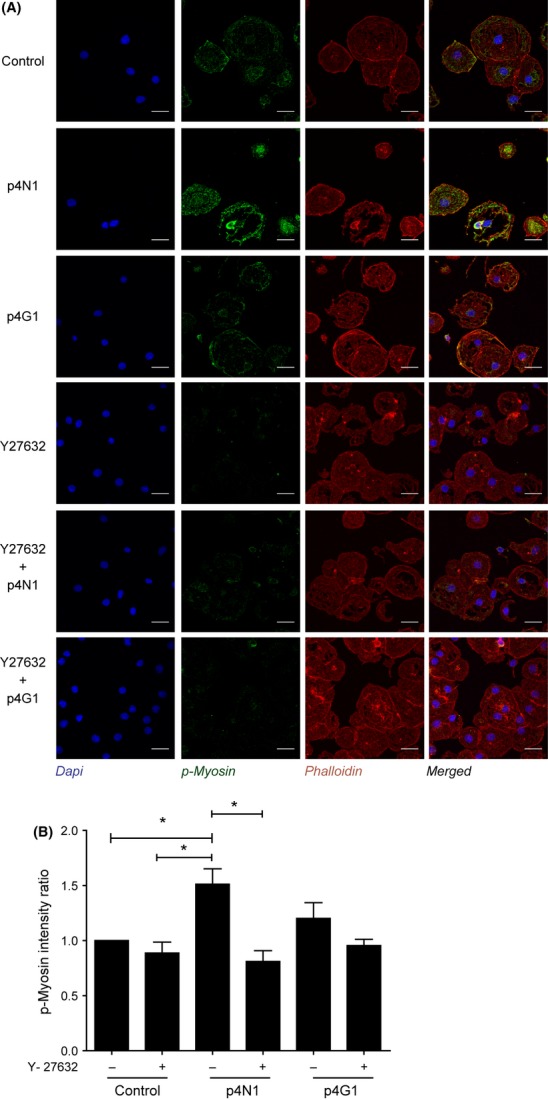
Simvastatin blocks p4N1-induced defenestration. Measurement of (A) porosity/μm2 and (B) fenestrae number/μm2 after p4N1, with or without simvastatin pretreatment. (C) Immuno-fluorescence of LSECs treated with p4N1, with or without simvastatin pretreatment (Nucleus –Blue; p-MLC –Green; F-Actin- red). (D) Quantification of p-MLC fluorescence, as a ratio vs. control. (* represents P < 0.05; for porosity/μm2, one-way anova was applied to Control, p4N1 and Simvastatin+ p4N1 treatments.)
Fig 2.

Thrombospondin 1 (TSP1)-induced defenestration was independent of TGF-β1 signalling. (A) SEM images of liver sinusoidal endothelial cells (LSEC)s pretreated with SB-431542 for 1 h before addition of TGF-β1 or TSP1 (a) Control; (b) SB-431542; (c) TSP1; (d) SB-431542+TSP1; (e) TGF-β1; (f) SB-431542+TGF-β1. All scale bars are 1 μm. (B) Porosity/μm2 and (C) Fenestrae number/μm2 after treatment with SB-431542, TSP1 and TGF-β1 (* represents P < 0.05 for one-way anova applied to the control, TSP1, and SB-431542+TSP1 treatments).
Fig 3.
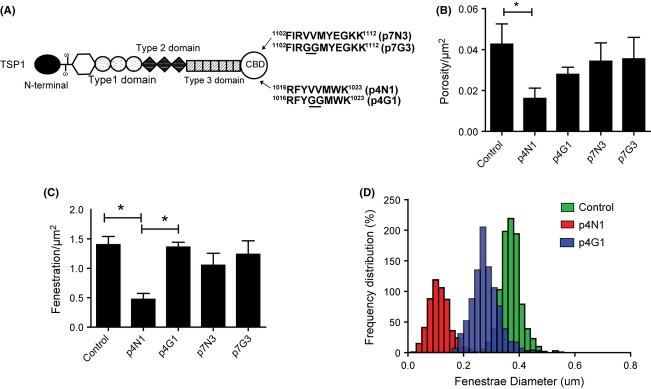
Defenestration was induced by a peptide from the Thrombospondin 1 (TSP)1 C-terminal domain. (A) Domains of TSP1 and the origins of the recombinant peptides from the C-terminal binding domain (CBD). Amino acid residues differing from the native TSP1 residues are underlined. (B) Porosity/μm2 and (C) Fenestrae number/μm2 of liver sinusoidal endothelial cells (LSEC) after treatment with CBD peptides. (D) Distribution of LSEC fenestrae diameter after peptide treatment. (* represents P < 0.05 for one-way anova applied to Control, p4N1 and p4G1 treatments).
Scanning electron microscopy
Liver sinusoidal endothelial cells were prepared for SEM following a previous protocol (28). Briefly, the samples were fixed in 2.5% glutaraldehyde solution (Sigma Aldrich) prepared in 0.1N sodium cacodylate (Sigma Aldrich) buffer for 12 h. The samples were treated with 1% osmium tetraoxide (Sigma Aldrich) solution for 1 h, followed by serial dilution in 25–100% ethanol and final drying in hexamethylene disilazane (Sigma Aldrich). Samples were sputter-coated with gold before visualization using JEOL 6701 at 10kV (JEOL, Tokyo, Japan).
Immunofluorescence
Liver sinusoidal endothelial cells were fixed with 4% paraformaldehyde (Sigma Aldrich) before permeabilization with 0.5% of 100% Triton-X (Sigma Aldrich). After blocking with 1% bovine serum albumin (Sigma Aldrich) for 1 h at room temperature, samples were incubated with p-myosin light chain antibody (Cell signalling, 1:100 dilution) overnight. DAPI (1:1000), phalloidin-633 (1:70) and secondary antibody (Alexa Fluor 488 (1:200), Invitrogen, CA, USA) were prepared in blocking buffer and incubated for 1 h. Samples were mounted with mounting medium and visualized using a Nikon A1R inverted confocal microscope with a Plan Apo VC 60× Oil 1.4NA objective lens.
Image analysis
Images were analysed using ImageJ software (http://rsb.info.nih.gov/ij/). Measurement of porosity (total fenestration area) and fenestrae number was done as previously (29). For each experiment at least five images were taken per sample, and a total of 400–2000 fenestrae were analysed per sample. To quantify porosity/μm2, the fenestrae diameters in each image were used individually to compute fenestrae areas, assuming fenestrae to be circular. The fenestrae areas were summed to give the total area occupied by fenestrae per image, which was normalized to the total area of the cell. The total number of fenestrae counted per image was also normalized to the cell area to give the fenestrae/μm2.
Statistical analysis
Unless otherwise stated, statistical significance was analysed for four treatment conditions (Control, Inhibitor, CD47-ligand, and Inhibitor+CD47-ligand) using 1-factorial analysis of variance (anova), followed by post-hoc comparison with Tukey's least significant difference (LSD) test. All tests were performed in MATLAB (Mathworks, Natick, MA, USA) using at least n = 3 independent replications, and P-values less than 0.05 were considered significant.
Results
TSP1 induced LSEC defenestration, regardless of TGF-β1 receptor inhibition
Thrombospondin 1 levels in plasma measured under different conditions vary between 14 μg/ml and 200 μg/ml (30–32). We treated primary cultures of LSEC with 0 ng/ml, 10 ng/ml or 100 ng/ml of TSP1 in basal M131 medium (M131 media+ 2% FBS) for 12 h (Fig. 1A). A quantity of 10 ng/ml of TSP1 did not affect the porosity/μm2 (Fig. 1B, black bars), nor the number of fenestrae (Fig. 1B, white bars), compared with untreated cells. A quantity of 100 ng/ml of TSP1 caused a significant decrease in both porosity and number of fenestrae.
Although there are no accepted biomarkers for fenestration, correlations have been observed between capillarization and the markers CD31 and CD44. CD31 [PECAM, marker for endothelial cells (33)] is low in normal LSEC and increases with capillarization. CD44 [Hyaluronic acid receptor (34,35)] is high in normal LSEC and decreases with capillarization. Immuno-staining for CD31 and CD44 showed that treatment of primary LSEC cells with 100 ng/ml TSP1 caused increased CD31 expression and decreased CD44 expression (Figure S1).
Because TSP1 is known to have potent effects on liver through activation of TGF-β1 (36), we next tested whether the effects of TSP1 on LSEC defenestration were dependent on TGF-β1. LSEC were pre-incubated with the small molecule inhibitor of TGF-β type II receptor, SB-431542 (37) (Fig. 2A). Cells pretreated with SB-431542 were not protected from defenestration induced by subsequent treatment with 100 ng/ml of TSP1. The doubly treated cells showed low porosity (Fig. 2B) and fenestrae number (Fig. 2C) similar to cells treated with 100 ng/ml TSP1 alone. There was no significant change in fenestration after treatment with TGF-β1 alone. Cells treated with SB-431542 showed a small increase in porosity that was not significant according to the one-way anova test, but would be significant as a categorical factor variable under a two-way anova test. Some effect from SB-431542 might be as a result of indirect inhibition of Rho by SB-431542 (38).
CD47-binding peptide induced LSEC defenestration
Anti-angiogenic effects of TSP1 have been characterized in detail using specific CD47-binding peptides p4N1 (RFYVVMWK) and p7N3 (FIRVVMYEGKK). The -VVM- motif, crucial for binding CD47, was changed to -GGM- for constructing negative control peptides p4G1 (RFYGGMWK) and p7G3 (FIRGGMYEGKK) (11,26,39,40). Treatment of LSEC with 1 μM of the p4N1 peptide significantly decreased the porosity (Fig. 3B) and number of fenestrae (Fig. 3C). The control peptide p4G1 did not cause any significant change in fenestration compared to untreated controls. The porosity and number of fenestrae were not significantly altered by the p7N3 treatment, nor by its control p7G3. To quantify changes in fenestrae size, we plotted the frequency distribution of fenestrae diameter (Fig. 3D) for cells treated with p4N1 and p7N3. The diameter of fenestrae significantly decreased after p4N1 treatment.
To further test the role of CD47 in inducing defenestration in the LSECs, cells were pretreated with a specific CD47-blocking antibody, B6H12 (41) (Fig. 4A). B6H12 alone was able to increase the porosity and the number of fenestrae (Fig.4B), suggesting that fenestration may be regulated by endogenous CD47-binding ligands. Pretreatment with B6H12 was able to protect the cells from defenestration by the p4N1 peptide. These results confirm the importance of CD47 in regulating sinusoidal endothelial cell fenestration.
Fig 4.

B6H12 blocked p4N1-induced defenestration. (A) SEM images of liver sinusoidal endothelial cells (LSEC) cultures treated with p4N1 peptide, with and without B6H12 pre-treatment. (a) Igg-isotype control; (b) B6H12; (c) Igg isotype+p4N1; (d) B6H12+p4n1; (e) Igg isotype +p4G1; (f) B6H12+ p4G1. All scale bars are 1 μm. (B) Porosity/μm2 and (C) fenestrae/μm2 of LSEC (* represents P < 0.05).
The role of Rho kinase in LSEC fenestration
Because the p4N1 peptide caused a significant decrease in fenestrae diameter (Fig. 3D), and because the diameter was previously shown to be regulated by actomyosin (21,42,43), we hypothesized that CD47-binding would cause cytoskeletal signalling to activate myosin through phosphorylation. Increased activation of Rho/ROCK signalling accompanies increased hepatic endothelial resistance with fibrosis, and intrahepatic resistance can be lowered by treatment with the ROCK inhibitor, Y-27632 (Ycmpd) (44). We treated the LSEC monocultures with 10 μM Ycmpd, prior to stimulation with p4N1 (Fig. 5). Treatment of LSEC cultures with Ycmpd alone significantly increased the number of fenestrae (Fig. 5C), indicating that Rho/ROCK signalling controls fenestration under basal conditions. The fenestration of LSECs after combined Ycmpd+p4N1 treatment was the same as after Ycmpd alone, indicating that Ycmpd protected the cells from p4N1-induced defenestration. The mutated p4G1 peptide did not significantly affect fenestration, with or without Ycmpd.
Fig 5.

Ycmpd prevents p4N1-induced LSEC defenestration. (A) SEM images of LSEC cultures treated with p4N1 peptide, with and without Ycmpd pretreatment. (a) control; (b) p4N1; (c) p4G1; (d) Ycmpd;(e) Ycmpd+p4n1; (f) Ycmpd+ p4G1. All scale bars are 1 μm. (B) Porosity/μm2 and (C) fenestrae/μm2 of LSEC after Ycmpd treatment (* represents P < 0.05).
Downstream of ROCK, to assess the activation of myosin, we measured the levels of phospho-myosin light chain (p-MLC) using immuno-fluorescence. Confocal images of LSECs treated with p4N1 alone showed approximately two-fold increase in the p-MLC content (Fig. 6A). Ycmpd alone decreased p-MLC levels slightly compared with untreated control (Fig. 6B). p4N1 plus Ycmpd caused p-MLC levels to remain similar to Ycmpd alone, indicating that Ycmpd protected myosin against the effects of p4N1. These results indicate that the CD47-binding peptide causes myosin activation through the Rho-ROCK pathway.
Fig 6.
Ycmpd blocks p4N1-induced myosin activation. (A) Immuno-fluorescence of LSECs treated with p4N1, with or without Ycmpd pretreatment (Nucleus–Blue; p-MLC–Green; F-Actin–Red.) (B) Quantification of p-MLC fluorescence intensity, as a ratio with respect to control untreated LSECs (* represents P < 0.05).
Effect of simvastatin on LSEC fenestration
Statin drugs have been shown to improve portal pressure in cirrhotic rats by inhibition of the Rho-ROCK pathway (45). Simvastatin improved liver perfusion and caused a decrease in the hepatic venous pressure gradient in patients with cirrhosis (46,47). We tested if simvastatin could protect LSEC from p4N1-induced defenestration. LSECs treated with 1 μM simvastatin alone showed an increase in porosity and fenestrae number (Fig 7). In LSEC cells incubated with simvastatin, subsequent p4N1 treatment caused no decrease in porosity nor in number of fenestrae. The defenestration effect of p4N1 was suppressed by simvastatin, because the p4N1+simvastatin combined treatment showed as much fenestration as simvastatin alone. Correlating with decreased fenestration, simvastatin caused more than two-fold decrease in p-MLC levels (Fig. 7C, Fig. 7D). In cells incubated with simvastatin, subsequent p4N1 treatment caused no increase in p-MLC levels. These results indicate that simvastatin protects LSEC from 4pN1-induced effects including myosin activation, loss of porosity and decreased fenestrae number.
Discussion
Thrombospondin 1 was sufficient to induce a defenestration response in LSECs (Fig. 1). LSECs are unique in having open fenestrae (12), but LSEC lose their fenestrated phenotype during fibrosis, in the process of capillarization (15). The TGF-β type II receptor inhibitor SB-21342 was unable to protect against TSP1-induced defenestration (Fig. 2), suggesting that TSP1-induced defenestration is independent of TGF-β1 activation. Likewise, addition of exogenous TGF-β1 had no effect on fenestration. Interestingly, SB-431542 caused a small increase in fenestration, consistently across the tested stimuli (control, TSP1 or TGF-β1), suggesting that endogenous TGF-β type-II receptor signalling does regulate fenestration. The matricellular protein TSP1 is known to increase during liver fibrosis (48,49) and it has pro-fibrotic effects by activating TGF-β1 and hepatic stellate cells (4). To the best of our knowledge, our results are the first to show that TSP1 can have pro-fibrotic effects on another non-parenchymal cell type, LSECs.
In addition to pro-fibrotic effects via TGF-β1, TSP1 also has important physiological functions as an inhibitor of angiogenesis. The C-terminal domain of TSP1 has -VVM- motifs that bind CD47. CD47 ligation inhibits angiogenesis at many levels including inhibition of VEGFR2 activation (9), NO-stimulated activation of sGC (10) and adenylate cyclase activation (11). We treated LSEC with -VVM- motif peptides p4N1 and p7N3 to test for CD47-mediated defenestration. p4N1 stimulation is an established model system for studying CD47 signalling in vitro, and has successfully elucidated endothelial cell effects of CD47 that were later confirmed in vivo using CD47-null mice. In LSEC cultures, p4N1 caused a significant decrease in porosity and fenestrae number (Fig 3), while the p7N3 peptide did not cause any significant change compared to untreated cells. The p4N1 and p7N3 peptides have different flanking sequences around the core CD47 binding motif, and future study should investigate how different flanking sequences are able to regulate the cellular response to CD47 ligands.
We confirmed that CD47 was indeed targeted by the p4N1 peptide, using a specific blocking antibody, B6H12 (50). In cells pretreated with B6H12, the addition of p4N1 peptide caused no further decrease in porosity or fenestrae number (Fig. 4). The B6H12 antibody to block CD47 is under active development for multiple cancers (51,52), and our experiments suggest that it may also have beneficial effects in fibrotic liver. Treatment with p4N1 also caused a significant decrease in the size of fenestrae (Fig. 3C), suggesting involvement of the actin cytoskeleton and/or myosin contraction. ROCK (Rho kinase) is a direct kinase activator of myosin, and intravenous administration of ROCK inhibitor Y-23763 (Ycmpd) has been shown to improve hepatic resistance in models of BDL fibrosis (53). In CCL4 models of fibrosis, Ycmpd has had anti-fibrotic effects by preventing the activation of hepatic stellate cells (44). In our study, addition of Ycmpd increased LSEC porosity and fenestrae number, confirming a role for the Rho-ROCK pathway in regulating fenestration (Fig.4). Pretreatment of LSECs with Ycmpd prevented p4N1 from inducing defenestration (Fig. 5) or myosin activation (Fig. 6). These results implicate the Rho-ROCK-myosin pathway in CD47-induced LSEC defenestration.
Simvastatin is known to be effective in resolving portal hypertension in patients with cirrhosis (46,47). In our experiments, simvastatin alone caused slight enhancement of fenestration. Pretreatment of LSECs with simvastatin provided full protection from p4N1-induced defenestration effects, including loss of porosity, decreased number of fenestrae and myosin activation. This suggests that simvastatin may have beneficial effects on liver vasculature via improved fenestration.
Many studies of vascular physiology have focused on the eNOS-NO-cGMP system. The NO system is a strong inducer of LSEC fenestration, and we suspect it is linked with many of the effects we have observed. CD47 is known to suppress the eNOS-NO-cGMP pathway through multiple redundant mechanisms (9–11), and our CD47-binding treatments (TSP1 or p4N1) could be suppressing the eNOS-NO-cGMP pathway as well. The Rho-ROCK pathway has complex interconnections with the NO system (54), as ROCK is capable of antagonizing the NO pathway (55), and the NO pathway is capable of antagonizing the Rho-ROCK pathway (56). Interplay between the NO pathway and the Rho-ROCK-myosin pathway might also provide a link between our simvastatin results and previous simvastatin studies that focused on the NO system (47,57,58). We have chosen to study the Rho-ROCK-myosin pathway because it offers additional insight into how therapeutic compounds might affect liver endothelium.
Future study should characterize the participation of eNOS, NO and cGMP in mediating the effects of CD47 ligation on fenestration. Another key priority for future study will be to target CD47 in vivo and assess its impact on fenestration during liver fibrosis. Our findings about the regulation of sinusoidal fenestrae in liver may also be applicable to the regulation of fenestrae in other endothelial cell types.
In conclusion, our study shows that CD47 is a potent regulator of fenestration in LSECs, and that simvastatin or Ycmpd can prevent CD47 ligation from inducing defenestration. We speculate that CD47 signalling may contribute to the therapeutic effectiveness of simvastatin and Ycmpd (Y-27632) towards liver endothelium.
Acknowledgments
We thank Rashidah Binte Sakban and Hanry Yu for the generous gift of non-parenchymal liver cells, Anilkumar M. Pillai and Gao Danmei for technical assistance and Filip Braet (Univ. of Sydney) for helpful conversations and SEM preparation protocols. LTK was supported by the Mechanobiology Institute, Singapore, and by an IUP grant from the Singapore-MIT Alliance, Computational Systems Biology Programme.
Supporting Information
Additional Supporting Information may be found in the online version of this article
The effect of Thrombospondin 1 (TSP1) on LSEC markers. LSECs were treated with 10 ng or 100 ng TSP1 overnight (scale bar, 10 μm). (A) Representative images of CD31 immuno-fluorescence (green) with increasing doses of TSP1. (B) Quantification of CD31 fluorescence intensity. There is significant increase in CD31 expression after TSP1 treatment. (C) Representative images of CD44 immuno-fluorescence (green) with increasing doses of TSP1. (D) Quantification of CD44 fluorescence intensity. There is significant decrease in CD44 expression after TSP1 treatment. (*represents P-value < 0.05, n = 3).
Supplementary
References
- 1.Kyriakides TR, Maclauchlan S. The role of thrombospondins in wound healing, ischemia, and the foreign body reaction. J cell commun signal. 2009;3:215–25. doi: 10.1007/s12079-009-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maile LA, Allen LB, Hanzaker CF, et al. Glucose regulation of thrombospondin and its role in the modulation of smooth muscle cell proliferation. Exp Diabetes Res. 2010;2010 doi: 10.1155/2010/617052. Article ID: 617052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myung SJ, Yoon JH, Gwak GY, et al. Bile acid-mediated thrombospondin-1 induction in hepatocytes leads to transforming growth factor-beta-dependent hepatic stellate cell activation. Biochem Biophys Res Commun. 2007;353:1091–6. doi: 10.1016/j.bbrc.2006.12.157. [DOI] [PubMed] [Google Scholar]
- 4.Breitkopf K, Sawitza I, Westhoff JH, et al. Thrombospondin 1 acts as a strong promoter of transforming growth factor beta effects via two distinct mechanisms in hepatic stellate cells. Gut. 2005;54:673–81. doi: 10.1136/gut.2004.042911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi H, Sakai K, Baba H, Sakai T. Thrombospondin-1 is a novel negative regulator of liver regeneration after partial hepatectomy through transforming growth factor-beta1 activation in mice. Hepatology. 2012;55:1562–73. doi: 10.1002/hep.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bornstein P. Thrombospondins function as regulators of angiogenesis. J cell commun signal. 2009;3:189–200. doi: 10.1007/s12079-009-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Short SM, Derrien A, Narsimhan RP, et al. Inhibition of endothelial cell migration by thrombospondin-1 type-1 repeats is mediated by beta1 integrins. J Cell Biol. 2005;168:643–53. doi: 10.1083/jcb.200407060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur S, Roberts DD. CD47 applies the brakes to angiogenesis via vascular endothelial growth factor receptor-2. Cell Cycle. 2011;10:10–2. doi: 10.4161/cc.10.1.14324. [DOI] [PubMed] [Google Scholar]
- 10.Isenberg JS, Annis DS, Pendrak ML, et al. Differential interactions of thrombospondin-1, -2, and -4 with CD47 and effects on cGMP signaling and ischemic injury responses. J Biol Chem. 2009;284:1116–25. doi: 10.1074/jbc.M804860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frazier WA, Gao AG, Dimitry J, et al. The thrombospondin receptor integrin-associated protein (CD47) functionally couples to heterotrimeric Gi. J Biol Chem. 1999;274:8554–60. doi: 10.1074/jbc.274.13.8554. [DOI] [PubMed] [Google Scholar]
- 12.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1:1. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaffner F, Poper H. Capillarization of hepatic sinusoids in man. Gastroenterology. 1963;44:239–42. [PubMed] [Google Scholar]
- 14.Xie G, Wang X, Wang L, et al. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology. 2012;142:918–27. doi: 10.1053/j.gastro.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48:920–30. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang BY, Ju XH, Fu BY, Zhang J, Cao YX. Effects of ethanol on liver sinusoidal endothelial cells-fenestrae of rats. Hepatobiliary Pancreat Dis Int. 2005;4:422–6. [PubMed] [Google Scholar]
- 17.McCuskey RS, Ito Y, Robertson GR, et al. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology. 2004;40:386–93. doi: 10.1002/hep.20302. [DOI] [PubMed] [Google Scholar]
- 18.Fraser R, Cogger VC, Dobbs B, et al. The liver sieve and atherosclerosis. Pathology. 2012;44:181–6. doi: 10.1097/PAT.0b013e328351bcc8. [DOI] [PubMed] [Google Scholar]
- 19.Narmada BC, Kang Y, Venkatraman L, et al. HSC-targeted delivery of HGF transgene via bile duct infusion enhances its expression at fibrotic foci to regress DMN-induced liver fibrosis. Hum Gene Ther. 2013;24:508–19. doi: 10.1089/hum.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oda M, Kazemoto S, Kaneko H, et al. Involvement of Ca++-calmodulin-actomyosin system in the contractility of hepatic sinusoidal endothelial fenestrae. In: Knook DL, Wisse E, editors. Cells of the Hepatic Sinusoid 4. Leiden: Kupffer Cell Foundation; pp. 174–8. [Google Scholar]
- 21.Yokomori H, Yoshimura K, Funakoshi S, et al. Rho modulates hepatic sinusoidal endothelial fenestrae via regulation of the actin cytoskeleton in rat endothelial cells. Lab Invest. 2004;84:857–64. doi: 10.1038/labinvest.3700114. [DOI] [PubMed] [Google Scholar]
- 22.Rombouts K, Kisanga E, Hellemans K, et al. Effect of HMG-CoA reductase inhibitors on proliferation and protein synthesis by rat hepatic stellate cells. J Hepatol. 2003;38:564–72. doi: 10.1016/s0168-8278(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 23.Braet F, De Zanger R, Sasaoki T, et al. Assessment of a method of isolation, purification, and cultivation of rat liver sinusoidal endothelial cells. Lab Invest. 1994;70:944–52. [PubMed] [Google Scholar]
- 24.Fleming YM, Ferguson GJ, Spender LC, et al. TGF-beta-mediated activation of RhoA signalling is required for efficient (V12)HaRas and (V600E)BRAF transformation. Oncogene. 2009;28:983–93. doi: 10.1038/onc.2008.449. [DOI] [PubMed] [Google Scholar]
- 25.Wakefield LM, Winokur TS, Hollands RS, et al. Recombinant latent transforming growth factor beta 1 has a longer plasma half-life in rats than active transforming growth factor beta 1, and a different tissue distribution. J Clin Investig. 1990;86:1976–84. doi: 10.1172/JCI114932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isenberg JS, Ridnour LA, Dimitry J, et al. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. 2006;281:26069–80. doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 27.Acquavella N, Quiroga MF, Wittig O, Cardier JE. Effect of simvastatin on endothelial cell apoptosis mediated by Fas and TNF-alpha. Cytokine. 2010;49:45–50. doi: 10.1016/j.cyto.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Wisse E, Braet F, Duimel H, et al. Fixation methods for electron microscopy of human and other liver. World J Gastroenterol. 2010;16:2851–66. doi: 10.3748/wjg.v16.i23.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheluvappa R, Shimmon R, Dawson M, Hilmer SN, Le Couteur DG. Reactions of Pseudomonas aeruginosa pyocyanin with reduced glutathione. Acta Biochim Pol. 2008;55:571–80. [PubMed] [Google Scholar]
- 30.Bergseth G, Lappegard KT, Videm V, Mollnes TE. A novel enzyme immunoassay for plasma thrombospondin Comparison with beta-thromboglobulin as platelet activation marker in vitro and in vivo. Thromb Res. 2000;99:41–50. doi: 10.1016/s0049-3848(00)00226-7. [DOI] [PubMed] [Google Scholar]
- 31.Switalska HI, Niewiarowski S, Tuszynski GP, et al. Radioimmunoassay of human platelet thrombospondin: different patterns of thrombospondin and beta-thromboglobulin antigen secretion and clearance from the circulation. J Lab Clin Med. 1985;106:690–700. [PubMed] [Google Scholar]
- 32.Dawes J, Clemetson KJ, Gogstad GO, et al. A radioimmunoassay for thrombospondin, used in a comparative study of thrombospondin, beta-thromboglobulin and platelet factor 4 in healthy volunteers. Thromb Res. 1983;29:569–81. doi: 10.1016/0049-3848(83)90212-8. [DOI] [PubMed] [Google Scholar]
- 33.Couvelard A, Scoazec JY, Feldmann G. Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am J Pathol. 1993;143:738–52. [PMC free article] [PubMed] [Google Scholar]
- 34.Tamaki S, Ueno T, Torimura T, Sata M, Tanikawa K. Evaluation of hyaluronic acid binding ability of hepatic sinusoidal endothelial cells in rats with liver cirrhosis. Gastroenterology. 1996;111:1049–57. doi: 10.1016/s0016-5085(96)70074-4. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki A, Angulo P, Lymp J, et al. Hyaluronic acid, an accurate serum marker for severe hepatic fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2005;25:779–86. doi: 10.1111/j.1478-3231.2005.01064.x. [DOI] [PubMed] [Google Scholar]
- 36.Kondou H, Mushiake S, Etani Y, et al. A blocking peptide for transforming growth factor-beta1 activation prevents hepatic fibrosis in vivo. J Hepatol. 2003;39:742–8. doi: 10.1016/s0168-8278(03)00377-5. [DOI] [PubMed] [Google Scholar]
- 37.Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia. 2005;7:509–21. doi: 10.1593/neo.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada H, Staten NR, Rajagopalan LE. TGF-beta1 mediated activation of Rho kinase induces TGF-beta2 and endothelin-1 expression in human hepatic stellate cells. J Hepatol. 2011;54:521–8. doi: 10.1016/j.jhep.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 39.Barazi HO, Li Z, Cashel JA, et al. Regulation of integrin function by CD47 ligands. Differential effects on alpha vbeta 3 and alpha 4beta1 integrin-mediated adhesion. J Biol Chem. 2002;277:42859–66. doi: 10.1074/jbc.M206849200. [DOI] [PubMed] [Google Scholar]
- 40.Gao AG, Lindberg FP, Finn MB, et al. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J Biol Chem. 1996;271:21–4. doi: 10.1074/jbc.271.1.21. [DOI] [PubMed] [Google Scholar]
- 41.Chao MP, Tang C, Pachynski RK, et al. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 2011;118:4890–901. doi: 10.1182/blood-2011-02-338020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braet F, De Zanger R, Baekeland M, et al. Structure and dynamics of the fenestrae-associated cytoskeleton of rat liver sinusoidal endothelial cells. Hepatology. 1995;21:180–9. [PubMed] [Google Scholar]
- 43.Yokomori H, Yoshimura K, Nagai T, et al. Sinusoidal endothelial fenestrae organization regulated by myosin light chain kinase and Rho-kinase in cultured rat sinusoidal endothelial cells. Hepatol Res. 2004;30:169–74. doi: 10.1016/j.hepres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Murata T, Arii S, Mori A, Imamura M. Therapeutic significance of Y-27632, a Rho-kinase inhibitor, on the established liver fibrosis. J Surg Res. 2003;114:64–71. doi: 10.1016/s0022-4804(03)00202-6. [DOI] [PubMed] [Google Scholar]
- 45.Trebicka J, Hennenberg M, Laleman W, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46:242–53. doi: 10.1002/hep.21673. [DOI] [PubMed] [Google Scholar]
- 46.Abraldes JG, Albillos A, Banares R, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology. 2009;136:1651–8. doi: 10.1053/j.gastro.2009.01.043. [DOI] [PubMed] [Google Scholar]
- 47.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Youssef M, Mu Y, Huang L, Stellmach V, Crawford SE. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J Pediatr Gastroenterol Nutr. 1999;28:386–92. doi: 10.1097/00005176-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 49.Hayashi K, Kurohiji T, Shirouzu K. Localization of thrombospondin in hepatocellular carcinoma. Hepatology. 1997;25:569–74. doi: 10.1002/hep.510250313. [DOI] [PubMed] [Google Scholar]
- 50.Azcutia V, Stefanidakis M, Tsuboi N, et al. Endothelial CD47 promotes vascular endothelial-cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J Immunol. 2012;189:2553–62. doi: 10.4049/jimmunol.1103606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chao MP, Alizadeh AA, Tang C, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71:1374–84. doi: 10.1158/0008-5472.CAN-10-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chao MP, Alizadeh AA, Tang C, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Q, Hennenberg M, Trebicka J, et al. Intrahepatic upregulation of RhoA and Rho-kinase signalling contributes to increased hepatic vascular resistance in rats with secondary biliary cirrhosis. Gut. 2006;55:1296–305. doi: 10.1136/gut.2005.081059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67:3823–36. doi: 10.1007/s00018-010-0460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ming XF, Viswambharan H, Barandier C, et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–77. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki H, Kimura K, Shirai H, et al. Endothelial nitric oxide synthase inhibits G12/13 and rho-kinase activated by the angiotensin II type-1 receptor: implication in vascular migration. Arterioscler Thromb Vasc Biol. 2009;29:217–24. doi: 10.1161/ATVBAHA.108.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brouet A, Sonveaux P, Dessy C, et al. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res. 2001;89:866–73. doi: 10.1161/hh2201.100319. [DOI] [PubMed] [Google Scholar]
- 58.Marrone G, Russo L, Rosado E, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J Hepatol. 2012;58:98–103. doi: 10.1016/j.jhep.2012.08.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of Thrombospondin 1 (TSP1) on LSEC markers. LSECs were treated with 10 ng or 100 ng TSP1 overnight (scale bar, 10 μm). (A) Representative images of CD31 immuno-fluorescence (green) with increasing doses of TSP1. (B) Quantification of CD31 fluorescence intensity. There is significant increase in CD31 expression after TSP1 treatment. (C) Representative images of CD44 immuno-fluorescence (green) with increasing doses of TSP1. (D) Quantification of CD44 fluorescence intensity. There is significant decrease in CD44 expression after TSP1 treatment. (*represents P-value < 0.05, n = 3).
Supplementary