

Abstract
Metamorphic proteins, including proteins with high levels of sequence identity but different folds, are exceptions to the long-standing rule-of-thumb that proteins with as little as 30% sequence identity adopt the same fold. Which topologies can be bridged by these highly identical sequences remains an open question. Here we bridge two 3-α-helix bundle proteins with two radically different folds. Using a straightforward approach, we engineered the sequences of one subdomain within maltose binding protein (MBP, α/β/α-sandwich) and another within outer surface protein A (OspA, β-sheet) to have high sequence identity (80 and 77%, respectively) with engineered variants of protein G (GA, 3-α-helix bundle). Circular dichroism and nuclear magnetic resonance spectra of all engineered variants demonstrate that they maintain their native conformations despite substantial sequence modification. Furthermore, the MBP variant (80% identical to GA) remained active. Thermodynamic analysis of numerous GA and MBP variants suggests that the key to our approach involved stabilizing the modified MBP and OspA subdomains via external interactions with neighboring substructures, indicating that subdomain interactions can stabilize alternative folds over a broad range of sequence variation. These findings suggest that it is possible to bridge one fold with many other topologies, which has implications for protein folding, evolution, and misfolding diseases.
Introduction
Decades of empirical data suggest that similar amino acid sequences encode similar folds. A predictive rule of thumb has resulted from these observations: sequences with as little as 30% aligned sequence identity typically adopt the same fold (1). Consequently, a change in fold topology appears extremely unlikely unless the majority of a protein’s residues are changed. This many-to-one relationship between sequence and structure tends to obscure the evolutionary pathway connecting one fold to another.
Recently, notable exceptions to this familiar relationship have been discovered, highly identical amino acid sequences that encode distinct folds. These exceptions have given new impetus to the hypothesis that novel protein folds can evolve via stepwise mutation (2). Over the last 10 years, seven natural and two engineered protein-fold switches have been identified (3–6). These shape-shifting polypeptides change conformation drastically in response to minor perturbations such as a single point mutation, a shift in pH, or addition of a metal. Additionally, a few earlier reports also demonstrated that proteins with >50% sequence identity can adopt different folds (7,8). In sum, these studies show that highly identical sequences can nevertheless bridge two different folds. One or several mutations to such a bridge sequence can result in a different fold.
We hypothesize that protein subdomains can be readily encoded by bridge sequences. Encompassed in a larger protein, these bridge sequences adopt a specific conformation through stabilizing contacts with neighboring subdomains. In isolation, i.e., absent these stabilizing interactions, bridge sequences adopt an alternative conformation.
Consistent with our hypothesis, six out of seven natural fold switches are protein subdomains that change conformations in response to an environmental change. These changes comprise a shift in pH (lymphotactin (9) and glycoprotein G (10)) or redox state (CLIC1 (11)), membrane release (P1 lysozyme (12)), or a change in binding mode (T7 RNA polymerase (13) and Mad2 (14)). Upon structural transformation, the fold-switching segment forms new stabilizing contacts with the structurally unchanged body of the protein, which functions as a supporting scaffold for both conformers. These interactions with neighboring subdomains play a central role in stabilizing both folds accessible to fold-switching segments of natural proteins. The seventh natural fold switch, the C-terminal domain of RfaH, also uses stabilizing external contacts to switch folds. It is a full protein domain that adopts an α-helical fold when interacting with the N-terminal RfaH domain but a β-barrel fold in isolation (4).
From a physicochemical perspective, engineering sequences that bridge the fold of a domain with the fold of a topologically distinct subdomain is advantageous. The sequence encoding the domain can fold cooperatively in isolation, but the subdomain-encoding sequence does not need to fold cooperatively. As a subdomain within a larger protein, this sequence relies on stabilizing interactions with neighboring substructures to assist cooperative folding into an accessible conformation that is not highly favored in isolation. This reasoning led to the successful engineering of an 11-residue peptide that switched conformations from an α-helix to a β-sheet depending on its context within the IgG binding domain of protein G (15).
By engineering bridge sequences that encode protein subdomains, we circumvent a major barrier common to previous comparable efforts to engineer bridge sequences. These efforts targeted protein domains exclusively. Protein domains are cooperative folding units (16). Therefore, previously engineered bridge sequences needed not only high levels of sequence identity but also enough self-contained information to encode two distinct, cooperatively-folding conformations. In order to switch folds, the conformational ensembles of these shape-shifting proteins needed both a primary fold and a latent state that would dominate the ensemble through a change in environment such as metal binding, a single amino-acid change, or oligomerization. Our approach obviates the need for this latent state by stabilizing a noncooperative member of the unfolded state through external subdomain interactions.
Backed by observations in nature and physicochemical principles, we test our hypothesis by engineering subsequences that bridge two topologically distinct subdomains with an unrelated protein domain. Contextualized within their parent protein, these subsequences, up to 80% identical with the unrelated domain, maintained their original fold. Furthermore, we found that well-established physicochemical principles were sufficient to guide the engineering of these highly identical sequences.
In further detail, we engineered two variants of GA, a 3-α-helix bundle protein, to have ≥50% sequence identity with subdomains of maltose binding protein (MBP, α/β/α-sandwich) and outer surface protein A (OspA, β-sheet). We solved the nuclear magnetic resonance (NMR) structure of a highly modified GA variant and found that it maintains its original 3-α-helix bundle topology. Circular dichroism (CD) and NMR evidence suggests the same for the other GA variants. In addition, we increased identity between the two fold pairs by changing amino acids within the corresponding MBP and OspA subdomains, raising sequence identity to 80 and 77%, respectively. The CD and NMR spectra of both modified proteins suggest that they adopt their original topologies. Furthermore, MBP remained active. These results support our hypothesis that interactions with neighboring subdomains play a central role in stabilizing alternative conformations accessible to a protein sequence.
Nomenclature
To distinguish between the different variants, we use the following nomenclature: %identityvariantreference sequence. All reference sequences for both pairs are the highest identity versions unless otherwise noted. As an example, 80GAMBP is the GA variant whose sequence is 80% identical to the highest identity subsequence in MBP, and 80MBPGA is the MBP variant whose subsequence is 80% identical to the highest identity GA sequence. Similarly, 77GAOspA is the GA sequence 77% identical to the highest identity OspA subsequence, and vice versa for 77OspAGA.
For the sake of brevity, we call GA a “source fold” and the MBP and OspA subdomains “destination folds”.
Materials and Methods
Fold pair selection
We aligned the sequence of PSD-1, a stable GA variant (17), with every register of every nonredundant (≤90% identical) protein sequence with an NMR structure or crystal structure of 2 Å or better (18). Structural similarity between PSD-1 and each sequence was quantified using the procedure detailed by Chellapa and Rose (19). Sequence diversity was determined using the CATH database (20). Proteins with sequences at least 15% identical to and structures distinct from GA were investigated. We selected MBP and OspA because of their high stabilities, abundant expression levels, and straightforward purification protocols. GA variant sequences replaced residues 12–67 in MBP and 24–79 in OspA.
Protein design
Putative mutations were manually selected using the mutation wizard in the software PYMOL (21). We ignored resulting clashes that minor backbone adjustments could alleviate. We avoided grouping three or more hydrophobic residues in close contact on the protein surface. Physical principles informing anticipated destabilizing effects are detailed in Table S2 in the Supporting Material.
Protein expression and purification
To facilitate their rapid purification, GA MBP, and OspA variants were cloned into a PPAL8 vector, which encodes an N-terminally His-tagged subtilisin prosequence at the N-terminus of the fusion protein. Mutations were made through Q5 mutagenesis (New England BioLabs, Ipswich, MA) or quick change reactions with PfuTurbo (Agilent Technologies, Santa Clara, CA). Fusion protein variants were expressed in BL-DE3 cells by autoinduction. Cells were lysed by sonication in 100 mM phosphate buffer (pH 6.8, 1 mM EDTA, 5 μM MgSO4, 15 μg DNAseI), one Complete Mini Protease pill (Roche, Basel, Switzerland), and fractionated by high-speed centrifugation (40,000 g for 45′). Soluble extract of the prodomain fusion protein was loaded on a Profinity Exact column (Bio-Rad, Hercules, CA (22)) at 1 mL/min, washed with 2 M NaOAC (at pH 6.6) in 2 mL aliquots at 2 mL/min, and eluted with 7 mL of 100 mM KPO4 (pH 6.8, 1 mM EDTA and 10 mM NaN3) at 0.2 mL/min. Buffer was exchanged at 2 mL/min to 100 mM KPO4 (at pH 6.8) with a 10-ml BioGel P-6 desalting cartridge (Bio-Rad). Before being loaded onto the subtilisin column, the 50GAOspA and 57GAOspA variants were loaded onto a Ni column in 100 mM phosphate and 10 mM imidazole (at pH 7.6), washed in 2-mL aliquots with the same buffer + 0.5 M NaCl, and eluted with 100 mM phosphate and 500 mM imidazole (at pH 8). Minimal medium (17) was used for 15N and 13C labeling. Labeled proteins were purified similarly, but on a 5-mL column and washed in 5-mL aliquots of NaOAc at 4 mL/min. All other rates and solutions were the same as for unlabeled proteins.
CD
CD measurements were performed with a spectropolarimeter (model J-720; JASCO, Easton, MD) using quartz cells with path lengths of 1 mm on protein concentrations from 20 to 30 μM. The ellipticity results were expressed as mean residue ellipticity, [θ], degrees per cm2/dmol with extinction coefficients estimated by EXPASY (23). Temperature-induced unfolding was performed in the temperature range between 25 and 95°C in 1-cm jacketed cuvettes. Ellipticities at 222 nm were continuously monitored at a scanning rate of 1°/min. The following equation was used to determine fraction-native:
Here, E is the experimentally measured CD signal at a given temperature, and mU/N ∗ T + bU/N values are the unfolded and native baselines, respectively, as a function of temperature (T).
NMR spectroscopy
NMR spectra were acquired on an Avance III 600 MHz spectrometer (Bruker, Billerica, MA) equipped with a z-gradient cryoprobe. A 13C-, 15N-labeled sample of 79GAMBP was prepared in 100 mM potassium phosphate buffer, pH 7.0, at a concentration of 0.27 mM. NMR assignments of backbone resonances were made utilizing the following three-dimensional experiments: HNCACB, CBCA(CO)NH, HNCO, and HNHA. All spectra were recorded at 10°C. Spectra were processed with the software NMRPIPE (24) and analyzed using the software SPARKY (25).
Structure calculation and analysis
The backbone chemical shifts of 80GAMBP were used in combination with the software CS-ROSETTA (26) to determine a three-dimensional structure. From 1000 calculated ROSETTA models, the 10 lowest energy structures formed an ensemble with backbone root-mean square deviation of 0.54 ± 0.16 Å over the ordered region. Structures were analyzed with the softwares PROCHECK-NMR (27), PYMOL (21), and MOLMOL (28).
Results
Fold pair selection
We selected a stable variant of GA (PSD-1 (17); Fig. 1, both red and purple), as our source fold. It adopts a 3-α-helix bundle fold, one of the most common small protein topologies adopted by multiple highly-dissimilar sequences (29). We hypothesized that this broad sequence diversity indicates a high mutational tolerance. Four additional properties of PSD-1 commended it as a source fold: its stability (5.6 kcal/mol (30)), multiple previous NMR studies (30–32), ability to switch folds in other contexts (5,33), and its length (56 residues). This shorter length is advantageous because smaller proteins tend to be less stable and therefore more amenable to adopting alternative topologies; they also require fewer amino acid substitutions to increase sequence identity. As of this writing, the longest fold-switching chain is 66 residues (4), and most known fold-switching segments are ≤50 residues long (3,6). This does not preclude longer protein chains from switching folds, especially intrinsically disordered proteins, whose flexibility may foster multiconformational behavior even at longer lengths (34). Still, we expect shorter protein chains to switch folds more frequently than longer ones.
Figure 1.
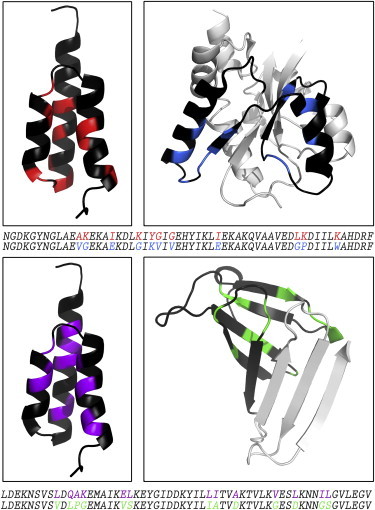
Engineered proteins with high levels of sequence identity but different folds. Out of 56 residues, 11 and 13 positions of nonidentity remain between the 80GAMBP-80MBPGA and 77GAOspA-77OspAGA fold pairs (top and bottom rows, respectively). (Black) Positions that are identical; colored positions denote remaining positions of nonidentity: (red) 80GAMBP; (blue) 80MBPGA; (purple) 77GAOspA; and (green) 77OspAGA. Color-coding of the sequence alignments corresponds to the coloring of the structures above. (Gray) Segments of MBP and OspA that lie outside of their high-identity subdomains and maintain their wild-type sequences. This figure shows truncated versions of MBP (N-domain only) and OspA (residues 23–116), although both experimental constructs are complete (N- and C-domains for MBP and residues 17–273 for OspA). PDB IDs are as follows: GA, 2FS1; MBP, 1N3X, chain A; OspA, 1OSP, chain O. To see this figure in color, go online.
Using the procedure below we selected two destination folds with different topologies: MBP (α/β/α-sandwich) and OspA (β-sheet). GA's sequence was aligned with every register of every nonredundant protein sequence (90% identity threshold) with a high-quality structure (2 Å or better). Potential destination folds were screened for structural dissimilarity using the method described by Chellapa and Rose (19) and selected by the following criteria.
-
1.
A frequently-occurring topology adopted by at least one other sequence with <25% identity, suggesting a high mutational tolerance.
-
2.
At least 15% sequence identity to GA.
-
3.
Stability ≥5 kcal/mol to allow a larger number of identity-increasing mutations.
-
4.
A topology different enough from a 3-α-helix bundle to demonstrate a substantial topological difference, as opposed to thermal fluctuations.
-
5.
Substantial tertiary interactions with neighboring substructures.
-
6.
Destination folds that must be monomeric.
-
7.
Previous NMR characterization.
Increasing sequence identity between the GA-MBP and GA-OspA fold pairs
We first engineered two series of GA variants with ever-increasing sequence identity to their corresponding MBP and OspA subdomains. These series resulted in GA variants with 59 and 57% sequence identity to the wild-type MBP and OspA destination folds, respectively: 59GAMBP and 57GAOspA. We then engineered the corresponding subsequences within MBP and OspA to further increase sequence identity to 80 and 77%, respectively, resulting in 80MBPGA and 77OspAGA. We used the following principles to design these variants.
Step 1. Align the two sequences and identify positions of nonidentity.
Step 2. Given a binary sequence space (choice of mutation restricted to either GA or destination fold residue), classify possible mutations by their anticipated destabilizing effects: minor, moderate, or significant (see Table S2). Additionally, all mutations must cause minimal clashes and avoid accumulating hydrophobic residues on the protein surface.
Step 3. Group similarly-classified mutations in a set, and make mutations in a given set.
Step 4. Experimentally screen the variant resulting from a set of mutations. If the variant appears folded by CD and cooperatively unfolds, then make the next set of mutations. For identity levels ≥50%, verify that the two-dimensional 15N HSQC spectrum is well dispersed to further confirm well-ordered structure.
Step 5. Repeat Steps 3 and 4 until any further mutations would be highly likely to destabilize the folds.
Remaining positions of nonidentity in GA-MBP and GA-OspA pairs are shown in Fig. 1. Most nonidentical positions in both GA variants lie within their cores, with the exception of a few surface residues whose mutational alternatives are proline, glycine, or hydrophobic residues likely to cause aggregation. Nonidentical positions in MBP also lie mostly in its core, forming hydrophobic contacts with neighboring local structures. Others constitute part of its sugar-binding site. Nonidentical positions in OspA comprise core residues, glycines, prolines, and surface hydrophilics that would likely cause aggregation if mutated to the corresponding hydrophobic residues in GA.
All designed variants maintain the same three-dimensional structures as their parent sequences
We determined the solution structure of 79GAMBP through NMR spectroscopy and found its structure was nearly identical to that of its parent, PSD-1 (Fig. 2). Triple-resonance methods were used to determine NMR assignments for all of the backbone atoms (1HN, 1Hα, 13Cα, 13Cβ, 13C′, 15N) in residues 2–56 of the polypeptide chain. These chemical shift assignments were then employed to determine a three-dimensional structure with the software CS-ROSETTA (26). We have previously established that CS-ROSETTA calculations provide structures that agree well with mainly nuclear Overhauser effect-derived conformations for similar types of proteins (35). The CS-ROSETTA structure of 79GAMBP displays a 3-α fold with α-helices at residues 8–23, 28–35, and 39–52, while residues 1–7 and 53–56 are disordered (Fig. 2 B). Comparison with the fold topology of the parent GA shows a backbone root-mean square deviation of 1.0 Å between the mean structures, indicating high overall similarity between the conformations of GA and 79GAMBP. Structure statistics are summarized in Table S3. The structure and NMR assignments have been deposited in the PDB and BMRB with accession codes 2MH8 and 19623, respectively.
Figure 2.
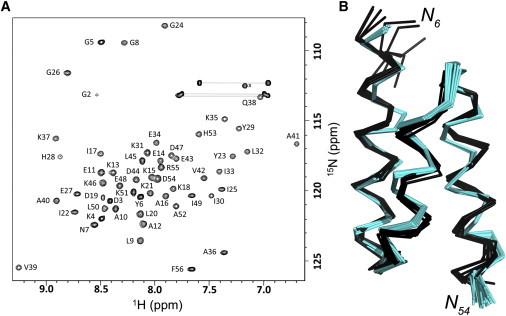
79GAMBP adopts a 3-α helix bundle topology. (A) Two-dimensional 15N HSQC spectrum of 79GAMBP. Main-chain NMR assignments are indicated. Side-chain amide signals are connected (horizontal lines); x denotes an aliased side-chain amino resonance. (B) NMR structure of 79GAMBP determined using CS-ROSETTA and the main-chain chemical shift assignments. The NMR ensemble of 10 lowest energy structures is shown for residues 6–54 in Cα ribbon representation (black). The parent GA structure, PDB 2FS1, is superimposed (cyan). To see this figure in color, go online.
CD and NMR studies of 80GAMBP and 77GAOspA indicate that they adopt the same 3-α helix bundle topology as 79GAMBP. The CD spectra of 80GAMBP, and77GAOspA are nearly identical to that of 79GAMBP, strongly suggesting a helical topology (see Fig. S1 a). Furthermore, thermal melting of 80GAMBP and 77GAOspA, monitoring the CD ellipticity at 222 nm, shows cooperative transitions (TM values of 57 and 58°C, respectively) that are consistent with a stably folded protein (see Fig. S1 b). Moreover, the two-dimensional 15N HSQC spectra of both 80GAMBP and 77GAOspA have well-dispersed main-chain amide signals, providing further evidence of stable structure (see Fig. S2).
The CD and NMR spectra of both 80MBPGA and77GAOspA suggest that they are folded in their parent conformations. The CD spectra of the two variants are consistent with their parent folds (see Fig. S3, a and b). Furthermore, the NMR spectra of both80MBPGA and 77OspAGA overlay well with the spectra of their parent proteins, suggesting that the engineered subsequences induced neither local nor global unfolding (Fig. 3, A and C). The variations in cross-peak positions are expected because the sequences of both 80MBPGA and 77OspAGA differ from their wild-type counterparts by 11 amino acids.
Figure 3.
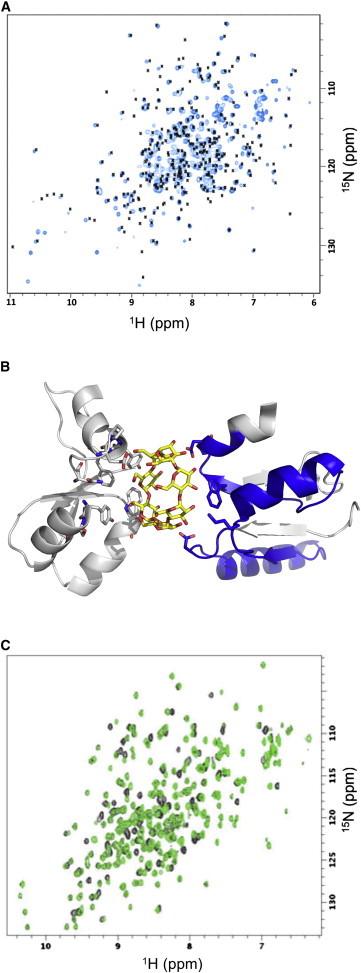
Both 80MBPGA and 77OspAGA fold similarly to their corresponding wild-type forms. (A) The two-dimensional 15N HSQC spectrum of 80GAMBP (blue) is overlaid with positions of assigned backbone resonances from the wild-type MBP spectrum (black). The 80MBPGA spectrum was recorded using similar conditions to those employed for the wild-type MBP (37°C, 20 mM sodium phosphate, pH 7.2, 3 mM sodium azide, 0.1 mg/mL Pefabloc, 2 mol equivalents of β-cyclodextrin). (B) MBP’s binding site complexed with β-cyclodextrin. (Sticks) β-cyclodextrin (yellow) and active-site residues in both the N- and C-domains. MBP’s modified subdomain (blue) contains four of the 14 active-site residues; all residues outside of this subdomain (gray) maintain their wild-type sequences. Only structures near the binding site are shown. PDB 1DMB. (C) The two-dimensional 15N HSQC spectrum of 77OspAGA (green) is consistent with that of wild-type (black). Both were acquired at 10°C in 100 mM of potassium phosphate buffer, pH 7.0. To see this figure in color, go online.
Furthermore, 80MBPGA remains active. Addition of β-cyclodextrin shifts the peaks of 80MBPGA’s two-dimensional 15N HSQC spectrum to locations like those of wild-type MBP under the same conditions (Fig. 3 A (36)). Accordingly, there is significant peak overlap between the two spectra, and the overall pattern of backbone amide shifts is very similar. These data therefore suggest that80MBPGA maintains the same general fold topology as its parent, wild-type MBP. In particular, the binding of β-cyclodextrin to80MBPGA strongly suggests that the α/β/α-sandwich topology of 80MBPGA’s N-domain is preserved because this domain comprises part of the binding epitope in wild-type MBP (Fig. 3 B).
Thermostability analysis of GA and MBP variants
After confirming that our high-identity GA-MBP and GA-OspA pairs adopted their parent conformations, we measured the thermostabilities of the GA-MBP variants and compared them to the anticipated destabilizing effects for each mutational set (Fig. 4). We found that changes in GA’s thermostabilities were generally well correlated with its anticipated mutational effects: minor mutations affected its TM value negligibly, while significant mutations affected its TM value the most substantially. This demonstrates that our method for classifying the destabilizing effects of mutations on GA was accurate. Because these classifications were based on experiments performed largely on small independently-folding protein domains, the consistency between predictions and experiments is expected. In contrast, little correlation was found between MBP’s changes in thermostability and the anticipated effects of its mutations. This demonstrates that these classifications were an inadequate predictor of mutational effects on MBP. Note, however, that small changes in TM values correspond to larger changes in ΔG for larger proteins, so the 2° decreases in MBP’s TM values are significant. We did not perform a similar analysis on the GA-OspA pair because no significant correlation would arise from the analysis: only four and three variants of each protein were produced, respectively.
Figure 4.
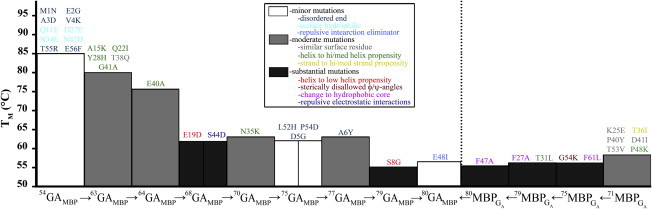
Thermostabilities of GA and MBP variants. Mutational series for GA and MBP begin on opposite sides of the figure (left and right, respectively; dotted line separation indicates that TM values on opposite sides cannot be compared meaningfully). TM values are color-coded by anticipated mutational effects on structure: minor (white), moderate (gray), and substantial (dark gray). The F27A T31L mutation is shown (dark gray) because the anticipated destabilization from the F27A mutation was expected to be significant. Mutations from parent proteins are listed above the TM values of each variant and are color-coded by their structural classification: disordered end (dark blue), surface hydrophilic (light blue), repulsive interaction eliminator (blue), similar noninteracting surface residue (gray), helix to residue with high/medium helix propensity (green), helix to residue with low helix propensity (dark orange-red), sterically disallowed ϕ/ψ angles (brown), change to hydrophobic core (magenta), and repulsive electrostatic interaction (purple). 68GAMBP and 75GAMBP are divided in half to show that two independent sets of mutations were made (see Table S1, A and B. Identity levels for both GA and MBP are with respect to the highest-identity partner. The initial level of identity between GA and MBP was 16%. To see this figure in color, go online.
The inconsistency between 80MBPGA’s experimentally determined TM values and its anticipated mutational effects suggested that interactions with neighboring substructures may be affecting its TM values. To test this hypothesis, we calculated the ΔΔG values of all GA and MBP variants from their TM values. We then compared these experimentally derived values with predicted ΔΔG values from POPMUSIC 2.1 (37), a web server that predicts the effects of point mutations on protein stability using statistical potentials.
Two conditions suggest that stabilizing interactions with neighboring subdomains are present.
-
1.
The experimentally derived ΔΔG values of the MBP variant must be significantly less than the predicted ΔΔG values. This indicates that the mutation was significantly less destabilizing than expected.
-
2.
Experimentally derived and predicted ΔΔG values should be consistent for all GA variants. This demonstrates that the predictions are generally good and, therefore, the inaccurate prediction of MBP’s ΔΔG values does not arise from a systematic error in the prediction algorithm.
Comparison of experimentally derived and predicted ΔΔG values for GA and MBP suggests that that interactions with neighboring substructures are likely to stabilize the native conformation of MBP’s engineered subdomain (see Table S4 and Fig. S4). Experimentally derived and predicted ΔΔG values were positively correlated for GA (R2 = 0.94, see Fig. S4), while MBP’s experimentally derived ΔΔG values were negatively correlated.
The negative correlation between experimentally derived and predicted ΔΔG values for MBP demonstrates that its predicted ΔΔG values are inconsistent with experiment. Specifically, the webserver predicted that four mutations would strongly destabilize its structure, but they were not nearly as destabilizing as expected (see Table S4). The most striking of these is the combined F27A and T31L mutation (Fig. 4 and see Table S4), which is predicted to destabilize MBP by 3.65 kcal/mol but, in fact, has no destabilizing effect. This discrepancy is surprising because F27 is a highly-conserved residue whose mutation to alanine is predicted to destabilize MBP by 3.43 kcal/mol because it is buried in the core of MBP with no solvent-accessible surface area.
Several factors are likely to contribute to this inconsistency.
-
1.
MBP’s backbone may have rearranged to allow stronger interactions between F27’s former hydrophobic contacts. These rearrangements could also foster new stabilizing interactions between nearby residues and the modified hydrophobic cluster.
-
2.
L31 could form compensatory hydrophobic contacts with the residues formerly interacting with F27. Inspection of the structure suggests, however, that this could only occur upon significant backbone rearrangement. In light of these two explanations, experiments are underway to determine the structure of this MBP variant and identify possible rearrangements.
-
3.
F27 could be buried in both the folded and unfolded states. If so, it would contribute no additional stabilizing free energy to MBP upon folding because it is buried in both states.
Regardless of the reason, however, all three of these explanations are likely to involve stabilizing interactions with neighboring substructures because four out of five residues interacting with residue 27 are >200 residues C-terminal to it. Regarding the mutations that did destabilize MBP significantly, we note that MBP’s free energy of unfolding is ∼10.2 kcal/mol (38). This high initial stability allows it to remain folded after more destabilizing mutations than a smaller protein, such as GA, could withstand.
The strong positive correlation (R2 = 0.94) between predicted and experimentally derived ΔΔG values for all GA variants demonstrates that the discrepancies between experimentally derived and predicted ΔΔG values in MBP are likely to be significant. POPMUSIC 2.1 predicted three substantially destabilizing mutations to GA, all of which correspond to the largest changes in its TM: 7–8°C each (Fig. 4 and see Table S4). All three mutations destabilize the structure of GA’s helices, but none of them are located within its hydrophobic core, nor are they glycine, which destabilizes helical structure more significantly. Furthermore, the effects of less-destabilizing mutations also correspond well with experimentally derived values.
Therefore, we conclude that the web server POPMUSIC failed to predict the ΔΔG values for MBP accurately because the effects of our mutations were outside of the scope of its predictive power. We hypothesize that MBP's conformation rearranged to optimize contacts with neighboring subdomains. Similar rearrangements would have had little effect on a small, independently-folding protein like GA because no similar contacts with neighboring substructures are available to stabilize it.
Discussion
We designed and produced two protein pairs with 80 and 77% sequence identity but different folds. One member of each fold pair was a protein subdomain stabilized by interactions with structural neighbors, and the other was an independently folding protein domain. Our design procedure was straightforward, involving manual designs based on well-established physicochemical principles. The key to our approach was using interactions with neighboring subdomains to stabilize the conformations of the protein subdomains with high levels of aligned sequence identity to GA (39). These interactions compensated for destabilizing effects of identity-increasing mutations to the subdomain.
In addition to stabilizing interactions from neighboring subdomains, two other factors bolstered our approach.
-
1.
Our selection criteria identified good candidates for design. All selected structures were stable, common, and highly sequence-diverse. The role of stability is clear: more robust proteins are more likely to withstand destabilizing mutations and maintain their folds. While less obvious, it is equally important for the folds to occur frequently in nature and be to encoded by diverse sequences, some of whose pairwise-aligned identities fall below the 25% threshold for possible homology. The importance is this: the protein backbone favorably adopts these folds, obviating the need for extensive sequence information to specify them (40).
-
2.
The second factor that aided our success was that there are more protein subdomains than domains. This larger pool of design candidates availed a more diverse population of sequences and structures than the comparatively limited set of independently stabilized domains.
Combining this work with an earlier study (5), we observe that GA variants can maintain their native 3-α-helix bundle topologies even after being engineered to extremely high sequence identity with several different folds. This is consistent with the network model of fold space (41). Applying this model, the 3-α-helix bundle is a hub fold connected to the alternative folds with which it has high sequence identity: α/β-grasp, α/β/α-sandwich, and β-sheet. Considering the topological diversity of these three folds, the 3-α-helix bundle is likely connected to other folds as well.
Are folds other than the 3-α-helix bundle equally amenable to bridging other topologies? One might argue that GA’s disordered terminal residues boost identity levels artificially because they have been mutated to several disparate sequences without significantly destabilizing the 3-α-helix bundle topology. Excluding them decreases overall identity only slightly, however, from 77–80 to 73–76%. These modified levels still exceed the 44% cutoff for inferring structural similarity between 45-residue protein chains (1), suggesting that other protein structures may also be hubs in fold space.
Further characterization of bridge sequences encoding protein subdomains promises insight into several other areas of study.
-
1.
It relates to protein misfolding diseases, which are associated with oligomerization of a particular protein subdomain, such as the amyloid-forming N-terminal segment of model protein Sup35 (42). Much like the protein substructures studied here, this subdomain changes conformation upon oligomerization, which provides intermolecular interactions that stabilize its disease-associated β-sheet structure. Proteins that shift conformations upon forming domain-swapped multimers behave similarly (43).
-
2.
Genes encoding large proteins could be spliced to express their fold-switching subdomains separately, allowing them to fold and function in isolation. Results from a 2008 genomic study are consistent with this idea. They suggest that alternatively spliced protein isoforms can adopt different folds (44).
-
3.
Additional study of bridge sequences will provide further insight into the role of epistatic mutations on protein evolution. A number of studies have shown that evolutionarily-conserved mutations outside of a protein’s active site can impact its function significantly (45). For example, changing a steroid receptor’s specificity from aldosterone to cortisol required two mutations, one of which had no apparent functional effect in isolation (46).
Similarly, the work we have presented focuses exclusively on changing residues within subdomains whose amino acid sequences are highly identical to GA’s sequence. Our design principles suggest that further identity-increasing mutations within the binary sequence spaces of the GA-MBP and GA-OspA fold pairs are likely to cause unfolding. Mutations outside of this binary sequence space along with residue changes outside of the MBP and OspA subdomains are probably required to engineer full protein switches—protein sequences that adopt one conformation in isolation and another when encompassed in a larger protein. We are currently designing experiments to investigate this possibility.
Conclusions
For this study, we designed two 3-α-helix bundle proteins with ∼80% sequence identity to two radically different folds: α/β/α-sandwich and β-sheet. NMR and CD evidence confirms that all four designed proteins maintain their parent folds. The α/β/α-sandwich and β-sheet folds were both subdomains within larger proteins. Using interactions with neighboring substructures to stabilize the folds of these subdomains was the key to our approach: these interactions eliminated the need for the α/β/α-sandwich and β-sheet to fold cooperatively in isolation. Thermodynamic measurements suggest that interactions with neighboring substructures may have also decreased the destabilizing effects of mutations to the subdomains. Given that 3-α-helix bundle variants have high levels of sequence identity with multiple different folds, it is possible for one protein fold to evolve into several—and possibly many—distinct conformations through stepwise mutation.
Acknowledgments
We thank Biao Ruan, Kathryn Fisher, Eun Jung Choi, and Dana Motabar for helpful experimental suggestions and scientific input, and George Rose, Aaron Robinson, Mike Harms, Rohit Pappu, and Joshua Porter for manuscript advice.
Support from the National Institutes of Health is gratefully acknowledged (grant No. R01GM062154 to J.O. and P.N.B.; grant No. R43CA163403 to P.N.B.; and grant No. F32GM10664901A1 to L.L.P.).
Supporting Material
References
- 1.Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 2.Cordes M.H.J., Stewart K.L. The porous borders of the protein world. Structure. 2012;20:199–200. doi: 10.1016/j.str.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Murzin A.G. Metamorphic proteins. Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- 4.Tomar S.K., Knauer S.H., Artsimovitch I. Interdomain contacts control folding of transcription factor RfaH. Nucl. Acids Res. 2013;41:10077–10085. doi: 10.1093/nar/gkt779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander P.A., He Y., Bryan P.N. A minimal sequence code for switching protein structure and function. Proc. Natl. Acad. Sci. USA. 2009;106:21149–21154. doi: 10.1073/pnas.0906408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambroggio X.I., Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J. Am. Chem. Soc. 2006;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 7.Dalal S., Balasubramanian S., Regan L. Protein alchemy: changing β-sheet into α-helix. Nature Struct. Mol. Biol. 1997;4:548–552. doi: 10.1038/nsb0797-548. [DOI] [PubMed] [Google Scholar]
- 8.Yuan S.M., Clarke N.D. A hybrid sequence approach to the paracelsus challenge. Proteins. 1998;30:136–143. doi: 10.1002/(sici)1097-0134(19980201)30:2<136::aid-prot3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 9.Tuinstra R.L., Peterson F.C., Volkman B.F. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc. Natl. Acad. Sci. USA. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roche S., Rey F.A., Bressanelli S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science. 2007;315:843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 11.Littler D.R., Harrop S.J., Curmi P.M.G. The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J. Biol. Chem. 2004;279:9298–9305. doi: 10.1074/jbc.M308444200. [DOI] [PubMed] [Google Scholar]
- 12.Xu M., Arulandu A., Young R. Disulfide isomerization after membrane release of its SAR domain activates P1 lysozyme. Science. 2005;307:113–117. doi: 10.1126/science.1105143. [DOI] [PubMed] [Google Scholar]
- 13.Tahirov T.H., Temiakov D., Yokoyama S. Structure of a T7 RNA polymerase elongation complex at 2.9 Å resolution. Nature. 2002;420:43–50. doi: 10.1038/nature01129. [DOI] [PubMed] [Google Scholar]
- 14.Mapelli M., Massimiliano L., Musacchio A. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–743. doi: 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 15.Minor D.L., Kim P.S. Context-dependent secondary structure formation of a designed protein sequence. Nature. 1996;380:730–734. doi: 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- 16.Porter L.L., Rose G.D. A thermodynamic definition of protein domains. Proc. Natl. Acad. Sci. USA. 2012;109:9420–9425. doi: 10.1073/pnas.1202604109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander P., Fahnestock S., Bryan P. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: why small proteins tend to have high denaturation temperatures. Biochemistry. 1992;31:3597–3603. doi: 10.1021/bi00129a007. [DOI] [PubMed] [Google Scholar]
- 18.Wang G., Dunbrack R.L. PISCES: recent improvements to a PDB sequence culling server. Nucl. Acids Res. 2005;33:W94–W98. doi: 10.1093/nar/gki402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chellapa G.D., Rose G.D. Reducing the dimensionality of the protein-folding search problem. Protein Sci. 2012;21:1231–1240. doi: 10.1002/pro.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orengo C.A., Michie A.D., Thornton J.M. CATH: a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1109. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 21.Delano W.L. Schrödinger; New York: 2002. The PYMOL Molecular Graphics System. [Google Scholar]
- 22.Ruan B., Fisher K.E., Bryan P.N. Engineering subtilisin into a fluoride-triggered processing protease useful for one-step protein purification. Biochemistry. 2004;43:14539–14546. doi: 10.1021/bi048177j. [DOI] [PubMed] [Google Scholar]
- 23.Gasteiger E., Hoogland C., Bairoch A. Chapter 52. Protein identification and analysis tools on the EXPASY server. In: Walker J., editor. The Proteomics Protocols Handbook. Humana Press; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]
- 24.Delaglio F., Grzesiek S., Bax A. NMRPIPE: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Goddard T.D., Kneller D.G. University of California at San Francisco; San Francisco, CA: 2004. SPARKY 3. [Google Scholar]
- 26.Shen Y., Lange O., Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laskowski R.A., Rullmann J.A., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 28.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 29.Frick I.M., Wikström M., Björck L. Convergent evolution among immunoglobulin G-binding bacterial proteins. Proc. Natl. Acad. Sci. USA. 1992;89:8532–8536. doi: 10.1073/pnas.89.18.8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Y., Rozak D.A., Orban J. Structure, dynamics, and stability variation in bacterial albumin binding modules: implications for species specificity. Biochemistry. 2006;45:10102–10109. doi: 10.1021/bi060409m. [DOI] [PubMed] [Google Scholar]
- 31.He Y., Chen Y., Orban J. NMR structures of two designed proteins with high sequence identity but different fold and function. Proc. Natl. Acad. Sci. USA. 2008;105:14412–14417. doi: 10.1073/pnas.0805857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Y., Chen Y., Orban J. Mutational tipping points for switching protein folds and functions. Structure. 2012;20:283–291. doi: 10.1016/j.str.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander P.A., He Y., Bryan P.N. The design and characterization of two proteins with 88% sequence identity but different structure and function. Proc. Natl. Acad. Sci. USA. 2007;104:11963–11968. doi: 10.1073/pnas.0700922104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.James L.C., Tawfik D.S. Conformational diversity and protein evolution: a 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 35.Shen Y., Bryan P.N., Bax A. De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds. Protein Sci. 2010;19:349–356. doi: 10.1002/pro.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardner K.H., Zhang X., Kay L.E. Solution NMR studies of a 42 KDa Escherichia coli maltose binding protein/β-cyclodextrin complex: chemical shift assignments and analysis. J. Am. Chem. Soc. 1998;120:11738–11748. [Google Scholar]
- 37.Dehouck Y., Kwasigroch J.M.M., Rooman M. POPMUSIC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011;12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganesh C., Shah A.N., Varadarajan R. Thermodynamic characterization of the reversible, two-state unfolding of maltose binding protein, a large two-domain protein. Biochemistry. 1997;36:5020–5028. doi: 10.1021/bi961967b. [DOI] [PubMed] [Google Scholar]
- 39.Best R.B. Bootstrapping new protein folds. Biophys. J. 2014;107:1040–1041. doi: 10.1016/j.bpj.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rose G.D., Fleming P.J., Maritan A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA. 2006;103:16623–16633. doi: 10.1073/pnas.0606843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyerguz L., Kleinberg J., Elber R. The network of sequence flow between protein structures. Proc. Natl. Acad. Sci. USA. 2007;104:11627–11632. doi: 10.1073/pnas.0701393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balbirnie M., Grothe R., Eisenberg D.S. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated β-sheet structure for amyloid. Proc. Natl. Acad. Sci. USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yadid I., Kirshenbaum N., Tawfik D.S. Metamorphic proteins mediate evolutionary transitions of structure. Proc. Natl. Acad. Sci. USA. 2010;107:7287–7292. doi: 10.1073/pnas.0912616107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birzele F., Csaba G., Zimmer R. Alternative splicing and protein structure evolution. Nucl. Acids Res. 2008;36:550–558. doi: 10.1093/nar/gkm1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harms M.J., Thornton J.W. Analyzing protein structure and function using ancestral gene reconstruction. Curr. Opin. Struct. Biol. 2010;20:360–366. doi: 10.1016/j.sbi.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bridgham J.T., Ortlund E.A., Thornton J.W. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–519. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.