

Abstract
Recent work has shown that engineered variants of cytochrome P450BM3 (CYP102A1) efficiently catalyze non-natural reactions, including carbene- and nitrene-transfer reactions. Given the broad substrate range of natural P450 enzymes, we set out to explore if this diversity could be leveraged to generate a broad panel of new catalysts for olefin cyclopropanation (i.e. carbene transfer). Here we take a step towards this goal by characterizing the carbene-transfer activities of four new wild-type P450s that have different native substrates. All four were active and exhibited a range of product selectivities in the model reaction, cyclopropanation of styrene using ethyl diazoacetate (EDA). Previous work on P450BM3 demonstrated that mutation of the axial coordinating cysteine, universally conserved among P450 enzymes, to serine, increased activity for this non-natural reaction. The equivalent mutation in the selected P450s was found to activate carbene-transfer chemistry both in vitro and in vivo. Furthermore, serum albumins complexed with hemin were also found to be efficient in vitro cyclopropanation catalysts.
Keywords: cytochrome P450, cyclopropanation, enzyme catalysis, metalloenzyme
Introduction
Recent years have seen a growing interest in developing more sustainable methods of chemical synthesis using biological catalysts.[1] The ability of enzymes to catalyze reactions that yield stereochemically complex products is unmatched. However, we lack enzymes that catalyze many useful reactions from the synthetic chemist’s repertoire. This laboratory has started to explore the ability of natural enzymes to catalyze non-natural reactions and the extent to which non-natural chemistry can be evolved in the laboratory. Cytochrome P450BM3 from Bacillus megaterium is a monooxygenase that is readily adapted for new applications by protein engineering.[2] We and others have expanded the catalytic scope of P450BM3 to include formal carbene and nitrene transfers.[3] Protein engineering can improve this non-natural reactivity to enable biocatalysis at preparatory scale, as recently demonstrated by the gram-scale synthesis of a chiral precursor to the antidepressant levomilnacipran.[4] Experience gathered in more than two decades of protein engineering has demonstrated that P450 enzymes can be engineered for new substrate and product selectivities.[2,5] We can use this experience to accelerate development of novel enzyme-catalyzed reactions.
Despite having low sequence identities, cytochrome P450s share a common fold and conserve a few key amino acid residues that are essential for catalytic activity and heme binding. Axial cysteine coordination to the heme iron is universally conserved and plays an essential role in P450 monooxygenase chemistry.[2] The coordinating cysteine is even conserved in P450 enzymes whose natural function is no longer oxygenation.[6] We found that mutation of this cysteine to serine in P450BM3 is highly activating of several non-natural reactions, especially cyclopropanation.[7] The activating effect of this mutation in P450BM3 is particularly apparent when the enzyme is used in the context of whole-cell catalysis. The enzyme binds non-natural substrates such as styrene with low affinity, resulting in poor substrate-induced transition of heme iron from low spin to high spin.[3a] As a result, NAD(P)H-driven reduction to Fe(II) is inefficient. We previously demonstrated that mutation of the axial ligand in P450BM3 from cysteine to serine facilitated NAD(P)H-dependent reduction of the heme-iron center, leading to high cyclopropanation catalytic efficiency and yield in vivo.[7]
Introduction of serine as the distal axial ligand shifts the P450BM3 CO ferrous Soret peak from 450 nm to 411 nm. Thus we refer to P450BM3 variants having this axial mutation as P411BM3 enzymes. We hypothesized that many potential cyclopropanation catalysts exist within the vast collection of naturally-occurring P450 enzymes, and that we should be able to generate a diverse set of equivalent serine-ligated ‘P411s’ for cyclopropanation and possibly other non-natural reactions. Here we report that other P450s indeed catalyze the model reaction of styrene cyclopropanation and that mutating the axial ligand increases this activity in vitro and in vivo. We also show that other hemoproteins, particularly heme-spiked serum albumins, are relatively efficient cyclopropanation catalysts in vitro.
Results
All catalysts were tested in the model cyclopropanation reaction of styrene using ethyl diazoacetate (EDA) under anaerobic, reducing conditions. This reaction creates two new stereocenters and hence four possible diastereomeric products (Figure 1). Previous work from our group showed that both free hemin and heat-inactivated P450BM3 catalyze this reaction to varying degrees, although with negligible enantioinduction and primarily yielding the trans products. This was also true for other heme proteins that were tested previously: catalase, horseradish peroxidase, cytochrome c and myoglobin.[3a] In contrast, the P450BM3 holoenzyme (heme domain and reductase) showed a slightly higher preference for the cis products and some enantioselectivity as well. We cautiously attributed this difference in selectivity to the unique ability of the intact P450 to direct the carbene transfer preferentially in the enzyme active site.
Figure 1.
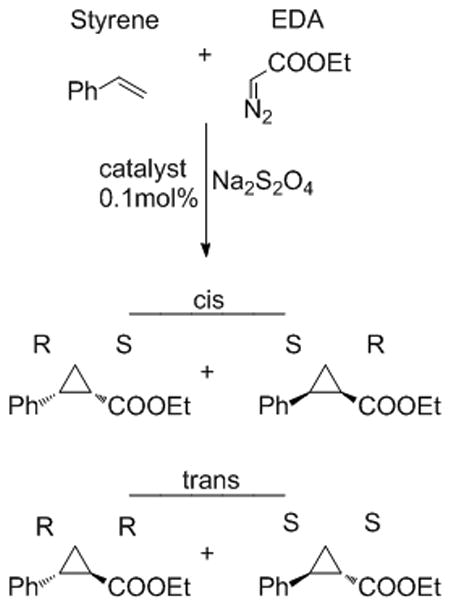
The model cyclopropanation reaction used to evaluate hemoproteins and cytochrome P450 enzymes. The reaction between styrene and ethyl diazoacetate generates four possible products.
We wanted to explore this non-natural activity with other hemoproteins and cytochrome P450s. Thus we began by investigating the natural heme-scavenging proteins human serum albumin (HSA) and bovine serum albumin (BSA)[8] both in the presence and absence of hemin. As a further control we included lysozyme, which has no known interaction with heme. None of the proteins catalyzed this reaction unless hemin was present (Table 1). HSA and BSA complexed with hemin exhibited significant activity and moderate enantioselectivity. Turnover numbers for serum albumins spiked with hemin increased 2 to 3 fold compared to free hemin. In contrast, adding lysozyme to hemin led to lower overall activity, with enantioinduction similar to free hemin.
Table 1.
Styrene cyclopropanation catalyzed by hemin and proteins spiked with hemin. 10 μM of hemin or equimolar concentrations of hemin and protein were mixed and incubated under anaerobic conditions at 24°C for 1h in 5% EtOH co-solvent, 20 mM styrene, 10 mM EDA under reducing conditions.
| Catalyst: | yield % | TTN | cis/trans | %cisee | % transee |
|---|---|---|---|---|---|
| Hemin | 18 | 175 | 14:86 | rac | rac |
| BSA | - | - | - | - | - |
| BSA + hemin | 35 | 349 | 9:91 | 7 | -10 |
| HSA | - | - | - | - | - |
| HSA + hemin | 57 | 567 | 9:91 | 15 | 17 |
| Lysozyme | - | - | - | - | - |
| Lysozyme + hemin | 8 | 78 | 13:87 | rac | rac |
The data represent triplicates with standard errors within the range of 20%. - denotes no product formation; rac indicates enantioselectivity below 5% ee.
To explore alternative P450 cyclopropanation catalysts we selected four previously untested P450 enzymes: P450cam, CYP153A6, TxtE and CYP119. These are all soluble, microbial enzymes that can be functionally expressed in E. coli at sufficient levels to study their non-natural activities. Their sequence identities range from 16% to 21% relative to the heme domain of P450BM3 (Supplementary Table 1). We tested the extensively characterized P450cam (CYP101) from Pseudomonas putida, whose native substrate is camphor,[9] as well as CYP153A6 from Mycobacterium sp. HXN-1500, a terminal alkane hydroxylase whose preferred substrate is octane.[10] We also investigated the recently discovered TxtE enzyme from Streptomyces scabies, which catalyzes the direct nitration of L-tryptophan,[6] and the highly thermostable CYP119 from Sulfolobus solfataricus, which has been shown to accept fatty acid substrates.[11]
All of the purified wild-type P450 enzymes catalyzed the cyclopropanation of styrene to some degree and exhibited different diastereo- and enantioselectivities (Table 2). P450cam exhibited the highest activity, measured in terms of total turnovers (452 TTN) in a 1-hour reaction. It also showed interesting selectivity, producing a slight excess of the (unfavored) cis diastereomers, with considerable enantioselectivity. The remaining wild-type enzymes, including the P450BM3 heme domain (P450BM3-h), were significantly less active than P450cam and produced primarily the trans products, with essentially no enantioinduction.
Table 2.
Activities, reported in terms of yield and total turnover number, of purified wild-type and axial Cys-Ser mutants of P450 enzymes for cyclopropanation of styrene using EDA under anaerobic conditions.
| Catalyst: | % yield | TTN | cis/trans | %cisee | % transee |
|---|---|---|---|---|---|
| P450BM3-h | 1 | 11 | 12:88 | -26 | rac |
| P450BM3-h-C400S | 41 | 411 | 9:91 | rac | -5 |
| CYP119 | 20 | 197 | 15:85 | 25 | rac |
| CYP119-C316S | 35 | 345 | 80:20 | 40 | 19 |
| CYP153A6 | 10 | 96 | 12:88 | -20 | rac |
| CYP153A6-C363S | 44 | 441 | 10:90 | rac | rac |
| TxtE | 18 | 178 | 30:70 | 36 | 12 |
| TxtE-C356S | n/a | n/a | n/a | n/a | n/a |
| P450cam | 45 | 452 | 57:43 | -43 | rac |
| P450cam-C357S | 49 | 487 | 9:91 | -7 | rac |
The data represent the average of triplicates with standard errors within the range of 20%. - denotes no product formation; rac indicates enantioselectivity below 5% ee. n/a : enzyme could not be purified.
We next introduced the known activating Ser mutation at the axial position of each wild-type P450 enzyme. Despite the low sequence identities, the conserved axial Cys residues were easily identified using a ClustalW2.1-generated sequence alignment (Supplementary Figure 1). All of the Ser mutants were purified using affinity chromatography. Under these standard expression and purification conditions we could not obtain purified TxtE-C356S and therefore did not investigate this variant further in vitro.
UV-visible absorption spectra of the purified Ser mutant enzymes revealed that CYP153A6-C363S has a ferrous CO Soret peak with a maximum at 413 nm, very similar to P450BM3-h-C400S (411 nm). The CYP119-C316S peak maximum was at 407 nm, whereas P450cam-C357S exhibited a peak maximum at 421 nm, possibly indicating a different ligation state (Supplementary Figure 2), as will be discussed below.
We compared the cyclopropanation activities of these mutant enzymes to the corresponding wild-type P450s (Table 2). Exchanging the axial Cys with Ser in P450cam had a negligible impact on the anaerobic styrene cyclopropanation activity of the purified enzyme. Introduction of Ser in the other P450s, however, significantly increased activity, with the largest increase observed for P450BM3-h-C400S (~40x). Although the thiolate (Cys) to alcohol (Ser) substitution tends to enhance the non-natural carbene transfer activity in the tested P450 enzymes, it also adversely affects diastereo- and enantioselectivity of the cyclopropanation in some cases. For example, whereas wild-type P450 enzymes gave moderate levels of enantioinduction, most Ser mutants exhibited significantly lower levels and gave hemin-like cis:trans ratios strongly favoring the trans products. The exception was CYP119: whereas the wild-type Cys-ligated enzyme gave a hemin-like cis:trans ratio (15:85), CYP119-C316S became highly cis selective (80:20). Enantioselectivity likewise increased from 25% eecis for CYP119-wt to 40% eecis for CYP119-C316S (Table 2).
In previous work we demonstrated that histidine ligation in P450BM3 facilitated synthesis of the chiral precursor to the antidepressant levomilnacipran even under aerobic conditions.[4] Here, we investigated whether the introduction of Ser at the axial position would also enable catalysis in the presence of oxygen, a strong inhibitor. P450BM3 and P450BM3-C400S show no activity in the presence of oxygen (Supplementary Table 3). In contrast, the Ser mutation in CYP153A6, CYP119 and P450cam led to an increase in activity under aerobic conditions, resulting in 117 TTN for CYP153A6-C363S and 71 TTN for CYP119-C316S. The highest activity (220 TTN) was observed for P450cam-C357S. The introduction of Ser at the axial position of P450cam, however, resulted in a loss of stereocontrol (Supplementary Table 3).
In previous work we demonstrated that serine ligation in an engineered P450BM3 variant improved NAD(P)H-driven catalysis in whole cells.[7] Strong reductants such as dithionite can be used when the reaction is carried out in vitro, but in vivo catalysis has to rely on endogenous reductants such as NAD(P)H. To determine whether introduction of the weaker alcohol ligand translates into higher yields for whole-cell catalysis for other enzymes, we also tested all the enzymes in vivo using the same substrate loading established for the in vitro experiments. First, however, we investigated the background reaction catalyzed by any free heme that accumulates in the E. coli cells. Previous work had shown that cells containing empty plasmid catalyzed some level of cyclopropanation (without enantioinduction), presumably due to free heme.[7] To determine the impact of heme accumulation on whole-cell catalysis we added different concentrations of the heme precursor δ-aminolevulinic acid (δ-ALA) to E. coli cultures lacking P450 expression (empty pET28 plasmid). We then measured the heme concentrations and the cyclopropanation activities of these cultures under anaerobic conditions. With increased concentrations of supplemented δ-ALA, we detected increased levels of intracellular heme (Supplementary Figure 3). As anticipated, this resulted in more non-specific catalysis of the model reaction between styrene and EDA in vivo. However, cells lacking P450 enzyme expression showed negligible enantioinduction and hemin-like diastereoselectivity (Table 3, Supplementary Table 2).
Table 3.
Styrene cyclopropanation yields and product distributions of wild-type and axial Cys-Ser mutants of P450 enzymes in E. coli whole cells. Control experiment was performed with empty pET28 plasmid.
| Catalyst: | yield % | cis/trans | %cisee | % transee |
|---|---|---|---|---|
| P450BM3-h | 14 | 12:88 | −8 | −19 |
| P450BM3-h-C400S | 69 | 10:90 | rac | −9 |
| CYP119 | 61 | 12:88 | rac | −6 |
| CYP119-C316S | 68 | 65:35 | 23 | rac |
| CYP153A6 | 10 | 13:87 | −8 | −25 |
| CYP153A6-C363S | 23 | 11:89 | rac | −13 |
| TxtE | 28 | 12:88 | rac | rac |
| TxtE-C356S | 47 | 12:88 | rac | −7 |
| P450cam | 43 | 81:19 | −38 | −11 |
| P450cam-C357S | 49 | 12:88 | −15 | rac |
| pET28-empty 250 μM δ-ALA | 28 | 14:86 | rac | −11 |
The data represent the average of triplicates with standard errors within the range of 20%. rac indicates enantioselectivity below 5% ee.
We then tested the selected enzymes in whole-cell catalysis supplementing the expression cultures with 250 μM δ-ALA to increase expression of the P450 enzymes. As anticipated, the axial ligand substitution generally had an activating effect, with the exception of P450cam where the yields from wild-type and its serine counterpart were similar (Table 3). TxtE-C356S, which could not be purified for in vitro experiments, showed higher activity in whole cells compared to TxtE wild-type, although the product distribution of both TxtE enzymes matched that of free hemin. The introduction of an axial serine ligand in P450BM3, CYP153A6, and CYP119 in contrast significantly increased activity, leading to yields of nearly 70% for whole-cell reactions with P450BM3-h-C400S and CYP119-C316S (Table 3).
Discussion
In light of recent results showing that engineered P450BM3 can be an excellent cyclopropanation catalyst,[3a, 4, 7] we investigated whether other cytochrome P450s also catalyze this reaction. We hypothesized that natural differences in substrate scope and active site shapes would contribute to differing reactivities and product selectivities. In fact all four wild-type P450 enzymes tested catalyzed the cyclopropanation of styrene, with notable differences in diastereo- and enantioselectivities. Furthermore, exchanging the axial ligand from Cys to Ser, a known activating mutation in P450BM3, tended to increase their activities, in vitro as well as in vivo. The magnitude of the activity increase depended on the parent enzyme: under anaerobic conditions P450BM3, CYP153A6 and CYP119 all showed strong gains in activity (2–40 fold increases), while the activity of P450cam was essentially unchanged upon axial mutation. The Ser mutation increased the catalytic activities sufficiently that Ser-substituted P450cam, CYP153A6 and CYP119 all catalyzed the cyclopropanation under aerobic conditions, although the activity was accompanied by a slight loss of stereocontrol in several cases.
In contrast to what was observed in vitro, the axial Ser mutant of TxtE gave a higher yield compared to its cysteine-ligated counterpart in whole-cell catalysis. The axial mutation also influenced product selectivity to varying degrees: P450cam‘s cis-selective catalysis shifted to a hemin-like predominantly trans product mixture, while the diastereoselectivities of P450BM3-h-C400S and CYP153A6-C363S remained comparable to their respective wild-type enzymes, with a slight decrease in enantioselectivity. CYP119 alone displayed increased enantio- and diastereoselectivity upon axial mutation, as we observed previously when the C400S mutation was introduced into the P450BM3-CIS variant.[7]
The reactivity trends of the axial mutants can be rationalized in part from the UV-visible absorption data: P450BM3-h-C400S, CYP153A6-C363S and CYP119-C316S all have similar ferrous-CO-bound spectra with Soret maxima at 407–413 nm. These spectral features suggest axial serine ligation, which is consistent with their significantly increased cyclopropanation activities. P450cam-C357S, however, has a distinctly different ferrous CO-bound spectrum with a peak maximum at 421 nm. It has been reported that substitution of the axial cysteine with a histidine or glycine ligand in P450cam results in a ferrous CO Soret peak maximum at 420 nm, which is indicative of histidine ligation.[12] Given this precedent, it is likely that the iron in the P450cam-C357S mutant is not ligated to serine but to an alternative histidine within the heme pocket (i.e. His 352, 355, or 361).[12] This implies a structural rearrangement in the heme pocket and could explain the loss in stereocontrol that occurs upon axial mutation as well as the greatly reduced inhibition of the activity by oxygen in this enzyme.
We also discovered that heme-complexed serum albumins possess significant carbene-transfer activity. Previous work has shown that HSA binds heme with high affinity in a hydrophobic pocket referred to as HSA subdomain IB.[13] Besides π-π stacking interactions of Tyr138 and Tyr161 with the porphyrin system, His146 and Lys190 form salt bridges with the heme propionates. Crystallography and resonance Raman spectroscopy identified Tyr161 as an oxygen donor for weak ligation of iron(III) heme.[8b, 14] His146 and both of these tyrosines are widely conserved among serum albumins, including BSA and HSA.[15] We compared the activities of the serum albumins to lysozyme (with no known hemin interaction) supplemented with an equimolar ratio of hemin. Whereas lysozyme lowered the activity of hemin, serum albumins loaded with hemin showed a significant increase in catalytic activity compared to the same concentration of free hemin. These results show that both axial ligation and a hydrophobic environment provided by a protein pocket contribute to catalysis.
One benefit of developing new genetically-encoded cyclopropanation catalysts is that the chemical transformation can be carried out in whole cells, without the need to purify the enzyme. In this vein, we have demonstrated that all of the tested P450s exhibited activity as whole-cell catalysts, including the TxtE-C356S variant, which we were unable to purify for in vitro experiments. That some enzymes such as TxtE (both wild-type and the axial serine mutant) gave hemin-like product distributions in vivo could be due to partial unfolding of the enzyme that exposes the heme and/or significant background from free heme relative to the enzyme, especially for enzymes that are unstable and/or poorly expressed.
The comparison of purified enzyme and whole-cell reactions reveals important points to consider when testing carbene-transfer biocatalysts. The model reaction between styrene and EDA used to evaluate cyclopropanation activity can be catalyzed by heme with and without a protein scaffold. Hence, to properly evaluate a catalyst, diastereo- and enantioselectivity are key parameters to distinguish between nonspecific background reactions catalyzed by free heme, which may occur in vitro as well as in host organisms such as E. coli. Artificially enriching intracellular heme levels in order to boost P450 expression may increase the background reactivity and interfere with a more selective enzyme-catalyzed reaction, especially if P450 expression levels are low. Introducing weaker axial ligands such as serine also may decrease the enzyme’s affinity for the heme prosthetic group and contribute to increased background reactivity from elevated free-heme levels in E. coli cells.
Not addressed in this study is the important effect of the distal heme environment in the binding pocket opposite the proximal axial ligand. We showed in previous work that mutations of P450BM3 in the distal active site pocket significantly influenced stereoselective catalysis for cyclopropanation.[3a] Hence, protein engineering of the distal pocket will increase efficiency and specificity of these and future non-natural reactions, as was recently demonstrated for cyclopropanation of N, N-diethyl-2-phenylacrylamide, yielding an intermediate en route to levomilnacipran.[4]
Conclusions
Olefin cyclopropanation represents a new addition to the already wide range of reactions catalyzed by members of the cytochrome P450 family. This non-natural reaction is enabled by the ability of EDA, a synthetic reagent not expected in the natural environment, to react with heme to generate the reactive iron-carbenoid intermediate that mediates styrene cyclopropanation. This non-natural reactivity can be accessed by various hemoproteins, including cytochrome P450s, and can be improved by protein engineering.
We hypothesized that different active site shapes would influence the stereochemical mixture of products formed. Consistent with this hypothesis, we find that both the choice of parent enzyme and the axial ligand influence the activity and stereoselectivity of cyclopropanation. Thus we propose that variants of different cytochrome P450s, and possibly other hemoproteins, will be interesting catalysts for the chemical synthesis of stereochemically complex cyclopropanated products.
Serum albumins show a high affinity for metal porphyrins and provide a hydrophobic pocket that allows them to catalyze this non-natural reaction as well as any of the wild-type P450s. The difference, we believe, lies in the potential for evolution. Albumins are expressed as insoluble aggregates in E. coli,[16] preventing simple evaluation of these proteins as whole-cell catalysts and making it impossible to engineer or direct their evolution in E. coli. Cytochrome P450s, in contrast, have a defined substrate binding pocket and are highly evolvable, features that lead to their ready adaptation to new catalytic demands.
Experimental Section
Unless otherwise noted, all chemicals and reagents were obtained from commercial suppliers (Sigma-Aldrich, Acros). Gas chromatography (GC) analyses were carried out using a Shimadzu GC-17A gas chromatograph, an FID detector, and J&W scientific cyclosil-B columns (30 m x 0.32 mm, 0.25 μm film and 30 m x 0.25 μm, 0.25 μm film). Plasmids pET22 and pET28 were used as cloning and expression vectors. All gene constructs except P450BM3 and CYP153A6 were generated from codon-optimized gBlocks® (IDT, San Diego, CA). Site-directed mutagenesis was accomplished by standard overlap mutagenesis using primers bearing desired mutations (IDT, San Diego, CA). Gene and primer sequences are available upon request. Electrocompetent BL21(DE3) E. cloni® (Lucigen) cells were prepared following the protocol of Sambrook et al.[17] Restriction enzymes, Phusion polymerase, and T4 ligase were purchased from New England Biolabs (NEB, Ipswich, MA). Alkaline phosphatase was obtained from Roche (Nutley, NJ). Gibson cloning reactions were performed according to manufacturer’s recommendations. Hyper Broth™ was obtained from AthenaES™ and used according to manufacturer’s recommendations.
Determination of enzyme concentration
Enzyme concentrations were determined from ferrous carbon monoxide binding difference spectra using previously reported extinction coefficients for cysteine-ligated (ε = 91,000 M−1 cm−1)[18] and serine-ligated enzymes (ε = 103,000 M−1 cm−1).[19] Concentrations of the serine-ligated mutants were also determined via hemochrome assay using the reported extinction coefficient (ε = 196,000 M−1 cm−1).[20]
Hemochrome assay
The final sample volume of purified protein samples was adjusted with H2O (to 735 μL). A solution of NaOH (1 M, 75 μL) was added and incubated for 5 min at RT. Pyridine (175 μL) was added and mixed via pipetting. A solution of sodium dithionite (100 mg/mL, 15 μL) was added and incubated for 10 min. Samples were measured using UV-VIS. Heme concentration was determined using the reported extinction coefficient (ε = 196,000 M−1 cm−1).[20]
Hemochrome assay of whole cells
For whole-cell reactions, hemochrome assays were performed as follows. Cells were normalized to OD600 = 30 in lysis buffer (25 mM TRIS-HCl, 10 mM NaCl, 0.5 mg/mL lysozyme and 0.02 mg/mL DNaseI). Cell lysis was performed for 2 h at 37 °C with an agitation rate of 200 rpm. The resulting lysate was centrifuged 10 min at 20000 x g and the supernatant was subjected to the hemochrome assay. Final sample volume was normalized with H2O (to 735 μL). A solution of NaOH (1 M, 75 μL) was added and incubated for 5 min at RT. Pyridine (175 μL) was added and mixed via pipetting. A solution of sodium dithionite (100 mg/mL, 15 μL) was added and incubated for 10 min. Samples were measured using UV-VIS. Heme concentration was determined using the reported extinction coefficient (ε = 196,000 M−1 cm−1).[20]
Protein expression
All enzymes were expressed in BL21(DE3) E. cloni® (Lucigen). Depending on the plasmid, cultivation was performed with kanamycin sulfate (50 μg/mL) for pET28 or ampicillin sodium salt (100 μg/mL) for pET22. LB medium (50 mL) was inoculated with a single clone and the overnight culture was used to inoculate Hyper Broth™ (final volume: 750 mL) with δ-ALA (250 μM final concentration) and incubated at 37°C with 225 rpm agitation rate. At OD600 = 1.5 cultures were induced with IPTG (500 μM final concentration), temperature was lowered to 20 °C, agitation rate was lowered to 120 rpm and the culture was incubated for 24 h. Following expression, cells were pelleted and frozen at −20 °C. For whole cell catalysis cells were resuspended in in nitrogen-free M9 medium (1 L contains 31 g Na2HPO4, 15 g KH2PO4, 2.5 g NaCl, 0.24 g MgSO4, 0.01 g CaCl2).
Protein purification
For purification, frozen cell pellets were resuspended (4 mL/g wet cell weight) in lysis buffer (25 mM TRIS-HCL, 100 mM NaCl, 10 mM imidazole, lysozyme (0.5 mg/mL), DNaseI (0.02 mg/mL), hemin (1 mg/g wet cell weight), pH 7.0). TxtE was resuspended (4 mL/g wet cell weight) in lysis buffer I (25 mM TRIS-HCL, 100 mM NaCl, 30 mM imidazole, pH 7.0). Cells were then disrupted by sonication (3 min, output control 1.5, duty cycle: 5 sec on / 10 sec off; Sonicator 3000, Misonix, Inc.). In the case of thermostable CYP119 the cell suspension was subsequently incubated for 30 min at 60 °C to precipitate E. coli proteins and improve purification. To pellet insoluble cell debris, lysates were centrifuged (30000 x g for 30 min at 4 °C). Cleared lysates were filtered using sterile Cellulose acetate syringe filters (0.22 μM, VWR) and then purified with Ni-NTA columns (GE Healthcare or Roche Applied Science) using an AKTAxpress purifier FPLC system (GE healthcare). All enzymes were eluted using a linear gradient from 100% buffer A (25 mM TRIS-HCL, 100 mM NaCl, 10 mM, pH 7.0) or buffer A I for TxtE (25 mM TRIS-HCL, 100 mM NaCl, 30 mM imidazole, pH 7.0), 0% buffer B (25 mM TRIS-HCL, 100 mM NaCl, 400 mM imidazole pH 7.0) or buffer B I for TxtE (25 mM TRIS-HCL, 100 mM NaCl, 300 mM imidazol, pH 7.0) to 100% buffer B over 10 column volumes. Fractions containing the respective enzymes were pooled, concentrated and exchanged into storage buffer (25 mM TRIS-HCL, 10 mM NaCl, pH 7.5) or (20 mM TRIS-HCL, pH 7.5) for TxtE. Concentrated proteins were aliquoted, flash-frozen on powdered dry ice, and stored at −20 °C. Enzyme concentrations were determined via CO binding difference spectra as described above from frozen aliquots.
In vitro small-scale bioconversions under anaerobic conditions
Small-scale reactions (400 μL, final enzyme concentration: 10 μM) were conducted in crimp vials (2 mL size, Agilent Technologies, San Diego, CA). A solution of the P450 enzyme (26.6 μM enzyme in storage buffer, 135 μL) and KPi buffer (1 M, pH 8.0, 15 μL) were added to the vial with a small stir bar before crimp sealing with a silicone septum. The reaction buffer (KPi 0.1 M, pH 8.0, 10 mM Na2S2O4) was sealed and degassed by bubbling argon through the solution for at least 5 min. The headspace of the reaction vial containing the P450 solution was flushed with argon over the protein solution without bubbling. Up to 8 reaction vials were degassed in series via cannulae. The buffer/reductant solution (230 μL) was added to the reaction vial with a syringe, while under argon purge. The gas lines were disconnected from the reaction vial before placing the vials on a stir plate. A 40x styrene (800 mM) solution in EtOH (10 μL) was added to the reaction vial via a glass syringe, and left to stir for about 30 sec. A 40x EDA (400 mM) solution in EtOH (10 μL) was then added and the reaction was left stirring for 1 h at 24°C. The final concentrations of the reagents were typically: styrene (20 mM), EDA (10 mM), Na2S2O4 (5.75 mM), and P450 (10 μM). The vials were opened, internal standard 2-phenylethanol (20 mM in EtOH, 20 μL,) was added followed by extraction with ethyl acetate (1 mL) immediately. This mixture was transferred to a 1.8 mL reaction tube, vortexed and centrifuged (16,000 x g, 10 min). The top organic layer was analyzed by chiral phase GC. Yields, diastereomeric ratios and enantiomeric excess were determined by GC analysis. Yields were based on EDA.
In vitro small-scale bioconversions under aerobic conditions
Small-scale reactions (400 μL, final enzyme concentration: 10 μM) were conducted in crimp vials (2 mL size, Agilent Technologies, San Diego, CA). A solution of the P450 enzyme (26.6 μM enzyme in storage buffer, 135 μL) and KPi buffer (1 M, pH 8.0, 15 μL) were added. The reaction buffer (230 μL, KPi 0.1 M, pH 8.0, 30 mM Na2S2O4) was added and the vials were crimp sealed with a silicon septum. A 40x styrene (800 mM) solution in EtOH (10 μL) was added to the reaction vial via a glass syringe and incubated for 5 min. A 40x EDA (400 mM) solution in EtOH (10 μL) was then added and the reaction was left for 1 h at RT with 400 rpm agitation rate. The final concentrations of the reagents were typically: styrene (20 mM), EDA (10 mM), Na2S2O4 (17.25 mM), and P450 (10 μM). The vials were opened, internal standard 2-phenylethanol (20 mM in EtOH, 20 μL,) was added followed by extraction with ethyl acetate (1 mL) immediately. This mixture was transferred to a 1.8 mL reaction tube, vortexed and centrifuged (16,000 x g, 10 min). The top organic layer was analyzed by chiral phase GC. Yields, diastereomeric ratios and enantiomeric excess were determined by GC analysis. Yields were based on EDA.
Whole-cell small-scale bioconversions under anaerobic conditions
All enzymes were expressed in E. coli BL21(DE3) E. cloni® (Lucigen). Overnight cultures were inoculated from a single clone. Depending on the respective plasmid, cultivation was performed with kanamycin sulfate (50 μg/mL) for pET28 or ampicillin sodium salt (100 μg/mL) for pET22. LB medium (3 mL) was inoculated with the respective clone and the overnight culture was used to inoculate Hyper Broth™ (final volume: 50 mL) with δ-ALA (1 M, 12.5 μL) and incubated at 37 °C, 225 rpm. At OD600=1.5 cultures were induced with IPTG (1 M, 50 μL), temperature was lowered to 20 °C, agitation rate was lowered to 120 rpm and the culture was incubated for 24 h. Cultures were harvested after 24 h and resuspended in nitrogen-free M9 medium (1 L contains 31 g Na2HPO4, 15 g KH2PO4, 2.5 g NaCl, 0.24 g MgSO4, 0.01 g CaCl2,) and the OD600 was adjusted to OD600 = 30.0. This E. coli cell suspension (2 mL) was bubbled with argon in a crimped 6 mL vial. Subsequently, the degassed cell suspension (430 μL) was added to a degassed 2 mL crimp vial containing glucose (250 mM, 50 μL). A 40x styrene solution in EtOH (800 mM, 10 μL) was added to the reaction vial via a glass syringe and incubated for 5 min. A 40x EDA solution in EtOH (400 mM, 10 μL) was then added and the reaction was shaken at 400 rpm for 20 h at RT. The final concentrations of the reagents were typically: styrene (16 mM) and EDA (8 mM). The internal standard 2-phenylethanol (20 mM, 20 μL) was added and reactions were subsequently extracted with ethyl acetate (1 mL). The top organic layer was analyzed by chiral phase GC. Yields, diastereomeric ratios and enantiomeric excess were determined by GC analysis. Yields were based on EDA.
Supplementary Material
Acknowledgments
The authors thank K. Rabe, N. E. Peck, and Z. J. Wang for helpful discussions and Y. Su for help with experiments. We also thank the Gordon and Betty Moore Foundation through grant GBMF2809 to the Caltech Programmable Molecular Technology Initiative. T. Heel is supported by a FWF Schroedinger fellowship (J3327-B21). S. C. Dodani and J. A. McIntosh are supported by Ruth L. Kirschstein NRSA postdoctoral fellowships from the National Institutes of Health (5F32GM106618 and F32GM101792, respectively). J. T. Meyerowitz is supported by the Department of Defense through the National Defense Science & Engineering Graduate Fellowship Program and by the National Science Foundation through the Graduate Research Fellowship Program. The content of this paper is solely the responsibility of the authors and does not represent the official views of any of the funding agencies.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- 1.a) Huisman GW, Collier SJ. Curr Opin Chem Biol. 2013;17:284–292. doi: 10.1016/j.cbpa.2013.01.017. [DOI] [PubMed] [Google Scholar]; b) Turner NJ, O'Reilly E. Nat Chem Biol. 2013;9:285–288. doi: 10.1038/nchembio.1235. [DOI] [PubMed] [Google Scholar]
- 2.a) Whitehouse CJ, Bell SG, Wong LL. Chem Soc Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]; b) Urlacher VB, Girhard M. Trends Biotechnol. 2012;30:26–36. doi: 10.1016/j.tibtech.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 3.a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH. Angew Chem Int Ed Engl. 2013;52:9309–9312. doi: 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Singh R, Bordeaux M, Fasan R. ACS Catal. 2014;4:546–552. doi: 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew Chem Int Ed Engl. 2014;53:6810–6813. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Jung ST, Lauchli R, Arnold FH. Curr Opin Biotechnol. 2011;22:809–817. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kumar S. Expert Opin Drug Metab Toxicol. 2010;6:115–131. doi: 10.1517/17425250903431040. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rabe KS, Gandubert VJ, Spengler M, Erkelenz M, Niemeyer CM. Anal Bioanal Chem. 2008;392:1059–1073. doi: 10.1007/s00216-008-2248-9. [DOI] [PubMed] [Google Scholar]
- 6.Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, Challis GL. Nat Chem Biol. 2012;8:814–816. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM. Nat Chem Biol. 2013;9:485–487. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Monzani E, Bonafe B, Fallarini A, Redaelli C, Casella L, Minchiotti L, Galliano M. Biochim Biophys Acta. 2001;1547:302–312. doi: 10.1016/s0167-4838(01)00192-3. [DOI] [PubMed] [Google Scholar]; b) Zunszain PA, Ghuman J, Komatsu T, Tsuchida E, Curry S. BMC Struct Biol. 2003;3:6. doi: 10.1186/1472-6807-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 10.Funhoff EG, Bauer U, Garcia-Rubio I, Witholt B, van Beilen JB. J Bacteriol. 2006;188:5220–5227. doi: 10.1128/JB.00286-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koo LS, Immoos CE, Cohen MS, Farmer PJ, Ortiz de Montellano PR. J Am Chem Soc. 2002;124:5684–5691. doi: 10.1021/ja017174g. [DOI] [PubMed] [Google Scholar]
- 12.Yoshioka S, Takahashi S, Hori H, Ishimori K, Morishima I. Eur J Biochem. 2001;268:252–259. doi: 10.1046/j.1432-1033.2001.01872.x. [DOI] [PubMed] [Google Scholar]
- 13.Simard JR, Zunszain PA, Hamilton JA, Curry S. J Mol Biol. 2006;361:336–351. doi: 10.1016/j.jmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Nicoletti FP, Howes BD, Fittipaldi M, Fanali G, Fasano M, Ascenzi P, Smulevich G. J Am Chem Soc. 2008;130:11677–11688. doi: 10.1021/ja800966t. [DOI] [PubMed] [Google Scholar]
- 15.Fanali G, Ascenzi P, Bernardi G, Fasano M. J Biomol Struct Dyn. 2012;29:691–701. doi: 10.1080/07391102.2011.672632. [DOI] [PubMed] [Google Scholar]
- 16.Latta M, Knapp M, Sarmientos P, Brefort G, Becquart J, Guerrier L, Jung G, Mayaux JF. Nat Biotechnol. 1987;5:1309–1314. [Google Scholar]
- 17.Sambrook EFJ, Maniatis T. Cold Spring Harbor Laboratory Press. 1989;2 [Google Scholar]
- 18.Omura T, Sato R. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 19.Vatsis KP, Peng HM, Coon MJ. J Inorg Biochem. 2002;91:542–553. doi: 10.1016/s0162-0134(02)00438-5. [DOI] [PubMed] [Google Scholar]
- 20.Berry EA, Trumpower BL. Anal Biochem. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.