

Abstract
The human breast cancer resistance protein (BCRP, gene symbol ABCG2) is an ATP-binding cassette (ABC) efflux transporter. It was so named because it was initially cloned from a multidrug-resistant breast cancer cell line where it was found to confer resistance to chemotherapeutic agents such as mitoxantrone and topotecan. Since its discovery in 1998, the substrates of BCRP have been rapidly expanding to include not only therapeutic agents but also physiological substances such as estrone-3-sulfate, 17β-estradiol 17-(β-d-glucuronide) and uric acid. Likewise, at least hundreds of BCRP inhibitors have been identified. Among normal human tissues, BCRP is highly expressed on the apical membranes of the placental syncytiotrophoblasts, the intestinal epithelium, the liver hepatocytes, the endothelial cells of brain microvessels, and the renal proximal tubular cells, contributing to the absorption, distribution, and elimination of drugs and endogenous compounds as well as tissue protection against xenobiotic exposure. As a result, BCRP has now been recognized by the FDA to be one of the key drug transporters involved in clinically relevant drug disposition. We published a highly-accessed review article on BCRP in 2005, and much progress has been made since then. In this review, we provide an update of current knowledge on basic biochemistry and pharmacological functions of BCRP as well as its relevance to drug resistance and drug disposition.
KEY WORDS: ABCG2, ATP-binding cassette, BCRP, drug transport, transporter
INTRODUCTION
The human ATP-binding cassette (ABC) proteins belong to a large protein superfamily that now comprises 48 members (http://nutrigene.4t.com/humanabc.htm). Many of the human ABC proteins are efflux transporters, and three of them, namely P-glycoprotein (P-gp, gene symbol ABCB1), the multidrug resistance protein 1 (MRP1, gene symbol ABCC1), and the breast cancer resistance protein (BCRP, gene symbol ABCG2), have been implicated to be the major efflux transporters responsible for multidrug resistance in cancer cells.
Human BCRP is encoded by the ABCG2 gene which is located on chromosome 4q22. Like P-gp and MRP1, BCRP possesses a very broad substrate and inhibitor specificity that is different from, but substantially overlaps with that of P-gp or MRP1. The role of BCRP in drug disposition has now also been appreciated because it highly resembles P-gp in tissue distribution and expression as well as the broad substrate and inhibitor specificity. Much progress has been made on substrates, inhibitors, and physiological and pharmacological roles of BCRP since we published the highly accessed review article on this subject in 2005 in the AAPS Journal (1). In the present review, we will provide an update of current knowledge on this topic.
BCRP IN HUMAN CANCERS
The majority of the work in this area has been done with leukemia, particularly acute myeloid leukemia (AML). Since this topic has been extensively reviewed elsewhere (2), here we only provide updates of most important findings. Several studies have shown a positive correlation between high levels of BCRP expression and poor clinical outcomes in AML, e.g., a relapsed or refractory disease state, lower response rate, shorter overall survival, and/or no complete remission; however, other studies reported no correlation of BCRP expression with clinical outcomes or no expression of BCRP in AML (see references provided in review by Natarajan et al. (2)). This discordance may be attributed, at least in part, to methodologies used to detect BCRP expression. Some studies analyzed BCRP messenger RNA (mRNA) expression, but others detected BCRP protein levels. A general caution is that mRNA levels may not reflect levels of protein expression or activity. Also, BCRP protein expression was generally detected by flow cytometry using BCRP-specific monoclonal antibodies, and BCRP activity was measured using a flow cytometric efflux assay. Such BCRP expression and activity assays may not be sensitive and accurate enough to quantify small differences in patient samples. Careful validation of BCRP expression and activity data is necessary. Another striking observation is that, even in some studies that showed a correlation between BCRP expression and clinical outcomes, the anticancer drugs used (e.g., anthracyclines and cytarabine) are generally poor substrates or even not substrates of BCRP. It has therefore been argued that BCRP could be a biomarker for, but not a mechanism of drug resistance in at least some AML patients (2). Lastly, characterization of the role of BCRP in clinical drug resistance of AML is further complicated by frequent co-expression with P-gp and MRP1 (3) which confounds the interpretation of the data.
The development of tyrosine kinase inhibitors (TKIs) such as imatinib, nilotinib, and dasatinib to inhibit the oncogenic tyrosine kinase BCR-ABL has revolutionized the therapy for chronic myeloid leukemia (CML). However, clinical resistance to these TKIs has already emerged. In vitro studies have demonstrated that these TKIs are substrates and/or inhibitors of the efflux transporters P-gp and BCRP (4) as well as the uptake transporter OCT1 (5). Therefore, contributions of these transporters to drug resistance in CML patients with clinical outcomes of TKI therapy were evaluated in several clinical studies. A recent study investigated the correlation between mRNA expression of various transporters (P-gp, BCRP, OCT1, and OATP1A2) in peripheral blood leukocytes and clinical outcomes (e.g., major and complete molecular responses as well as drug resistance) in 118 chronic-phase CML patients receiving a standard dose of imatinib mesylate (6). They found that BCRP mRNA expression in non-responders was higher than that in responders before and during imatinib therapy. Furthermore, BCRP was overexpressed in those who did not achieve major molecular response. In the responder group, patients who achieved major molecular response had higher mRNA expression of OCT1. These data suggest that higher BCRP expression may be associated with imatinib resistance, and higher OCT1 expression could be associated with a successful imatinib therapy, in CML patients.
BCRP expression has also been detected in a variety of solid tumors (7). The correlation between BCRP expression and clinical outcomes has primarily been evaluated in breast cancer and non-small cell lung cancer (NSCLC). In breast cancer, only one study reported a correlation between BCRP mRNA expression and response in a subgroup of patients receiving anthracycline-based chemotherapy (5-fluorouracil, adriamycin/epirubicin, and cyclophosphamide), and such a correlation did not exist in the cyclophosphamide, methotrexate, and 5-fluorouracil-treated group of patients (8). However, whether BCRP plays a role in drug resistance in these breast cancer patients is not known because anthracyclines are poor substrates of wild-type BCRP that is detected in cancer patients. A more recent study examined BCRP expression (mRNA and immunohistochemistry) and resistance to 5-fluorouracil (a BCRP substrate) in 140 breast cancer tissues specimens, and found that resistance to 5-fluorouracil was significantly correlated with the levels of BCRP expression; however, no outcome data were reported (9). In NSCLC, one earlier study reported a strong correlation between BCRP expression in tumor samples from 72 untreated stage IIIB or IV NSCLC patients and the response rate to platinum-based chemotherapy, and expression of other transporters including P-gp, MRP1, MRP2, and MRP3 was not significantly associated with response or survival (10). A more recent study showed that high BCRP expression determined by immunohistochemistry in biopsy specimens predicts short survival for advanced NSCLC patients treated with platinum-based chemotherapy (11). Since platinum compounds are not known to be BCRP substrates, the mechanisms by which BCRP expression is associated with clinical outcomes in lung cancer patients are not clear. Most recently, BCRP expression in 67 surgically resected pancreatic ductal adenocarcinoma samples determined using immunohistochemistry was reported to be a significant prognostic factor for early tumor recurrence and poor survival (12). Overall, the role of BCRP in drug resistance in cancers has not been well established. There are currently no clinical studies aimed at overcoming cancer drug resistance by inhibiting BCRP.
BCRP SUBSTRATES
Substrates of BCRP initially were reported to be a wide range of chemotherapeutics such as mitoxantrone, camptothecin derivates, flavopiridol, and methotrexate (1). Notably, several TKIs such as imatinib, gefitinib, and nilotinib are BCRP substrates (1,13). A variety of photosensitizers including pheophorbide A, protoporphyrin IX, and related compounds are also BCRP substrates, suggesting that BCRP is a possible cause of cellular resistance to photodynamic therapy (14).
Other classes of anticancer drugs including vinblastine, cisplatin, and paclitaxel are not BCRP substrates (13). BCRP substrates are not limited to chemotherapeutics. Drugs that have been shown to be BCRP substrates include, among others, prazosin, glyburide, cimetidine, sulfasalazine, and rosuvastatin (1,13). Nucleoside and nucleotide analogs such as AZT and lamivudine are also BCRP substrates (1).
Fluorescent probes have proven to be useful reagents for analysis of cellular expression and function of ABC transporters. Fluorescent compounds that are commonly used as BCRP probe substrates include BODIPY-prazosin, Hoechst 33342, and pheophorbide A (1). Rhodamine 123 and Lyso-Tracker Green are substrates of the mutants, R482G and R482T, but not substrates of wild-type BCRP (15).
BCRP also transports conjugated organic anions, particularly sulfated and glucuronide conjugates, such as estrone-3-sulfate, dehydroepiandrosterone (DHEAS), and 17β-estradiol 17-(β-d-glucuronide) (1). Organic conjugates of drugs, xenobiotics, and endogenous substances all could be BCRP substrates (1). In general, sulfated conjugates seem to be better BCRP substrates than glutathione and glucuronide conjugates. In addition, phosphorylated nucleosides and nucleotides, particularly their monophosphates such as AZT 5′-monophosphate, are also BCRP substrates (1).
Other BCRP substrates include chemical toxicants such as the carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), phototoxic compounds such as protoporphyrin IX, the lipid phosphatidylserine, flavonoids such as genistein, uric acid, and vitamins (1,13). Collectively, BCRP has a very broad substrate specificity that is substantially overlapping, but distinct from that of P-gp or MRP1 (1,13). Selected substrates of wild-type BCRP that are therapeutic agents are shown in Table I. Although the number of known BCRP substrates is now over 200, not much work has been done to analyze structure-activity relationship (SAR). The only SAR study was for camptothecin analogs, and the authors found that BCRP preferentially transports the camptothecin analogs with high polarity at carbon positions 10 and 11 over those with low polarity (41). We have recently developed a predictive model for BCRP substrates using a support vector machine (SVM) method based on 263 known BCRP substrates and non-substrates (42). This SVM model has an overall prediction accuracy of ∼76%, and thus would be useful for prediction and screening of new BCRP substrates.
Table I.
Selected Drugs That Are Substrates of BCRP
| Drug | Reference |
|---|---|
| Anthracenes | |
| Mitoxantrone | (16,17) |
| Bisantrene | (17) |
| Aza-anthrapyrazole (BBR3390) | (18,19) |
| Camptothecin derivates | |
| Topotecan | (17) |
| SN-38 | (20) |
| Irinotecan | (21,22) |
| Diflomotecan | (23) |
| Polyglutamates | |
| Methotrexate | (24,25) |
| Methotrexate-Glu2 | (24,26) |
| Methotrexate-Glu3 | (24,26) |
| Nucleoside analogs | |
| AZT | (27,28) |
| AZT 5′-monophosphate | (27,28) |
| Lamivudine (3TC) | (27,28) |
| Other drugs | |
| Prazosin | (17) |
| Indolocarbazole | (29) |
| Flavopiridol | (30) |
| Canertinib (CI1033) | (31) |
| Imatinib mesylate (STI571) | (32) |
| Gefitinib (ZD1839) | (33) |
| Nilotinib | (34) |
| Glyburide | (35) |
| Cimetidine | (36) |
| Sulfasalazine | (37) |
| Nitrofurantoin | (38) |
| Rosuvastatin | (39) |
| Pantoprazole | (40) |
Substrates listed in this table are only for wild-type BCRP
BCRP INHIBITORS
A large number of BCRP inhibitors with diverse chemical structures have been identified. Some selected BCRP inhibitors that are therapeutics are shown in Table II. First of all, many P-gp inhibitors are also excellent BCRP inhibitors. The first example of such BCRP inhibitors is GF120918 with an IC50 value of ∼50 nM for BCRP (58). We were the first to report that the HIV protease inhibitors ritonavir, saquinavir, and nelfinavir are effective BCRP inhibitors (48). TKIs such as imatinib, nilotinib, and apatinib (4) and inhibitors for other type kinases such as the serine/threonine Polo-like kinase 1 inhibitor BI2536 (59) are also potent dual P-gp/BCRP inhibitors.
Table II.
Selected Drugs That Are Inhibitors of BCRP
| Drug | IC50 (nM) | Reference |
|---|---|---|
| Tyrosine kinase inhibitors | ||
| Gefitinib | 300 | (43) |
| Imatinib mesylate | 170 | (44) |
| Erlotinib | ND | (45) |
| Nilotinib | ND | (4,46) |
| Lapatinib | ND | (47) |
| HIV protease inhibitors | ||
| Ritonavir | 19,500 | (48) |
| Saquinavir | 19,500 | (48) |
| Nelfinavir | 12,500 | (48) |
| Lopinavir | 7660 | (49) |
| HCV protease inhibitors | ||
| Boceprevir | 81,000 | (50) |
| Telaprevir | 30,000 | (51) |
| Calcium channel blockers | ||
| Dipyridamole | 6400 | (52) |
| Nicardipine | 4800 | (52) |
| Nimodipine | 13,700 | (52) |
| Nitrendipine | ND | (52) |
| Antifungal azoles | ||
| Ketoconazole | 15,300 | (53) |
| Itraconazole | ND | (53) |
| Fluoconazole | ND | (53) |
| Immunosuppressants | ||
| Cyclosporin A | 4300 | (53) |
| Tacrolimus | 3600 | (53) |
| Sirolimus | 1900 | (53) |
| Other drugs | ||
| Novobiocin | 50–100 | (54,55) |
| Tamoxifen | ND | (56) |
| Reserpine | ND | (57) |
| Omeprazole | 10,000–50,000 | (40) |
| Pantoprazole | ND | (40) |
ND not determined
There are highly selective BCRP inhibitors. The typical example is fumitremorgin C (FTC) secreted from the fungi Aspergillus fumigatus with an IC50 value of ∼1 μM. FTC did not inhibit P-gp or MRP1 (21). Neurotoxicity of FTC precludes its use in in vivo studies. Several FTC analogues including Ko132, Ko134, and Ko143 with a much more potent inhibitory effect (with IC50 values of 100–200 nM) and a high selectivity while displaying low in vivo neurotoxicity have been developed (60).
Other notable BCRP inhibitors include, among others, novobiocin (54), tamoxifen, and its derivatives TAG-11 and TAG-139 (56), reserpine (57), the pipecolinate derivatives VX-710 (Biricodar) (61), tryprostatin A (a A. fumigatus second metabolite) (62), and dietary flavonoids such as chrysin and biochanin A (63).
Besides the abovementioned inhibitors, derivatives of a variety of known BCRP inhibitors such as resveratrol, tariquidar, chromone, and chalcone were synthesized and tested in the past 5 years. Several of the derivatives have been shown to be potent and highly specific BCRP inhibitors. For example, placement of the quinolone-2-carboxamido group to position 3 of the benzamide moiety of tariquidar resulted in a highly potent and selective BCRP inhibitor with an IC50 value of 60 nM for BCRP, of >29,000 nM for P-gp, and of >20,000 nM for MRP2 (64). Likewise, a chromone derivative was found to be one of the most active, selective, and non-toxic BCRP inhibitors reported ever (with an IC50 value of 110 nM) (65).
The molecular mechanisms of BCRP inhibition have not been fully understood, but could be diverse. For example, some inhibitors such as FTC and Ko143 are considered “general” inhibitors as they inhibit ATPase activity of BCRP. Other inhibitors are BCRP substrates, and as such, can act as competitive inhibitors. In this regard, some inhibitors may interact with BCRP on binding sites of one class of substrates but not others, and hence only inhibit efflux of one particular class of substrates. It is also possible that some inhibitors interact with BCRP on sites other than substrate binding sites and induce conformational changes in the large binding pocket, and thus allosterically affect transport of some substrates. All these can result in substrate-dependent inhibition. For example, Giri et al. (66) performed transport inhibition studies and found that nelfinavir effectively inhibited efflux of the nucleoside substrates zidovudine and abacavir, but had no effect on efflux of prazosin and imatinib, suggesting that zidovudine and abacavir possibly interact with BCRP at sites that do not overlap with those for prazosin and imatinib. We therefore hypothesize that BCRP possesses multiple substrate sites (see details in the “STRUCTURE AND FUNCTION section”), and as such, inhibition of BCRP can be substrate-dependent. Such complex inhibition mechanisms remain a challenge for predicting and screening BCRP inhibitors in drug discovery. To address this issue, it would be highly valuable to develop ligand-based computational methods for predicting general and substrate-dependent inhibitors of BCRP. A recent study using the Bayesian classification method developed predictive models for BCRP inhibitors with an overall prediction accuracy of ∼70% (67), suggesting that development of classification methods for virtual screening of novel BCRP inhibitors is possible. Other classification methods such as the support vector machine (SVM) may prove valuable too. We used the SVM method to predict P-gp and BCRP substrates (42,68). Such classification methods would predict BCRP inhibitors purely based on structural features of known BCRP inhibitors and non-inhibitors, regardless of their mechanisms of inhibition.
A large number of structure-activity relationship (SAR) and quantitative SAR (QSAR) studies have been done for structurally related or diverse BCRP inhibitors to understand the structural features of compounds critical for an inhibitor. This topic has been extensively reviewed elsewhere (69–71). Lipophilicity seems to be a significant determinant for BCRP inhibition for some compounds including flavonoids and FTC analogs (60,72), but not for other compounds such as the tariquidar analogs (73). Planar structure, amine bonded to a carbon of a heterocyclic ring, and hydrogen bonding potential may also be important for certain inhibitors (13,71). At present, SAR and QSAR models based on one set of inhibition data generally cannot be extrapolated to a different set of data. This limitation cannot be resolved until we fully understand the mechanism by which BCRP interacts with substrates and inhibitors.
STRUCTURE AND FUNCTION
To understand the mechanism by which BCRP interacts with substrates and inhibitors, it is essential to first understand the structure and function of the transporter. BCRP is a polytopic transmembrane (TM) protein with 655 amino acids. It is the second member of the subfamily G of the large human ABC transporter superfamily, and hence also named as ABCG2. Two unique features in BCRP distinguish it from most other ABC transporters including P-gp and MRP1. First, BCRP is a half ABC transporter with only one nucleotide-binding domain (NBD) and one membrane-spanning domain (MSD) (2,74). In comparison, P-gp and MRP1 contain two tandem repeated halves. Second, the NBD in BCRP precedes the MSD, a domain organization that is opposite to that of P-gp and MRP1 (2,74). Such unique structural features imply that BCRP may operate quite differently in transport mechanism compared to P-gp and MRP1. Key knowledge and recent progress regarding structure and function of BCRP are summarized below.
Membrane Topology
An accurate membrane topology is essential for reliable homology modeling and mechanistic understanding of drug transporters. For example, a recent three-dimensional (3D) model of BCRP predicted based on evolutionary sequence information cannot be correct because the topology of BCRP (with 7 TM α-helices) that these authors used is incorrect (75). We have recently determined the topology of BCRP using HA epitope insertion and immunofluorescence (76) (Fig. 1). This topology suggests that BCRP contains 6 TM α-helices; however, the computer-predicted TM2 and TM5 are shifted to the extracellular and intracellular loops, respectively, in the experimental topology (76). Such a significant shift of TM helices would result in a drastic change in helical packing of BCRP in its 3D structure. It is worth noting that the only known N-glycosylation site at position 596 (Asn596) (77) in the experimental topology is located in the extracellular loop connecting TM5 and TM6. According to the so-called “12 + 14 rule”; that is, the acceptor site Asn in extracellular loops must be spaced at least 12 residues from the proximal and 14 residues from the distal TM segments to be efficiently glycosylated (78), Asn596 in the experimental topology can be glycosylated, which is consistent with experimental findings. While this is a novel topology, people continue debating on whether this topology is accurate due to the concern that insertion of HA tags may distort helical packing and hence overall structure of BCRP. Therefore, this new experimental topology requires further validation using other appropriate biochemical and biophysical methods.
Fig. 1.
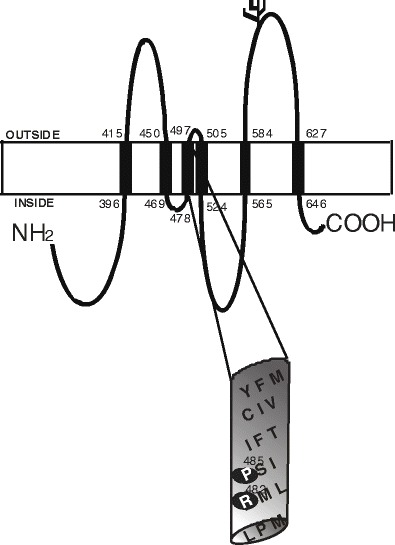
A membrane topology model of BCRP. BCRP contains one NBD and one MSD with six TM α-helices. The boundary of TM α-helices is approximate and based on our experimentally determined membrane topology. The N-glycosylation site (Asn596) is indicated in the extracellular loop connecting TM5 and TM6. The putative TM3 is shown in an expanded view. Two residues in TM3 important for substrate selectivity (Arg482 and Pro485) are indicated by shaded cycles in the expanded view of TM3
Homodimerization or Homooligomerization
Using sucrose density gradient sedimentation and non-denaturing gel electrophoresis, Xu et al. (79) demonstrated that detergent-solubilized BCRP may form homotetramers. Likewise, the electron microscopy (EM) analysis revealed that BCRP existed as a tetramer of dimers in detergent solutions (80) or a tetramer in 2D crystals (81). Recently, evidence that BCRP can form homodimers or homooligomers in intact cells has been obtained by using techniques such as fluorescence resonance energy transfer (FRET) (82) or bimolecular fluorescence complementation (83). The mechanism by which BCRP forms homodimers or homooligomers is not clear. The Cys residue at position 603 seems to be involved in intermolecular disulfide bond formation (84); however, substitutions of Cys603 had no effects on either dimer/oligomer formation or activity of BCRP in intact cells (82,83). It is possible that, in addition to intermolecular disulfide bonds formed by Cys603, intermolecular disulfide bonds formed by other Cys residues and/or non-covalent protein-protein interactions could also be crucial for BCRP dimer/oligomer formation. Indeed, Mitomo et al. (85) found that BCRP fully retained transport activity in the presence of 2-mercatoethanol at 10 mM, a concentration that is sufficient to break down disulfide bonds between BCRP monomers. These data suggest that intermolecular disulfide bonds alone are not essential for BCRP dimer/oligomer formation and function. One possibility is that BCRP dimers are assembled in the membrane also through protein-protein interactions, and therefore can still be fully maintained even if disulfide bond formation is diminished.
Structure Determination and Homology Modeling
The first structural study by McDevitt et al. (80) illustrated that BCRP protein particles in a detergent solution formed a higher order oligomeric complex that was organized as a tetramer of BCRP dimers. A 3D structure at 18-Å resolution was constructed, which allowed visualization of an overall shape and an oligomeric state for BCRP. More recently, Rosenberg et al. (81) reported the first projection structures of BCRP determined by cryo-EM of well-diffracting 2D crystals. The 2D crystals showed a p121b symmetry and the projection structures were determined to 5-Å resolution. At this resolution, ring-shaped high-density features in the projection maps were visualized, probably representing TM α-helices. There were four BCRP monomers (two BCRP dimers) in one unit cell of the 2D crystals, indicating the existence of an oligomeric complex of BCRP. This study also illustrated a significant conformational change upon mitoxantrone binding; that is, BCRP had a more closed and symmetric configuration in the presence of mitoxantrone than that with no mitoxantrone bound. A 3D structure of BCRP at a medium resolution based on these 2D crystals has now been constructed (Rosenberg et al., manuscript submitted).
Without high-resolution 3D structures, homology models play an important role in interpretation of experimental data and in providing guidance for future studies. Several earlier studies developed homology models of BCRP based on computer-predicted topology which is now known to be different from the experimentally determined topology (76). Here, we briefly discuss the homology models of BCRP developed in our laboratory based on the experimental topology. We first refined sequence alignment for TM segments between BCRP and the templates (MsbA, the first and second halves of mouse P-gp and Sav1866) by comparing the experimentally determined TM segments of BCRP with those of the templates observed in crystal structures. Next, the templates were edited to reflect the same domain organization in BCRP by “cutting” the linker regions between the MSD and the NBD of the templates. Three homology models of BCRP representing different conformational states have been generated. The first model based on MsbA (PDB code 3B5W) represents the substrate-unbound nucleotide-free inward-facing open apo conformation. The second model based on mouse P-gp (PDB code 3G60) represents the substrate-bound nucleotide-free inward-facing closed apo conformation (Fig. 2). The third model based on Sav1866 (PDB code 2HYD) represents the nucleotide-bound outward-facing conformation. The inward-facing states display a wide separation of the two NBDs that is open to the intracellular side of the plasma membrane (Fig. 2). The distance between the two NBDs in the open apo state is greater than that in the closed apo state. On the other hand, the extracellular side of the inward-facing conformations is closed by joining together of two large extracellular loops connecting TMs 1 and 2 and TMs 5 and 6. In contrast, the outward-facing conformation is exactly opposite with the extracellular side open and the intracellular side closed. These atomic homology models have already been published (74,81).
Fig. 2.
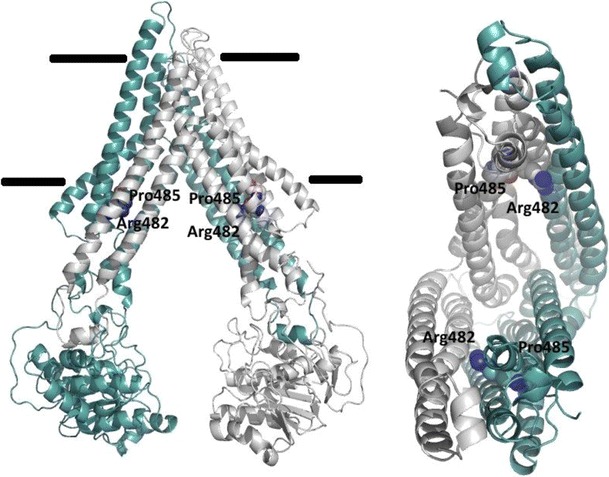
A homology model of BCRP based on the mouse P-gp structure representing a nucleotide-free inward-facing “closed apo” conformation. Two BCRP monomers in a dimer are shown in different colors. The internal cavity formed by TMs is open to the intracellular space. Arg482 and Pro485 in TM3 are shown in blue and red colors. The right panel only shows TM helices
The homology models are consistent with biochemical data published to date. First, the intracellular entry of the inward-facing models is spacious enough to allow access of a bulk of BCRP substrates from the inner lipid leaflet of the plasma membrane or cytoplasm. Molecular docking of several BCRP substrates to the closed apo model indeed suggests the existence of multiple binding sites in the large central pocket primarily formed by TM α-helices (86). Second, Arg482 has been extensively studied by site-directed mutagenesis and found to be crucial for substrate specificity and transport activity (see details in Mutagenesis analysis below). In the models, Arg482 in TM3 is located in the central binding pocket at a position near the cytosolic membrane interface (Fig. 2). Docking calculations indicate that Arg482 directly interacts with mitoxantrone and Hoechst33342, but not with prazosin and SN-38 (86). This is consistent with previous findings that resistance to mitoxantrone was increased, but resistance to SN-38 or efflux of prazosin was not affected, by mutations of Arg482 (16,87). This also is in agreement with the studies showing that prazosin binds to a site that does not fully overlap with that for mitoxantrone or Hoechst33342 (88) and the binding of a prazosin derivative to BCRP was relatively unaffected by mutations of Arg482 (89). Molecular docking also has provided explanations for the unique role of Arg482 in determining transport selectivity for methotrexate and derivatives. Molecular docking using the closed apo model suggests that methotrexate can directly associate with the positively charged Arg482 by direct salt-bridge interactions via their negatively charged carboxylate or sulfate groups (90). However, such electrostatic interactions do not occur for mitoxantrone, prazosin, or Hoechst33342, explaining why mutations of Arg482 do not significantly affect efflux of the three substrates (16), but abolish transport of methotrexate (24). In summary, the homology models could be used to interpret biochemical data. More studies are needed to further refine and validate these models, given the slow pace in determining high-resolution 3D structures of BCRP.
Mutagenesis Analysis
Spontaneous mutations of Arg482 were initially discovered in BCRP from drug-selected drug-resistant cancer cell lines (91). This residue was found to be a critical determinant of substrate selectivity and immediately became the subject of extensive mutagenesis studies. Wild-type BCRP with Arg482 does not transport daunorubicin, rhodamine 123, and Lyso-Tracker Green; however, these compounds are excellent substrates of the BCRP mutants R482T and R482G (16,91). Methotrexate is a substrate of wild-type BCRP only (24). Mitoxantrone, BODIPY-prazosin, and Hoechst 33342 are substrates of both wild-type BCRP and the two mutants (16). Arg482 is predicted to be located in TM3 α-helix near the cytoplasmic interface (Fig. 1), and is likely part of the large drug binding pocket. As discussed above, Arg482, a positively charged residue, is possibly involved in salt-bridge interactions with some substrates (90). It should be emphasized that mutations of Arg482 have never been identified in human subjects or in DNA samples from cancer patients.
The realization that Arg482 is located in a TM α-helix triggered mutagenesis studies on other residues in TM α-helices (Fig. 2). We identified a polar residue, Thr402 in TM1, which is important for overall transport activity. Ala or Arg substitution of Thr402 caused a significant reduction by 50–90% in efflux of mitoxantrone, BODIPY-prazosin, and Hoechst33342 as well as its ability to confer resistance to mitoxantrone and SN-38 (92). According to the closed apo model, Thr402 is not directly involved in substrate binding, but participates in interhelical interactions that are functionally important. Consistent with this observation, Thr402 was proposed to be near or part of the GXXXG motif that may play an important role in dimerization or helical interactions (93). Pro residues in TM α-helices often form flexible hinges and can play a key role in dynamic conformational changes. We found that Ala substitution of Pro485 in TM3 significantly reduced efflux of BODIPY-prazosin by 70%, but had no effect on efflux of mitoxantrone and Hoechst 33342 (94). Homology modeling suggests that Pro485 can introduce a flexible hinge in TM3, and as such, making the drug binding pocket more structurally dynamic. Mutation of Pro485 would eliminate conformational flexibility of the drug binding pocket, thus affecting binding of some drugs, but not others. Thus, structural flexibility introduced by Pro485 in TM3 may contribute to substrate specificity. Notably, both Arg482 and Pro485 are located within TM3 (Figs. 1 and 2). Based on these findings, we hypothesize that TM3 is part of the translocation pathway that encompasses multiple substrate binding sites, and as such, TM3 is critical for conformational dynamics of the binding pocket and plays a crucial role in determining substrate specificity.
Other residues that are functionally important include Glu446 in the extracellular loop connecting TMs 1 and 2 (87), Leu554 in the intracellular loop connecting TMs 4 and 5 (95), and Lys86 and Glu211 in the NBD (96–98). Mutations of these residues resulted in either significantly impaired or no transport activity. Mutations of Lys86 and Glu211 have been shown to cause a complete loss of ATPase activity (96,98). However, another study showed that the activity loss caused by mutations of Lys86 was possibly due to altered subcellular localization and cell surface targeting of BCRP (97). More information about mutations and their effects on function and expression of BCRP can be found in an open access database (http://abcmutations.hegelab.org/). More mutagenesis studies should be carried out, particularly in the MSD, to expand the scope of our understanding of critical residues important for substrate selectivity and overall transport activity.
Multiple Substrate Binding Sites
It is generally believed that ABC transporters possess multiple drug binding sites in a large pocket formed by TM α-helices. Although high-resolution 3D structures of BCRP have not been available, several lines of biochemical evidence support the existence of multiple binding sites. First of all, as we discussed earlier, mutations of some residues such as Arg482 and Pro485 that are likely located in the drug binding pocket affect efflux of some substrates, but not others. Whether these residues directly interact with substrates or play an indirect but critical role in maintaining the architecture or inducing conformational changes of binding sites requires further investigation. Additional evidence comes from direct binding or transport studies. For example, photolabeling of BCRP with the substrate [125I]iodoaryl azidoprazosin was inhibited by some compounds, but not others (99). Clark et al. conducted direct binding kinetic studies and showed that there are possibly two distinct binding sites in BCRP, one for mitoxantrone and Hoechst33342 and another for prazosin (88). Giri et al. performed transport inhibition studies and found that the nucleoside analog substrates zidovudine and abacavir seem to interact with BCRP at sites that do not overlap with those for prazosin or imatinib (66). Lastly, our homology models have been used to interpret mutagenesis and transport data of our own (92,94) and others studies (90). The models suggest a large internal cavity formed by two bundles of six TMs that is spacious enough to accommodate multiple drugs. The exact locations of binding sites in BCRP are still not known, and this awaits the determination of high-resolution 3D structures of the transporter complexed with one or more substrates or inhibitors.
SINGLE NUCLEOTIDE POLYMORPHISMS
A large number (>80) of single nucleotide polymorphisms (SNPs) of the ABCG2 gene have been identified in DNA samples of ethnically diverse origins. Of these SNPs, 34G>A (V12M) and 421C>A (Q141K) occur most frequently in East Asians (∼30–60%) and with relatively low allele frequencies in Caucasians and African-American populations (∼5–10%). All other SNPs generally have allele frequencies of ∼1% or less. Notably, two SNPs, 376C>T (Q126stop) and 1000G>T (E334stop) result in substitutions of a stop code in the ABCG2 gene. In vitro expression and functional studies generally support the conclusion that the Q141K variant resulting from the 421C>A SNP has reduced cell surface expression in transfected cells and therefore cells expressing Q141K display lower efflux activities compared to those expressing wild-type BCRP (100). V12M resulting from the 34G>A SNP and other variants (e.g., I206L, F208S, N590Y, and D620N) display expression levels and drug resistance profiles comparable to wild-type BCRP (100,101). The SNPs 114T>C, 369C>T, 474C>T, 564A>G, 1098G>A, and 1425A>G do not cause amino acid changes; but whether they can alter BCRP expression and activity by affecting protein translation is not known. SNPs in the promoter region such as −15622C>T and −1379A>G have also been reported and may affect transcriptional regulation of the ABCG2 gene (102).
TISSUE LOCALIZATION AND ROLE IN DRUG DISPOSITION
Among normal human tissues, the highest expression of BCRP was observed on the apical membrane of the placental syncytiotrophoblasts (103). In addition, BCRP is prominently expressed on the apical membrane of the epithelium in the small intestine and colon and on the canalicular membrane of hepatocytes (103). BCRP is also expressed on the apical membrane of human kidney proximal tubular cells (104); however, its level of expression in human kidney is relatively low compared to that in the liver and small intestine. BCRP is also on the apical (luminal) membrane of the microvessel endothelial cells in human brain (105) and the retinal capillary endothelial cells (106) as well as in the blood-testis (107) and blood-spinal cord (108) barriers. The tissue localization of Bcrp1, the rodent homolog of human BCRP in mice and rats, is similar to that in humans.
This strategic localization and substantial expression of BCRP in human and rodent tissues implies that BCRP can play a crucial role in limiting absorption (in the small intestine), mediating distribution (e.g., in the blood–brain and blood–placental barriers), and facilitating biliary and renal elimination (in the liver and kidney) of drugs or xenobiotics that are BCRP substrates. The role of BCRP in drug disposition was first appreciated in studies using Bcrp1-knockout mice and will be briefly discussed below.
In the liver and kidney, BCRP facilitates biliary and renal elimination of drugs and xenobiotics. Breedveld et al. (40) demonstrated that the area under the concentration-time curve (AUC) of intravenously administered methotrexate in wild-type mice was increased ∼twofold by co-administration of intravenous (IV) pantoprazole (a Bcrp1 inhibitor); however, the AUC of IV methotrexate in Bcrp1-knockout mice was not affected. IV pantoprazole also decreased the systemic clearance of IV methotrexate in wild-type mice to the same level as in Bcrp1-knockout mice, but had no effect on methotrexate clearance in Bcrp1-knockout mice. Further analysis confirmed that pantoprazole reduced methotrexate clearance by predominantly inhibiting biliary excretion of methotrexate mediated by Bcrp1 (40). Many similar studies can be found in an excellent review by Vlaming et al. (109).
In the blood–brain barrier, numerous studies have now confirmed that BCRP significantly limits brain penetration of drugs or xenobiotics, which is consistent with its high levels of expression on the luminal side of brain endothelial cells. For example, Agarwal et al. (110) showed that the steady-state brain-to-plasma concentration ratio of sorafenib in Bcrp1-knockout mice was increased ∼fourfold compared to that in wild-type mice. Since P-gp and Bcrp1 are co-localized to the same site in the blood–brain barrier, a synergistic effect between the two transporters was observed in many studies, that is, the brain exposure (brain-to-plasma AUC or concentration ratio) of a P-gp/BCRP dual substrate in P-gp/Bcrp1 double-knockout mice is much greater than additive of brain exposure to the drug in P-gp and Bcrp1 single-knockout mice (111). This synergistic effect does not seem due to direct biological or physical interactions between the two transporters, and can be explained by pharmacokinetic theory (112). That is, such apparent “synergy” would be expected if P-gp and BCRP are the principal pathways of clearance of the drug from the brain. This situation is analogous to fraction of a drug metabolized by two enzymes. Theoretically, complete inhibition of both enzymes could lead to an infinite increase in the plasma concentration ratio (in the presence of inhibitor vs. in the absence of inhibitor) of the drug.
In the blood–placental barrier, BCRP expels drugs or xenobiotics from the fetal compartment back to the maternal circulation, thus limiting their fetal exposure. We found that fetal exposure (fetal-to-maternal plasma AUC ratios) to nitrofurantoin and glyburide in Bcrp1-knockout mice was increased ∼five- and twofold, respectively, compared to those in wild-type mice (35,113). The role of BCRP in determining fetal exposure to drugs and xenobiotics can also be studied using other methods such as human placenta perfusion, and this topic has been extensively reviewed elsewhere (114).
BCRP is also expressed in the mammary gland and is strongly induced in the lactating breast of mice, cows, and humans (115). In the mammary gland, Bcrp1 has been shown to actively transport drugs (e.g., topotecan and cimetidine), xenobiotics (e.g., 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine or PhIP), and vitamins (e.g., riboflavin) into breast milk (115,116). Although pharmacological or toxicological implications of BCRP-mediated milk secretion of drugs and xenobiotics are not known, caution should be taken when lactating women take medications that are BCRP substrates so that undesired side-effects or toxicity for their breast-feeding babies may be avoided.
The role of BCRP in drug disposition in humans has been demonstrated in clinical studies. One early study revealed that co-administration of oral GF120918 significantly enhanced oral bioavailability of topotecan in cancer patients from 40 to 97% (117), suggesting that inhibition of BCRP in the small intestine increased the absorption of topotecan. Subsequent studies investigated the correlation between ABCG2 SNPs and pharmacokinetics (PK) of drugs that are BCRP substrates. The most extensively studied drugs are statins and anti-cancer drugs, and the most extensively analyzed SNP is ABCG2 421C>A. Most of the clinical studies published thus far investigating the impact of ABCG2 421C>A SNP on drug PK are summarized in Table III. Overall, the impact of the ABCG2 421C>A SNP on drug PK seems to depend on the drug tested, the genotype that the subjects carry (421CA heterozygous or 421AA homozygous), and the route of drug administration. As shown in Table III, even for well-established BCRP substrates such as topotecan, irinotecan, and nitrofurantoin, clinical studies have not revealed statistically significant effects of the ABCG2 421C>A SNP on PK of these drugs. For drugs, particularly the statins such as rosuvastatin, fluvastatin, simavastatin, and atorvastatin on which the ABCG2 421C>A SNP had an effect, it is generally the 421AA homozygous genotype, but not the 421CA heterozygous genotype, that was associated with significantly higher plasma AUC or Cmax of orally administered drugs. Most of the clinical studies used oral administration with only a few that utilized intravenous (IV) administration. We notice that the route of drug administration could also have an effect. For example, the 421CA genotype was shown to be associated with significantly increased plasma AUC or Cmax only after single IV administration of diflomotecan (23).
Table III.
Clinical Studies Investigating Impact of the ABCG2 421C>A SNP on Pharmacokinetics of Drugs Compared to Subjects Carrying the Wild-Type ABCB2 Gene
| Tested drug | Drug dosing | # of subjecta | 421C>A SNPb | Results | Impactd | Ref | |
|---|---|---|---|---|---|---|---|
| AUC Δc (%) | Cmax or C Δc (%) | ||||||
| Diflomotecan | IV single dosing | 5 Caucasian | CA | 199.35 | 168.24 | Yes | (23) |
| Teriflunomide | Oral single dosing | 8 | CA | 52.79 | 28.86 | Yes | (118) |
| Teriflunomide | Oral single dosing | 9 | CA and AA | 82.72 | 29.85 | Yes | (118) |
| 9-aminocamptothecin | Oral multiple dosing | 2 | CA | 258.04 | Yes | (119) | |
| Sulfasalazine | Oral single dosing | 16 Japanese | CA | 92.98 | 71.43 | Yes | (120) |
| Sulfasalazine | Oral single dosing | 9 Japanese | AA | 246.2 | 164.29 | Yes | (120) |
| Sulfasalazine | Oral single dosing | 5 | CA | 137.46 | 70.79 | Yes | (121) |
| Gefitinib | Oral multiple dosing | 7 | CA | 17.58 | Yes | (122) | |
| Sunitinib | Oral retrospective | 8 | CA | 78.57 | Yes | (123) | |
| Sunitinib | Oral retrospective | 1 | AA | 221.43 | Yes | (123) | |
| Imatinib | Oral multiple dosing | 25 Japanese | CA and AA | Yes | (124) | ||
| Sunitinib | Oral retrospective | 1 | AA | 221.43 | Yes | (123) | |
| Rosuvastatin | Oral single dosing | 7 Chinese | CA and AA | 78.22 | 94.12 | Yes | (125) |
| Rosuvastatin | Oral single dosing | 6 Chinese | AA | 57.96 | 41.04 | Yes | (126) |
| Rosuvastatin | Oral multiple dosing | 39 Chinese | AA | 119.5 | Yes | (127) | |
| Rosuvastatin | Oral single dosing | 4 Caucasian | AA | 144.3 | 131.3 | Yes | (128) |
| Fluvastatin | Oral single dosing | 5 Caucasian | AA | 72.55 | 87.4 | Yes | (129) |
| Simvastatin | Oral single dosing | 5 Caucasian | AA | 110.59 | 62.5 | Yes | (129) |
| Atorvastatin | Oral single dosing | 4 Caucasian | AA | 71.54 | 46.36 | Yes | (128) |
| Irinotecan | IV multiple dosing | 23 Asian | CA | 28 | No | (130) | |
| Irinotecan | IV single dosing | 16 Caucasian | CA | −3.37 | No | (131) | |
| Irinotecan | IV multiple dosing | 42 | CA | −0.85 | No | (132) | |
| Irinotecan | IV multiple dosing | 4 | AA | −19.17 | No | (132) | |
| Diflomotecan | Oral multiple dosing | 5 Caucasian | CA | 15.1 | 2.95 | No | (23) |
| Topotecan | Oral single dosing | 2 Caucasian | CA | 30.26 | No | (133) | |
| Topotecan | IV multiple dosing | 2 Caucasian | CA | −0.97 | No | (133) | |
| Erlotinib | Oral multiple dosing | 13 | CA | 19.46 | 12.45 | No | (134) |
| Telatinib | Oral multiple dosing | 6 Caucasian | CA | 17.09 | No | (135) | |
| Docetaxel | IV multiple dosing | 21 Asian | CA | 10.89 | 3.81 | No | (136) |
| Docetaxel | IV multiple dosing | 5 Asian | AA | 5.94 | −20.95 | No | (136) |
| Imatinib | Oral multiple dosing | 13 Japanese | CA | No | (137) | ||
| Imatinib | Oral multiple dosing | 32 Korean | CA | No | (138) | ||
| Imatinib | Oral multiple dosing | 8 Korean | AA | No | (138) | ||
| Danusertib | IV single dosing | 11 | CA | No | (139) | ||
| Fluvastatin | Oral single dosing | 4 Caucasian | CA | −12.41 | −32.28 | No | (129) |
| Fluvastatin | Oral single dosing | 7 Chinese | CA | 9.02 | 16.34 | No | (140) |
| Pravastatin | Oral single dosing | 4 Caucasian | CA | 26.09 | 53.02 | No | (129) |
| Pravastatin | Oral single dosing | 5 Caucasian | AA | −12.32 | 0.35 | No | (129) |
| Simvastatin | Oral single dosing | 4 Caucasian | CA | 60.17 | 30.0 | No | (129) |
| Simvastatin | Oral single dosing | 9 Chinese | CA and AA | 7.07 | 3.42 | No | (141) |
| Atorvastatin | Oral single dosing | 12 Caucasian | CA | 20.38 | 2.55 | No | (128) |
| Rosuvastatin | Oral single dosing | 12 Caucasian | CA | 22.31 | 11.19 | No | (128) |
| Rosuvastatin | Oral single dosing | 15 Chinese | CA | 7.89 | 1.27 | No | (126) |
| Rosuvastatin | Oral multiple dosing | 108 Chinese | CA | No | (127) | ||
| Pitavastatin | Oral single dosing | 7 Japanese | CA | 19.24 | 33.65 | No | (142) |
| Pitavastatin | Oral single dosing | 21 Chinese | CA | 54.02 | 34.38 | No | (143) |
| Pitavastatin | Oral single dosing | 16 Korean | CA | −1.84 | −1.73 | No | (144) |
| Pitavastatin | Oral single dosing | 3 Japanese | AA | −3.58 | 34.94 | No | (142) |
| Pitavastatin | Oral retrospective | 5 Korean | AA | 9.28 | −39.19 | No | (144) |
| Nitrofurantoin | Oral single dosing | 12 Chinese | CA | 9.5 | 9.83 | No | (145) |
| Nitrofurantoin | Oral single dosing | 12 Chinese | AA | 4.98 | 10.06 | No | (145) |
| Sulfasalazine | Oral single dosing | 12 Chinese | CA | −47.66 | −57.61 | No | (146) |
| Sulfasalazine | Oral single dosing | 12 Chinese | AA | 95.33 | 71.07 | No | (146) |
| Telmisartan | Oral single dosing | 24 | CA | 29.37 | No | (147) | |
| Telmisartan | Oral single dosing | 15 Chinese | CA | 2.18 | 15.4 | No | (148) |
| Telmisartan | Oral single dosing | 2 | AA | 8.82 | No | (147) | |
| Telmisartan | Oral single dosing | 3 Chinese | AA | 36.58 | −27.72 | No | (148) |
| Olmesartan | Oral single dosing | 25 Korean | CA | 3.49 | 7.02 | No | (149) |
| Olmesartan | Oral single dosing | 3 Korean | AA | 13.57 | 21.49 | No | (149) |
| Tacrolimus | Oral multiple dosing | 17 | CA and AA | 37.12 | No | (150) | |
| Lamivudine | Oral single dosing | 6 Korean | AA | 5.45 | 1.36 | No | (151) |
aThese are the numbers of subjects carrying the indicated ABCG2 SNP. The numbers of subjects carrying wild-type ABCG2 gene vary from studies to studies, but are usually larger than the numbers of subjects carrying the indicted ABCG2 SNP
b“CA” or “AA” means that subjects carried the 421C/A heterozygous genotype or the 421A/A homozygous genotype, respectively. “CA and AA” means that subjects were a group of individuals combining both the heterozygous and homozygous genotypes
cΔ (%) indicates percentage changes in AUC or Cmax. The AUC data include percentage changes in AUC(0-t) or AUC(0-∞)
dImpact (Yes) indicates that there are statistically significant changes in at least one of the PK parameters: plasma AUC, CL, plasma Cmax, and plasma Cmin. Impact (No) indicates that there are no statistically significant changes in any of the PK parameters. For some drugs, PK changes in AUC or Cmax were not reported and therefore are not shown in this table
We see contradictory results from different studies for the same drugs. The typical example of such drugs is sulfasalazine. Sulfasalazine is a BCRP substrate and was suggested to be used as an in vivo BCRP probe based on the finding that the AUC of oral sulfasalazine in subjects carrying the 421CA genotype was 2.5-times greater than that in subjects carrying the 421CC genotype (121). The observation was initially confirmed by a second study with Japanese subjects showing that the AUC of oral sulfasalazine in subjects carrying the 421CA or 421AA genotype was 1.9- or 3.5-times greater, respectively, than that in subjects carrying the 421CC genotype (120). However, these findings were not reproduced in a third study with Chinese subjects (146). Reasons for such contradictory data are not known, but might be related to the relatively small sample size of these studies (usually <20 subjects for a specific genotype). On the other hand, three different studies have consistently shown that pitavastatin PK is not associated with either the 421CA or the 421AA genotype (142–144). Changes in rosuvastatin PK have been consistently shown to be associated with the 421AA, but not the 421CA genotype (125–128). The other common ABCG2 SNP, 34G>A, does not significantly affect drug PK.
Due to its importance in drug disposition, BCRP has been recognized by the FDA to be one of the key drug transporters involved in clinically relevant drug disposition and drug-drug interactions (DDIs) (152). As discussed above, the co-administration of oral GF120918 and topotecan increased the oral bioavailability of topotecan (117). More recently, several BCRP substrate drugs were shown to significantly interact with other drugs that are BCRP inhibitors in humans, implying that BCRP may play a crucial role in such DDIs. These DDIs include the interactions between atorvastatin and tipranavir/ritonavir (153), rosuvastatin and tipranavir/ritonavir (153), rosuvastatin and atazanavir/ritonavir (154), rosuvastatin and lopinavir/ritonavir (155), rosuvastatin and cyclosporine (156), rosuvastatin and eltrombopag (157,158), rosuvastatin and GSK1292263 (159), simvastatin and GSK1292263 (159), sulfasalazine and curcumin (160), and methotrexate and the proton pump inhibitor omeprazole, lansoprazole, or pantoprazole (161,162). Such DDIs all resulted in at least 20% significant increase in plasma AUC, Cmax, and/or clearance of the BCRP substrate drugs particularly rosuvastatin. Changes in AUC and/or Cmax of these DDIs are summarized in Table IV. It should be pointed out that the above DDIs could also be caused by inhibiting OATP-mediated uptake of drugs into hepatocytes, thus increasing plasma AUC and/or Cmax (see below). We also note that, except for methotrexate, the above DDIs were observed almost exclusively with oral administration of BCRP substrates and inhibitors. It is not clear whether the observed DDIs are caused by inhibiting BCRP in the small intestine or in the liver or both. For the DDIs involving statins, inhibiting BCRP in the small intestine leading to greater oral absorption seems to be more likely. This is because, inhibiting BCRP in the liver may increase accumulation in hepatocytes, but does not necessarily affect plasma AUC or Cmax for the stains which have relatively low membrane permeability and require transporters to cross the cell membrane of hepatocytes. This concept has been illustrated by PBPK modeling using scaling factors obtained by comparing in vitro and in vivo parameters of pravastatin in rats for hepatic uptake and canalicular efflux (163), and is consistent with the fact that almost all of the clinical studies that show impact of the ABCG2 421C>A SNP on drug PK were carried out with oral drug administration (Table III). More studies are needed to elucidate the exact mechanisms of the DDIs.
Table IV.
Clinical Drug-Drug Interactions Potentially Involving BCRP
| Affected drug | Interacting compound | Drug dosing | AUC Δ (%) | Cmax Δ (%) | Ref |
|---|---|---|---|---|---|
| Rosuvastatin | Atazanavir/Ritonavir | Oral | 213 | 600 | (154) |
| Rosuvastatin | Cyclosporine | Oral | 610 | 960 | (156) |
| Rosuvastatin | Lopinavir/Ritonavir | Oral | 110 | 370 | (155) |
| Rosuvastatin | Tipranavir/Ritonavir | Oral | 37 | 123 | (153) |
| Atorvastatin | Tipranavir/Ritonavir | Oral | 836 | 761 | (153) |
| Rosuvastatin | Eltrombopag | Oral | 55 | 103 | (157) |
| Rosuvastatin | GSK1292263 | Oral | 39 | (159) | |
| Simvastatin | GSK1292263 | Oral | 34 | (159) | |
| Sulfasalazine | Curcumin | Oral | 220 | 180 | (160) |
Δ (%) indicates percentage changes in AUC or Cmax. Studies showing interactions of IV methotrexate with proton pump inhibitors did not report changes in AUC or Cmax, and therefore are not included in this table
At present, there is not a reliable in vivo clinical probe substrate for BCRP. Since many of the statins examined in the DDI studies are also OATP substrates and the inhibitors used can also inhibit OATPs, the exact contribution of BCRP to the DDIs cannot be readily quantified. Because rosuvastatin is the most extensively analyzed drug in numerous clinical studies which consistently pinpoint a role of BCRP in disposition of the drug, it may be an appropriate clinical probe for BCRP. Rosuvastatin is minimally metabolized and is not a P-gp substrate (164). However, since it is also a substrate of OATP1B1, OATP1B3, and OATP2B1 (39), it could be suitable for assessing BCRP activity in organs with low OATP expression such as the small intestine and the blood–brain barrier. Indeed, the strong interaction between oral eltrombopag and rosuvastatin in human subjects has been suggested to be caused by complete inhibition of BCRP in the small intestine by eltrombopag (158). Sulfasalazine has previously been suggested to be used as an in vivo probe for assessing the role of BCRP in oral drug bioavailability. This requires further validation because of the report of contradictory results. Another issue for evaluation and reliable prediction of in vivo BCRP activity is how to accurately determine the absolute amount of BCRP expressed in tissues and cells and the intracellular drug concentrations directly exposed to BCRP in tissues and cells. For the former, absolute quantification of BCRP by liquid chromatography mass spectrometry may provide a solution (165). For the latter, there has not been an ideal solution yet.
Similarly, although a large number of BCRP inhibitors have been discovered, a highly selective and potent BCRP inhibitor suitable for clinical studies that specifically target BCRP has yet to be established. Most of the drugs that have been shown to interact with BCRP substrate drugs in clinical studies are also inhibitors of other transporters. GF120918 (a dual P-gp/BCRP inhibitor) was used in one clinical study to evaluate the role of BCRP in limiting oral absorption of topotecan (117). This is because topotecan is possibly a good BCRP substrate, but a relatively poor P-gp substrate. However, since GF120918 is not an approved drug, it generally cannot be used in humans.
PHYSIOLOGICAL AND PATHOPHYSIOLOGICAL FUNCTIONS
Due to its wide tissue and cellular distribution, BCRP is believed to have important physiological and pathophysiological functions in tissue and cellular protection and in mediating homeostasis of physiological substrates. Jonker et al. (166) fed Bcrp1-knockout and wild-type mice a particular diet enriched in alfalfa-derived chlorophyll and observed phototoxicity in Bcrp1-knockout mice, but not in wild-type mice. It turned out that Bcrp1-knockout mice had much increased absorption and cellular accumulation of the chlorophyll degradation product, pheophorbide A, resulting in severe phototoxic lesions on light-exposed skin (166). Bcrp1 is also expressed on the plasma membrane of mature erythrocytes and was shown to reduce cellular protoporphyrin IX levels (167). Protoporphyrin IX is an important precursor to biologically essential prosthetic groups such as heme, cytochrome c, and chlorophylls. Because elevated cellular accumulation of heme and protoporphyrin IX is associated with formation of membrane lipid-damaging reactive oxygen species, it is speculated that BCRP plays an important role in protecting cells from oxidative damage by decreasing cellular accumulation of porphryins (167).
Zhou et al. (57) showed that the “side-population” (SP) cells in mouse blood that are enriched for stem cells expressed the highest level of Bcrp1 mRNA compared with other cell populations. This is the first demonstration that Bcrp1 is highly expressed in hematopoietic stem cells. Subsequently, BCRP expression was observed in stem cells from a variety of human tissues including blood (168), pancreas islets (169), and liver (170). Thus, BCRP is considered a stem cell marker. It was natural to speculate that BCRP provides cellular protection for stem cells. Indeed, Krishnamurthy et al. (171) have demonstrated that BCRP protects hematopoietic stem cells under hypoxic conditions by preventing the accumulation of heme that causes mitochondrial death and that BCRP expression is upregulated in stem cells under hypoxic conditions. BCRP is highly expressed in cancer stem cells. Cancer stem cells are inherently present in the tumor cell populations. Due to high expression of BCRP (and possibly other ABC transporters such as P-gp as well), cancer stem cells are resistant to chemotherapeutics. After chemotherapy, these cells survive and ultimately differentiate to mature tumor cells (13). This is a new theory about the development of multidrug resistance in cancers which will possibly impact the practice of chemotherapy by selectively targeting cancer stem cells.
BCRP is most abundantly expressed in the placenta among normal human tissues and has been proposed to protect the placental trophoblasts from oxidative damage (172). Intrauterine growth restriction (IUGR) is a condition that is known to be associated with excessive oxidative stress (173). BCRP was found to have significantly reduced expression in the placenta from IUGR pregnancies compared to normal pregnancies, suggesting that the capacity of BCRP in protecting placental cells is possibly diminished in subjects with IUGR (172).
BCRP has recently been implicated for the development of gout (174). The common ABCG2 421C>A SNP has been shown to be significantly associated with elevated serum uric levels and the onset of gout. In vitro transport studies confirmed that uric acid is a BCRP substrate, and cells expressing the Q141K variant resulting from the ABCG2 421C>A SNP had lower uric acid efflux activity than cells expressing wild-type BCRP. Subjects carrying the ABCG2 421C>A SNP are likely at an increased risk for developing gout due to reduced activity of BCRP for renal elimination of uric acid.
BCRP has also been implicated in the development of Alzheimer’s disease (AD). AD is possibly caused by abnormal accumulation of amyloid-beta (Abeta) peptides in the brain, leading to neurotoxicity. It has been shown that Abeta peptides are likely BCRP substrate, and that brain accumulation of Abeta1–40 in Bcrp1−/− mice is much higher than that in wild-type mice (175). Furthermore, BCRP expression in AD brain with cerebral amyloid angiopathy is induced (175). BCRP may therefore play a role in protecting the brain from developing the AD.
Lastly, it has recently been shown that ABCG2 null alleles define a new blood type, the Jr(a-) blood type, and red blood cells from the individuals with the Jr(a-) blood type do not express BCRP (176). However, the Jr(a-) individuals with ABCG2 null alleles appear phenotypically normal (176). The role of BCRP in determining the Jr(a-) blood type and pharmacological implications of this observation are currently not known. It would be important to know if and how drug disposition is altered in this unique natural BCRP knockout population.
REGULATION OF BCRP EXPRESSION
BCRP expression can be regulated at the transcriptional level. In humans, the promoter region of the ABCG2 gene is designated E1A and E1B/C. The predominant BCRP promoter is E1B/C which was initially characterized by Bailey-Dell et al. (177). This promoter is TATA-less, contains several SP1 sites, and is downstream of a putative CpG island (177). To date, the cis regulatory elements identified in the BCRP promoter include an estrogen response element (ERE), a progesterone response element (PRE), a hypoxia response element (HRE), an antioxidant response element (ARE), an aryl hydrocarbon response element (AhRE), and the active nuclear factor kB subunit (NFkB) response element (2). Thus, the ABCG2 gene is upregulated under hypoxic conditions via the hypoxia-inducible factor 1α (HIF-1α) (171), by estradiol through estrogen receptor α (ERα) (178), by progesterone via progesterone receptor B (PRB) (179), and by aryl hydrocarbon receptor agonists through the aryl hydrocarbon receptor (AhR) (180). BCRP expression has also been shown to be induced via the peroxisome proliferators-activated receptor gamma (PPARγ) (181) or downregulated by dexamethasone possibly via glucocorticoid receptor (GR) (182). Nevertheless, regulation of the ABCG2 gene and/or BCRP protein expression seems quite complicated. There are controversial reports particularly for regulation of the ABCG2 gene and/or BCRP protein expression by steroid hormones. There are studies showing upregulation of the ABCG2 gene and induction of BCRP protein by estradiol (178,183,184); however, other studies showed downregulation of BCRP by estradiol possibly via posttranscriptional regulation (185–187). Likewise, we reported induction of BCRP by progesterone in human placental BeWo cells (179,186), but others showed downregulation of BCRP by progesterone in human breast cancer cells (188). Reasons of such contradicting data are not known, but may be related to cell- or organ-specific regulation (e.g., organ-specific promoters) or whether transcriptional or posttranscriptional regulation plays a predominant role in experimental systems used in these studies or if there are cooperative interactions between hormones and other regulatory factors. Binding of ER has been shown to enhance transcriptional regulation of the ABCG2 gene by cytokines through p65 or NFkB (189).
Epigenetic regulation of the ABCG2 gene has also been reported. In drug-resistant cancer cell lines, elevated BCRP levels were reported to be associated with hypomethylation or unmethylation of the CpG island (190,191) and with histone hyperacetylation of the ABCG2 promoter (191). BCRP expression can be downregulated by microRNAs by binding to the 3′ UTR of the ABCG2 gene and negative modulation of transcript stability and protein translation (192). Several such microRNAs including hsa-miR-519c, hsa-miR520h, and hsa-miR328 have been identified (193–195).
Posttranslational regulation can affect translocation and expression of BCRP on cell surface. As stated earlier, estradiol was shown to downregulate BCRP expression in ERα-positive cancer cells by decreasing BCRP protein synthesis and maturation (185) or in brain capillaries through a nongenomic pathway (187). Pim-1 kinase phosphorylates BCRP and promotes its dimerization and plasma membrane trafficking (196). BCRP protein expression can also be decreased by long-term exposure (>24 h) of cells to certain compounds (197). For a more extensive and comprehensive review on regulation of BCRP, please refer to the excellent review by Natarajan et al. (2).
CONCLUSION
In the past several years, we have seen significant progress in understanding the role of BCRP in drug transport. Our knowledge about drugs and xenobiotics as BCRP substrates and/or inhibitors has increased dramatically. However, the mechanism by which BCRP acts to transport drugs or xenobiotics and is inhibited is still poorly understood at the molecular level. This awaits further biochemical, biophysical, and computational studies on BCRP, including mutational analyses to enhance the scope of our understanding of amino acids involved in drug interaction and transport selectivity, determination of high-resolution 3D structures, identification of drug binding sites, and computational prediction of BCRP substrates and inhibitors. Such studies will provide the molecular basis for developing new ways to circumvent drug resistance in cancers as well as predict and modulate drug disposition such as increasing brain drug penetration.
There is mounting evidence now to support the notion that BCRP plays an important role in drug disposition. Hence, to predict the impact of BCRP on drug pharmacokinetics and drug-drug interactions in humans, it is essential to understand how BCRP expression is regulated by xenobiotics or physiological and pathological conditions, and how to extrapolate BCRP activity from in vitro or animal data to in vivo data in humans. In this regard, it is critical to identify an appropriate in vivo clinical probe substrate and inhibitor for BCRP and develop methods to accurately quantify the absolute amount of BCRP expressed in tissues and cells as well as the intracellular concentrations of drugs and xenobiotics exposed to BCRP in tissues and cells.
Owing to the importance of BCRP in drug disposition and in developing gout, caution should be taken when a BCRP substrate drug with narrow therapeutic window or a potent BCRP inhibitor is to be administered to patients carrying the ABCG2 421C>A SNP so that undesired toxicities or side effects of medications may be avoided.
Acknowledgments
This work is supported in part by the NIH Grant DA032507. We gratefully thank Dr. Isabelle Ragueneau-Majlessi and Sophie Argon for search of the UW Metabolism and Transport Drug Interaction Database (DIDB) and the UW ePKGene Database for the impact of BCRP and ABCG2 SNPs on drug PK. We greatly acknowledge Dr. Zsolt Bikadi for preparing Fig. 2. Due to a limitation in the number of references imposed by the journal, many excellent studies cannot be cited in this review article. We appreciate the contributions of all of the authors to this important field of research.
References
- 1.Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005;7:E118–E133. doi: 10.1208/aapsj070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol. 2012;83:1084–1103. doi: 10.1016/j.bcp.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benderra Z, Faussat AM, Sayada L, Perrot JY, Tang R, Chaoui D, et al. MRP3, BCRP, and P-glycoprotein activities are prognostic factors in adult acute myeloid leukemia. Clin Cancer Res. 2005;11:7764–7772. doi: 10.1158/1078-0432.CCR-04-1895. [DOI] [PubMed] [Google Scholar]
- 4.Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, et al. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab Dispos. 2010;38:1371–1380. doi: 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–935. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 6.de Lima LT, Vivona D, Bueno CT, Hirata RD, Hirata MH, Luchessi AD, et al. Reduced ABCG2 and increased SLC22A1 mRNA expression are associated with imatinib response in chronic myeloid leukemia. Med Oncol. 2014;31:851. doi: 10.1007/s12032-014-0851-5. [DOI] [PubMed] [Google Scholar]
- 7.Diestra JE, Scheffer GL, Catala I, Maliepaard M, Schellens JH, Scheper RJ, et al. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol. 2002;198:213–219. doi: 10.1002/path.1203. [DOI] [PubMed] [Google Scholar]
- 8.Burger H, Foekens JA, Look MP, Meijer-van Gelder ME, Klijn JG, Wiemer EA, et al. RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: correlation with chemotherapeutic response. Clin Cancer Res. 2003;9:827–836. [PubMed] [Google Scholar]
- 9.Yuan JH, Cheng JQ, Jiang LY, Ji WD, Guo LF, Liu JJ, et al. Breast cancer resistance protein expression and 5-fluorouracil resistance. Biomed Environ Sci. 2008;21:290–295. doi: 10.1016/S0895-3988(08)60044-6. [DOI] [PubMed] [Google Scholar]
- 10.Yoh K, Ishii G, Yokose T, Minegishi Y, Tsuta K, Goto K, et al. Breast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer. Clin Cancer Res. 2004;10:1691–1697. doi: 10.1158/1078-0432.CCR-0937-3. [DOI] [PubMed] [Google Scholar]
- 11.Ota S, Ishii G, Goto K, Kubota K, Kim YH, Kojika M, et al. Immunohistochemical expression of BCRP and ERCC1 in biopsy specimen predicts survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Lung Cancer. 2009;64:98–104. doi: 10.1016/j.lungcan.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Lee SH, Kim H, Hwang JH, Lee HS, Cho JY, Yoon YS, et al. Breast cancer resistance protein expression is associated with early recurrence and decreased survival in resectable pancreatic cancer patients. Pathol Int. 2012;62:167–175. doi: 10.1111/j.1440-1827.2011.02772.x. [DOI] [PubMed] [Google Scholar]
- 13.Stacy AE, Jansson PJ, Richardson DR. Molecular pharmacology of ABCG2 and its role in chemoresistance. Mol Pharmacol. 2013;84:655–669. doi: 10.1124/mol.113.088609. [DOI] [PubMed] [Google Scholar]
- 14.Robey RW, Steadman K, Polgar O, Bates SE. ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther. 2005;4:187–194. doi: 10.4161/cbt.4.2.1440. [DOI] [PubMed] [Google Scholar]
- 15.Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, et al. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2) Biochim Biophys Acta. 2001;1512:171–182. doi: 10.1016/S0005-2736(01)00308-X. [DOI] [PubMed] [Google Scholar]
- 16.Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–1978. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Litman T, Brangi M, Hudson E, Fetsch P, Abati A, Ross DD, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113(Pt 11):2011–2021. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 18.Hazlehurst LA, Foley NE, Gleason-Guzman MC, Hacker MP, Cress AE, Greenberger LW, et al. Multiple mechanisms confer drug resistance to mitoxantrone in the human 8226 myeloma cell line. Cancer Res. 1999;59:1021–1028. [PubMed] [Google Scholar]
- 19.Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000;60:47–50. [PubMed] [Google Scholar]
- 20.Nakatomi K, Yoshikawa M, Oka M, Ikegami Y, Hayasaka S, Sano K, et al. Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commun. 2001;288:827–832. doi: 10.1006/bbrc.2001.5850. [DOI] [PubMed] [Google Scholar]
- 21.Rabindran SK, He H, Singh M, Brown E, Collins KI, Annable T, et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998;58:5850–5858. [PubMed] [Google Scholar]
- 22.Maliepaard M, van Gastelen MA, Tohgo A, Hausheer FH, van Waardenburg RC, de Jong LA, et al. Circumvention of breast cancer resistance protein (BCRP)-mediated resistance to camptothecins in vitro using non-substrate drugs or the BCRP inhibitor GF120918. Clin Cancer Res. 2001;7:935–941. [PubMed] [Google Scholar]
- 23.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther. 2004;76:38–44. doi: 10.1016/j.clpt.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y, et al. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63:4048–4054. [PubMed] [Google Scholar]
- 25.Volk EL, Farley KM, Wu Y, Li F, Robey RW, Schneider E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002;62:5035–5040. [PubMed] [Google Scholar]
- 26.Volk EL, Schneider E. Wild-type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res. 2003;63:5538–5543. [PubMed] [Google Scholar]
- 27.Wang X, Furukawa T, Nitanda T, Okamoto M, Sugimoto Y, Akiyama S, et al. Breast cancer resistance protein (BCRP/ABCG2) induces cellular resistance to HIV-1 nucleoside reverse transcriptase inhibitors. Mol Pharmacol. 2003;63:65–72. doi: 10.1124/mol.63.1.65. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Nitanda T, Shi M, Okamoto M, Furukawa T, Sugimoto Y, et al. Induction of cellular resistance to nucleoside reverse transcriptase inhibitors by the wild-type breast cancer resistance protein. Biochem Pharmacol. 2004;68:1363–1370. doi: 10.1016/j.bcp.2004.05.052. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa R, Hara Y, Arakawa H, Nishimura S, Komatani H. ABCG2 confers resistance to indolocarbazole compounds by ATP-dependent transport. Biochem Biophys Res Commun. 2002;299:669–675. doi: 10.1016/S0006-291X(02)02712-2. [DOI] [PubMed] [Google Scholar]
- 30.Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, et al. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001;7:145–152. [PubMed] [Google Scholar]
- 31.Erlichman C, Boerner SA, Hallgren CG, Spieker R, Wang XY, James CD, et al. The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res. 2001;61:739–748. [PubMed] [Google Scholar]
- 32.Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- 33.Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- 34.Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21:1267–1275. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- 35.Zhou L, Naraharisetti SB, Wang H, Unadkat JD, Hebert MF, Mao Q. The breast cancer resistance protein (Bcrp1/Abcg2) limits fetal distribution of glyburide in the pregnant mouse: an Obstetric-Fetal Pharmacology Research Unit Network and University of Washington Specialized Center of Research Study. Mol Pharmacol. 2008;73:949–959. doi: 10.1124/mol.107.041616. [DOI] [PubMed] [Google Scholar]
- 36.Pavek P, Merino G, Wagenaar E, Bolscher E, Novotna M, Jonker JW, et al. Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J Pharmacol Exp Ther. 2005;312:144–152. doi: 10.1124/jpet.104.073916. [DOI] [PubMed] [Google Scholar]
- 37.van der Heijden J, de Jong MC, Dijkmans BA, Lems WF, Oerlemans R, Kathmann I, et al. Development of sulfasalazine resistance in human T cells induces expression of the multidrug resistance transporter ABCG2 (BCRP) and augmented production of TNFalpha. Ann Rheum Dis. 2004;63:138–143. doi: 10.1136/ard.2002.005249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merino G, Jonker JW, Wagenaar E, van Herwaarden AE, Schinkel AH. The breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion, and milk secretion of the antibiotic nitrofurantoin. Mol Pharmacol. 2005;67:1758–1764. doi: 10.1124/mol.104.010439. [DOI] [PubMed] [Google Scholar]
- 39.Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008;36:2014–2023. doi: 10.1124/dmd.108.021410. [DOI] [PubMed] [Google Scholar]
- 40.Breedveld P, Zelcer N, Pluim D, Sonmezer O, Tibben MM, Beijnen JH, et al. Mechanism of the pharmacokinetic interaction between methotrexate and benzimidazoles: potential role for breast cancer resistance protein in clinical drug-drug interactions. Cancer Res. 2004;64:5804–5811. doi: 10.1158/0008-5472.CAN-03-4062. [DOI] [PubMed] [Google Scholar]
- 41.Yoshikawa M, Ikegami Y, Hayasaka S, Ishii K, Ito A, Sano K, et al. Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells. Int J Cancer. 2004;110:921–927. doi: 10.1002/ijc.20216. [DOI] [PubMed] [Google Scholar]
- 42.Hazai E, Hazai I, Ragueneau-Majlessi I, Chung SP, Bikadi Z, Mao Q. Predicting substrates of the human breast cancer resistance protein using a support vector machine method. BMC Bioinforma. 2013;14:130. doi: 10.1186/1471-2105-14-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ozvegy-Laczka C, Hegedus T, Varady G, Ujhelly O, Schuetz JD, Varadi A, et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol. 2004;65:1485–1495. doi: 10.1124/mol.65.6.1485. [DOI] [PubMed] [Google Scholar]
- 44.Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–2337. doi: 10.1158/0008-5472.CAN-03-3344. [DOI] [PubMed] [Google Scholar]
- 45.Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–11020. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 46.Hegedus C, Ozvegy-Laczka C, Apati A, Magocsi M, Nemet K, Orfi L, et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br J Pharmacol. 2009;158:1153–1164. doi: 10.1111/j.1476-5381.2009.00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gupta A, Zhang Y, Unadkat JD, Mao Q. HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2) J Pharmacol Exp Ther. 2004;310:334–341. doi: 10.1124/jpet.104.065342. [DOI] [PubMed] [Google Scholar]
- 49.Weiss J, Rose J, Storch CH, Ketabi-Kiyanvash N, Sauer A, Haefeli WE, et al. Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J Antimicrob Chemother. 2007;59:238–245. doi: 10.1093/jac/dkl474. [DOI] [PubMed] [Google Scholar]
- 50.Chu X, Cai X, Cui D, Tang C, Ghosal A, Chan G, et al. In vitro assessment of drug-drug interaction potential of boceprevir associated with drug metabolizing enzymes and transporters. Drug Metab Dispos. 2013;41:668–681. doi: 10.1124/dmd.112.049668. [DOI] [PubMed] [Google Scholar]
- 51.Fujita Y, Noguchi K, Suzuki T, Katayama K, Sugimoto Y. Biochemical interaction of anti-HCV telaprevir with the ABC transporters P-glycoprotein and breast cancer resistance protein. BMC Res Notes. 2013;6:445. doi: 10.1186/1756-0500-6-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Gupta A, Wang H, Zhou L, Vethanayagam RR, Unadkat JD, et al. BCRP transports dipyridamole and is inhibited by calcium channel blockers. Pharm Res. 2005;22:2023–2034. doi: 10.1007/s11095-005-8384-4. [DOI] [PubMed] [Google Scholar]
- 53.Gupta A, Unadkat JD, Mao Q. Interactions of azole antifungal agents with the human breast cancer resistance protein (BCRP) J Pharm Sci. 2007;96:3226–3235. doi: 10.1002/jps.20963. [DOI] [PubMed] [Google Scholar]
- 54.Yang CH, Chen YC, Kuo ML. Novobiocin sensitizes BCRP/MXR/ABCP overexpressing topotecan-resistant human breast carcinoma cells to topotecan and mitoxantrone. Anticancer Res. 2003;23:2519–2523. [PubMed] [Google Scholar]
- 55.Shiozawa K, Oka M, Soda H, Yoshikawa M, Ikegami Y, Tsurutani J, et al. Reversal of breast cancer resistance protein (BCRP/ABCG2)-mediated drug resistance by novobiocin, a coumermycin antibiotic. Int J Cancer. 2004;108:146–151. doi: 10.1002/ijc.11528. [DOI] [PubMed] [Google Scholar]
- 56.Sugimoto Y, Tsukahara S, Imai Y, Ueda K, Tsuruo T. Reversal of breast cancer resistance protein-mediated drug resistance by estrogen antagonists and agonists. Mol Cancer Ther. 2003;2:105–112. [PubMed] [Google Scholar]
- 57.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 58.de Bruin M, Miyake K, Litman T, Robey R, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999;146:117–126. doi: 10.1016/S0304-3835(99)00182-2. [DOI] [PubMed] [Google Scholar]
- 59.Wu CP, Hsiao SH, Sim HM, Luo SY, Tuo WC, Cheng HW, et al. Human ABCB1 (P-glycoprotein) and ABCG2 mediate resistance to BI 2536, a potent and selective inhibitor of Polo-like kinase 1. Biochem Pharmacol. 2013;86:904–913. doi: 10.1016/j.bcp.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Loevezijn A, Allen JD, Schinkel AH, Koomen GJ. Inhibition of BCRP-mediated drug efflux by fumitremorgin-type indolyl diketopiperazines. Bioorg Med Chem Lett. 2001;11:29–32. doi: 10.1016/S0960-894X(00)00588-6. [DOI] [PubMed] [Google Scholar]
- 61.Minderman H, O’Loughlin KL, Pendyala L, Baer MR. VX-710 (biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res. 2004;10:1826–1834. doi: 10.1158/1078-0432.CCR-0914-3. [DOI] [PubMed] [Google Scholar]
- 62.Woehlecke H, Osada H, Herrmann A, Lage H. Reversal of breast cancer resistance protein-mediated drug resistance by tryprostatin A. Int J Cancer. 2003;107:721–728. doi: 10.1002/ijc.11444. [DOI] [PubMed] [Google Scholar]
- 63.Zhang S, Yang X, Morris ME. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol Pharmacol. 2004;65:1208–1216. doi: 10.1124/mol.65.5.1208. [DOI] [PubMed] [Google Scholar]
- 64.Kuhnle M, Egger M, Muller C, Mahringer A, Bernhardt G, Fricker G, et al. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the p-glycoprotein (ABCB1) modulator tariquidar. J Med Chem. 2009;52:1190–1197. doi: 10.1021/jm8013822. [DOI] [PubMed] [Google Scholar]
- 65.Valdameri G, Genoux-Bastide E, Peres B, Gauthier C, Guitton J, Terreux R, et al. Substituted chromones as highly potent nontoxic inhibitors, specific for the breast cancer resistance protein. J Med Chem. 2012;55:966–970. doi: 10.1021/jm201404w. [DOI] [PubMed] [Google Scholar]
- 66.Giri N, Agarwal S, Shaik N, Pan G, Chen Y, Elmquist WF. Substrate-dependent breast cancer resistance protein (Bcrp1/Abcg2)-mediated interactions: consideration of multiple binding sites in in vitro assay design. Drug Metab Dispos. 2009;37:560–570. doi: 10.1124/dmd.108.022046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pan Y, Chothe PP, Swaan PW. Identification of novel breast cancer resistance protein (BCRP) inhibitors by virtual screening. Mol Pharm. 2013;10:1236–1248. doi: 10.1021/mp300547h. [DOI] [PubMed] [Google Scholar]
- 68.Bikadi Z, Hazai I, Malik D, Jemnitz K, Veres Z, Hari P, et al. Predicting P-glycoprotein-mediated drug transport based on support vector machine and three-dimensional crystal structure of P-glycoprotein. PLoS One. 2011;6:e25815. doi: 10.1371/journal.pone.0025815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishikawa T, Hirano H, Saito H, Sano K, Ikegami Y, Yamaotsu N, et al. Quantitative structure-activity relationship (QSAR) analysis to predict drug-drug interactions of ABC transporter ABCG2. Mini-Rev Med Chem. 2012;12:505–514. doi: 10.2174/138955712800493825. [DOI] [PubMed] [Google Scholar]
- 70.Gandhi YA, Morris ME. Structure-activity relationships and quantitative structure-activity relationships for breast cancer resistance protein (ABCG2) AAPS J. 2009;11:541–552. doi: 10.1208/s12248-009-9132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nicolle E, Boumendjel A, Macalou S, Genoux E, Ahmed-Belkacem A, Carrupt PA, et al. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliv Rev. 2009;61:34–46. doi: 10.1016/j.addr.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 72.Zhang S, Yang X, Coburn RA, Morris ME. Structure activity relationships and quantitative structure activity relationships for the flavonoid-mediated inhibition of breast cancer resistance protein. Biochem Pharmacol. 2005;70:627–639. doi: 10.1016/j.bcp.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 73.Pick A, Muller H, Wiese M. Structure-activity relationships of new inhibitors of breast cancer resistance protein (ABCG2) Bioorg Med Chem. 2008;16:8224–8236. doi: 10.1016/j.bmc.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 74.Ni Z, Bikadi Z, Rosenberg MF, Mao Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2) Curr Drug Metab. 2010;11:603–617. doi: 10.2174/138920010792927325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marks DS, Hopf TA, Sander C. Protein structure prediction from sequence variation. Nat Biotechnol. 2012;30:1072–1080. doi: 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang H, Lee EW, Cai X, Ni Z, Zhou L, Mao Q. Membrane topology of the human breast cancer resistance protein (BCRP/ABCG2) determined by epitope insertion and immunofluorescence. Biochemistry. 2008;47:13778–13787. doi: 10.1021/bi801644v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mohrmann K, van Eijndhoven MA, Schinkel AH, Schellens JH. Absence of N-linked glycosylation does not affect plasma membrane localization of breast cancer resistance protein (BCRP/ABCG2) Cancer Chemother Pharmacol. 2005;56:344–350. doi: 10.1007/s00280-005-1004-5. [DOI] [PubMed] [Google Scholar]
- 78.Popov M, Tam LY, Li J, Reithmeier RA. Mapping the ends of transmembrane segments in a polytopic membrane protein. Scanning N-glycosylation mutagenesis of extracytosolic loops in the anion exchanger, band 3. J Biol Chem. 1997;272:18325–18332. doi: 10.1074/jbc.272.29.18325. [DOI] [PubMed] [Google Scholar]
- 79.Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem. 2004;279:19781–19789. doi: 10.1074/jbc.M310785200. [DOI] [PubMed] [Google Scholar]
- 80.McDevitt CA, Collins RF, Conway M, Modok S, Storm J, Kerr ID, et al. Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure. 2006;14:1623–1632. doi: 10.1016/j.str.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 81.Rosenberg MF, Bikadi Z, Chan J, Liu X, Ni Z, Cai X, et al. The human breast cancer resistance protein (BCRP/ABCG2) shows conformational changes with mitoxantrone. Structure. 2010;18:482–493. doi: 10.1016/j.str.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ni Z, Mark ME, Cai X, Mao Q. Fluorescence resonance energy transfer (FRET) analysis demonstrates dimer/oligomer formation of the human breast cancer resistance protein (BCRP/ABCG2) in intact cells. Int J Biochem Mol Biol. 2010;1:1–15. doi: 10.9735/0976-0482.1.2.1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haider AJ, Briggs D, Self TJ, Chilvers HL, Holliday ND, Kerr ID. Dimerization of ABCG2 analysed by bimolecular fluorescence complementation. PLoS One. 2011;6:e25818. doi: 10.1371/journal.pone.0025818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kage K, Fujita T, Sugimoto Y. Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci. 2005;96:866–872. doi: 10.1111/j.1349-7006.2005.00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mitomo H, Kato R, Ito A, Kasamatsu S, Ikegami Y, Kii I, et al. A functional study on polymorphism of the ATP-binding cassette transporter ABCG2: critical role of arginine-482 in methotrexate transport. Biochem J. 2003;373:767–774. doi: 10.1042/BJ20030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cai X, Bikadi Z, Ni Z, Lee EW, Wang H, Rosenberg MF, et al. Role of basic residues within or near the predicted transmembrane helix 2 of the human breast cancer resistance protein in drug transport. J Pharmacol Exp Ther. 2010;333:670–681. doi: 10.1124/jpet.109.163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. Int J Cancer. 2003;107:757–763. doi: 10.1002/ijc.11484. [DOI] [PubMed] [Google Scholar]
- 88.Clark R, Kerr ID, Callaghan R. Multiple drugbinding sites on the R482G isoform of the ABCG2 transporter. Br J Pharmacol. 2006;149:506–515. doi: 10.1038/sj.bjp.0706904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ejendal KF, Diop NK, Schweiger LC, Hrycyna CA. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006;15:1597–1607. doi: 10.1110/ps.051998406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li L, Sham YY, Bikadi Z, Elmquist WF. pH-dependent transport of pemetrexed by breast cancer resistance protein. Drug Metab Dispos. 2011;39:1478–1485. doi: 10.1124/dmd.111.039370. [DOI] [PubMed] [Google Scholar]
- 91.Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–6639. [PubMed] [Google Scholar]
- 92.Ni Z, Bikadi Z, Cai X, Rosenberg MF, Mao Q. Transmembrane helices 1 and 6 of the human breast cancer resistance protein (BCRP/ABCG2): identification of polar residues important for drug transport. Am J Physiol Cell Physiol. 2010;299:C1100–C1109. doi: 10.1152/ajpcell.00160.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Polgar O, Ierano C, Tamaki A, Stanley B, Ward Y, Xia D, et al. Mutational analysis of threonine 402 adjacent to the GXXXG dimerization motif in transmembrane segment 1 of ABCG2. Biochemistry. 2010;49:2235–2245. doi: 10.1021/bi902085q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ni Z, Bikadi Z, Shuster DL, Zhao C, Rosenberg MF, Mao Q. Identification of proline residues in or near the transmembrane helices of the human breast cancer resistance protein (BCRP/ABCG2) that are important for transport activity and substrate specificity. Biochemistry. 2011;50:8057–8066. doi: 10.1021/bi200573t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kage K, Tsukahara S, Sugiyama T, Asada S, Ishikawa E, Tsuruo T, et al. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int J Cancer. 2002;97:626–630. doi: 10.1002/ijc.10100. [DOI] [PubMed] [Google Scholar]
- 96.Ozvegy C, Varadi A, Sarkadi B. Characterization of drug transport, ATP hydrolysis, and nucleotide trapping by the human ABCG2 multidrug transporter. Modulation of substrate specificity by a point mutation. J Biol Chem. 2002;277:47980–47990. doi: 10.1074/jbc.M207857200. [DOI] [PubMed] [Google Scholar]
- 97.Henriksen U, Gether U, Litman T. Effect of walker A mutation (K86M) on oligomerization and surface targeting of the multidrug resistance transporter ABCG2. J Cell Sci. 2005;118:1417–1426. doi: 10.1242/jcs.01729. [DOI] [PubMed] [Google Scholar]
- 98.Hou YX, Li CZ, Palaniyandi K, Magtibay PM, Homolya L, Sarkadi B, et al. Effects of putative catalytic base mutation E211Q on ABCG2-mediated methotrexate transport. Biochemistry. 2009;48:9122–9131. doi: 10.1021/bi900675v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shukla S, Robey RW, Bates SE, Ambudkar SV. The calcium channel blockers, 1,4-dihydropyridines, are substrates of the multidrug resistance-linked ABC drug transporter, ABCG2. Biochemistry. 2006;45:8940–8951. doi: 10.1021/bi060552f. [DOI] [PubMed] [Google Scholar]
- 100.Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–616. [PubMed] [Google Scholar]
- 101.Vethanayagam RR, Wang H, Gupta A, Zhang Y, Lewis F, Unadkat JD, et al. Functional analysis of the human variants of breast cancer resistance protein: I206L, N590Y, and D620N. Drug Metab Dispos. 2005;33:697–705. doi: 10.1124/dmd.105.003657. [DOI] [PubMed] [Google Scholar]
- 102.Noguchi K, Katayama K, Sugimoto Y. Human ABC transporter ABCG2/BCRP expression in chemoresistance: basic and clinical perspectives for molecular cancer therapeutics. Pharmgenomics Pers Med. 2014;7:53–64. doi: 10.2147/PGPM.S38295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH, et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001;61:3458–3464. [PubMed] [Google Scholar]
- 104.Huls M, Brown CD, Windass AS, Sayer R, van den Heuvel JJ, Heemskerk S, et al. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008;73:220–225. doi: 10.1038/sj.ki.5002645. [DOI] [PubMed] [Google Scholar]
- 105.Cooray HC, Blackmore CG, Maskell L, Barrand MA. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport. 2002;13:2059–2063. doi: 10.1097/00001756-200211150-00014. [DOI] [PubMed] [Google Scholar]
- 106.Asashima T, Hori S, Ohtsuki S, Tachikawa M, Watanabe M, Mukai C, et al. ATP-binding cassette transporter G2 mediates the efflux of phototoxins on the luminal membrane of retinal capillary endothelial cells. Pharm Res. 2006;23:1235–1242. doi: 10.1007/s11095-006-0067-2. [DOI] [PubMed] [Google Scholar]
- 107.Robillard KR, Hoque T, Bendayan R. Expression of ATP-binding cassette membrane transporters in rodent and human Sertoli cells: relevance to the permeability of antiretroviral therapy at the blood-testis barrier. J Pharmacol Exp Ther. 2012;340:96–108. doi: 10.1124/jpet.111.186916. [DOI] [PubMed] [Google Scholar]
- 108.Jablonski MR, Jacob DA, Campos C, Miller DS, Maragakis NJ, Pasinelli P, et al. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol Dis. 2012;47:194–200. doi: 10.1016/j.nbd.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vlaming ML, Lagas JS, Schinkel AH. Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev. 2009;61:14–25. doi: 10.1016/j.addr.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 110.Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336:223–233. doi: 10.1124/jpet.110.175034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethy l]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metab Dispos. 2009;37:439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- 112.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333:788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Y, Wang H, Unadkat JD, Mao Q. Breast cancer resistance protein 1 limits fetal distribution of nitrofurantoin in the pregnant mouse. Drug Metab Dispos. 2007;35:2154–2158. doi: 10.1124/dmd.107.018044. [DOI] [PubMed] [Google Scholar]
- 114.Ni Z, Mao Q. ATP-binding cassette efflux transporters in human placenta. Curr Pharm Biotechnol. 2011;12:674–685. doi: 10.2174/138920111795164057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jonker JW, Merino G, Musters S, van Herwaarden AE, Bolscher E, Wagenaar E, et al. The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat Med. 2005;11:127–129. doi: 10.1038/nm1186. [DOI] [PubMed] [Google Scholar]
- 116.van Herwaarden AE, Wagenaar E, Merino G, Jonker JW, Rosing H, Beijnen JH, et al. Multidrug transporter ABCG2/breast cancer resistance protein secretes riboflavin (vitamin B2) into milk. Mol Cell Biol. 2007;27:1247–1253. doi: 10.1128/MCB.01621-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 118.Kim KA, Joo HJ, Park JY. Effect of ABCG2 genotypes on the pharmacokinetics of A771726, an active metabolite of prodrug leflunomide, and association of A771726 exposure with serum uric acid level. Eur J Clin Pharmacol. 2011;67:129–134. doi: 10.1007/s00228-010-0916-0. [DOI] [PubMed] [Google Scholar]
- 119.Zamboni WC, Ramanathan RK, McLeod HL, Mani S, Potter DM, Strychor S, et al. Disposition of 9-nitrocamptothecin and its 9-aminocamptothecin metabolite in relation to ABC transporter genotypes. Investig New Drugs. 2006;24:393–401. doi: 10.1007/s10637-006-6335-5. [DOI] [PubMed] [Google Scholar]
- 120.Yamasaki Y, Ieiri I, Kusuhara H, Sasaki T, Kimura M, Tabuchi H, et al. Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin Pharmacol Ther. 2008;84:95–103. doi: 10.1038/sj.clpt.6100459. [DOI] [PubMed] [Google Scholar]
- 121.Urquhart BL, Ware JA, Tirona RG, Ho RH, Leake BF, Schwarz UI, et al. Breast cancer resistance protein (ABCG2) and drug disposition: intestinal expression, polymorphisms and sulfasalazine as an in vivo probe. Pharmacogenet Genomics. 2008;18:439–448. doi: 10.1097/FPC.0b013e3282f974dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li J, Cusatis G, Brahmer J, Sparreboom A, Robey RW, Bates SE, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–438. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- 123.Mizuno T, Fukudo M, Terada T, Kamba T, Nakamura E, Ogawa O, et al. Impact of genetic variation in breast cancer resistance protein (BCRP/ABCG2) on sunitinib pharmacokinetics. Drug Metab Pharmacokinet. 2012;27:631–639. doi: 10.2133/dmpk.DMPK-12-RG-026. [DOI] [PubMed] [Google Scholar]
- 124.Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H, et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet. 2010;55:731–737. doi: 10.1038/jhg.2010.98. [DOI] [PubMed] [Google Scholar]
- 125.Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ, et al. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006;373:99–103. doi: 10.1016/j.cca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 126.Zhou Q, Ruan ZR, Yuan H, Xu DH, Zeng S. ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421c>A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Die Pharmazie. 2013;68:129–134. [PubMed] [Google Scholar]
- 127.Lee HK, Hu M, Lui S, Ho CS, Wong CK, Tomlinson B. Effects of polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on plasma concentrations of rosuvastatin and lipid response in Chinese patients. Pharmacogenomics. 2013;14:1283–1294. doi: 10.2217/pgs.13.115. [DOI] [PubMed] [Google Scholar]
- 128.Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86:197–203. doi: 10.1038/clpt.2009.79. [DOI] [PubMed] [Google Scholar]
- 129.Keskitalo JE, Pasanen MK, Neuvonen PJ, Niemi M. Different effects of the ABCG2 c.421C>A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics. 2009;10:1617–1624. doi: 10.2217/pgs.09.85. [DOI] [PubMed] [Google Scholar]
- 130.Jada SR, Lim R, Wong CI, Shu X, Lee SC, Zhou Q, et al. Role of UGT1A1*6, UGT1A1*28 and ABCG2 c.421C>A polymorphisms in irinotecan-induced neutropenia in Asian cancer patients. Cancer Sci. 2007;98:1461–1467. doi: 10.1111/j.1349-7006.2007.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.de Jong FA, Marsh S, Mathijssen RH, King C, Verweij J, Sparreboom A, et al. ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin Cancer Res. 2004;10:5889–5894. doi: 10.1158/1078-0432.CCR-04-0144. [DOI] [PubMed] [Google Scholar]
- 132.Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY, et al. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer. 2007;110:138–147. doi: 10.1002/cncr.22760. [DOI] [PubMed] [Google Scholar]
- 133.Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, et al. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol Ther. 2005;4:650–658. doi: 10.4161/cbt.4.6.1731. [DOI] [PubMed] [Google Scholar]
- 134.Rudin CM, Liu W, Desai A, Karrison T, Jiang X, Janisch L, et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol. 2008;26:1119–1127. doi: 10.1200/JCO.2007.13.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Steeghs N, Gelderblom H, Wessels J, Eskens FA, de Bont N, Nortier JW, et al. Pharmacogenetics of telatinib, a VEGFR-2 and VEGFR-3 tyrosine kinase inhibitor, used in patients with solid tumors. Investig New Drugs. 2011;29:137–143. doi: 10.1007/s10637-009-9347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chew SC, Singh O, Chen X, Ramasamy RD, Kulkarni T, Lee EJ, et al. The effects of CYP3A4, CYP3A5, ABCB1, ABCC2, ABCG2 and SLCO1B3 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of docetaxel in nasopharyngeal carcinoma patients. Cancer Chemother Pharmacol. 2011;67:1471–1478. doi: 10.1007/s00280-011-1625-9. [DOI] [PubMed] [Google Scholar]
- 137.Yamakawa Y, Hamada A, Nakashima R, Yuki M, Hirayama C, Kawaguchi T, et al. Association of genetic polymorphisms in the influx transporter SLCO1B3 and the efflux transporter ABCB1 with imatinib pharmacokinetics in patients with chronic myeloid leukemia. Ther Drug Monit. 2011;33:244–250. doi: 10.1097/FTD.0b013e31820beb02. [DOI] [PubMed] [Google Scholar]
- 138.Seong SJ, Lim M, Sohn SK, Moon JH, Oh SJ, Kim BS, et al. Influence of enzyme and transporter polymorphisms on trough imatinib concentration and clinical response in chronic myeloid leukemia patients. Ann Oncol. 2013;24:756–760. doi: 10.1093/annonc/mds532. [DOI] [PubMed] [Google Scholar]
- 139.Steeghs N, Mathijssen RH, Wessels JA, de Graan AJ, van der Straaten T, Mariani M, et al. Influence of pharmacogenetic variability on the pharmacokinetics and toxicity of the aurora kinase inhibitor danusertib. Investig New Drugs. 2011;29:953–962. doi: 10.1007/s10637-010-9405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhou Q, Ruan ZR, Yuan H, Zeng S. CYP2C9*3(1075A>C), MDR1 G2677T/A and MDR1 C3435T are determinants of inter-subject variability in fluvastatin pharmacokinetics in healthy Chinese volunteers. Arzneimittelforschung. 2012;62:519–524. doi: 10.1055/s-0032-1323696. [DOI] [PubMed] [Google Scholar]
- 141.Zhou Q, Ruan ZR, Jiang B, Yuan H, Zeng S. Simvastatin pharmacokinetics in healthy Chinese subjects and its relations with CYP2C9, CYP3A5, ABCB1, ABCG2 and SLCO1B1 polymorphisms. Die Pharmazie. 2013;68:124–128. [PubMed] [Google Scholar]
- 142.Ieiri I, Suwannakul S, Maeda K, Uchimaru H, Hashimoto K, Kimura M, et al. SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther. 2007;82:541–547. doi: 10.1038/sj.clpt.6100190. [DOI] [PubMed] [Google Scholar]
- 143.Zhou Q, Chen QX, Ruan ZR, Yuan H, Xu HM, Zeng S. CYP2C9*3(1075A>C), ABCB1 and SLCO1B1 genetic polymorphisms and gender are determinants of inter-subject variability in pitavastatin pharmacokinetics. Die Pharmazie. 2013;68:187–194. [PubMed] [Google Scholar]
- 144.Oh ES, Kim CO, Cho SK, Park MS, Chung JY. Impact of ABCC2, ABCG2 and SLCO1B1 polymorphisms on the pharmacokinetics of pitavastatin in humans. Drug Metab Pharmacokinet. 2013;28:196–202. doi: 10.2133/dmpk.DMPK-12-RG-068. [DOI] [PubMed] [Google Scholar]
- 145.Adkison KK, Vaidya SS, Lee DY, Koo SH, Li L, Mehta AA, et al. The ABCG2 C421A polymorphism does not affect oral nitrofurantoin pharmacokinetics in healthy Chinese male subjects. Br J Clin Pharmacol. 2008;66:233–239. doi: 10.1111/j.1365-2125.2008.03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Adkison KK, Vaidya SS, Lee DY, Koo SH, Li L, Mehta AA, et al. Oral sulfasalazine as a clinical BCRP probe substrate: pharmacokinetic effects of genetic variation (C421A) and pantoprazole coadministration. J Pharm Sci. 2010;99:1046–1062. doi: 10.1002/jps.21860. [DOI] [PubMed] [Google Scholar]
- 147.Yamada A, Maeda K, Ishiguro N, Tsuda Y, Igarashi T, Ebner T, et al. The impact of pharmacogenetics of metabolic enzymes and transporters on the pharmacokinetics of telmisartan in healthy volunteers. Pharmacogenet Genomics. 2011;21:523–530. doi: 10.1097/FPC.0b013e3283482502. [DOI] [PubMed] [Google Scholar]
- 148.Chen WQ, Shu Y, Li Q, Xu LY, Roederer MW, Fan L, et al. Polymorphism of ORM1 is associated with the pharmacokinetics of telmisartan. PLoS One. 2013;8:e70341. doi: 10.1371/journal.pone.0070341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim CO, Cho SK, Oh ES, Park MS, Chung JY. Influence of ABCC2, SLCO1B1, and ABCG2 polymorphisms on the pharmacokinetics of olmesartan. J Cardiovasc Pharmacol. 2012;60:49–54. doi: 10.1097/FJC.0b013e3182576098. [DOI] [PubMed] [Google Scholar]
- 150.Ogasawara K, Chitnis SD, Gohh RY, Christians U, Akhlaghi F. Multidrug resistance-associated protein 2 (MRP2/ABCC2) haplotypes significantly affect the pharmacokinetics of tacrolimus in kidney transplant recipients. Clin Pharmacokinet. 2013;52:751–762. doi: 10.1007/s40262-013-0069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim HS, Sunwoo YE, Ryu JY, Kang HJ, Jung HE, Song IS, et al. The effect of ABCG2 V12M, Q141K and Q126X, known functional variants in vitro, on the disposition of lamivudine. Br J Clin Pharmacol. 2007;64:645–654. doi: 10.1111/j.1365-2125.2007.02944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pham PA, la Porte CJ, Lee LS, van Heeswijk R, Sabo JP, Elgadi MM, et al. Differential effects of tipranavir plus ritonavir on atorvastatin or rosuvastatin pharmacokinetics in healthy volunteers. Antimicrob Agents Chemother. 2009;53:4385–4392. doi: 10.1128/AAC.00449-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Busti AJ, Bain AM, Hall RG, 2nd, Bedimo RG, Leff RD, Meek C, et al. Effects of atazanavir/ritonavir or fosamprenavir/ritonavir on the pharmacokinetics of rosuvastatin. J Cardiovasc Pharmacol. 2008;51:605–610. doi: 10.1097/FJC.0b013e31817b5b5a. [DOI] [PubMed] [Google Scholar]
- 155.Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr. 2008;47:570–578. doi: 10.1097/QAI.0b013e318160a542. [DOI] [PubMed] [Google Scholar]
- 156.Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004;76:167–177. doi: 10.1016/j.clpt.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 157.Allred AJ, Bowen CJ, Park JW, Peng B, Williams DD, Wire MB, et al. Eltrombopag increases plasma rosuvastatin exposure in healthy volunteers. Br J Clin Pharmacol. 2011;72:321–329. doi: 10.1111/j.1365-2125.2011.03972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Takeuchi K, Sugiura T, Matsubara K, Sato R, Shimizu T, Masuo Y, et al. Interaction of novel platelet-increasing agent eltrombopag with rosuvastatin via breast cancer resistance protein in humans. Drug Metab Dispos. 2014;42:726–734. doi: 10.1124/dmd.113.054767. [DOI] [PubMed] [Google Scholar]
- 159.Polli JW, Hussey E, Bush M, Generaux G, Smith G, Collins D, et al. Evaluation of drug interactions of GSK1292263 (a GPR119 agonist) with statins: from in vitro data to clinical study design. Xenobiotica. 2013;43:498–508. doi: 10.3109/00498254.2012.739719. [DOI] [PubMed] [Google Scholar]
- 160.Kusuhara H, Furuie H, Inano A, Sunagawa A, Yamada S, Wu C, et al. Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. Br J Pharmacol. 2012;166:1793–1803. doi: 10.1111/j.1476-5381.2012.01887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Suzuki K, Doki K, Homma M, Tamaki H, Hori S, Ohtani H, et al. Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br J Clin Pharmacol. 2009;67:44–49. doi: 10.1111/j.1365-2125.2008.03303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ranchon F, Vantard N, Gouraud A, Schwiertz V, Franchon E, Pham BN, et al. Suspicion of drug-drug interaction between high-dose methotrexate and proton pump inhibitors: a case report—should the practice be changed? Chemotherapy. 2011;57:225–229. doi: 10.1159/000327372. [DOI] [PubMed] [Google Scholar]
- 163.Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther. 2009;328:652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 164.Huang L, Wang Y, Grimm S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab Dispos. 2006;34:738–742. doi: 10.1124/dmd.105.007534. [DOI] [PubMed] [Google Scholar]
- 165.Prasad B, Lai Y, Lin Y, Unadkat JD. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J Pharm Sci. 2013;102:787–793. doi: 10.1002/jps.23436. [DOI] [PubMed] [Google Scholar]
- 166.Jonker JW, Buitelaar M, Wagenaar E, Van Der Valk MA, Scheffer GL, Scheper RJ, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA. 2002;99:15649–15654. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhou S, Zong Y, Ney PA, Nair G, Stewart CF, Sorrentino BP. Increased expression of the Abcg2 transporter during erythroid maturation plays a role in decreasing cellular protoporphyrin IX levels. Blood. 2005;105:2571–2576. doi: 10.1182/blood-2004-04-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Naylor CS, Jaworska E, Branson K, Embleton MJ, Chopra R. Side population/ABCG2-positive cells represent a heterogeneous group of haemopoietic cells: implications for the use of adult stem cells in transplantation and plasticity protocols. Bone Marrow Transplant. 2005;35:353–360. doi: 10.1038/sj.bmt.1704762. [DOI] [PubMed] [Google Scholar]
- 169.Lechner A, Leech CA, Abraham EJ, Nolan AL, Habener JF. Nestin-positive progenitor cells derived from adult human pancreatic islets of Langerhans contain side population (SP) cells defined by expression of the ABCG2 (BCRP1) ATP-binding cassette transporter. Biochem Biophys Res Commun. 2002;293:670–674. doi: 10.1016/S0006-291X(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 170.Shimano K, Satake M, Okaya A, Kitanaka J, Kitanaka N, Takemura M, et al. Hepatic oval cells have the side population phenotype defined by expression of ATP-binding cassette transporter ABCG2/BCRP1. Am J Pathol. 2003;163:3–9. doi: 10.1016/S0002-9440(10)63624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–24225. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 172.Evseenko DA, Murthi P, Paxton JW, Reid G, Emerald BS, Mohankumar KM, et al. The ABC transporter BCRP/ABCG2 is a placental survival factor, and its expression is reduced in idiopathic human fetal growth restriction. FASEB J. 2007;21:3592–3605. doi: 10.1096/fj.07-8688com. [DOI] [PubMed] [Google Scholar]
- 173.Biri A, Bozkurt N, Turp A, Kavutcu M, Himmetoglu O, Durak I. Role of oxidative stress in intrauterine growth restriction. Gynecol Obstet Invest. 2007;64:187–192. doi: 10.1159/000106488. [DOI] [PubMed] [Google Scholar]
- 174.Woodward OM, Kottgen A, Coresh J, Boerwinkle E, Guggino WB, Kottgen M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci USA. 2009;106:10338–10342. doi: 10.1073/pnas.0901249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xiong H, Callaghan D, Jones A, Bai J, Rasquinha I, Smith C, et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood–brain barrier for Abeta(1–40) peptides. J Neurosci. 2009;29:5463–5475. doi: 10.1523/JNEUROSCI.5103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Saison C, Helias V, Ballif BA, Peyrard T, Puy H, Miyazaki T, et al. Null alleles of ABCG2 encoding the breast cancer resistance protein define the new blood group system Junior. Nat Genet. 2012;44:174–177. doi: 10.1038/ng.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bailey-Dell KJ, Hassel B, Doyle LA, Ross DD. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim Biophys Acta. 2001;1520:234–241. doi: 10.1016/S0167-4781(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 178.Ee PL, Kamalakaran S, Tonetti D, He X, Ross DD, Beck WT. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 2004;64:1247–1251. doi: 10.1158/0008-5472.CAN-03-3583. [DOI] [PubMed] [Google Scholar]
- 179.Wang H, Lee EW, Zhou L, Leung PC, Ross DD, Unadkat JD, et al. Progesterone receptor (PR) isoforms PRA and PRB differentially regulate expression of the breast cancer resistance protein in human placental choriocarcinoma BeWo cells. Mol Pharmacol. 2008;73:845–854. doi: 10.1124/mol.107.041087. [DOI] [PubMed] [Google Scholar]
- 180.To KK, Robey R, Zhan Z, Bangiolo L, Bates SE. Upregulation of ABCG2 by romidepsin via the aryl hydrocarbon receptor pathway. Mol Cancer Res. 2011;9:516–527. doi: 10.1158/1541-7786.MCR-10-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Szatmari I, Vamosi G, Brazda P, Balint BL, Benko S, Szeles L, et al. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23823. doi: 10.1074/jbc.M604890200. [DOI] [PubMed] [Google Scholar]
- 182.Honorat M, Mesnier A, Di Pietro A, Lin V, Cohen P, Dumontet C, et al. Dexamethasone down-regulates ABCG2 expression levels in breast cancer cells. Biochem Biophys Res Commun. 2008;375:308–314. doi: 10.1016/j.bbrc.2008.07.149. [DOI] [PubMed] [Google Scholar]
- 183.Yasuda S, Itagaki S, Hirano T, Iseki K. Expression level of ABCG2 in the placenta decreases from the mid stage to the end of gestation. Biosci Biotechnol Biochem. 2005;69:1871–1876. doi: 10.1271/bbb.69.1871. [DOI] [PubMed] [Google Scholar]
- 184.Li W, Jia M, Qin X, Hu J, Zhang X, Zhou G. Harmful effect of ERbeta on BCRP-mediated drug resistance and cell proliferation in ERalpha/PR-negative breast cancer. FEBS J. 2013;280:6128–6140. doi: 10.1111/febs.12533. [DOI] [PubMed] [Google Scholar]
- 185.Imai Y, Ishikawa E, Asada S, Sugimoto Y. Estrogen-mediated post transcriptional down-regulation of breast cancer resistance protein/ABCG2. Cancer Res. 2005;65:596–604. doi: 10.1158/0008-5472.CAN-05-1894. [DOI] [PubMed] [Google Scholar]
- 186.Wang H, Zhou L, Gupta A, Vethanayagam RR, Zhang Y, Unadkat JD, et al. Regulation of BCRP/ABCG2 expression by progesterone and 17beta-estradiol in human placental BeWo cells. Am J Physiol Endocrinol Metab. 2006;290:E798–E807. doi: 10.1152/ajpendo.00397.2005. [DOI] [PubMed] [Google Scholar]
- 187.Hartz AM, Mahringer A, Miller DS, Bauer B. 17-beta-Estradiol: a powerful modulator of blood–brain barrier BCRP activity. J Cereb Blood Flow Metab. 2010;30:1742–1755. doi: 10.1038/jcbfm.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wu X, Zhang X, Sun L, Zhang H, Li L, Wang X, et al. Progesterone negatively regulates BCRP in progesterone receptor-positive human breast cancer cells. Cell Physiol Biochem. 2013;32:344–354. doi: 10.1159/000354442. [DOI] [PubMed] [Google Scholar]
- 189.Pradhan M, Bembinster LA, Baumgarten SC, Frasor J. Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NF{kappa}B cooperativity at adjacent response elements. J Biol Chem. 2010;285:31100–31106. doi: 10.1074/jbc.M110.155309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Turner JG, Gump JL, Zhang C, Cook JM, Marchion D, Hazlehurst L, et al. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood. 2006;108:3881–3889. doi: 10.1182/blood-2005-10-009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.To KK, Zhan Z, Bates SE. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Mol Cell Biol. 2006;26:8572–8585. doi: 10.1128/MCB.00650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.To KK, Zhan Z, Litman T, Bates SE. Regulation of ABCG2 expression at the 3′ untranslated region of its mRNA through modulation of transcript stability and protein translation by a putative microRNA in the S1 colon cancer cell line. Mol Cell Biol. 2008;28:5147–5161. doi: 10.1128/MCB.00331-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.To KK, Robey RW, Knutsen T, Zhan Z, Ried T, Bates SE. Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol Cancer Ther. 2009;8:2959–2968. doi: 10.1158/1535-7163.MCT-09-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang F, Xue X, Wei J, An Y, Yao J, Cai H, et al. hsa-miR-520h downregulates ABCG2 in pancreatic cancer cells to inhibit migration, invasion, and side populations. Br J Cancer. 2010;103:567–574. doi: 10.1038/sj.bjc.6605724. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 195.Pan YZ, Morris ME, Yu AM. MicroRNA-328 negatively regulates the expression of breast cancer resistance protein (BCRP/ABCG2) in human cancer cells. Mol Pharmacol. 2009;75:1374–1379. doi: 10.1124/mol.108.054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H, et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. 2008;283:3349–3356. doi: 10.1074/jbc.M707773200. [DOI] [PubMed] [Google Scholar]
- 197.Peng H, Qi J, Dong Z, Zhang JT. Dynamic vs static ABCG2 inhibitors to sensitize drug resistant cancer cells. PLoS One. 2010;5:e15276. doi: 10.1371/journal.pone.0015276. [DOI] [PMC free article] [PubMed] [Google Scholar]