

Abstract
Summary: k–Top Scoring Pairs (kTSP) is a classification method for prediction from high-throughput data based on a set of the paired measurements. Each of the two possible orderings of a pair of measurements (e.g. a reversal in the expression of two genes) is associated with one of two classes. The kTSP prediction rule is the aggregation of voting among such individual two-feature decision rules based on order switching. kTSP, like its predecessor, Top Scoring Pair (TSP), is a parameter-free classifier relying only on ranking of a small subset of features, rendering it robust to noise and potentially easy to interpret in biological terms. In contrast to TSP, kTSP has comparable accuracy to standard genomics classification techniques, including Support Vector Machines and Prediction Analysis for Microarrays. Here, we describe ‘switchBox’, an R package for kTSP-based prediction.
Availability: The ‘switchBox’ package is freely available from Bioconductor: http://www.bioconductor.org.
Contact: bahman@jhu.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Finding ‘omics’-based biomarkers for clinical use has been extensively studied in numerous diseases. However, mature clinical applications of these biomarkers are scarce due to technological, mathematical and translational barriers (Winslow et al., 2012). Basing the prediction solely on the ordering of a small number of features (e.g. gene expression ranks within profiles) may overcome such barriers to clinical translation (Eddy et al., 2010).
Rank-based classifiers are robust to data normalization and yield transparent decision rules (Eddy et al. 2010). The first and simplest of these rank-based methods, the Top Scoring Pair (TSP) classifier, in which the decision rule is entirely determined by the ordering of two features (i.e. the relative expression of two genes), was introduced in Geman et al. (2004). Multiple extensions to TSP were proposed [e.g. Lin et al. (2009); Tan et al. (2005)] and some have been successfully applied to human cancer such as simplifying clinical biomarkers (Marchionni et al., 2013). A theoretical analysis, R implementation and description of the TSP algorithm can be found in Denis (2013; http://hal.archives-ouvertes.fr/docs/00/78/48/69/PDF/Article.pdf), Leek (2009) and Leek (2014), respectively. One powerful successor of TSP is k–Top Scoring Pairs (kTSP; Tan et al., 2005), which applies majority voting among multiple pairs of features. kTSP has outperformed Support Vector Machines in an open challenge for cancer classification (Geman et al., 2008) and yielded comparable accuracy to the Mammaprint breast cancer assay (Marchionni et al., 2013).
Here, we introduce an R package, ‘switchBox’, for kTSP. This package selects the gene pairs for the kTSP decision rule. The package also implements a new approach to choose the number of pairs, k, based on the analysis of variance introduced in Afsari et al. (2014), which is less computationally intensive and less prone to overfitting than the original method introduced in Tan et al. (2005) and implemented in the R ‘ktspair’ package (Damond, 2011). In addition, ‘switchBox’ provides more flexibility in the selection of candidate ranges of k, as well as alternative strategies for pair votes aggregation compared with the previous R implementation (Damond, 2011). Finally, ‘switchBox’ has a method for calculating sample-specific scores based on the pairs (see Methods), which can be extended beyond classification to class discovery problems.
2 METHODS
kTSP decision is based on k feature (e.g. gene) pairs, denoted by . We also denote the feature profile by . The particular decision rule using the k comparisons is simply determined by the aggregate vote statistic
| (1) |
where I is the logical indicator function. The kTSP classification decision is based on thresholding κ, i.e. provided the labels . The standard threshold is , equivalent to majority voting. The only parameters required for calculating κ are the number and choice of feature pairs. In the introductory paper to TSP (Geman et al., 2004), the authors proposed a score for each pair of features, which measures the discriminative power of a two-feature comparison. The score assigned to genes i and j was defined as
The first training algorithm proposed for training kTSP, i.e. for finding Θ, was an ad hoc method based on the score (Tan et al., 2005).
The ‘switchBox’ package implements a formal method of feature selection based on analysis of variance (Afsari et al., 2014). Briefly, this method selects the feature pairs maximizing the distance between the expectation of κ in each group normalized by the variance. The target set of feature pairs is then
| (2) |
This method as implemented in ‘switchBox’ uses a greedy search for for computational efficiency. This search process simultaneously selects the optimal number of features, requiring only an upper bound on the number of feature pairs as input. To find , we optimize Equation (2) greedily and with empirical estimates from the data.
3 IMPLEMENTATION
For computational efficiency and speed, ‘switchBox’ calculates the score between all feature pairs using C routines. The user can directly calculate the score of a desired set of features or feature pairs by invoking the SWAP.CalculateSignedScore function.
The package provides a training function (SWAP.KTSP.Train) for the classifier and a function (SWAP.KTSP.Classify) for predicting the label of an unseen sample. The training function allows the user to filter either the individual features or the feature pairs, thereby reducing the variability in the learned decision rules. The package also provides a function (SWAP.CalculateSignedScore) to calculate the pairwise scores from any subset of features or subset of feature pairs.
Below we briefly show how to train a kTSP classifier for breast cancer recurrence within 5 years using gene expression data from Marchionni et al. (2013), described in further detail in the ‘switchBox’ package vignette. First, we load the example training and testing gene expression data contained in the ‘switchBox’ package. We then train the classifier and compute the confusion matrix for predictions on the test samples as follows:
###Loading training and test data
data(matTraining)
data(matTesting)
### Training kTSP and classifying new samples
classifier <- SWAP.KTSP.Train(matTraining, trainingGroup)
testPrediction <- SWAP.KTSP.Classify(matTesting, classifier)
### Making confusion matrix
table(testPrediction, testingGroup)
We have found that the kTSP classifier from ‘switchBox’ is more robust and inferred at greater computational speed than that from the ‘ktspair’ R package (Supplementary Document). In addition, ‘switchBox’ provides an additional function SWAP.KTSP.Statistics to calculate kTSP statistics, i.e. κ in Equation (1). This function is useful for generating ROC curves and for calculating ranked-based statistics from TSPs found in the classifier. For example, the code below generates a heatmap to depict classification results for each pair in the classifier (Fig. 1).
kappa <- SWAP.KTSP.Statistics( matTraining, classifier)
heatmap(1*kappa$comparisons, scale=“none”, labRow= trainingGroup)
Fig. 1.
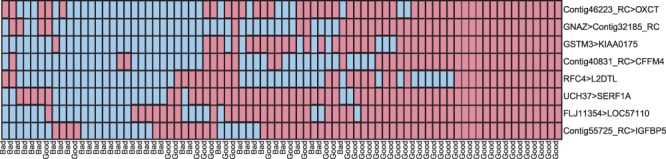
The comparisons votes (y-axis) versus samples (x-axis). The samples are labeled either good prognosis or bad prognosis for breast cancer. Truth and falsehood of the comparisons are indicated by blue (lighter shade) and red (darker shade), respectively. The combination of the votes can be used to classify, illustrated by requiring at least three votes for declaring bad prognosis. More explanation and code for this figure can be found in the Supplementary Document
4 CONCLUSION
We introduced ‘switchBox’, an R package for kTSP classifier with a robust procedure for pair selection as previously described in Afsari et al. (2014). As mentioned in Afsari et al. (2014), the procedure requires less computation and is less prone to overfitting than the one described in Tan et al. (2005) and implemented in ‘ktspair’ package (Damond, 2011). Moreover, we provide functions for calculating auxiliary statistics as well as any user-defined combination of the comparisons.
Funding: L.M. was supported by the National Institutes of Health (NIH-)NCI [P30 CA006973]; L.M. and E.J.F. by the Cleveland Foundation The Helen Masenhimer Fellowship Award; B.A. and D.G. by the NIH-NCRR [UL1 RR 025005]. E.J.F. and B.A. by NIH-NCI [K25 CA141053].
Conflict of interest: none declared.
Supplementary Material
REFERENCES
- Afsari B, et al. Rank discriminants for predicting phenotypes from RNA expression. Ann. Appl. Stat. 2014 [Google Scholar]
- Damond J. ktspair: k-Top Scoring Pairs for Microarray Classification. 2011. R package version 1.0, CRAN. [Google Scholar]
- Denis C. Top Scoring Pair Classifiers: Asymptotics and Applications. 2013. in archive. [Google Scholar]
- Eddy JA, et al. Relative expression analysis for molecular cancer diagnosis, prognosis. Technol. Cancer Res. Treat. 2010;9:149–159. doi: 10.1177/153303461000900204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geman D, et al. Classifying gene expression profiles from pairwise mrna comparisons. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1071. Article19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geman D, et al. Microarray Classification from Several Two-gene Experssion Comparisons. 2008. ICMLA, San Diego, CA, IEEE (Winner, ICMLA Microarray Classification Algorithm Competition) [Google Scholar]
- Leek J. tspair: top scoring pairs for microarray classification. 2014. R package version 1.22.0, Bioconductor. [Google Scholar]
- Leek JT. The tspair package for finding top scoring pair classifiers in r. Bioinformatics. 2009;25:1203–1204. doi: 10.1093/bioinformatics/btp126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, et al. The ordering of expression among a few genes can provide simple cancer biomarkers and signal brca1 mutations. BMC Bioinformatics. 2009;10:256. doi: 10.1186/1471-2105-10-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionni L, et al. A simple and reproducible breast cancer prognostic test. BMC Genomics. 2013;14:336. doi: 10.1186/1471-2164-14-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AC, et al. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics. 2005;21:3896–3904. doi: 10.1093/bioinformatics/bti631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow R, et al. The emerging discipline of computational medicine. Sci. Transl. Med. 2012;4:158rv11. doi: 10.1126/scitranslmed.3003528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.