

Abstract
Capnocytophaga canimorsus, a dog mouth commensal and a member of the Bacteroidetes phylum, causes rare but often fatal septicemia in humans that have been in contact with a dog. Here, we show that C. canimorsus strains isolated from human infections grow readily in heat-inactivated human serum and that this property depends on a typical polysaccharide utilization locus (PUL), namely, PUL3 in strain Cc5. PUL are a hallmark of Bacteroidetes, and they encode various products, including surface protein complexes that capture and process polysaccharides or glycoproteins. The archetype system is the Bacteroides thetaiotaomicron Sus system, devoted to starch utilization. Unexpectedly, PUL3 conferred the capacity to acquire iron from serotransferrin (STF), and this capacity required each of the seven encoded proteins, indicating that a whole Sus-like machinery is acting as an iron capture system (ICS), a new and unexpected function for Sus-like machinery. No siderophore could be detected in the culture supernatant of C. canimorsus, suggesting that the Sus-like machinery captures iron directly from transferrin, but this could not be formally demonstrated. The seven genes of the ICS were found in the genomes of several opportunistic pathogens from the Capnocytophaga and Prevotella genera, in different isolates of the severe poultry pathogen Riemerella anatipestifer, and in strains of Bacteroides fragilis and Odoribacter splanchnicus isolated from human infections. Thus, this study describes a new type of ICS that evolved in Bacteroidetes from a polysaccharide utilization system and most likely represents an important virulence factor in this group.
INTRODUCTION
Capnocytophaga canimorsus is a commensal bacterium from the oral cavity of dogs that is regularly isolated, since its description in 1989, from extremely severe human infections worldwide (1, 2). Following contact with a dog, these infections generally start with vague influenza symptoms, and patients enter the hospital with fulminant septicemia often associated with peripheral gangrene. Mortality is as high as 40% in spite of adequate antibiotherapy and frequent amputations (1, 3–7). Infections do not necessarily occur after severe injuries, which generally are followed by a preventive antibiotic treatment, but rather after small bites, scratches, or even licks (8). Many cases involve splenectomized, alcoholic, or immunocompromised patients, but more than 40% of the cases concern healthy people with no obvious risk factors (5, 8–12), indicating that C. canimorsus infections are not restricted to immunocompromised individuals. It is worth noting that there is no report of a dog having been infected by C. canimorsus, although 74% of the dogs carry it (13–15). Thus, evolution shaped these bacteria essentially as commensals of the mouth and not as pathogens. Besides C. canimorsus, the oral cavity of dogs also harbors Capnocytophaga cynodegmi (14), the species most closely related to C. canimorsus, with a difference in the 16S RNA sequence only in the range of 1.5% (13). Interestingly, C. cynodegmi is not reported to cause human infections (13, 16). Other bacteria from the genus Capnocytophaga colonize the oral cavity of diverse mammals, including humans (17, 18). Capnocytophaga are fastidious capnophilic (i.e., CO2 loving) Gram-negative bacteria that belong to the family of Flavobacteriaceae in the phylum Bacteroidetes. Flavobacteriaceae include a variety of environmental and marine bacteria, such as Flavobacterium johnsoniae (19), and a few severe animal pathogens, like Flavobacterium psychrophilum, the causative agent of cold water disease in salmonid fish (20), and Riemerella anatipestifer, which causes duckling disease in waterfowl and turkeys (21, 22). Besides the Flavobacteriaceae, the phylum Bacteroidetes includes the Bacteroidaceae, which contain many anaerobic commensals of the mammalian intestinal flora, such as Bacteroides thetaiotaomicron and Bacteroides fragilis (23). The phylum Bacteroidetes is taxonomically remote from the Proteobacteria group, including most studied human pathogens, and the biology of these bacteria reveals a number of original features. One of these features is the presence of many systems resembling the archetypal starch utilization system (Sus) discovered in B. thetaiotaomicron (24). The Sus system is a cell envelope-associated multiprotein complex characterized by the coordinated action of several proteins and lipoproteins involved in substrate binding, degradation, and internalization into the periplasm (14, 24–31). Subsequent microbial genome sequencing projects revealed the presence of many polysaccharide utilization loci (PUL) encoding Sus-like systems in the genome of B. thetaiotaomicron and other saccharolytic Bacteroidetes (26, 31, 32), targeting all major classes of host and dietary glycans (33). The genome of saprophytic Bacteroidetes like F. johnsoniae also contains a large number of PUL (34), indicating that they are a hallmark of the Bacteroidetes phylum rather than of the commensal Bacteroides only. The genome of the clinical isolate type strain C. canimorsus 5 (also called strain Cc5) (35, 36) contains 13 such PUL, which may encode surface feeding machineries (37). At least 10 of them are expressed, accounting for more than half of the surface-exposed proteins, when Cc5 bacteria are grown on HEK293 cells. All of these findings indicate that surface-exposed complexes specialized in foraging complex glycans or other macromolecules play a central role in the biology of C. canimorsus (37). Indeed, C. canimorsus has the unusual property of harvesting N-linked glycan chains of soluble proteins like immunoglobulins and even of surface glycoproteins from animal cells, including phagocytes. This capacity depends on a Sus-like complex encoded by PUL5 (38). However, the function of the other PUL is not known yet, and their impact on pathogenicity is unclear. In the present study, we aimed at identifying C. canimorsus virulence factors implicated in septicemia, and we demonstrate that PUL3 encodes a Sus-like system devoted to the acquisition of iron from transferrins, including human serotransferrin (STF).
MATERIALS AND METHODS
Ethics statement.
Blood samples from healthy volunteers who had signed a written informed consent were provided by the Blutspendezentrum SRK Beider Basel. The experiments were approved by the Ethikkommission Beider Basel EKBB (no. EK398/11).
Bacterial strains.
This study was carried out with C. canimorsus strains isolated from human infections (35, 36) and C. canimorsus and C. cynodegmi strains isolated from dogs in two areas of Switzerland. One strain of C. cynodegmi was purchased from the ATCC. Escherichia coli S17-1, Pseudomonas aeruginosa PAO1, and the C. canimorsus mutant strains are described in Table S1 in the supplemental material.
Conventional bacterial growth conditions and selective agents.
C. canimorsus bacteria were routinely grown on heart infusion agar (Difco) supplemented with 5% sheep blood (Oxoid) (SB plates) for 2 days at 37°C in the presence of 5% CO2. Escherichia coli strains were grown routinely in lysogeny broth (LB) at 37°C. Pseudomonas aeruginosa PAO1 (39) was grown on SB plates at 37°C in the presence of 5% CO2. To select for plasmids, antibiotics were added at the following concentrations: 10 μg · ml−1 erythromycin, 10 μg · ml−1 cefoxitin, 20 μg · ml−1 gentamicin.
Mutagenesis by allelic exchange and trans-complementation.
Mutagenesis of the Cc5 wild type (wt) was performed as described in reference (40), with slight modifications. Briefly, replacement cassettes with flanking regions spanning approximately 500 bp homologous to regions directly framing targeted genes were constructed with a three-fragment overlapping PCR strategy. First, two PCRs using Phusion polymerase (M0530S; New England BioLabs) were performed on 100 ng of Cc5 genomic DNA with primers for the upstream (oligonucleotides 1.1 and 1.2) and downstream (oligonucleotides 2.1 and 2.2) regions flanking the sequence targeted for deletion. Primers 1.2 and 2.1 included a 20-bp extension at their 5′ extremities corresponding to both ends of the ermF gene (including the promoter). The ermF resistance gene was amplified from pMM13 with primers 3.1 and 3.2, which included approximately 20-bp extensions for further annealing to amplify homologous regions. All three PCR products were cleaned and then mixed in equal amounts for PCR using Phusion polymerase. The initial denaturation was at 98°C for 2 min, followed by 10 cycles without primers to allow annealing and extension of the overlapping fragments (98°C for 30 s, 50°C for 40 s, and 72°C for 2 min). After the addition of external primers (1.1 and 2.2), the program was continued for 20 cycles (98°C for 30 s, 50°C for 40 s, and 72°C for 2 min 30 s) and finally for 10 min at 72°C. Final PCR products consisted of locus::ermF insertion cassettes and were digested with PstI and SpeI for cloning into the appropriate sites of the C. canimorsus suicide vector, pMM25 (40). The resulting plasmids were transferred by RP4-mediated conjugative DNA transfer from E. coli S17-1 to Cc5 to allow the integration of the insertion cassette. Transconjugants then were selected for the presence of the ermF resistance cassette and checked for sensitivity to cefoxitin, indicating the loss of the pMM25 backbone, and the mutated regions were sequenced with primers 1.1 and 2.2. Trans-complementation of the different knockouts was done by introducing the relevant genes cloned in the C. canimorsus expression vector pPM5. Mutant strains are listed in Table S1 in the supplemental material, primers are in Table S2, and plasmids are in Table S3.
PCR screen for PUL3.
For PCR screen of PUL3 genes, strains were grown for 2 days on SB plates, collected, and resuspended in 400 μl phosphate-buffered saline (PBS) at an OD600 of 2. Bacterial suspensions then were centrifuged at 6,000 relative centrifugal forces (RCF) for 5 min and resuspended in 400 μl H2O. Five-μl aliquots of bacterial suspensions then were used in 35-cycle PCRs as described in reference 13. Primers used for the amplification of genes Ccan_03640 (icsA), Ccan_03650 (icsC), Ccan_03680 (icsD), Ccan_03690 (icsE), Ccan_03700 (icsF), Ccan_03710 (icsG), and Ccan_03720 (icsH) are listed in Table S2 in the supplemental material. 16S rRNA genes were amplified as a control (see Table S2).
Sera and protein-depleted serum derivatives.
Batches of fresh human blood pooled from 20 individuals were collected at the University Hospital of Basel (Blutspendezentrum). The pooled blood was clotted and centrifuged for 10 min at 6,000 RCF, and the supernatant (serum) was collected for further analyses. Alternatively, human serum collected off the clot from healthy normal humans was purchased from EMD Millipore (S1-liter; Billerica, MA, USA). Serum then was heat inactivated (HIHS) at 55°C for 1 h when required. Protein-depleted human serum (PDHS) was obtained by collecting the flowthrough of 15 ml human serum passed through a single Amicon filter unit with a nominal molecular mass limit of 50 kDa (UFC905024; Millipore) by spinning at 4,000 RCF for 40 min at 20°C. Protein depletion then was monitored by SDS-PAGE (41) and silver staining (42). Transferrin depletion was checked by anti-STF immunoblotting (goat anti-human transferrin; T2027; Sigma-Aldrich). Filtered serum then was heat inactivated as described above.
Growth in heat-inactivated and protein-depleted human sera.
Growth assays were performed in 96-well plates. Inocula were prepared from cultures grown on SB plates, set to an OD600 of 0.2, and serially diluted 1:10 four times. Twenty-two- and 10-μl bacterial suspensions then were used to inoculate 200 μl of HIHS and 50 μl of PDHS, respectively. HIHS was supplemented with iron (III) chloride (FeCl3; 0.25 mM), iron (III) citrate (FeC6H5O7; 0.25 mM), or iron (II) sulfate (FeSO4; 0.25 mM) if required. PDHS was supplemented with iron (III) chloride (0.25 mM), human STF (3 g · liter−1; 16-16-032001; Athens Research), human ApoSTF (3 g · liter−1; 16-16-A32001; Athens Research), human lactoferrin (1.5 g · liter−1; 30-1147; Fitzgerald), bovine STF (3 g · liter−1; PRO-510; Prospecbio), hemin (0.25 mM; H9039; Sigma-Aldrich), or hemoglobin (0.1 mM; H7379; Sigma-Aldrich) if required. Equivalent volumes of inocula also were plated in order to precisely determine bacterial concentrations by CFU counting at the inoculation time point. Infections then were incubated statically for 23 h at 37°C in the presence of 5% CO2. Serial dilutions were plated on SB plates, and CFU were determined. The number of generations was calculated according to the following formula: CFU in the well = inoculum × 2Number of generations. Cocultures were performed essentially in same the way, except that 200 μl of HIHS was inoculated with both 22 μl of wild-type C. canimorsus 5 and 22 μl of the deletion strain set at an OD600 of 0.2 and serially diluted 1:10 four times. In addition, serial dilutions following incubation were plated in parallel on SB plates and on SB plates containing erythromycin for the selection of deletion strains. The total growth of the deletion strains corresponded to the CFU on erythromycin-containing plates, while the total growth of the wt was determined by subtracting the CFU counts on erythromycin-containing plates from the CFU counts on SB plates.
Protein concentrations were checked using a Bio-Rad protein assay kit (500-0002; Bio-Rad), and iron concentrations, except for hemoglobin, were checked using the ferrozine assay (43). The iron concentration of hemoglobin was specifically determined using a modified ferrozine assay (44). Protein and iron concentrations are given in Table S4 in the supplemental material.
Monitoring of transcription by real-time RT-PCR.
The Cc5 wt was inoculated at a density of 5 × 105 bacteria · ml−1 in 15 ml of HIHS at 37°C in the presence of 5% CO2 with or without 0.25 mM iron (III) citrate. Bacteria were harvested after 6 h (corresponding to the mid-log growth phase) by centrifugation at 7,000 RCF at 4°C for 5 min. The pellet was resuspended in RNAprotect bacterial reagent (76506; Qiagen) and centrifuged again at 5,000 RCF for 10 min. The ΔfurA deletion strain was grown under the same conditions without the addition of iron. Bacteria were lysed in 200 μl Tris-EDTA (TE) buffer containing proteinase K (60 mAU · ml−1; 19131; Qiagen) and lysozyme (1 mg · ml−1; 10837059001; Roche) for 10 min at 25°C on a shaker. RNA was extracted with the miRNeasy minikit (217004; Qiagen). One ml of QIAzol reagent was heated up to 65°C and added to each sample. Samples were vortexed for 3 min and incubated for 5 min at room temperature. Two hundred μl chloroform was added. The following steps were performed according to the manufacturer's instructions. To remove genomic DNA, an on-column DNase digestion and an additional DNase digestion postextraction were performed using an RNase-free DNase set (79254; Qiagen). RNA was purified with the RNeasy MinElute cleanup kit (74204; Qiagen). RNA integrity was verified by nondenaturing agarose gel electrophoresis (1% agarose [EP-0010-05; Eurogentec] in Tris-acetate-EDTA [TAE]). The absence of genomic DNA was tested by PCR for 16S rRNA. One hundred to 500 ng of RNA was reverse transcribed using Superscript II reverse transcriptase (200 U) (18064-014; Invitrogen) and random primers (100 ng · ml−1) (48190011; Invitrogen) according to the manufacturer's instructions. A no-enzyme control was included for all RNA samples to confirm the absence of genomic DNA. Quantitative PCR (qPCR) was performed using FastStart Universal SYBR Master (Rox) (04913850001; Roche) and primers at 0.3 μM. Primers were designed with NCBI primer-BLAST. Three technical replicates were run for each target and condition. Before performing the actual qPCR, serial cDNA dilutions were amplified, and PCR and primer efficiencies were evaluated by means of a standard curve. All qPCRs were performed on a StepOne machine (Applied Biosystems) using the following thermal cycling conditions: 2 min at 50°C, 10 min at 95°C, 40 cycles 15 s at 95°C, and 1 min at 60°C. Fold change was calculated as described in reference 45, with the ΔΔCT method (where CT is threshold cycle) considering the efficiency of the PCR for each target. 16S rRNA served as a reference gene.
Transferrin deglycosylation analyses and lectin stainings.
For the assessment of the deglycosylation of STF by Cc5, bacteria were collected from SB plates and resuspended in PBS at an OD600 of 1. One hundred microliters of bacterial suspensions then was incubated with 100 μl of a transferrin (16-16-032001; Athens Research) solution (0.2 g · liter−1) for 180 min at 37°C. As a negative control, 200 μl of a 1:2-diluted transferrin solution alone was incubated for 180 min at 37°C. Samples then were centrifuged for 5 min at 13,000 RCF, supernatant was collected, and a 12-μl aliquot was loaded in a 12% SDS-PAGE gel. Samples were analyzed by Coomassie brilliant blue R250 (B0149; Sigma) and lectin stainings with Sambucus nigra lectin (SNA) according to the manufacturer's recommendations (digoxigenin glycan differentiation kit; 11210238001; Roche). For the deglycosylation of human STF by PNGase F, 9 μl of human STF (2 g · liter−1; 16-16-032001; Athens Research) was incubated with 2 μl of either fresh or heat-inactivated (10 min at 75°C) enzyme (P0704L; New England BioLabs) in the presence of 1.2 μl of 10× G7 buffer (B3704; New England BioLabs) for 2 h at 37°C. Deglycosylation then was monitored by immunoblotting and lectin stainings with SNA as described above. For subsequent growth assays, PDHS was supplemented with 4 μl of deglycosylated STF for a minimal final concentration required for growth of 0.1 g · liter−1.
Siderophore detection assay.
Siderophore production was assayed using a modified chrome azurol S (CAS) procedure (46, 47). CAS reagent was prepared as described in reference 46. In order to reach the same final count, C. canimorsus 5 and Pseudomonas aeruginosa PAO1 were inoculated at approximately 104 and 107 bacterial cells, respectively, in 1 ml HIHS in 12-well plates and incubated for 23 h at 37°C in the presence of 5% CO2. Serial dilutions were plated on SB plates to determine the final growth by CFU counting. Bacterial cells were removed by two successive centrifugation steps at 12,000 RCF for 5 min at 20°C. Supernatants then were dialyzed overnight at 4°C (3,500 molecular weight cutoff [MWCO]; 133110; Spectra/Por Biotech) against 4 ml double-distilled water (ddH2O) containing 0.02% sodium azide. An uninfected control sample of HIHS was treated in parallel. Finally, dialysates were concentrated for approximately 8 h at 37°C to 200 to 250 μl using a Concentrator plus centrifuge (Eppendorf). Fifty μl of dialysate was mixed with an equal volume of CAS solution in a 96-well plate and incubated for 4 h at 37°C. Absorbance at 630 nm was measured using an xMark microplate spectrophotometer (Bio-Rad) and Microplate Manager 6 software (version 6.0; Bio-Rad), ddH2O containing 0.02% sodium azide serving as a blank, and uninfected HIHS serving as the reference. All measurements were realized in duplicates. Siderophore production was estimated by comparing the ratio [(A630 of sample)/(A630 of reference)] of Cc5 and PAO1 dialysates to a desferrioxamine mesylate (Desferal) (252750; Calbiochem) standard curve in ddH2O containing 0.02% sodium azide.
Uptake of iron from transferrin by C. canimorsus.
55Fe-transferrin was prepared according to references 48 and 49. ApoSTF at 1 mg · ml−1 in 40 mM Tris-HCl buffer (pH 7.4) containing 2 mM sodium carbonate was mixed with 0.075 μmol of sodium citrate and 0.0075 μmol of 55Fe-Cl3 (Perkin-Elmer) and incubated at room temperature for 30 min. The solution then was transferred into dialysis tubing (6,000 to 8,000 MWCO; 132665; Spectra/Por Biotech) and dialyzed four times against 250 ml 40 mM Tris-HCl buffer (pH 7.4) containing 2 mM sodium carbonate for 16 h. The final protein and iron concentrations were evaluated as described above, and transferrin was found to be 20% iron saturated. Five hundred μl 55Fe-STF (3.25 μM) was mixed with 500 μl of bacterial suspension in RPMI (R8758; Sigma-Aldrich) with 2.5% HIHS set to an OD600 of 1. The mixture was incubated statically at 37°C for 24 h, and a control sample was incubated in parallel on ice. Cells then were harvested by centrifugation (6,000 RCF, 3 min), washed four times with 1 ml PBS, and resuspended in a final volume of 1 ml of PBS. The OD600 was measured for each sample, and equivalent amounts of bacteria were transferred into scintillation vials. Four ml of scintillation liquid (Ultima Gold; 6013329; Perkin-Elmer) was added, and vials were incubated overnight in the dark. Radioactivity associated with bacteria was quantified with a Beckman LS6500 liquid scintillation counter (Beckman Coulter, Fullerton, CA).
Identification of PUL3 gene members in the genome of other organisms.
icsA (Ccan_03640; gi|340621142; YP_004739593.1), icsC (Ccan_03650; gi|340621143; YP_004739594), Ccan_03660 (gi|340621144; YP_004739595.1), Ccan_03670 (gi|340621145; YP_004739596.1), icsD (Ccan_03680; gi|340621146; YP_004739597.1), icsE (Ccan_03690; gi|340621147; YP_004739598.1), icsF (Ccan_03700; gi|340621148; YP_004739599.1), icsG (Ccan_03710; gi|340621149; YP_004739600.1), and icsH (Ccan_03720; gi|340621150; YP_004739601.1) from C. canimorsus 5 were blasted against the nr database and clustered at 70% identity (50). Hits above the threshold (high-scoring segment pair E value of <10−5) were aligned with ClustalW (default settings) (51). Alignments were used to build hidden Markov models with HMMER.3 (http://hmmer.org/). Models were calibrated and searched against a local copy of the microbial complete genome database, including approximately 2,100 genomes (NCBI) with HMMER.3 and an E value cutoff of 0.0001. A series of Perl scripts was used to sort the outputs and to count occurrences of complete or partial systems. Occurrences of homologous systems then were reported on an illustrative phylogenetic tree based on 16S rRNA sequences from the ribosomal database project (RDP; http://rdp.cme.msu.edu/index.jsp). All sequences were 1,200 nucleotides long and tagged as good quality according to RDP. Type strains and isolated samples were preferred. At least two sequences per genus were downloaded as alignment files from RDP. Consensus was inferred using EMBOSS (http://www.sanger.ac.uk/Software/EMBOSS). Genus consensuses were aligned with ClustalW (default settings), and phylogenetic analyses were conducted in MEGA4 (52). Evolutionary history was inferred using unweighted-pair group method using average linkages (UPGMA), and evolutionary distances were computed using the maximum composite likelihood method. All positions containing gaps and missing data were eliminated (complete deletion option), leaving a total of 1,143 positions in the final data set. Further searches for PUL3 genes involved in iron acquisition in organisms absent from the complete genome database were based on PSIBLAST searches with default parameters at the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with two reiterations in total. Only the first 500 hits below an E value of 0.05 were considered. The computations were performed on the CPU cluster of the [BC]2 Basel Computational Biology Center (http://www.bc2.ch/center/index.htm).
Accession numbers for relevant genes and proteins mentioned in the text.
The sequences of icsA (Ccan_03640; gi|340621142; YP_004739593.1), icsC (Ccan_03650; gi|340621143; YP_004739594), icsD (Ccan_03680; gi|340621146; YP_004739597.1), icsE (Ccan_03690; gi|340621147; YP_004739598.1), icsF (Ccan_03700; gi|340621148; YP_004739599.1), icsG (Ccan_03710; gi|340621149; YP_004739600.1), and icsH (Ccan_03720; gi|340621150; YP_004739601.1) were deposited in GenBank previously.
RESULTS
C. canimorsus strains isolated from human infections grow readily in heat-inactivated human serum.
While Cc5 bacteria survived in 10% fresh human serum (53), they were killed in 100% fresh human serum (FHS) (data not shown). In contrast, they grew readily in 100% heat-inactivated human serum (HIHS) (Fig. 1), reaching after 23 h a density of about 15 × 109 CFU · ml−1 irrespective of the inoculum. In order to assess the relevance of this observation for pathogenesis of human infection, we monitored the growth of 78 different Capnocytophaga strains in HIHS. Nine strains of C. canimorsus isolated from human infections (referred to as clinical isolates), 62 strains of C. canimorsus isolated from dog mouth (dog isolates), and 7 strains of oral canine C. cynodegmi were inoculated in HIHS, and colonies were counted after 23 h of incubation. All clinical isolates grew readily, achieving 19 ± 3.7 generations (Fig. 1). In contrast, dog isolates fell into two groups, a first group of strains (31 strains, 50%) performed 18.4 ± 3.2 generations, similar to the clinical isolates, while a second group of 31 strains either did not grow or produced fewer than 8 generations (Fig. 1). The very different proportions of C. canimorsus strains able to grow in HIHS among clinical isolates and dog strains strongly suggests that this capacity correlates with pathogenicity and that clinical isolates originate from a subpopulation of dog strains. The strains of C. cynodegmi that were tested performed 9.3 ± 4 generations in HIHS (Fig. 1), which is significantly different from both groups of C. canimorsus isolated from dogs (P values below 10−3). This is somewhat surprising, given that C. cynodegmi is not reported to cause systemic human infections. However, the differences between C. cynodegmi and the two groups of C. canimorsus suggest that several factors can influence the growth of bacteria from this taxon in HIHS.
FIG 1.

Growth of C. canimorsus and C. cynodegmi strains in heat-inactivated human serum. The number of generations achieved after 23 h in HIHS for individual Capnocytophaga species strains are graphed. Black, clinical isolates of C. canimorsus; the diamond shape indicates strain Cc5; gray, dog isolates of C. canimorsus; white, dog isolates of C. cynodegmi. The best significant expectation-maximization clustering of the C. canimorsus dog isolates is reached when clustering the isolates into the two groups (growing and nongrowing) separated by the dotted line. Solid lines indicate the average number of generations for each group (averages from 3 experiments). ***, t test error probability below 0.001. The clinical isolates and the growing dog isolates of C. canimorsus cannot be discriminated by a t test on the sole basis of their growth scores (n.s.). The group of C. cynodegmi strains displays intermediate growth.
PUL3 is crucial for growth in HIHS.
In order to identify the genes underlying the capacity to grow in HIHS, we compared the genomes of Cc5 (35), three additional clinical isolates of C. canimorsus (Cc2, Cc11, and Cc12) (36), three C. canimorsus dog strains that failed to grow in HIHS (CcD38, CcD93, and CcD95), and three strains of C. cynodegmi that displayed moderate growth levels (Ccyn2B, Ccyn49044, and Ccyn74). The genome sequences and their annotations will be described in detail elsewhere. This comparative analysis identified 97 orthologous groups of genes whose presence correlates with the capacity to grow in HIHS. Only 54 of these orthologous clusters included genes with a predicted function. Thirty-eight were involved in a variety of processes, but 16 encoded Sus-like feeding complexes (data not shown). The latter 16 genes belong to only 3 polysaccharide utilization loci, namely PUL3 (9 genes), PUL7 (6 genes), and PUL11 (one gene) (37). Because PUL genes represent 16.5% of those differentiating strains that can or cannot grow in HIHS while all of the PUL genes represent only about 4% of the Cc5 complete genome, we first tested the PUL3, PUL7, and PUL11 knockout mutants for growth in HIHS. In good agreement with the prediction based on genomics, bacteria deprived of the PUL3 locus were dramatically impaired in their capacity to grow in HIHS, while bacteria deprived of PUL7 or PUL11 did not show any significant growth reduction compared to the wt (see Fig. S1 in the supplemental material). We also tested the 10 Cc5 knockout mutants deprived of the other PUL genes (37). Not surprisingly, PUL5 mutants had a moderate growth defect in HIHS compared to the wt (see Fig. S1). This is consistent with the fact that PUL5 encodes the Gpd glycoprotein deglycosylation system that is essential for aminosugar scavenging (37, 54). The deletion of PUL1 also led to a moderate growth defect, but this was not investigated further.
PUL3 has a unique genetic organization compared to the other PUL genes of C. canimorsus 5.
PUL3 was annotated as a large locus of 15 genes sharing the same transcriptional orientation (Ccan_03600 to Ccan_03740) (37) (Fig. 2). PUL3 has two major features that make it different from the other PUL of Cc5. First, it is the only PUL where the susC-like gene Ccan_03650 is separated from the susD-like lipoprotein gene by other genes. However, these intervening genes (Ccan_03660 and Ccan_03670) have a functional annotation that is unusual for PUL genes (Fig. 2A), suggesting that they have inserted within an ancestral canonical PUL. The second unusual feature of PUL3 is the presence of two susC-like genes instead of a single one (Ccan_03640 and Ccan_03650). Ccan_03640 is 378 amino acids smaller than Ccan_03650 and shares some remote similarities with the iron (III) dicitrate transporter FecA of E. coli (Uniprot accession number P13036). Significant intergenic regions of around 400 bp frame each susC-like gene (Fig. 2A), while in most PUL there is only one large noncoding sequence with promoter activity located upstream from the single susC homologue (37). As for most other PUL, the last genes from the putative main operon (Ccan_03690 to Ccan_03720) encode conserved hypothetical lipoproteins for which no function could be assigned (Fig. 2A). Genes at both ends of the locus (Ccan_03610, Ccan_03620, and Ccan_03730) seem to lie outside the putative main operon; nevertheless, their predicted localization and function is compatible with a role in glycan or glycoprotein degradation at the bacterial surface (Fig. 2A). A bias in the DNA K-mer composition, as detected by the Alien_hunter software (55), can be observed from Ccan_03640 to Ccan_03720 with respect to the rest of the chromosome (Fig. 2A), suggesting that the central region of PUL3 has been acquired more recently than the other genes at the periphery.
FIG 2.

Functional characterization of PUL3. (A) Genetic organization and functional annotation of PUL3. Genes likely involved in the capture of iron by C. canimorsus in human serum are labeled icsA-H. Gray-delineated white arrows indicate genes whose deletion had no effect on iron acquisition. The two genes marked by dashed white arrows were not knocked out in this study. White and black circles at the N terminus of the coding sequences indicate that the protein has a type I or type II (lipoprotein) signal peptide, respectively. The numbers under the arrows correspond to the Ccan_ gene references of strain Cc5. The black double arrow indicates the span of the deletion in the ΔPUL3 mutant used throughout this study. The gray double arrow shows the range of the region of PUL3 exhibiting a DNA composition bias with respect to the rest of the chromosome, as computed by Alien_Hunter with a local score of 34.589 (default significance cutoff, 18). (B) Number of generations achieved by the wt and single-gene mutants in HIHS (white bars) and in HIHS supplemented with 0.25 mM iron (III) chloride (FeCl3) (black bars). Gray bars indicate the growth of mutants trans-complemented with a plasmid expressing the corresponding deleted gene. Error bars indicate standard deviations (averages from 3 experiments). All differences above 7 generations have t test-based error probabilities below 0.008.
The sus-like genes of PUL3 are required for iron scavenging in human serum.
Since the annotation of Ccan_03640 pointed to an iron transporter, we tested whether the addition of various iron sources to the HIHS could rescue the growth of the ΔPUL3 mutant bacteria. When HIHS was supplemented with different iron salts at a concentration of 250 μM, the growth of ΔPUL3 mutant bacteria was fully restored to the wt level (data not shown).
In order to investigate whether the whole Sus-like apparatus or only one of the SusC-like proteins was involved in iron uptake, we performed a systematic replacement of each of the 13 genes, ranging from Ccan_03610 to Ccan_03730, by an erythromycin resistance cassette. Interestingly, the substitution of each of the seven typical PUL genes had a drastic effect on the growth capacity in HIHS (Fig. 2B). Indeed, the deletion of each of the two susC homologs (Ccan_03640 and Ccan_03650), the susD homolog (Ccan_03680), and each of the four uncharacterized lipoprotein genes (Ccan_03690, Ccan_03700, Ccan_03710, and Ccan_03720) reduced the number of generations per 23 h from 22.5 ± 0.8 to an average of 4.5 ± 1.1 (Fig. 2B). As expected, the addition of iron (III) chloride to the HIHS restored the growth capacity of all the mutant strains (19.4 ± 1.7 generations) (Fig. 2B). Trans-complementation of the seven individual mutants restored the growth capacity, indicating that each of these genes is involved in the growth process in HIHS (Fig. 2B). These results lead to the conclusion that iron uptake requires not only a putative TonB-dependent outer membrane transporter but also a multiprotein Sus-like complex.
The strains deleted of the two genes with an unusual functional annotation for PUL genes (Ccan_03660 and Ccan_03670) and the deletion mutants for upstream (Ccan_03610, Ccan_03620, and Ccan_03630) and downstream (Ccan_03730) genes in the locus were able to grow normally in HIHS (Fig. 2B). Thus, the locus encoding the iron capture system (ICS) (gray double arrow in Fig. 2A) is smaller than the whole of PUL3, as initially described by Manfredi et al. (37), and corresponds to the genes sharing a similar K-mer bias in their DNA content (55), as mentioned above. We named the seven genes required for iron acquisition ics. We called Ccan_03640 and Ccan_03650, the two putative TonB-dependent porins (SusC-like), icsA and icsC, respectively, and the gene encoding a homolog of susD (Ccan_03680) was named icsD. The four additional lipoproteins were named according to their order in the putative operon of icsE, icsF, icsG, and icsH (Ccan_03690, Ccan_03700, Ccan_03710, and Ccan_03720, respectively) (Fig. 2A). We suggest limiting PUL3 to the genes forming an iron capture system that has been acquired at once by horizontal transfer.
PUL3 expression is regulated by iron and FurA.
If PUL3 was indeed devoted to iron capture, its expression probably would be modulated by iron. To assess this, we monitored the expression by real-time PCR of three PUL3 genes (Ccan_03640, Ccan_03650, and Ccan_03680) as representatives of the PUL3 locus, comparing the expression of these genes in Cc5 bacteria grown in HIHS to those of bacteria grown in HIHS supplemented with iron (III) citrate as a source of free iron. The addition of iron (III) citrate led to a ca. 2-fold decrease in the expression of all three PUL3 genes, indicating that PUL3 expression is modulated by the presence of free iron in the serum (see Fig. S2 in the supplemental material).
In many bacteria, the expression of genes involved in iron storage and iron uptake, such as iron channels, as well as transferrin and hemoglobin binding proteins and siderophores, is regulated by the transcriptional regulator FurA. Upon increasing the concentration of free iron, Fe2+ cations may bind to FurA, which then activates or represses gene transcription (56). Since the genome of Cc5 encodes a FurA-like protein (Ccan_15860), we generated a furA deletion mutant. We then quantified Ccan_03640 (icsA), Ccan_03650 (icsC), and Ccan_03680 (icsD) mRNA levels by real-time PCR, in the wt and the furA mutant, during growth in HIHS. The expression of PUL3 genes increased by about 2-fold in the furA mutant strain compared to wt levels (see Fig. S2 in the supplemental material). These results suggest that furA regulates PUL3 and reinforces the previous results showing that PUL3 is modulated by iron.
PUL3 encodes a system capturing iron from human transferrin.
In order to identify the source of iron exploited by C. canimorsus in human serum, we first depleted the HIHS of most of its protein content until the growth of C. canimorsus became dependent on the supply of iron (III) chloride. Protein depletion was monitored by silver-stained SDS-PAGEs and mass spectrometry analysis (Fig. 3A). With the exception of small amounts of human serum albumin (Uniprot accession number P02768), only trace amounts of other proteins could be detected in the PDHS. Depletion of STF, the major iron-binding protein in human serum, was confirmed specifically by Western blotting (Fig. 3B). We then tested whether the addition of human serotransferrin could restore growth in this PDHS. As shown in Fig. 3C, human iron-bound STF could restore the growth of wt Cc5 bacteria but not of ΔPUL3 mutant bacteria. In contrast, when human ApoSTF was used instead of its iron-loaded counterpart, neither wt bacteria nor the ΔPUL3 bacteria grew. Additionally, we monitored the uptake of iron from 55Fe-loaded transferrin by wt and ΔPUL3 mutant Cc5 bacteria. As shown in Fig. 3D, over a period of 24 h, wt Cc5 bacteria assimilated around 200-fold more 55Fe at 37°C than on ice, indicating that the capture mechanism is an active mechanism. In good agreement with the previous data, at 37°C, ΔPUL3 mutant bacteria captured around 80-fold less iron than did wt bacteria. Together, these data demonstrate that PUL3 encodes an ICS that allows iron scavenging from transferrin.
FIG 3.
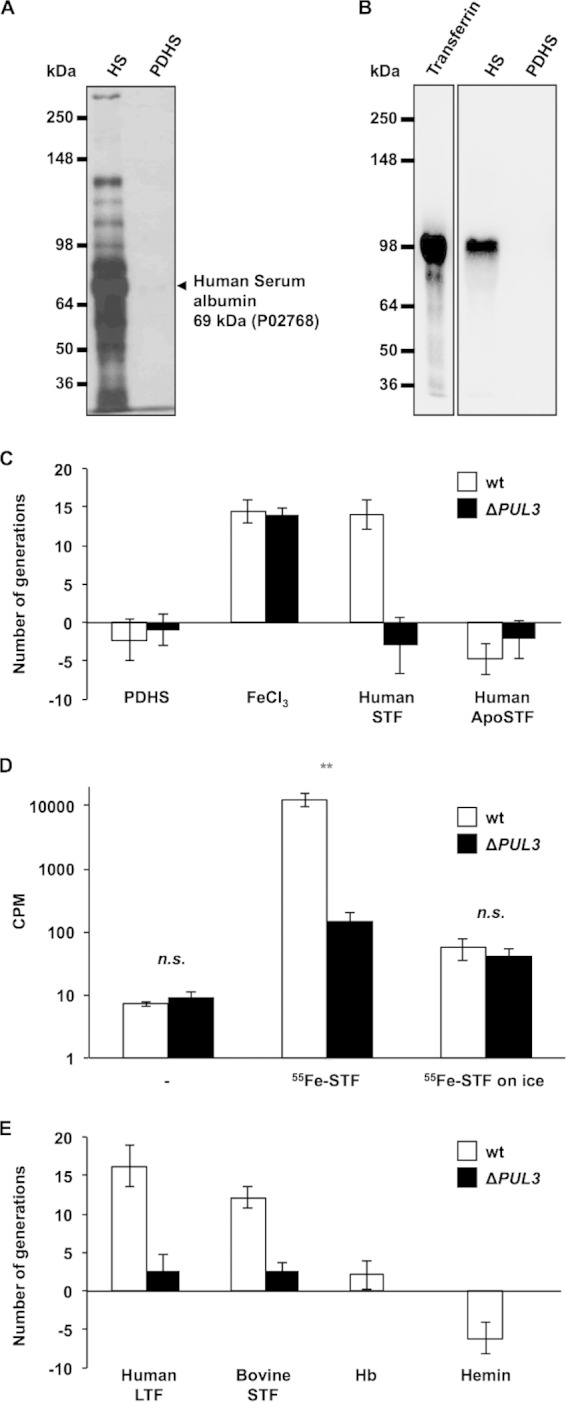
PUL3 encodes an iron capture system targeting transferrins. (A) Silver-stained SDS-PAGE of normal human serum (HS) (0.1 μl) and protein-depleted human serum (PDHS) (10 μl). The arrow indicates traces of serum albumin as identified by mass spectrometry. Numbers on the left indicate the protein masses of the references in kDa. (B) Anti-transferrin Western blot. The first lane corresponds to purified human serotransferrin (0.3 μg). Lanes two and three were loaded as described for lanes one and two of panel A. Numbers on the left indicate the protein masses of the references in kDa. (C) Number of generations achieved by wt (white bars) and ΔPUL3 mutant (black bars) bacteria after 23 h in PDHS supplemented with 0.25 mM iron (III) chloride (FeCl3) and human serotransferrin at 3 g · liter−1 (human STF) or human apo-serotransferrin at 3 g · liter−1 (human ApoSTF). (D) Uptake of iron from transferrin by C. canimorsus cells. Number of cpm (counts per minute) measured for wt (white bars) and ΔPUL3 mutant (black bars) Cc5 bacteria incubated without (−) or with 55Fe-labeled serotransferrin (55Fe-STF) for 24 h at 37°C. Bacteria incubated on ice in the presence of 55Fe-labeled STF serve as the control. Error bars indicate standard deviations (averages from 3 experiments). **, t test error probability of <0.01. (E) Number of generations achieved by wt (white bars) and ΔPUL3 mutant (black bars) bacteria after 23 h in PDHS supplemented with human lactoferrin at 1.5 g · liter−1 (human LTF), bovine serotransferrin at 3 g · liter−1 (bovine STF), hemoglobin at 0.1 mM (Hb), and hemin at 0.25 mM. Error bars represent standard deviations (averages from 3 experiments). All differences above 9 generations have t test-based P values below 0.0034.
Given the oral ecology of C. canimorsus, we tested whether lactoferrin (LTF), which is abundant in saliva and body fluids, also could serve as an iron source. Like human STF, human LTF could restore the growth of the wt but not of the ΔPUL3 mutant bacteria in PDHS (Fig. 3E). Since humans are not a natural host for C. canimorsus, we suspected that the ICS would not be human specific. Indeed, bovine STF could serve as an iron source in a PUL3-dependent manner (Fig. 3E). Despite the broad recognition spectrum among members of the transferrin family, other iron-binding molecules found in the human body, such as hemoglobin or hemin, could not restore the growth defect of wt bacteria in PDHS, indicating that C. canimorsus is not able to directly take up heme or to secrete hemophores (Fig. 3E). This suggests that the PUL3-encoded system is specific for proteins of the transferrin family.
Iron capture from transferrin does not involve soluble factors.
Several attempts to demonstrate the direct binding of transferrin to the ICS turned out to be unsuccessful. Hence, we had to exclude that the ICS could be involved in the synthesis, the release, or the capture of an intermediary siderophore. To do this, we performed a series of cross-feeding experiments between wt and individual PUL3 gene mutants. We first confirmed that the growth defect of the ΔPUL3 mutant bacteria in HIHS still could be rescued by the addition of iron in the presence of wt bacteria, indicating that there is no competition between the strains (see Fig. S3A in the supplemental material). We then tested whether the mutants lacking a single ics gene could grow in HIHS in the presence of wt bacteria. As shown in Fig. S3B in the supplemental material, the presence of wt bacteria did not allow the growth of any ics mutant. Thus, we can exclude that PUL3 gene products serve to export or synthesize a soluble siderophore.
We then examined the genome of Cc5 to detect genes involved in siderophore synthesis. The search included genes encoding the synthesis of enterochelin, vibriobactin, pyochelin, yersiniabactin, mycobactin, corynebactin, bacillibactin, myxochelin A or B, and, more generally, carboxylate, catecholate, and hydroxamate siderophores. No homologs were detected, suggesting that C. canimorsus does not produce already-known siderophores, but one cannot exclude that C. canimorsus synthesizes a totally new and unknown class of iron-fetching molecules. Therefore, we attempted to detect a siderophore in the concentrated HIHS culture supernatant of Cc5, taking Pseudomonas aeruginosa PAO1 (57, 58) as a control. While the chrome azurol technique (46, 47) detected a siderophore in the culture supernatant of PAO1, even after a 10-fold dilution, it gave a negative result for the undiluted Cc5 culture supernatant at a comparable biomass (see Fig. S3C and D in the supplemental material).
Although these observations do not formally rule out that C. canimorsus secretes a siderophore that would be captured by the ICS, they make it unlikely.
Iron capture occurs independently of the N-glycosylation of transferrin.
Since C. canimorsus has been shown to deglycosylate N-linked glycoproteins through the PUL5-encoded GpdG complex (54), we investigated whether the glycosylation state of transferrin plays a role in iron capture. We first monitored the glycosylation state of the protein prior to and after incubation with C. canimorsus. Not surprisingly, we observed a strong deglycosylation of the N-linked glycan chains of human STF by wild-type C. canimorsus, and this deglycosylation turned out to be dependent on PUL5 (see Fig. S4A and B in the supplemental material). However, deletion of PUL5 had only a slight effect on growth in HIHS (see Fig. S1), suggesting that the iron capture system is not acting downstream of the PUL5-encoded Gpd complex (54). In addition, nondenaturing removal of N-linked glycan chains from human STF with a PNGase F treatment prior to PDHS supplementation (Fig. 4A) did not alter iron chelation by STF, as indicated by the low growth level of the ΔPUL3 mutant, or prevent the ICS activity in the case of wt bacteria (Fig. 4A and B). These observations indicate that N-linked glycans of human transferrin do not play any determinant role in the process of iron extraction from STF.
FIG 4.

Process of iron capture from STF is independent from N-linked glycan chains. (A) Sambucus nigra lectin (SNA) staining (top) and anti-serotransferrin immunoblot (bottom) of human serotransferrin (STF) after treatment with fresh (lane 1) and heat-inactivated (lane 2) PNGase F. The black arrow corresponds to the position of the intact protein, while the gray arrow indicates the faint shifted band of the N-deglycosylated STF. Numbers on the left indicate the protein mass of the references in kDa. (B) Number of generations achieved by wt (white bars) and PUL3-deleted (black bars) bacteria after 23 h in PDHS supplemented with human STF (120 mg · liter−1) treated with either fresh or heat-inactivated PNGase F. Error bars represent standard deviations (averages from 3 experiments). For comparisons to wt values, t test-based error probabilities were <0.01 (**) and <0.001 (***), respectively.
In C. canimorsus and C. cynodegmi, the capacity to grow in HIHS correlates with the presence of ics genes.
We mentioned before that among the strains for which the full genome was sequenced, there was a perfect correlation between growth in HIHS and the presence of PUL3. We then sought to further validate the hypothesis that growth in HIHS depends on the capacity to acquire iron by testing the effect of iron supplementation on the growth of 15 strains otherwise unable to grow on HIHS. These 15 strains were known to be devoid of PUL3 because their full genome was sequenced (CcD38, CcD93, and CcD95) or because the individual ics genes could not be amplified by PCR (12 strains) (data not shown). As expected, the addition of an excess of free iron strongly enhanced the growth of all of these strains in HIHS (Fig. 5).
FIG 5.

Dog strains unable to grow in HIHS are rescued by the addition of iron. Shown are the number of generations achieved by the wt and ΔPUL3 isolates, three sequenced dog isolates (CcD95, CcD93, and CcD38), and 12 other randomly picked dog isolates after 23 h in HIHS alone (white bars) or supplemented with 0.25 mM iron (III) citrate (FeC6H5O7) (black bars). Error bars indicate standard deviations (averages from 3 experiments). An asterisk indicates sequenced dog strains.
In conclusion, growth in HIHS globally correlates with the presence of PUL3, and the absence of growth in HIHS correlates with the absence of PUL3 genes. All of this suggests that the ICS, encoded within the accessory genome of Capnocytophaga, is a major factor responsible for iron capture and, by extension, for growth in HIHS.
The complete ICS is found in Bacteroidetes species most frequently isolated from human infections.
Each of the 9 genes of PUL3 (Ccan_03640-Ccan_03720) was considered to assess the occurrence of the ICS within the bacterial kingdom. Search models for each gene of PUL3 were built and screened against the complete genome database (http://www.ncbi.nlm.nih.gov/genome/browse/). Out of the 2,100 genomes screened, the two genes which were not involved in the ICS (Ccan_03660 and Ccan_03670) were found in a large taxonomic range and frequently were independent of the occurrence of the other genes of PUL3 (data not shown). In contrast, the seven ics genes were identified only in synteny in the complete genomes of three other Bacteroidetes isolated from infected humans: Bacteroides fragilis YCH46 (NC_006347), isolated from a human septicemia, Bacteroides fragilis NCTC9343 uid57639 (NC_003228), isolated from an abdominal infection, and Odoribacter splanchnicus DSM20712 uid63397 (NC_015160), isolated from an abdominal abscess (see Fig. S5A in the supplemental material). In addition, Riemerella anatipestifer DSM15868 uid60727 (NC_014738), isolated from a duck infectious serositis, also possesses the seven ics genes, although the synteny is not entirely conserved (see Fig. S5A).
Additional PSI-BLAST searches for the ics genes were carried out against the nonredundant database. The complete set of genes required for the ICS again was exclusively identified in organisms implicated in human or animal infections. These include several Capnocytophaga and Prevotella species, diverse Riemerella anatipestifer isolates, and several additional Bacteroides fragilis isolates, Ornithobacterium rhinotracheale DSM 15997, Odoribacter splanchnicus DSM 20712, and Porphyromonas sp. strain F0450, oral taxon 279 (see Fig. S5B in the supplemental material). Thus, the ICS described here is present in a number of Bacteroidetes species with pathogenic potential. Interestingly, PUL3 occurs in bacteria that are able to infect not only mammals but also birds.
DISCUSSION
Here, we showed that nine C. canimorsus strains out of nine isolates from human infections grow and survive in HIHS, while only half of the strains isolated from the oral cavity of dogs do so. By genome comparison of representative isolates from groups with distinct growth capacities in HIHS, we could delimit a subset of 97 genes from the Capnocytophaga accessory genome potentially involved in growth and survival in human serum. Interestingly, this pool of genes was enriched in genes of the so-called polysaccharide utilization loci of Bacteroidetes (16 genes) (30). Out of the 13 PUL knockout mutants (37), two of them showed a moderate growth defect, while the deletion of PUL3 led to a dramatic impairment in the capacity to replicate in HIHS. As suggested by the functional annotation of IcsA, a FecA homologue (59), the PUL3-encoded machinery was found to be responsible for the acquisition of iron in human serum. Importantly, iron acquisition did not require IcsA only but also six other ics-encoded proteins (IcsC to IcsH). Consistent with its role in iron scavenging in human serum, the PUL3-encoded system proved to be essential for fetching iron ions from serotransferrin. Acquisition of iron via heme utilization has been described previously for Porphyromonas and Bacteroides (60–62); however, in the case of C. canimorsus, neither hemin nor hemoglobin was able to rescue iron deprivation in the PDHS, indicating that C. canimorsus is not able to directly take up heme or secrete hemophores. Additionally, the hypothesis that PUL3 is involved in the release of a siderophore was investigated through different approaches and no evidence could be gained, suggesting that iron capture from transferrin does not involve soluble factors. Thus, by analogy with the systems encoded by other PUL, we hypothesize that the iron capture system (ICS) directly interacts with STF, but this could not be formally demonstrated because of the existence of another receptor, still unidentified, that binds many glycoproteins, including STF. Clearly, further work is needed to decipher the mechanism by which the PUL3-encoded Sus-like machinery captures iron from transferrin.
Evolutionarily distant from the Tbp or Lbp system of pathogenic Neisseriaceae and Pasteurellaceae (63) or from the staphylococcal transferrin receptor (64), the C. canimorsus ICS initially had been annotated as a polysaccharide utilization system. Indeed, like the canonical starch utilization system (Sus), it consists of SusC and SusD homologs and additional lipoproteins, coencoded within a single putative operon. Despite these classical features of typical polysaccharide-degrading complexes of Bacteroidetes, the ICS was shown to function independently from the presence or absence of N-linked transferrin glycan moieties. Whether this capture involves some glycan chains of transferrin still needs to be clarified. Another point that requires further clarification is the requirement of the two different SusC-like (putative TonB-dependent porins) proteins IcsA and IcsC.
The presence of a partially conserved PUL3 devoid of the fecA-like transporter gene (icsA) in several environmental and plant-associated Bacteroidetes spp. suggests the existence of an ancestral version of PUL3 possibly devoted to a classical carbohydrate substrate. On the other hand, with 341 genome hits, icsA is the ics gene with the broadest taxonomic occurrence. It can be identified in genomes from diverse taxonomic groups, including Proteobacteria, Spirochetes, Bacteroidetes, or green sulfur bacteria. This contrasts with the occurrence of the other SusC-like gene, icsC, which was identified in only 54 genomes, including 48 from the Bacteroidetes phylum (data not shown). This taxonomic restriction to the Bacteroidetes phylum is typical of PUL genes and suggests that icsA has been integrated into a classical PUL, which then evolved as a complex iron acquisition system. The other genes of PUL3 essential for iron capture (icsD, icsE, icsF, icsG, and icsH) were exclusively identified among Bacteroidetes, with icsG being found exclusively in genomes including the six other ics genes and representing a good marker for the presence of the ICS in other organisms.
Strikingly, the correlation between the occurrence of PUL3 genes in C. canimorsus and the capacity to grow in HIHS strongly suggests a crucial role of the ICS in the process of converting harmless commensal C. canimorsus into potential pathogens. The deep compositional DNA bias (55) shared by the genes of PUL3 (from Ccan_03640 [icsA] to Ccan_03720 [icsH]) with respect to the chromosomal backbone indicates that they were acquired from another organism at the same time. Thus, it is not surprising to repeatedly find a conserved version of PUL3 in the genome of Bacteroidetes species most frequently isolated from human infections (e.g., for many clinical isolates of B. fragilis). To our knowledge, this is the first report of a PUL-encoded system serving a purpose other than glycan chain degradation and, by extension, iron acquisition. Besides, the ICS is unique among Gram-negative bacteria in that it can handle a wide range of transferrin isomers, including paralogic (e.g., human STF and human LTF) and orthologic (e.g., human STF and bovine STF) variants, potentially allowing growth in different host environments. This feature consequently explains the taxonomic spread of the ICS among pathogens, which can be considered a key virulence factor of Bacteroidetes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Paul Jenö and Suzanne Moes for their help with mass spectrometry and Simon Ittig, Ulrich Wiesand, and Klaus Handloser for their technical support. We also thank Urs Jenal and Peter Broz for sharing laboratory resources.
This work was supported by grant 3100A0-128659 from the Swiss National Science Foundation and ERC 2011-ADG 20110310 from the European Research Council to G.R.C.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02042-14.
REFERENCES
- 1.Bobo RA, Newton EJ. 1976. A previously undescribed gram-negative bacillus causing septicemia and meningitis. Am J Clin Pathol 65:564–569. [DOI] [PubMed] [Google Scholar]
- 2.Brenner DJ, Hollis DG, Fanning GR, Weaver RE. 1989. Capnocytophaga canimorsus sp. nov. (formerly CDC group DF-2), a cause of septicemia following dog bite, and C. cynodegmi sp. nov., a cause of localized wound infection following dog bite. J Clin Microbiol 27:231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pers C, Gahrn-Hansen B, Frederiksen W. 1996. Capnocytophaga canimorsus septicemia in Denmark, 1982-1995: review of 39 cases. Clin Infect Dis 23:71–75. doi: 10.1093/clinids/23.1.71. [DOI] [PubMed] [Google Scholar]
- 4.Le Moal G, Landron C, Grollier G, Robert R, Burucoa C. 2003. Meningitis due to Capnocytophaga canimorsus after receipt of a dog bite: case report and review of the literature. Clin Infect Dis 36:e42–46. doi: 10.1086/345477. [DOI] [PubMed] [Google Scholar]
- 5.Janda JM, Graves MH, Lindquist D, Probert WS. 2006. Diagnosing Capnocytophaga canimorsus infections. Emerg Infect Dis 12:340–342. doi: 10.3201/eid1202.050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westwell AJ, Kerr K, Spencer MB, Hutchinson DN. 1989. DF-2 infection. BMJ 298:116–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailie WE, Stowe EC, Schmitt AM. 1978. Aerobic bacterial flora of oral and nasal fluids of canines with reference to bacteria associated with bites. J Clin Microbiol 7:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tierney DM, Strauss LP, Sanchez JL. 2006. Capnocytophaga canimorsus mycotic abdominal aortic aneurysm: why the mailman is afraid of dogs. J Clin Microbiol 44:649–651. doi: 10.1128/JCM.44.2.649-651.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lion C, Escande F, Burdin JC. 1996. Capnocytophaga canimorsus infections in human: review of the literature and cases report. Eur J Epidemiol 12:521–533. doi: 10.1007/BF00144007. [DOI] [PubMed] [Google Scholar]
- 10.Hantson P, Gautier PE, Vekemans MC, Fievez P, Evrard P, Wauters G, Mahieu P. 1991. Fatal Capnocytophaga canimorsus septicemia in a previously healthy woman. Ann Emerg Med 20:93–94. doi: 10.1016/S0196-0644(05)81130-8. [DOI] [PubMed] [Google Scholar]
- 11.Saab M, Corcoran JP, Southworth SA, Randall PE. 1998. Fatal septicaemia in a previously healthy man following a dog bite. Int J Clin Pract 52:205. [PubMed] [Google Scholar]
- 12.Deshmukh PM, Camp CJ, Rose FB, Narayanan S. 2004. Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee. Am J Med Sci 327:369–372. doi: 10.1097/00000441-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 13.Mally M, Paroz C, Shin H, Meyer S, Soussoula LV, Schmiediger U, Saillen-Paroz C, Cornelis GR. 2009. Prevalence of Capnocytophaga canimorsus in dogs and occurrence of potential virulence factors. Microbes Infect 11:509–514. doi: 10.1016/j.micinf.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki M, Kimura M, Imaoka K, Yamada A. 2010. Prevalence of Capnocytophaga canimorsus and Capnocytophaga cynodegmi in dogs and cats determined by using a newly established species-specific PCR. Vet Microbiol 144:172–176. doi: 10.1016/j.vetmic.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Umeda K, Hatakeyama R, Abe T, Takakura KI, Wada T, Ogasawara J, Sanada SI, Hase A. 2014. Distribution of Capnocytophaga canimorsus in dogs and cats with genetic characterization of isolates. Vet Microbiol 171:153–159. 10.1016/j.vetmic.2014.03.023 [DOI] [PubMed] [Google Scholar]
- 16.Blanche P, Bloch E, Sicard D. 1998. Capnocytophaga canimorsus in the oral flora of dogs and cats. J Infect 36:134. [DOI] [PubMed] [Google Scholar]
- 17.Kolenbrander PE, Palmer RJ Jr, Periasamy S, Jakubovics NS. 2010. Oral multispecies biofilm development and the key role of cell-cell distance. Nat Rev Microbiol 8:471–480. doi: 10.1038/nrmicro2381. [DOI] [PubMed] [Google Scholar]
- 18.Frandsen EV, Poulsen K, Kononen E, Kilian M. 2008. Diversity of Capnocytophaga species in children and description of Capnocytophaga leadbetteri sp. nov. and Capnocytophaga genospecies AHN8471. Int J Syst Evol Microbiol 58:324–336. doi: 10.1099/ijs.0.65373-0. [DOI] [PubMed] [Google Scholar]
- 19.McBride MJ. 2004. Cytophaga-flavobacterium gliding motility. J Mol Microbiol Biotechnol 7:63–71. doi: 10.1159/000077870. [DOI] [PubMed] [Google Scholar]
- 20.Duchaud E, Boussaha M, Loux V, Bernardet JF, Michel C, Kerouault B, Mondot S, Nicolas P, Bossy R, Caron C, Bessieres P, Gibrat JF, Claverol S, Dumetz F, Le Henaff M, Benmansour A. 2007. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat Biotechnol 25:763–769. doi: 10.1038/nbt1313. [DOI] [PubMed] [Google Scholar]
- 21.Segers P, Mannheim W, Vancanneyt M, De Brandt K, Hinz KH, Kersters K, Vandamme P. 1993. Riemerella anatipestifer gen. nov., comb nov, the causative agent of septicemia anserum exsudativa, and its phylogenetic affiliation within the Flavobacterium-Cytophaga rRNA homology group. Int J Syst Bacteriol 43:768–776. doi: 10.1099/00207713-43-4-768. [DOI] [PubMed] [Google Scholar]
- 22.Subramaniam S, Huang B, Loh H, Kwang J, Tan HM, Chua KL, Frey J. 2000. Characterization of a predominant immunogenic outer membrane protein of Riemerella anatipestifer. Clin Diagn Lab Immunol 7:168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coyne MJ, Chatzidaki-Livanis M, Paoletti LC, Comstock LE. 2008. Role of glycan synthesis in colonization of the mammalian gut by the bacterial symbiont Bacteroides fragilis. Proc Natl Acad Sci U S A 105:13099–13104. doi: 10.1073/pnas.0804220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reeves AR, Wang GR, Salyers AA. 1997. Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron. J Bacteriol 179:643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho KH, Salyers AA. 2001. Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron. J Bacteriol 183:7224–7230. doi: 10.1128/JB.183.24.7224-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. 2009. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem 284:24673–24677. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves AR, D'Elia JN, Frias J, Salyers AA. 1996. A Bacteroides thetaiotaomicron outer membrane protein that is essential for utilization of maltooligosaccharides and starch. J Bacteriol 178:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koropatkin NM, Martens EC, Gordon JI, Smith TJ. 2008. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16:1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shipman JA, Berleman JE, Salyers AA. 2000. Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J Bacteriol 182:5365–5372. doi: 10.1128/JB.182.19.5365-5372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shipman JA, Cho KH, Siegel HA, Salyers AA. 1999. Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron. J Bacteriol 181:7206–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martens EC, Chiang HC, Gordon JI. 2008. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 33.Bjursell MK, Martens EC, Gordon JI. 2006. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem 281:36269–36279. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- 34.McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, Goltsman E, Wang W, Xu J, Hunnicutt DW, Staroscik AM, Hoover TR, Cheng YQ, Stein JL. 2009. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl Environ Microbiol 75:6864–6875. doi: 10.1128/AEM.01495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manfredi P, Pagni M, Cornelis GR. 2011. Complete genome sequence of the dog commensal and human pathogen Capnocytophaga canimorsus strain 5. J Bacteriol 193:5558–5559. doi: 10.1128/JB.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin H, Mally M, Kuhn M, Paroz C, Cornelis GR. 2007. Escape from immune surveillance by Capnocytophaga canimorsus. J Infect Dis 195:375–386. doi: 10.1086/510243. [DOI] [PubMed] [Google Scholar]
- 37.Manfredi P, Renzi F, Mally M, Sauteur L, Schmaler M, Moes S, Jeno P, Cornelis GR. 2011. The genome and surface proteome of Capnocytophaga canimorsus reveal a key role of glycan foraging systems in host glycoproteins deglycosylation. Mol Microbiol 81:1050–1060. doi: 10.1111/j.1365-2958.2011.07750.x. [DOI] [PubMed] [Google Scholar]
- 38.Mally M, Shin H, Paroz C, Landmann R, Cornelis GR. 2008. Capnocytophaga canimorsus: a human pathogen feeding at the surface of epithelial cells and phagocytes. PLoS Pathog 4:e1000164. doi: 10.1371/journal.ppat.1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 40.Mally M, Cornelis GR. 2008. Genetic tools for studying Capnocytophaga canimorsus. Appl Environ Microbiol 74:6369–6377. doi: 10.1128/AEM.01218-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schagger H, von Jagow G. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 42.Rabilloud T, Carpentier G, Tarroux P. 1988. Improvement and simplification of low-background silver staining of proteins by using sodium dithionite. Electrophoresis 9:288–291. doi: 10.1002/elps.1150090608. [DOI] [PubMed] [Google Scholar]
- 43.Valcour AA, Krzymowski G, Onoroski M, Bowers GN Jr, McComb RB. 1990. Proposed reference method for iron in serum used to evaluate two automated iron methods. Clin Chem 36:1789–1792. [PubMed] [Google Scholar]
- 44.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. 2004. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem 331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 45.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexander DB, Zuberer DA. 1991. Use of chrome azurol S reagents to evaluate siderophore production by rhizosphere bacteria. Biol Fertil Soils 12:39–45. doi: 10.1007/BF00369386. [DOI] [Google Scholar]
- 47.Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal Biochem 160:47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
- 48.Grenier D, Tanabe S. 2011. Transferrin as a source of iron for Campylobacter rectus. J Oral Microbiol 2011:3. doi: 10.3402/jom.v3i0.5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goulet V, Britigan B, Nakayama K, Grenier D. 2004. Cleavage of human transferrin by Porphyromonas gingivalis gingipains promotes growth and formation of hydroxyl radicals. Infect Immun 72:4351–4356. doi: 10.1128/IAI.72.8.4351-4356.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Jaroszewski L, Godzik A. 2002. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18:77–82. doi: 10.1093/bioinformatics/18.1.77. [DOI] [PubMed] [Google Scholar]
- 51.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 53.Shin H, Mally M, Meyer S, Fiechter C, Paroz C, Zaehringer U, Cornelis GR. 2009. Resistance of Capnocytophaga canimorsus to killing by human complement and polymorphonuclear leukocytes. Infect Immun 77:2262–2271. doi: 10.1128/IAI.01324-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Renzi F, Manfredi P, Mally M, Moes S, Jeno P, Cornelis GR. 2011. The N-glycan glycoprotein deglycosylation complex (Gpd) from Capnocytophaga canimorsus deglycosylates human IgG. PLoS Pathog 7:e1002118. doi: 10.1371/journal.ppat.1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vernikos GS, Parkhill J. 2006. Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22:2196–2203. doi: 10.1093/bioinformatics/btl369. [DOI] [PubMed] [Google Scholar]
- 56.Carpenter BM, Whitmire JM, Merrell DS. 2009. This is not your mother's repressor: the complex role of fur in pathogenesis. Infect Immun 77:2590–2601. doi: 10.1128/IAI.00116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cornelis P. 2010. Iron uptake and metabolism in pseudomonads. Appl Microbiol Biotechnol 86:1637–1645. doi: 10.1007/s00253-010-2550-2. [DOI] [PubMed] [Google Scholar]
- 58.Cornelis P, Dingemans J. 2013. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front Cell Infect Microbiol 3:75. doi: 10.3389/fcimb.2013.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hussein S, Hantke K, Braun V. 1981. Citrate-dependent iron transport system in Escherichia coli K-12. Eur J Biochem 117:431–437. doi: 10.1111/j.1432-1033.1981.tb06357.x. [DOI] [PubMed] [Google Scholar]
- 60.Olczak T, Sroka A, Potempa J, Olczak M. 2008. Porphyromonas gingivalis HmuY and HmuR: further characterization of a novel mechanism of heme utilization. Arch Microbiol 189:197–210. doi: 10.1007/s00203-007-0309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Otto BR, Kusters JG, Luirink J, de Graaf FK, Oudega B. 1996. Molecular characterization of a heme-binding protein of Bacteroides fragilis BE1. Infect Immun 64:4345–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otto BR, Sparrius M, Wors DJ, de Graaf FK, MacLaren DM. 1994. Utilization of haem from the haptoglobin-haemoglobin complex by Bacteroides fragilis. Microb Pathog 17:137–147. doi: 10.1006/mpat.1994.1060. [DOI] [PubMed] [Google Scholar]
- 63.Fuller CA, Yu R, Irwin SW, Schryvers AB. 1998. Biochemical evidence for a conserved interaction between bacterial transferrin binding protein A and transferrin binding protein B. Microb Pathog 24:75–87. doi: 10.1006/mpat.1997.0174. [DOI] [PubMed] [Google Scholar]
- 64.Modun B, Morrissey J, Williams P. 2000. The staphylococcal transferrin receptor: a glycolytic enzyme with novel functions. Trends Microbiol 8:231–237. doi: 10.1016/S0966-842X(00)01728-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.