

Abstract
Since the discovery of phenothiazines as tau protein aggregation inhibitors, many additional small molecule inhibitors of diverse chemotype have been discovered and characterized in biological model systems. Although direct inhibition of tau aggregation has shown promise as a potential treatment strategy for depressing neurofibrillary lesion formation in Alzheimer’s disease, the mechanism of action of these compounds has been unclear. However, recent studies have found that tau aggregation antagonists exert their effects through both covalent and non-covalent means, and have identified associated potency and selectivity driving features. Here we review small-molecule tau aggregation inhibitors with a focus on compound structure and inhibitory mechanism. The elucidation of inhibitory mechanism has implications for maximizing on-target efficacy while minimizing off-target side effects.
Keywords: Alzheimer’s disease, tau, neurofibrillary tangle, paired helical filaments, aggregation
INTRODUCTION
Neurofibrillary lesions composed of tau protein are a defining pathology of AD [1, 2]. The most commonly analyzed and modeled neurofibrillary lesion is the neurofibrillary tangle (NFT), which corresponds to tau deposited in cell bodies. However, the overwhelming majority of cortical tau deposits in AD appear within neuronal processes in the form of dystrophic neurites and neuropil threads [3]. The substantial increase in bulk tau levels that accompanies lesion formation results primarily from accumulation of insoluble tau aggregates [4], most likely because they or their misfolded precursors evade endogenous clearance mechanisms [5, 6]. In addition to serving as markers for differential diagnosis and staging of disease [7], tau aggregates can foster disease propagation (reviewed in [8]) and serve as direct sources of toxicity [9, 10]. Although the tau species that mediate toxicity and the mechanisms through which they act are not established (reviewed in [11]), work in model systems suggests several possibilities. First, mathematical modeling experiments predict that bulk accumulation of cytoplasmic aggregates in cell bodies can depress neuronal energy metabolism through molecular crowding effects once a critical threshold level is exceeded [12]. The effect of molecular crowding on the physiology of neuronal processes (where most tau deposition occurs) is unknown. Second, in addition to homotypic aggregation, tau can co-aggregate with other proteins [13], including microtubule associated proteins [14, 15], potentially depressing their levels. Although loss of normal tau protein is well tolerated in animal models (reviewed in [16]), simultaneous depletion of different classes of microtubule associated proteins has severe consequences [17, 18]. Finally, certain tau aggregates can directly disrupt membrane integrity [19]. For these reasons, and because tau aggregation is a purely pathological process unrelated to normal tau function, diverse strategies for inhibiting tau misfolding and aggregation are being investigated as potential therapies against neurofibrillary lesion formation and disease progression (reviewed in [20]).
Here we focus on direct inhibition of tau aggregation with small molecules as one approach for depressing and potentially reversing neurofibrillary lesion formation. The strategy is limited to early stages of disease before aggregates become irreversibly insoluble through crosslinking [21]. It also faces technological hurdles, including the natively unfolded structure of the tau target, which limits the potential for identifying high-affinity ligand binding sites, and the relatively large surface areas that mediate tau-tau interactions in disease, which could require impractically large molecules for direct antagonism (reviewed in [22]). Nonetheless, since the feasibility of inhibiting tau aggregation was established [23], hundreds of seemingly unrelated small-molecule inhibitors of tau aggregation have been disclosed [24-31]. One inhibitor, methylene blue (a phenothiazine), has shown promise for slowing dementia progression in a clinical study [32]. But methylene blue has diverse biological activities, and the role of tau aggregation inhibition in its clinical profile is not established (reviewed in [33]). To clarify the utility of tau aggregation inhibition in clinical or biological studies, a new generation of high-affinity and selective compounds are needed. The challenge is to elucidate mechanisms through which tau protein-protein interactions can be inhibited while preserving molecular characteristics of inhibitors appropriate for brain uptake, and ideally, oral administration.
THE TAU TARGET
Tau protein is abundant in brain tissue (low micromolar bulk concentration) where it functions in monomeric form as a microtubule-associated protein (reviewed in [34]). However, in AD brain it dissociates from microtubules and forms filamentous, cytoplasmic inclusions. Part of the attraction of tau aggregation as a drug discovery target stems from its selective association with disease and its irrelevance for normal function. Nonetheless, tau is an unusually complicated drug target owing to its structural features. First, monomeric tau is not a single entity, but a mixture of six isoforms that arise from alternative splicing of exons 2, 3, and 10 from MAPT gene transcripts (reviewed in [35]). Exons 2 and 3 encode 29-residue acidic inserts in the N-terminal projection domain of tau, whereas exon 10 encodes a 31-residue microtubule binding repeat in the C-terminal domain (Fig. 1). An efficacious aggregation inhibitor should interact with all six tau species ranging from 352 – 441 amino acids in length.
Fig. 1.
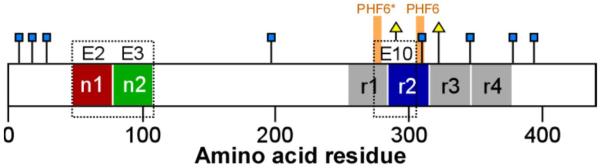
Tau protein primary structure. Human central nervous system tau is composed of six isoforms derived from alternatively splicing of exons E2, E3 and E10. Each isoform consists of an N-terminal projection domain containing up to two alternative segments (n1 and n2), and a C-terminal microtubule binding domain containing three or four imperfect repeats (r1 – r4; mapped on the basis of sequence homology as in [126]). They also contain one or two cysteine residues (triangles) that are prone to oxidation and adduct formation. Tau aggregation leading to filament formation is mediated by hexapeptide motifs (PHF6 and PHF6*) located in the microtubule binding domain. All tau isoforms also contain eight aromatic residues (squares), five of which (Y197, Y310, F346, F378, and Y394) engage in complex formation with non-covalent aggregation inhibitor PcTS [67] . Sequence numbering corresponds to the human 2N4R isoform (National Center for Biotechnology Information accession number NP_005901).
Second, tau is an intrinsically disordered protein that lacks the higher order structure normally associated with high-affinity ligand binding. In addition to fostering conformational flexibility, disordered structure exposes the majority of serine, threonine, and lysine residues to solvent where they can engage modification enzymes. For example, post-translational modification of tau in the form of phosphorylation exceeds 9 mol/mol stoichiometry in AD tissue distributed over dozens of sites (reviewed in [36]). Acetylation and methylation of lysine residues also has been reported although the stoichiometry of these modifications has not [37-40]. Disordered structure also exposes hydrophobic and nucleophilic cysteine residues to solvent (one in 3R forms, two in 4R forms; Fig. 1) where they can oxidize to form adducts with electrophilic compounds or disulfide cross links among or within tau proteins. In fact, both tau cysteines are flanked by basic lysine residues that maximize sulfhydryl reactivity [41]. The combination of differing post-translational modifications and sulfhydryl contents confer additional structural heterogeneity on tau proteins.
Finally, in disease, tau enters aggregation pathways that yield mature filaments containing cross-β-sheet structure as well as a range of smaller aggregate forms. The core of AD-tissue derived tau filaments is composed of at least three microtubule binding repeats [42], which in synthetic filaments adopts parallel in-register β-sheet structure [43, 44]. The repeat region contains two hexapeptide motifs, 275VQIINK280 (termed PHF6*) and 306VQIVYK311 (termed PHF6) that are essential for fibril formation [45] (Fig. 1). In vitro, one pathway to mature filaments leverages a nucleation-elongation mechanism [46, 47], where the rate-limiting nucleation event corresponds to dimerization (Fig. 2A), and the efficient elongation phase corresponds to addition of monomers to the ends of growing polymers. This mechanism is ideal for generating a limited number of aggregates that achieve long length. A pathway involving isodesmic assembly of small aggregates constitutes an alternative route to these stable structures [48-50] (Fig. 2B). Small aggregates can be SDS-stable, with some forms involving disulfide bonds, and others that do not [51]. Regardless of aggregation mechanism, the toxicity associated with aggregates in biological models correlates inversely with aggregate size (reviewed in [52]). Although the structural basis of aggregate toxicity is not clear, these data indicate that small-molecule antagonists of aggregation ideally should depress the formation of a range of aggregated species associated with tau misfolding in disease.
Fig. 2.

Aggregation pathways leading to tau filament formation in vitro. A, nucleation – dependent pathway, where natively unfolded monomer (1) adopts an aggregation competent conformation (2) in the presence of aggregation inducer. For heparin inducer, this conformation is associated with monomer compaction [127]. Nucleation corresponds to dimerization of this species (3), whereas elongation is mediated by monomer addition to yield mature filaments (4). This pathway dominates in vitro for certain tau constructs and inducers [46, 47, 49]. B, oligomerization pathway, where natively unfolded monomer (1) interacts with aggregation inducer to form oligomers (3a) that can self associate isodesmically to form mature filaments (4). This pathway dominates in vitro for certain tau truncation constructs in the presence of heparin inducer [49]. Secondary pathways involving filament fragmentation and secondary nucleation that can contribute to aggregation kinetic profile [128] are not shown.
AGGREGATION ASSAY METHODS
Identification and analysis of tau aggregation inhibitors relies primarily on assays conducted in vitro with recombinant human tau proteins. These assays leverage exogenous anionic inducers such as heparin [53] or anionic surfactants [54] to increase the rate and extent of tau aggregation. Aggregation propensity can be further increased by employing tau fragments comprising the microtubule binding repeat region instead of full-length protein [55-57]. Nonetheless, aggregation assays still require 2 – 20 micromolar tau protein to support measurable aggregate products over tractable incubation times. Primary assays that directly detect aggregation products include ultracentrifugation [25], thioflavin dye-based fluorescence [25, 58], ultrafiltration [59, 60], solid-phase immunoassay [26], and electron microscopy [61, 62] approaches. Recently developed cell-based methods will likely prove useful as well [63, 64]. Fluorescence-based assays have the greatest throughput, and so are well suited for primary screens, whereas electron microscopy approaches provide more detailed information regarding product morphology, quantity, and composition [62]. Regardless of modality, tau aggregation assays have limited ability to refine structure activity relationships because of the high tau concentrations needed to support aggregation. For inhibitors that interact stoichiometrically with tau monomers, it is not possible to resolve inhibitory potency below the concentration of target [65]. As a result, more progress has been made in identifying scaffold classes and mechanism of action than in optimizing inhibitor potency to the levels of traditional, receptor-targeted agents (reviewed in [66]).
Secondary assay methods detect tau-ligand interactions rather than inhibitory activity, and can in principle be applied over a wider range of tau concentrations and employ higher resolution methodology than primary assays. For example, NMR spectroscopy can detect direct interactions between ligand and tau protein at amino acid resolution [67]. Interactions between small molecules and monomeric β-amyloid [68] and α-synuclein [69-71] have been reported as well. However, this approach requires high-micromolar to low-millimolar concentrations of ligand, and therefore is limited to highly soluble ligands. Also, it is not clear that the detected interactions are maintained at pharmacologically relevant concentrations. In contrast, centrifugation assays coupled with immunoblot detection have much lower information content, but detect complex formation at significantly lower tau concentrations [72]. Intrinsic fluorescence methods have been applied to α-synuclein for detection of conformational changes accompanying complex formation [73-75] and may be applicable to tau as well. However, these approaches have the same limitations for structure activity relationship refinement as the primary assays discussed above.
Finally, in silico methods leverage three-dimensional models of tau aggregates elucidated by X-ray crystallographic or NMR spectroscopic methods to discover candidate inhibitor binding sites. One approach involves co-crystallizing tau fragments with ligands, then computationally refining resolved binding pockets or surfaces [76]. Novel ligands can be docked into the binding site identified in the model and optimized for interaction energy [77, 78]. The approach requires high-resolution atomic models, which for tau is limited to short peptide fragments that form steric zippers [79]. Also, the current binding poses appear heavily influenced by crystal contacts (i.e., the packing of ligand with protein during crystallization), and so may not recapitulate interactions with full-length tau under more physiological conditions.
A second in silico method again leverages three-dimensional tau aggregate models, but focuses on computational detection of solvent-exposed hydrogen bonds termed dehydrons (reviewed in [80]). Dehydrons are markers for protein-protein interactions because their sequestration from surrounding water can strengthen and stabilize otherwise exposed H-bonds by nearly two orders of magnitude. Therefore, ligand-mediated shielding of dehydrons is predicted to decrease the driving force for protein-protein interactions and also foster disaggregation. Potential interactions between dehydrons of a β-amyloid dimer and curcumin (a naturally occurring phenol in the Indian spice turmeric [81]) have been investigated by molecular dynamics simulations [82]. Application of this discovery approach to tau aggregates will depend on the availability of atomic models. However, unlike other computational approaches, its structural needs can be satisfied with models of tau aggregates alone rather than of aggregate-ligand complexes.
COVALENT TAU AGGREGATION INHIBITORS
Tau aggregation inhibitors identified to date fall into two broad mechanistic classes. The first class corresponds to agents that either covalently modify tau directly or foster formation of covalent bonds within or between tau proteins to yield aggregation-incompetent products. Covalent inhibitors can attack any or all species in an aggregation pathway, but appear to be especially efficacious modifiers of tau monomer, from which all aggregated species ultimately derive. For example, oleocanthal (Fig. 3), a natural product aldehyde, reacts with epsilon amino groups of lysine residues [83-85], including residues residing in the microtubule binding repeat region, to form imines (i.e., Schiff base formation). In contrast, α,β-unsaturated aldehydes such as cinnamaldehyde (Fig. 3) are electrophilic Michael acceptors that undergo nucleophilic attack by cysteine residues of tau [86]. Other α,β-unsaturated aldehydes, such as asperbenzaldehyde [60], may interact with tau in a similar manner. The electrophile need not be an aldehyde, as this moiety can be replaced with other electron withdrawing groups (reviewed in [87]). Nor is electrophilic character limited to Michael acceptors, as shown by the anti-aggregation activity of compounds containing electrophilic chloroacetyl moieties [88]. Furthermore, the electrophilic character of ligands can change during the course of experimentation, and unmask latent inhibitory activity. For example, baicalein, a polyalcohol flavonoid, can oxidize to quinone form, which then dominates the reaction as a covalent inhibitor [89] (Fig. 3). Generation of electrophiles through oxidation is of special concern because tau aggregation conditions used in many laboratories employ protracted incubation (≥8 h) in the absence of reducing agents that could otherwise slow compound oxidation and electrophilic addition [24, 90, 91]. Covalent inhibitors may be expected to nonspecifically interact with and disrupt the normal functions of off-target proteins as well. Nonetheless, dimethylfumarate, an electrophile capable of reacting covalently with cysteine sulfhydryls, recently was approved for oral treatment of multiple sclerosis [92], indicating that electrophilic compounds acting through covalent inhibitory mechanisms can be useful therapeutic agents.
Fig. 3.

Covalent tau aggregation inhibitors. These typically undergo either imine formation, where tau lysines attack their electrophilic aldehyde or ketone moieties (e.g, 5), or Michael addition, where tau cysteines attack their α,β-unsaturated electrophilic moieties (e.g., 6; box). Conversion of baicalein (7) to its quinone (8) through oxidation exemplifies how electrophlic character can change with protracted incubation. Arrows indicate potential sites of nucleophilic attack.
When left unprotected during incubation (i.e., by avoiding addition of exogenous reducing agents), the cysteine sulfhydryls of tau spontaneously oxidize to form inter- and intra-molecular disulfide bonds [51]. Depending on tau isoform, these products can support or inhibit tau aggregation [56]. Therefore, compounds that accelerate disulfide bond formation in the absence of reducing agent can appear to possess tau aggregation inhibitory activity. Aminothienopyridazines (ATPZs) are thought to inhibit tau aggregation through this mechanism [93]. Other redox-active compounds, including the phenothiazine methylene blue, also can modulate cysteine oxidation when incubated in the absence of exogenous reducing agents for extended incubation periods [93, 94]. Because a reducing intracellular environment is normally maintained by high concentrations of reduced sulfhydryl groups in the form of glutathione [95], it is likely that compounds acting solely through this mechanism will have low potency and efficacy in vivo.
NON-COVALENT TAU AGGREGATION INHIBITORS
The second broad class of aggregation inhibitor interacts with tau species noncovalently at various points in the aggregation pathway. These inhibitors are structurally diverse [66, 96, 97] and appear to act through multiple mechanisms that alone or in concert depress the aggregation propensity of tau-ligand complexes. For example, small molecules can interact directly with tau monomers. It has been proposed that even transient interactions could depress entry into aggregation pathways by altering the rate at which natively unfolded polypeptides adopt aggregation competent conformations [98]. The latter are characterized in part by solvent-exposed patches of hydrophobic residues. Theoretically, a rapid rate of interconversion between aggregation competent and incompetent conformations (i.e., the reconfiguration rate) should depress formation of stable intermolecular interactions with partner peptides by minimizing solvent exposure of hydrophobic residues that favor intermolecular association. The concept has been demonstrated with α-synuclein as the natively unfolded protein substrate and curcumin as the aggregation inhibitor [74]. Curcumin increased α-synuclein reconfiguration rate 15-fold at stoichiometric ligand concentration and 30-fold at 1.5:1 curcumin:peptide ratio while completely depressing stable protein-protein interactions. This mechanism of inhibition has the potential to halt aggregation at its earliest stages (i.e., before the formation of oligomers or mature fibrils) while favoring maintenance of soluble monomer. Because tau aggregation is sensitive to curcumin conjugates [99], this mechanism may be relevant for tau as well. However, it is unclear whether curcumin or other ligand could be optimized to interact with a specific molecular target relative to other natively unfolded peptides. In fact, ~50% of proteins contain long stretches of unfolded structure (reviewed in [100]), which could potentially cross react with inhibitors that target such regions.
Another class of ligand, termed molecular tweezers, interacts with natively unfolded tau monomers by selectively binding lysine side chains [101]. The ability of stoichiometric levels of lead molecular tweezer CLR01 to inhibit tau fibril formation has been shown in vitro [102]. Lower CLR01:protein ratios were required for inhibition of tau versus Aβ aggregation, suggesting that some target selectivity can be achieved. Although inhibition of tau aggregation may be direct through binding and sterically blocking lysine residues in the microtubule binding repeat region, recent experiments with α-synuclein indicate that CLR01 also may lower aggregation propensity by increasing reconfiguration rate [75]. CLR01 was tested in a triple-transgenic mouse model of early-onset AD (PSEN1 mutant M146V; APP mutant KM670/671NL; tau mutant P301L) with 40mg/kg/d CLR01 in saline for 28 days [102]. Despite its large size and fixed negative charge, CLR01 crossed the blood brain barrier. Immunohistochemistry showed a 33% decrease in amyloid as well as phospho-tau, however bulk tau levels were unchanged. It remains to be seen whether molecular tweezers can be optimized to interact with specific targets relative to other lysine-rich proteins such as histones.
Noncovalent tau aggregation inhibitors also may act by blocking formation of steric zipper structures common to cross-β-sheet forming peptides. Short segments of amyloidogenic sequences have been crystallized in forms that exhibit similar properties as their full-length counterparts, including diameter and helical pitch of fibril, diffraction pattern, and ability to bind dyes [78]. Co-crystals grown from KLVFFA segment of Aβ protein (10 mM) along with the small dye molecule Orange-G (1 mM) diffract to 1.8 Å resolution. The resulting three dimensional model revealed that the dye bound within the steric zipper of KLVFFA while making electrostatic interactions with lysine in an adjacent zipper. Orange-G as well as 1,1-dicyano-2-[6-(dimethylamino)naphthalen-2-yl]propene (DDNP) and curcumin also were co-crystallized with a peptide corresponding to the PHF6 motif of tau. Orange-G wedged itself into the steric zipper by shifting the interdigitation of the peptide side chains whereas DDNP and curcumin bound to cylindrical cavities formed by crystal contacts. This structure shows that small molecules could potentially interfere with stabilizing protomer interactions occurring perpendicular to fibril axes. It has been proposed that interference with π-stacking of aromatic side chains resident in many aggregation motifs, including PHF6 and PHF6* of tau protein, may inhibit protomer interactions occurring parallel to the fibril axis as well [103]. It is not clear whether such interactions occur in dilute solution in the absence of crystal contacts.
Finally, tau filament formation can be inhibited by sequestering tau in the form of stable off-pathway oligomers. For example, phthalocyanine tetrasulfonate (PcTS; a cyclic tetrapyrrole) interacts directly with tau monomers to form SDS-stable oligomers [67]. At high ligand and protein concentrations, binding is mediated by aromatic residues Y310, F346, and F378 (Fig. 1) with lesser involvement of Y197 and Y394, suggesting that interactions between the aromatic rings of inhibitor and amino acid sidechains play an important role in PcTS binding. Despite conversion into an oligomeric state, tau protomers did not adopt appreciable secondary structure in the form of α-helix or β-sheet. This was consistent with further EPR data showing that the nature of this PcTS-tau oligomeric complex was dynamic in nature. The size of the oligomeric complex increased with PcTS:tau ratio, varying from 5-11 tau molecules at equal ratios to >20 tau molecules at 15:1 PcTS:tau ratios. PcTS inhibited tau aggregation in neuroblastoma N2a cells almost completely at 50 μM concentration, suggesting that the oligomeric products are more efficiently cleared from cells than filamentous tau.
SDS-stable oligomers composed of full-length tau also rapidly form at low micromolar concentrations in the presence of cyanine, triarylmethine, rhodanine, and phenothiazine aggregation inhibitors [72, 90]. Although some triarylmethines and rhodanines have electrophilic character [104, 105], the rank order of tau aggregation inhibitory potency in the presence of excess sulfhydryl reagent (i.e., dithiothreitol) does not correlate well with electrophilicity. For example, the triarylmethine crystal violet is a weak electrophile [104], but an efficacious aggregation inhibitor [27]. At low micromolar tau concentrations, oligomer formation required the presence of aggregation inducer, suggesting that species residing along or off the aggregation pathway are substrates for inhibitor binding [72]. Furthermore, intact PHF6 and PHF6* hexapeptide motifs were not required for interaction with cyanine, indicating that species formed subsequent to filament nucleation (Fig. 2A) are not required for inhibitory activity. Brain permeable compounds acting through this mechanism could have therapeutic utility. First, cyanine inhibitors depress tau aggregation in ex vivo mouse models of tauopathy without inducing apoptotic responses associated with toxicity [27, 106]. Although oligomer formation has been linked to toxicity in some biological models [107], the physical characteristics of inhibitor-stabilized oligomers differ [67]. Second, the mechanism supports interaction with both 3R and 4R isoforms as well as missense tauopathy mutants, suggesting it could be broadly applicable to both AD and frontotemporal lobar degeneration diseases. Third, its depletion of aggregation competent monomers fosters endwise disaggregation of mature filaments [108], including those formed from filament-stabilizing pseudophosphorylation mutants [109]. Finally, it does not interact with natively unfolded tau monomer [72], which is an important binding partner of microtubules. In terms of compound optimization, tau oligomerization provides a secondary assay for interaction at the low tau concentrations and short incubation times needed to refine the structure activity relationships of high-potency inhibitors.
STRUCTURAL COMMONALITIES AMONG NONCOVALENT INHIBITORS
Despite commonalities in inhibitory mechanism, noncovalent inhibitors differ markedly in structure, and traditional structure activity analysis focused on sterics has identified only those features that drive potency within a series [91, 110]. However, many aggregation inhibitors share the absorbance characteristics of dyes (i.e., they absorb electromagnetic radiation in the visible spectrum), a property that stems from delocalized π-electron distribution [111]. For example, certain thiacarbocyanine, phenothiazine, and triarylmethine derivatives are among the most potent tau aggregation inhibitors reported [27], and all share a centrosymmetric structure with extensive delocalization of their fixed cationic charge (Fig. 4). The resulting structures are planar and highly “polarizable”, meaning that electron density can easily shift about the molecule when exposed to an external electric field, such as an adjacent dipole or ion. These properties are ideal for supporting van der Waals interactions with protein binding partners containing their own highly polarizable moieties, such as aromatic side chains [112, 113]. The resulting interactions are nearly electrostatic in strength [114]. In particular, they rationalize the selective association of ligands with the aromatic amino acid side chains of tau [67] and other aggregating proteins [82]. Although such interactions may be nonspecific at high tau and ligand concentrations, their dependence on inducer at low tau concentrations [72] suggests that protein conformation also may contribute to binding affinity. Identification of these conformations in the context of tau protein could help speed optimization of aggregation inhibitors.
Fig. 4.

Non-covalent tau aggregation inhibitors. Models of representative members of cyanine (9), phenothiazine (10), and triarylmethine (11) inhibitor chemotypes were generated at the B3LYP/6-311++G(d,p) level of theory and visualized in GaussView 4.1.2 as described previously [129]. The semi-transparent electrostatic potential surface, color-coded red (−33.2 kcal/mol) to blue (33.2 kcal/mol), overlays compound ball and stick atoms (carbon, grey; hydrogen, white; nitrogen, blue; oxygen, red; and sulfur, yellow). Calculated dipole moments are depicted as arrows. All three compounds share delocalized electronic structure.
High polarizability is not limited to diffuse lipophilic cations such as cyanines, phenothiazines, and triarylmethines: it is a property shared by compounds containing highly conjugated π-electron networks, and includes uncharged aggregation inhibitors such as curcumin. As noted above, certain curcumin derivatives are capable of directly inhibiting tau aggregation [99], and curcumin itself has been investigated extensively as an inhibitor of Aβ and α-synuclein aggregation where it binds protein monomers directly [115], acts on monomers to increase reconfiguration rate [74], stabilizes oligomeric species [116], and shields dehydrons potentially involved in amyloidogenic interactions [82]. Thus curcumin can act at least in part through the noncovalent inhibitory mechanisms summarized above. Although structure activity relationship analysis of curcumin analogs has been interpreted in terms of sterics [117] or electrophilicity [118], it is also consistent with ligand polarizability as being a descriptor of binding affinity. For example, curcumin can adopt keto or enol tautomeric forms (Fig. 5A). The enol form, which is more stable than the keto form by ~20 kJ/mol [119], is more polarizable in the ground state and presents a flat surface for interaction with binding partners. In contrast, the keto form has reduced resonance owing to breakage of electronic delocalization [120]. These characteristics appear as a more even distribution of surface electrostatic potential and electronic density in the enol form versus the keto form (Fig. 5BC). The enol form of curcumin strongly binds Aβ aggregates, whereas the keto form binds only weakly [121, 122]. The enol form also binds prion fibrils as evidenced by an induced circular dichroism signal that does not appear upon curcumin interaction with α-helical intermediates [123]. In this example, binding is proposed to inhibit aggregation by blocking fibril elongation. The various mechanisms of action reported for curcumin likely results from the electronic configuration and geometry of its tautomeric species.
Fig. 5.

Electronic structures of curcumin tautomers. A, the enol and keto tautomers of curcumin are both electrophiles (electrophilic α,β-unsaturated ketone moieties are marked by boxes, whereas the points of nucleophilic attack are marked with arrows) that can react with tau sulfhydryls. The enol tautomer binds Aβ aggregates more avidly then does the keto form [121] despite being a weaker electrophile. B, electrostatic surface potential maps for enol (12a) and keto (12b) tautomers were generated at the B3LYP/6-311++G(d,p) level of theory and visualized in GaussView 4.1.2. The semi-transparent electrostatic potential surface, color-coded red (−33.2 kcal/mol) to blue (33.2 kcal/mol), overlays compound ball and stick atoms. Calculated dipole moments are depicted as black arrows. In both tautomers, methoxy groups serve as electron donors whereas the bridge oxygens serve as electron acceptors. C, maps of electron density differences between the ground (S0) and lowest identified singlet excited states (S1), where green contours represent the accumulation of electron density in the S1 excited state and red contours depict loss of electron density from the S0 ground state. Gas-phase calculations were performed in Turbomole and visualized in UCSF Chimera Alpha Version 1.5 (build 31329) as described previously [129] with iso-contour values normalized to ± 0.002 a.u. Relative to the keto form, the planar enol form of curcumin is more polarizable (81.1 Å3 versus 57.7 Å3), presents a more even distribution of surface electrostatic potential in the ground state (reflected in a lower dipole moment), and more evenly redistributes electron density across the length of the molecule on excitation to the S1 state.
Structure activity relationship analysis indicates that ligand polarizability can be maximized in two ways. The first is to position electron donating and withdrawing groups adjacent to the conjugated π-electron network. For example, the rank order of inhibitory potency of diffuse lipophilic cations parallels the strengths of constituent electron donor moieties [27, 72]. This strategy has the potential to maximize polarizability while minimizing ligand size, and therefore to facilitate blood-brain barrier penetration [124]. High polarizability can also be supported by increasing the size of the conjugated π-electron network. This approach may rationalize the reported activities of polyene, porphyrin, PcTS, and other large inhibitors of tau aggregation [67, 90]. It also illustrates how a common descriptor of potency can rationalize the activity of seemingly disparate structures.
CONCLUSIONS
Recent mechanistic insights have helped categorize the many small-molecule tau aggregation inhibitors reported in the literature and have provided insights into directions for their future development. Covalent inhibition is mediated primarily by electrophilic moieties resident in compounds or generated by them during extended incubation periods. These can foster adduct formation with the cysteine residues found in all tau isoforms. Whereas nucleophlic cysteine residues are buried in typical globular proteins [125], they appear to be unusually accessible in tau protein owing to its natively unfolded structure. Noncovalent inhibition is mediated in part by interaction with highly polarizable phenylalanine and tyrosine residues that can influence aggregation phenomena in multiple ways, including the stabilization of off-aggregation pathway oligomers. The potential contribution of tau conformation to inhibitory potency suggests a route toward selectivity, and an important target for future structural studies. Finally, identification of descriptors of inhibitory potency may provide a rational approach to compound optimization. Combined with new sensitive assay methods, this approach could extend structure activity relationship analysis to better identify high-affinity inhibitors for biological testing.
ACKNOWLEDGMENTS
Authors were supported by funding from the National Institutes of Health (AG14452) and the Alzheimer’s Drug Discovery Foundation.
ABBREVIATIONS USED
- Aβ
β-amyloid peptide
- AD
Alzheimer’s disease
- NFT
Neurofibrillary tange
REFERENCES
- [1].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mitchell TW, Nissanov J, Han LY, Mufson EJ, Schneider JA, Cochran EJ, et al. Novel method to quantify neuropil threads in brains from elders with or without cognitive impairment. J Histochem Cytochem. 2000;48:1627–38. doi: 10.1177/002215540004801206. [DOI] [PubMed] [Google Scholar]
- [4].Khatoon S, Grundke-Iqbal I, Iqbal K. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 1994;351:80–84. doi: 10.1016/0014-5793(94)00829-9. [DOI] [PubMed] [Google Scholar]
- [5].Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–30. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- [6].Wang Y, Kruger U, Mandelkow E, Mandelkow EM. Generation of tau aggregates and clearance by autophagy in an inducible cell model of tauopathy. Neurodegener Dis. 2010;7:103–7. doi: 10.1159/000285516. [DOI] [PubMed] [Google Scholar]
- [7].Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- [8].Holmes BB, Diamond MI. Prion-like Properties of tau protein: The importance of extracellular tau as a therapeutic target. J Biol Chem. 2014;289:19855–61. doi: 10.1074/jbc.R114.549295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–48. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bandyopadhyay B, Li G, Yin H, Kuret J. Tau aggregation and toxicity in a cell culture model of tauopathy. J Biol Chem. 2007;282:16454–64. doi: 10.1074/jbc.M700192200. [DOI] [PubMed] [Google Scholar]
- [11].Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT. Are tangles as toxic as they look? J Mol Neurosci. 2011;45:438–44. doi: 10.1007/s12031-011-9566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vazquez A. Metabolic states following accumulation of intracellular aggregates: implications for neurodegenerative diseases. PLoS One. 2013;8:e63822. doi: 10.1371/journal.pone.0063822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and α-synuclein. Science. 2003;300:636–40. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- [14].Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–66. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ke YD, Suchowerska AK, van der Hoven J, De Silva DM, Wu CW, van Eersel J, et al. Lessons from tau-deficient mice. Int J Alzheimers Dis. 2012:873270. doi: 10.1155/2012/873270. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Teng J, Takei Y, Harada A, Nakata T, Chen J, Hirokawa N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol. 2001;155:65–76. doi: 10.1083/jcb.200106025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lasagna-Reeves CA, Sengupta U, Castillo-Carranza D, Gerson JE, Guerrero-Munoz M, Troncoso JC, et al. The formation of tau pore-like structures is prevalent and cell specific: possible implications for the disease phenotypes. Acta neuropathologica communications. 2014;2:56. doi: 10.1186/2051-5960-2-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Iqbal K, Gong CX, Liu F. Microtubule-associated protein tau as a therapeutic target in Alzheimer’s disease. Expert Opin Ther Targets. 2014;18:307–18. doi: 10.1517/14728222.2014.870156. [DOI] [PubMed] [Google Scholar]
- [21].Selkoe DJ, Ihara Y, Salazar FJ. Alzheimer’s disease: insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Science. 1982;215:1243–5. doi: 10.1126/science.6120571. [DOI] [PubMed] [Google Scholar]
- [22].Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev Mol Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213–18. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pickhardt M, von Bergen M, Gazova Z, Hascher A, Biernat J, Mandelkow EM, et al. Screening for inhibitors of tau polymerization. Curr Alzheimer Res. 2005;2:219–26. doi: 10.2174/1567205053585891. [DOI] [PubMed] [Google Scholar]
- [25].Crowe A, Huang W, Ballatore C, Johnson RL, Hogan AM, Huang R, et al. Identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry. 2009;48:7732–45. doi: 10.1021/bi9006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wischik CM, Edwards PC, Harrington CR, Roth M, Klug A. Inhibition of tau-tau association. 6,953,794 U.S. Patent. 2005
- [27].Chang E, Congdon EE, Honson NS, Duff KE, Kuret J. Structure-activity relationship of cyanine tau aggregation inhibitors. J Med Chem. 2009;52:3539–47. doi: 10.1021/jm900116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, et al. Phenylthiazolyl-hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry. 2007;46:10016–23. doi: 10.1021/bi700878g. [DOI] [PubMed] [Google Scholar]
- [29].Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, et al. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed Engl. 2007;46:9215–9. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- [30].Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, et al. Anthraquinones inhibit tau aggregation and dissolve alzheimer paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–35. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- [31].Viayna E, Sola I, Bartolini M, De Simone A, Tapia-Rojas C, Serrano FG, et al. Synthesis and multitarget biological profiling of a novel family of rhein derivatives as disease-modifying anti-Alzheimer agents. J Med Chem. 2014;57:2549–67. doi: 10.1021/jm401824w. [DOI] [PubMed] [Google Scholar]
- [32].Wischik CM, Bentham P, Wischik DJ, Seng KM. Tau aggregation inhibitor (TAI) therapy with rember(tm) arrests disease progression in mild and moderate Alzheimer’s disease over 50 weeks. Alzheimers Dement. 2008;4:T167. [Google Scholar]
- [33].Wischik CM, Harrington CR, Storey JM. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88:529–39. doi: 10.1016/j.bcp.2013.12.008. [DOI] [PubMed] [Google Scholar]
- [34].Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–26. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Andreadis A. Tau splicing and the intricacies of dementia. J Cell Physiol. 2012;227:1220–5. doi: 10.1002/jcp.22842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–9. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- [37].Funk KE, Thomas SN, Schafer KN, Cooper GL, Liao Z, Clark DJ, et al. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem J. 2014;462:77–88. doi: 10.1042/BJ20140372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thomas SN, Funk KE, Wan Y, Liao Z, Davies P, Kuret J, et al. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. 2012;123:105–17. doi: 10.1007/s00401-011-0893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lutolf MP, Tirelli N, Cerritelli S, Cavalli L, Hubbell JA. Systematic modulation of Michael-type reactivity of thiols through the use of charged amino acids. Bioconjug Chem. 2001;12:1051–6. doi: 10.1021/bc015519e. [DOI] [PubMed] [Google Scholar]
- [42].Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993;12:365–70. doi: 10.1002/j.1460-2075.1993.tb05665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Daebel V, Chinnathambi S, Biernat J, Schwalbe M, Habenstein B, Loquet A, et al. β-Sheet core of tau paired helical filaments revealed by solid-state NMR. J Am Chem Soc. 2012;134:13982–9. doi: 10.1021/ja305470p. [DOI] [PubMed] [Google Scholar]
- [44].Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci USA. 2004;101:10278–83. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc Natl Acad Sci USA. 2000;97:5129–34. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Congdon EE, Kim S, Bonchak J, Songrug T, Matzavinos A, Kuret J. Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J Biol Chem. 2008;283:13806–16. doi: 10.1074/jbc.M800247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA. 1998;95:15712–17. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, et al. Granular tau oligomers as intermediates of tau filaments. Biochemistry. 2007;46:3856–61. doi: 10.1021/bi061359o. [DOI] [PubMed] [Google Scholar]
- [49].Ramachandran G, Udgaonkar JB. Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by Tau protein. J Biol Chem. 2011;286:38948–59. doi: 10.1074/jbc.M111.271874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pastor MT, Kummerer N, Schubert V, Esteras-Chopo A, Dotti CG, Lopez de la Paz M, et al. Amyloid toxicity is independent of polypeptide sequence, length and chirality. J Mol Biol. 2008;375:695–707. doi: 10.1016/j.jmb.2007.08.012. [DOI] [PubMed] [Google Scholar]
- [51].Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, et al. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007;25:3020–29. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- [52].Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol. 2013;4:114. doi: 10.3389/fneur.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–53. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- [54].Chirita CN, Necula M, Kuret J. Anionic Micelles and Vesicles Induce tau Fibrillization in vitro. J Biol Chem. 2003;278:25644–50. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- [55].Yao J, Gao X, Sun W, Yao T, Shi S, Ji L. Molecular hairpin: a possible model for inhibition of tau aggregation by tannic acid. Biochemistry. 2013;52:1893–902. doi: 10.1021/bi400240c. [DOI] [PubMed] [Google Scholar]
- [56].Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41:14885–96. doi: 10.1021/bi026469j. [DOI] [PubMed] [Google Scholar]
- [57].Yao TM, Tomoo K, Ishida T, Hasegawa H, Sasaki M, Taniguchi T. Aggregation analysis of the microtubule binding domain in tau protein by spectroscopic methods. J Biochem (Tokyo) 2003;134:91–99. doi: 10.1093/jb/mvg116. [DOI] [PubMed] [Google Scholar]
- [58].Pouplana S, Espargaro A, Galdeano C, Viayna E, Sola I, Ventura S, et al. Thioflavin-s staining of bacterial inclusion bodies for the fast, simple, and inexpensive screening of amyloid aggregation inhibitors. Curr Med Chem. 2014;21:1152–9. doi: 10.2174/09298673113206660256. [DOI] [PubMed] [Google Scholar]
- [59].Chang E, Kuret J. Detection and quantification of tau aggregation using a membrane filter assay. Anal Biochem. 2008;373:330–6. doi: 10.1016/j.ab.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Paranjape SR, Chiang YM, Sanchez JF, Entwistle R, Wang CC, Oakley BR, et al. Inhibition of Tau aggregation by three Aspergillus nidulans secondary metabolites: 2,omega-dihydroxyemodin, asperthecin, and asperbenzaldehyde. Planta Med. 2014;80:77–85. doi: 10.1055/s-0033-1360180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Necula M, Kuret J. Electron microscopy as a quantitative method for investigating tau fibrillization. Anal Biochem. 2004;329:238–46. doi: 10.1016/j.ab.2004.02.023. [DOI] [PubMed] [Google Scholar]
- [62].Huseby CJ, Kuret J. Analyzing tau aggregation with electron microscopy. Methods Mol Biol. 2014 doi: 10.1007/978-1-4939-2978-8_7. in press. [DOI] [PubMed] [Google Scholar]
- [63].Chun W, Waldo GS, Johnson GV. Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3β activation and caspase 3 cleavage. J Neurochem. 2007;103:2529–39. doi: 10.1111/j.1471-4159.2007.04941.x. [DOI] [PubMed] [Google Scholar]
- [64].Tak H, Haque MM, Kim MJ, Lee JH, Baik JH, Kim Y, et al. Bimolecular fluorescence complementation; lighting-up tau-tau interaction in living cells. PLoS One. 2013;8:e81682. doi: 10.1371/journal.pone.0081682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Copeland RA. Evaluation of enzyme inhibitors in drug discovery : a guide for medicinal chemists and pharmacologists. Wiley; Hoboken, N.J.: 2005. [PubMed] [Google Scholar]
- [66].Bulic B, Pickhardt M, Mandelkow E. Progress and developments in tau aggregation inhibitors for Alzheimer disease. J Med Chem. 2013;56:4135–55. doi: 10.1021/jm3017317. [DOI] [PubMed] [Google Scholar]
- [67].Akoury E, Gajda M, Pickhardt M, Biernat J, Soraya P, Griesinger C, et al. Inhibition of tau filament formation by conformational modulation. J Am Chem Soc. 2013;135:2853–62. doi: 10.1021/ja312471h. [DOI] [PubMed] [Google Scholar]
- [68].Ono K, Li L, Takamura Y, Yoshiike Y, Zhu L, Han F, et al. Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site-specific binding. J Biol Chem. 2012;287:14631–43. doi: 10.1074/jbc.M111.325456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rao JN, Dua V, Ulmer TS. Characterization of α-synuclein interactions with selected aggregation-inhibiting small molecules. Biochemistry. 2008;47:4651–6. doi: 10.1021/bi8002378. [DOI] [PubMed] [Google Scholar]
- [70].Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, et al. Small molecule inhibitors of α-synuclein filament assembly. Biochemistry. 2006;45:6085–94. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- [71].Lamberto GR, Binolfi A, Orcellet ML, Bertoncini CW, Zweckstetter M, Griesinger C, et al. Structural and mechanistic basis behind the inhibitory interaction of PcTS on α-synuclein amyloid fibril formation. Proc Natl Acad Sci U S A. 2009;106:21057–62. doi: 10.1073/pnas.0902603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Schafer KN, Cisek K, Huseby CJ, Chang E, Kuret J. Structural determinants of Tau aggregation inhibitor potency. J Biol Chem. 2013;288:32599–611. doi: 10.1074/jbc.M113.503474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ahmad B, Chen Y, Lapidus LJ. Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc Natl Acad Sci U S A. 2012;109:2336–41. doi: 10.1073/pnas.1109526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ahmad B, Lapidus LJ. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J Biol Chem. 2012;287:9193–9. doi: 10.1074/jbc.M111.325548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Acharya S, Safaie BM, Wongkongkathep P, Ivanova MI, Attar A, Klarner FG, et al. Molecular basis for preventing α-synuclein aggregation by a molecular tweezer. J Biol Chem. 2014;289:10727–37. doi: 10.1074/jbc.M113.524520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Eisenberg DS, Jiang L, Meytal L, Liu C. Pharmacophores for amyloid fibers involved in alzheimer’s disease. 2013;A2 WO2013010176. [Google Scholar]
- [77].Mohamed T, Hoang T, Jelokhani-Niaraki M, Rao PP. Tau-derived-hexapeptide 306VQIVYK311 aggregation inhibitors: nitrocatechol moiety as a pharmacophore in drug design. ACS Chem Neurosci. 2013;4:1559–70. doi: 10.1021/cn400151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Landau M, Sawaya MR, Faull KF, Laganowsky A, Jiang L, Sievers SA, et al. Towards a pharmacophore for amyloid. PLoS Biol. 2011;9:e1001080. doi: 10.1371/journal.pbio.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- [80].Fernandez A, Crespo A. Protein wrapping: a molecular marker for association, aggregation and drug design. Chem Soc Rev. 2008;37:2373–82. doi: 10.1039/b804150b. [DOI] [PubMed] [Google Scholar]
- [81].Kim J, Lee HJ, Lee KW. Naturally occurring phytochemicals for the prevention of Alzheimer’s disease. J Neurochem. 2010;112:1415–30. doi: 10.1111/j.1471-4159.2009.06562.x. [DOI] [PubMed] [Google Scholar]
- [82].Zhao LN, Chiu SW, Benoit J, Chew LY, Mu Y. The effect of curcumin on the stability of Aβ dimers. J Phys Chem B. 2012;116:7428–35. doi: 10.1021/jp3034209. [DOI] [PubMed] [Google Scholar]
- [83].Li W, Sperry JB, Crowe A, Trojanowski JQ, Smith AB, 3rd, Lee VM. Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J Neurochem. 2009;110:1339–51. doi: 10.1111/j.1471-4159.2009.06224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Monti MC, Margarucci L, Riccio R, Casapullo A. Modulation of tau protein fibrillization by oleocanthal. J Nat Prod. 2012;75:1584–8. doi: 10.1021/np300384h. [DOI] [PubMed] [Google Scholar]
- [85].Monti MC, Margarucci L, Tosco A, Riccio R, Casapullo A. New insights on the interaction mechanism between tau protein and oleocanthal, an extra-virgin olive-oil bioactive component. Food Funct. 2011;2:423–8. doi: 10.1039/c1fo10064e. [DOI] [PubMed] [Google Scholar]
- [86].George RC, Lew J, Graves DJ. Interaction of cinnamaldehyde and epicatechin with tau: implications of beneficial effects in modulating Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2013;36:21–40. doi: 10.3233/JAD-122113. [DOI] [PubMed] [Google Scholar]
- [87].Gersch M, Kreuzer J, Sieber SA. Electrophilic natural products and their biological targets. Nat Prod Rep. 2012;29:659–82. doi: 10.1039/c2np20012k. [DOI] [PubMed] [Google Scholar]
- [88].Haque MM, Kim D, Yu YH, Lim S, Kim DJ, Chang YT, et al. Inhibition of tau aggregation by a rosamine derivative that blocks tau intermolecular disulfide cross-linking. Amyloid. 2014;21:185–90. doi: 10.3109/13506129.2014.929103. [DOI] [PubMed] [Google Scholar]
- [89].Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL. The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J Biol Chem. 2004;279:26846–57. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- [90].Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, et al. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–23. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- [91].Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophys Res Comm. 2007;358:1–6. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kees F. Dimethyl fumarate : a Janus-faced substance? Expert Opin Pharmacother. 2013;14:1559–67. doi: 10.1517/14656566.2013.804912. [DOI] [PubMed] [Google Scholar]
- [93].Crowe A, James MJ, Lee VM, Smith AB, 3rd, Trojanowski JQ, Ballatore C, et al. Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J Biol Chem. 2013;288:11024–37. doi: 10.1074/jbc.M112.436006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, Zweckstetter M. Mechanistic basis of phenothiazine-driven inhibition of Tau aggregation. Angew Chem Int Ed Engl. 2013;52:3511–5. doi: 10.1002/anie.201208290. [DOI] [PubMed] [Google Scholar]
- [95].Morris G, Anderson G, Dean O, Berk M, Galecki P, Martin-Subero M, et al. The Glutathione System: A New Drug Target in Neuroimmune Disorders. Mol Neurobiol. 2014;50 doi: 10.1007/s12035-014-8705-x. in press. [DOI] [PubMed] [Google Scholar]
- [96].Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007;282:10311–24. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- [97].Stefani M, Rigacci S. Protein folding and aggregation into amyloid: the interference by natural phenolic compounds. Int J Mol Sci. 2013;14:12411–57. doi: 10.3390/ijms140612411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lapidus LJ. Understanding protein aggregation from the view of monomer dynamics. Molecular bioSystems. 2013;9:29–35. doi: 10.1039/c2mb25334h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Dolai S, Shi W, Corbo C, Sun C, Averick S, Obeysekera D, et al. “Clicked” sugar-curcumin conjugate: modulator of amyloid-β and tau peptide aggregation at ultralow concentrations. ACS Chem Neurosci. 2011;2:694–9. doi: 10.1021/cn200088r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010:568068. doi: 10.1155/2010/568068. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sinha S, Lopes DH, Du Z, Pang ES, Shanmugam A, Lomakin A, et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc. 2011;133:16958–69. doi: 10.1021/ja206279b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Attar A, Ripoli C, Riccardi E, Maiti P, Li Puma DD, Liu T, et al. Protection of primary neurons and mouse brain from Alzheimer’s pathology by molecular tweezers. Brain. 2012;135:3735–48. doi: 10.1093/brain/aws289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- [104].Eldem Y, Ozer I. Electrophilic reactivity of cationic triarylmethane dyes towards proteins and protein-related nucleophiles. Dyes Pigments. 2004;60:49–54. [Google Scholar]
- [105].Carlson EE, May JF, Kiessling LL. Chemical probes of UDP-galactopyranose mutase. Chem Biol. 2006;13:825–37. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- [106].Congdon EE, Figueroa YH, Wang L, Toneva G, Chang E, Kuret J, et al. Inhibition of tau polymerization with a cyanine dye in two distinct model systems. J Biol Chem. 2009;284:20830–9. doi: 10.1074/jbc.M109.016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–41. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- [108].Necula M, Chirita CN, Kuret J. Cyanine dye n744 inhibits tau fibrillization by blocking filament extension: implications for the treatment of tauopathic neurodegenerative diseases. Biochemistry. 2005;44:10227–37. doi: 10.1021/bi050387o. [DOI] [PubMed] [Google Scholar]
- [109].Necula M, Kuret J. Site-specific pseudophosphorylation modulates the rate of tau filament dissociation. FEBS Lett. 2005;579:1453–57. doi: 10.1016/j.febslet.2005.01.047. [DOI] [PubMed] [Google Scholar]
- [110].Bulic B, Pickhardt M, Mandelkow EM, Mandelkow E. Tau protein and tau aggregation inhibitors. Neuropharmacology. 2010;59:276–89. doi: 10.1016/j.neuropharm.2010.01.016. [DOI] [PubMed] [Google Scholar]
- [111].Dähne S. Color and Constitution: One Hundred Years of Research. Science. 1978;199:1163–67. doi: 10.1126/science.199.4334.1163. [DOI] [PubMed] [Google Scholar]
- [112].Dwyer DS. Electronic properties of amino acid side chains: quantum mechanics calculation of substituent effects. BMC Chem Biol. 2005;5:2. doi: 10.1186/1472-6769-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Nayeem A, Krystek S, Jr., Stouch T. An assessment of protein-ligand binding site polarizability. Biopolymers. 2003;70:201–11. doi: 10.1002/bip.10434. [DOI] [PubMed] [Google Scholar]
- [114].Parsegian VA. Van der Waals forces : a handbook for biologists, chemists, engineers, and physicists. Cambridge University Press; New York: 2006. [Google Scholar]
- [115].Ngo ST, Li MS. Curcumin binds to Aβ1-40 peptides and fibrils stronger than ibuprofen and naproxen. J Phys Chem B. 2012;116:10165–75. doi: 10.1021/jp302506a. [DOI] [PubMed] [Google Scholar]
- [116].Singh PK, Kotia V, Ghosh D, Mohite GM, Kumar A, Maji SK. Curcumin modulates α-synuclein aggregation and toxicity. ACS Chem Neurosci. 2013;4:393–407. doi: 10.1021/cn3001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Reinke AA, Gestwicki JE. Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: influence of linker length and flexibility. Chem Biol Drug Des. 2007;70:206–15. doi: 10.1111/j.1747-0285.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- [118].Lopez-Lazaro M. Anticancer and carcinogenic properties of curcumin: considerations for its clinical development as a cancer chemopreventive and chemotherapeutic agent. Mol Nutr Food Res. 2008;52(Suppl 1):S103–27. doi: 10.1002/mnfr.200700238. [DOI] [PubMed] [Google Scholar]
- [119].Kolev TM, Velcheva EA, Stamboliyska BA, Spiteller M. DFT and experimental studies of the structure and vibrational spectra of curcumin. Int J Quantum Chem. 2005;102:1069–79. [Google Scholar]
- [120].Lopez-Tobar E, Blanch GP, del Castillo MLR, Sanchez-Cortes S. Encapsulation and isomerization of curcumin with cyclodextrins characterized by electronic and vibrational spectroscopy. Vib Spectrosc. 2012;62:292–98. [Google Scholar]
- [121].Yanagisawa D, Shirai N, Amatsubo T, Taguchi H, Hirao K, Urushitani M, et al. Relationship between the tautomeric structures of curcumin derivatives and their Aβ-binding activities in the context of therapies for Alzheimer’s disease. Biomaterials. 2010;31:4179–85. doi: 10.1016/j.biomaterials.2010.01.142. [DOI] [PubMed] [Google Scholar]
- [122].Asti M, Ferrari E, Croci S, Atti G, Rubagotti S, Iori M, et al. Synthesis and characterization of 68Ga-labeled curcumin and curcuminoid complexes as potential radiotracers for imaging of cancer and Alzheimer’s disease. Inorg Chem. 2014;53:4922–33. doi: 10.1021/ic403113z. [DOI] [PubMed] [Google Scholar]
- [123].Hafner-Bratkovic I, Gaspersic J, Smid LM, Bresjanac M, Jerala R. Curcumin binds to the α-helical intermediate and to the amyloid form of prion protein - a new mechanism for the inhibition of PrPSc accumulation. J Neurochem. 2008;104:1553–64. doi: 10.1111/j.1471-4159.2007.05105.x. [DOI] [PubMed] [Google Scholar]
- [124].Liu X, Tu M, Kelly RS, Chen C, Smith BJ. Development of a computational approach to predict blood-brain barrier permeability. Drug Metab Dispos. 2004;32:132–9. doi: 10.1124/dmd.32.1.132. [DOI] [PubMed] [Google Scholar]
- [125].Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–8. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- [126].Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–9. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Elbaum-Garfinkle S, Rhoades E. Identification of an aggregation-prone structure of tau. J Am Chem Soc. 2012;134:16607–13. doi: 10.1021/ja305206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Ramachandran G, Udgaonkar JB. Evidence for the existence of a secondary pathway for fibril growth during the aggregation of tau. J Mol Biol. 2012;421:296–314. doi: 10.1016/j.jmb.2012.01.007. [DOI] [PubMed] [Google Scholar]
- [129].Cisek K, Jensen JR, Honson NS, Schafer KN, Cooper GL, Kuret J. Ligand electronic properties modulate tau filament binding site density. Biophys Chem. 2012;170:25–33. doi: 10.1016/j.bpc.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]