

Abstract
Nephrotic syndrome (NS), the association of gross proteinuria, hypoalbuminaemia, edema, and hyperlipidemia, can be clinically divided into steroid-sensitive (SSNS) and steroid-resistant (SRNS) forms. SRNS regularly progresses to end-stage renal failure. By homozygosity mapping and whole exome sequencing, we here identify recessive mutations in Crumbs homolog 2 (CRB2) in four different families affected by SRNS. Previously, we established a requirement for zebrafish crb2b, a conserved regulator of epithelial polarity, in podocyte morphogenesis. By characterization of a loss-of-function mutation in zebrafish crb2b, we now show that zebrafish crb2b is required for podocyte foot process arborization, slit diaphragm formation, and proper nephrin trafficking. Furthermore, by complementation experiments in zebrafish, we demonstrate that CRB2 mutations result in loss of function and therefore constitute causative mutations leading to NS in humans. These results implicate defects in podocyte apico-basal polarity in the pathogenesis of NS.
Main Text
Podocytes are highly specialized and polarized epithelial cells that are critical for renal glomerular filtration via their interdigitated foot processes connected by the slit diaphragm.1 Accordingly, disruption of foot process organization inevitably results in nephrotic syndrome (NS).2 Steroid-resistant NS (SRNS) leads to end-stage renal disease.3–5 We have recently shown in a cohort of families affected by SRNS that 33% of all cases are caused by mutation in 1 of 21 different genes described in Mendelian forms of SRNS.6 However, a large percentage of cases remain molecularly unsolved. To identify additional genes mutated in SRNS in humans, we obtained blood samples and pedigrees after acquiring informed consent from individuals with SRNS and their family members. Approval for human subject research was obtained from the institutional review boards at the University of Michigan and the Boston Children’s Hospital. We performed homozygosity mapping (HM)7 followed by whole exome sequencing (WES) in these families affected by SRNS. In a family (A1968) of Turkish origin, two siblings had SRNS with renal histology of focal segmental glomerulosclerosis (FSGS) (Table 1). HM in both affected siblings yielded five regions of homozygosity by descent with a cumulative genomic length of ∼106 Mb. None of the homozygous peaks coincided with any of seven common recessive causes of SRNS (Figure 1A), suggesting that genes known to be mutated in SRNS were not likely to be involved. By WES in one of the affected siblings from family A1968, we detected a homozygous missense mutation: c.1859G>C (p.Cys620Ser) in exon 7 of CRB2 (crumbs family member 2; RefSeq accession number NM_173689 [MIM 609720]) on chromosome 9 (Figures 1B–1F). This variant was the only homozygous variant remaining from the variant filtering process (Table S1 available online). The mutation alters an evolutionarily conserved cysteine residue within the tenth EGF-like repeat (Figures 1C–1F). It segregated with the affected status in this family and was absent from >190 ethnically matched healthy control individuals and from >6,500 European controls in the Exome Variant Server (Table 1).
Table 1.
Recessive CRB2 Mutations Detected in Four Families Affected by SRNS
| Family and Individual | Ethnic Origin | Parental Consanguinity | Nucleotide Alteration(s)a | Alteration(s) in Coding Sequence | Exon (Segregation) | Amino Acid Sequence Conservationb | PolyPhen-2 Score | Age at Onset | Kidney Disease | Histology (at Age) |
|---|---|---|---|---|---|---|---|---|---|---|
| A1968-21, A1968-22 | Turkey | yes | c.1859G>C | p.Cys620Ser | 7 (HOM, M, P) | C. elegans | 0.989 | 6 years, 4 years | SRNS, SRNS | FSGS (6 years), FSGS (4 years) |
| S1232 | Europe | no | c.1882C>T (c.3089_3104dup) | p.Arg628Cys (p.Gly1036Alafs∗43) | 7 (het, M), 10 (het, de novo in child) | C. intestinalis, NA | 0.549, NA | 9 mo | SRNS | ND |
| A3893-21 | Turkey | yes | c.1886G>C | p.Cys629Ser | 7 (HOM, M, P) | C. elegans | 0.997 | 3 years | SRNS | FSGS (3 years) |
| A2222-21 | Western Europe | yes | c.3746G>A | p.Arg1249Gln | 13 (HOM, M, P) | C. elegans | 0.998 | ND | SRNS | FSGS (ND) |
Abbreviations are as follows: FSGS, focal segmental glomerulosclerosis; het, heterozygous in affected individual; HOM, homozygous in affected individual; M, heterozygous mutation identified in mother; NA, not applicable; ND, no data or DNA available; P, heterozygous mutation identified in father; SRNS, steroid-resistant nephrotic syndrome.
All mutations were absent from >190 ethnically matched healthy control individuals and from >6,500 European control individuals in the EVS server.
CRB2 has been evolutionarily conserved through evolution to C. intestinalis (sea squirt) (RefSeq XP_002124076.1), D. melanogaster (RefSeq AAA28428.1), and C. elegans (RefSeq NP_510822.1).
Figure 1.
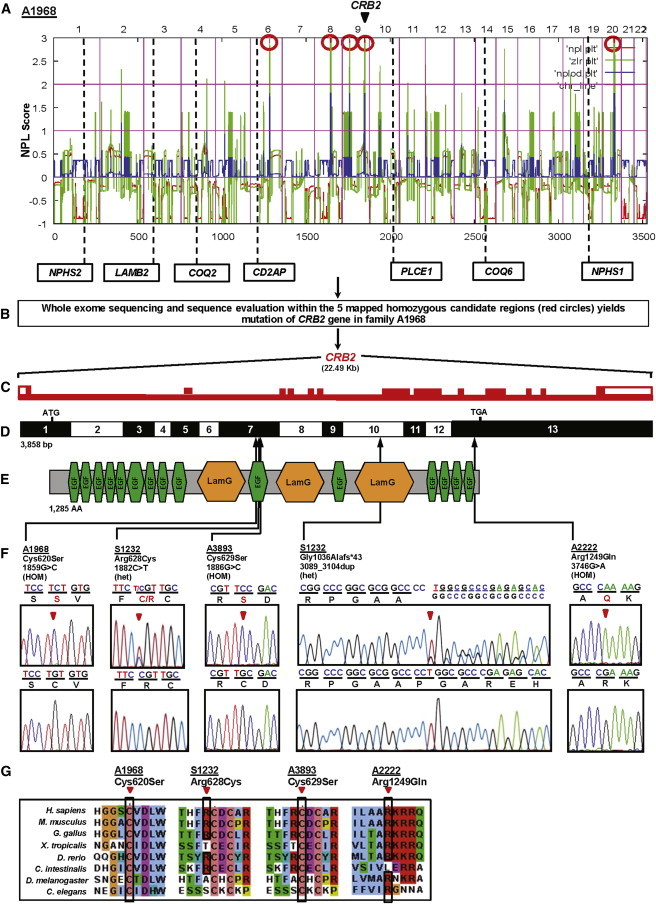
Homozygosity Mapping and WES Identifies CRB2 Mutations as Causing Steroid-Resistant Nephrotic Syndrome in Humans
(A) Nonparametric LOD score (NPL) profile across the human genome in two sibs with SRNS of consanguineous family A1968. SNP mapping was performed with the Affymetrix 250 StyI array. SNP positions on human chromosomes are concatenated from p-ter (left) to q-ter (right) on the x axis. Genetic distance is given in cM. Five maximum NPL peaks (red circles) indicate candidate regions of homozygosity by descent. Note that none of the peaks overlap with any of the seven known recessive NS loci.
(B) WES of one of the affected siblings from family A1968 and sequence evaluation within the five mapped homozygous candidate regions (red circles in A) yields mutation of CRB2 in A1968.
(C) The CRB2 gene extends over 22.49 kb and contains 13 exons (vertical hatches).
(D) Exon structure of human CRB2 cDNA. Positions of start codon (ATG) at nt +1 and of stop codon (TGA) are indicated. For the mutations detected (see F), arrows indicate positions in relation to exons and protein domains (see E).
(E) Domain structure of the CRB2 protein. 15 EGF-like; calcium-binding domains (green) and 3 Laminin G-like domains (orange) are predicted.
(F) CRB2 mutations detected in four families affected by SRNS. Family number and predicted translational change are indicated (see Table 1). Sequence traces are shown for homozygous mutations above normal controls, and mutated nucleotides are indicated by arrowheads. “HOM” denotes homozygous and “het” denotes heterozygous mutations.
(G) The conservation across evolution of altered amino acid residues is shown for all four missense variants (p.Cys620Ser, p.Arg628Cys, p.Cys629Ser, and p.Arg1249Gln).
By WES in another family (S1232) with an individual affected with SRNS, we identified compound heterozygous mutations: c.1882C>T (p.Arg628Cys) and c.3089_3104dup (p.Gly1036Alafs∗43) in CRB2 (Figure 1F, Table 1). The heterozygous mutation c.1882C>T (p.Arg628Cys) altered an amino acid residue that was conserved from C. intestinalis to humans and was inherited from the mother (Figure 1G, Table 1). The other heterozygous mutation in this individual was a deleterious duplication of 16 bases c.3089_3104dup (p.Gly1036Alafs∗43) in exon 10 of CRB2 (Figure 1F). This variant occurred de novo in the affected individual (Table 1). The duplication was confirmed by PCR amplification, cloning, and sequencing of the genomic DNA from the affected individual (Figure S1).
To discover additional mutations in CRB2, we then performed array-based multiplex barcoded PCR amplification and next-generation sequencing8 in an additional 1,010 families with SRNS. In an individual from Turkey with SRNS (A3893-21), we detected a homozygous missense mutation (c.1886G>C [p.Cys629Ser]) in exon 7 of CRB2 (Figure 1F, Table 1). In another individual from an unrelated family (A2222-21), we identified a third homozygous missense mutation: c.3746G>A (p.Arg1249Gln) in CRB2 (Figure 1F, Table 1). The missense mutation c.1886G>C (p.Cys629Ser) also alters a conserved cysteine within the tenth EGF-like repeat, whereas c.3746G>A (p.Arg1249Gln) changes a conserved arginine in the cytoplasmic tail of CRB2 (Figure 1F). Renal biopsy revealed FSGS in four of the five individuals (Table 1).
CRB2 spans 22.49 kb on chromosome 9q33.4 (Figure 1C). The longest transcript of CRB2 (RefSeq NM_173689 [MIM 609720]) has 13 coding exons (Figure 1D). As a result of alternative splicing, CRB2 encodes two isoforms: isoform 1, a putative type I transmembrane protein of 1,285 amino acids (Figure 1E), and isoform 2, a secreted protein of 1,176 amino acids.9 CRB2 is known to contain 15 extracellular EGF-like domains and 3 extracellular laminin G-like domains (Figure 1E). Interestingly, three of the identified missense mutations (p.Cys620Ser, p.Arg628Cys, and p.Cys629Ser) occur within exon 7 of CRB2, which encodes the extracellular tenth EGF-like domain of this protein. This suggests that the tenth EGF-like domain might play an important role in CRB2 function in podocytes. Interestingly, many other disease-associated missense mutations affect amino acids in the well-conserved EGF-like repeats and laminin A domains of the paralog CRB1, implying an important function for the extracellular region of CRB1 in human retinal dystrophies.10,11
We performed immunofluoresence staining in rat kidneys and demonstrated that CRB2 is expressed in podocytes in adult rat glomeruli (Figure 2). CRB2-positive staining was seen in cells positive for the podocyte markers WT1, GLEPP1, SYNAPTOPODIN, and PODOCALYXIN (Figure 2). CRB2 colocalizes most tightly with GLEPP1 among podocytic markers used in the immunofluorescence, consistent with the localization of CRB2 at the slit diaphragms of podocytes (Figure 2C).
Figure 2.
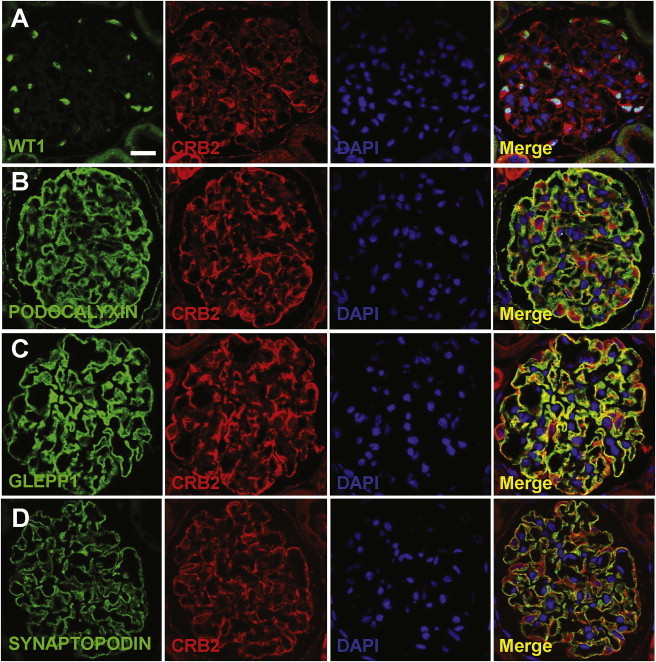
Localization of CRB2 in Adult Rat Kidney
(A) Coimmunofluorescence of CRB2 (Abgent) with WT1 (Santa Cruz Biotech). CRB2 localizes to podocytes, the nuclei of which are marked by WT1.
(B–D) Coimmunofluorescence of CRB2 with podocytic markers PODOCALYXIN (B), GLEPP1 (C), and SYNAPTOPODIN (D) (American Research Products). CRB2 colocalizes most tightly with GLEPP1 among podocytic markers used in immunofluorescence. Note that PODOCALYXIN and GLEPP1 mark the apical podocyte foot process domain, and GLEPP1 is next to the slit membrane adherens junctions. SYNAPTOPODIN marks podocyte processes distal of the slit membrane.
Scale bar represents 10 μm. PODOCALYXIN and GLEPP1 antibodies were kindly provided by Roger C. Wiggins at the University of Michigan.
In an earlier study, we reported that morpholino-induced knockdown of zebrafish crb2b resulted in podocyte foot process defects with ensuing proteinuria.12 To genetically define crb2b function in podocyte differentiation, we now obtained a stable heritable loss-of-function mutation in crb2b. The crb2b mutant allele was caused by a retroviral murine leukemia virus (MLV) insertion in the crb2b locus and is transmitted to offspring as a recessive mutation in Mendelian ratios (see Supplemental Methods).13,14 crb2b−/− homozygous embryos are indistinguishable from crb2b+/− sibs up to 4 days postfertilization (dpf), after which they show pronephric cysts and pericardial edema, both indicators of kidney dysfunction (Figures 3A and 3B). By 5 dpf, crb2b−/− embryos have smaller eyes, consistant with requirement in photoreceptor differentiation.15,16 The pronephric and eye phenotypes are due to specific loss of crb2b gene function, as shown by the fact that both can be rescued by injection of full-length zebrafish Crb2b mRNA (Figure 3C).
Figure 3.
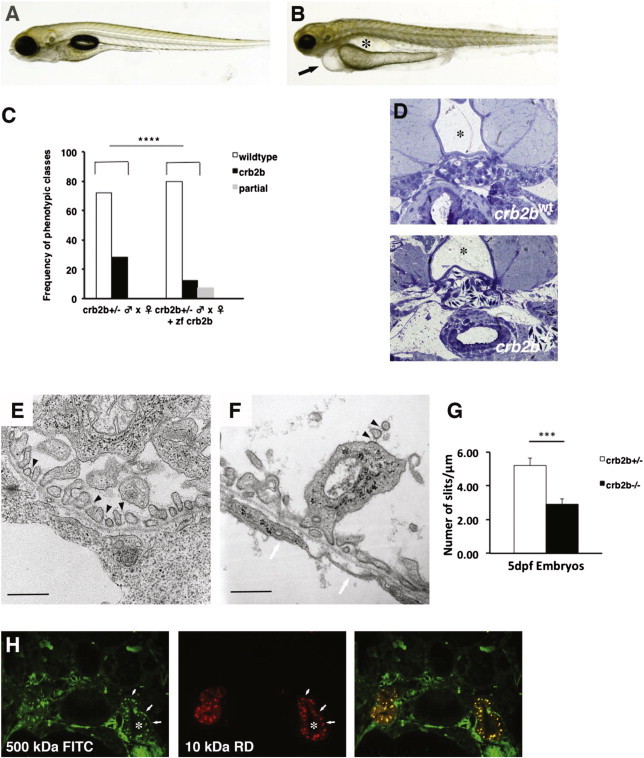
Zebrafish crb2b−/− Mutants Have Morphologically Defective Podocytes
(A and B) Brightfield images of zebrafish 5 days postfertilization (dpf): crb2bwt (A) and crb2b−/− larvae (B). crb2b−/− homozygous mutants show reduced eye size, pericardial effusion (arrow), and pronephric cysts (asterisk).
(C) Rescue of the crb2b−/− eye, pronephric, and pericardial effusion phenotypes by injection of full-length zebrafish Crb2b mRNA. (crb2b+/− ♂ × ♀, n = 75; crb2b+/− ♂ × ♀ + zebrafish Crb2b, n = 183 embryos, p < 0.0001). Complete rescue (black bars) and partial rescue (gray bars) frequencies are shown. Phenotypes were scored at 4.5 dpf.
(D) Transverse sections at the level of the glomerulus in crb2bwt and crb2b−/− 5 dpf larvae. In controls, the glomerulus is directly ventral to the notochord (asterisk) and dorsal aorta. In crb2bwt, capillary loops are densely packed and covered with podocytes. In crb2b−/− glomeruli, capillary loops are fused together and podocytes are attached to the loops.
(E and F) Electron microsopic analysis of podocyte foot process organization in control crb2bwt (E) and crb2b−/− (F) homozygotes at 5 dpf. In crb2bwt, slit diaphragms are visible (black arrowheads in E). The crb2b−/− mutant podocytes show disorganized foot process formation, apical membrane projections containing slit diaphragms (black arrowheads in F) in the urinary space, and a rarefaction of slit diaphragms. A glomerular basement membrane is visible. The endothelium lacks membrane fenestrations in crb2b−/− mutants (white arrowheads in F). Scale bars represent 500 nm.
(G) Quantification of slit diaphragm defects in crb2b−/− mutants (p < 0.001, n = 3 regions/glomerulus from 3 different glomeruli). Data represent the mean ± SEM.
(H) Dye filtration assay shows that FITC-labeled 500 kDa (green) and rhodamine-labeled 10 kDa dextran (red) dyes injected into living 4.5 dpf crb2b−/− mutants are both passed into and endocytosed (arrows) by the pronephric proximal tubules. The asterisk marks the tubule lumen.
Histological sectioning showed glomerular morphogenesis defects in crb2b−/− homozygotes (Figure 3D). We next performed electron microscopic analysis of crb2b−/− mutant pronephric glomeruli to assess podocyte structure. Ultrastructurally, the crb2b−/− homozygotes show disruption of the regular array of patterned podocyte foot processes, which represents the disapearance of slit diaphragms (Figures 3E–3G). Interestingly, crb2b−/− foot processes contain vesicular-like structures not observed in control crb2b+/− sibs. In addition, the apical membranes of crb2b−/− podocytes show membrane projections that reach into Bowman’s space (Figures 3E, 3F, and S2). In crb2b−/− glomeruli, the glomerular basement membrane (GBM) is present but capillary endothelia lack membrane fenestrations (Figure 3F). In control phenotypically wild-type 5 dpf crb2bwt embryos, we counted 2.67 ± 0.71 fenestrations/μm (n = 5 capillary loops from 3 glomeruli). However, in 5 dpf crb2b−/− embryos, we found no glomerular capillary endothelial fenestrations at all (Figure 3G). In order to determine whether glomerular filtration function was affected in crb2b−/− mutants, we performed a dye filtration assay in living 4.5 dpf larvae. Both 500 kDa FITC-labeled and 10 kDa rhodamine-labeled dextrans colocalized within the pronephric proximal tubules, indicating compromised size selectivity in the glomerular filtration barrier (Figure 3H). We conclude that crb2b is genetically required for correct foot process arborization and podocyte morphological differentiation.
Because Crb proteins are required for epithelial apical basal differentiation, we examined whether cell polarity might be affected in crb2b−/− podocytes. Phalloidin labels the F-actin network of podocyte foot processes. In phenotypically wild-type crb2bwt 4.5 dpf larvae, phalloidin labeled the basal F-actin rich podocyte processes that cover the outer aspect of glomerular capillaries. We found that in crb2b−/−, phalloidin is basally concentrated and seen outlining large fused capillaries, indicating that capillary morphogenesis is affected (Figure 4A). Podocyte apical membranes are rich in podocalyxin.17 In both crb2bwt sibs and crb2b−/− embryos, α-Pdxl218 staining is present in podocyte membranes, indicating the presence of apical membranes. However, in crb2b−/− podocytes, ectopic α-Pdxl2 membrane extensions are seen in the Bowman’s space (insets, Figures 4B, 4C, and 4F), suggesting apical membrane defects. Nephrin is a transmembrane protein component of the podocyte slit diaphragms and basally localized in crb2bwt podocytes.19–21 In contrast, we found apical α-nephrin12 localization in crb2b−/− podocytes, indicating defects in nephrin trafficking (insets, Figures 4D and 4G). ZO-1 is a tight junction protein and also found in podocyte slit diaphragms.22,23 In crb2b−/− podocytes, α-ZO-1 staining was found to be generally reduced (Figure 4E). These results indicate that apical membrane differentiation and protein trafficking of slit components are affected in crb2b mutants.
Figure 4.
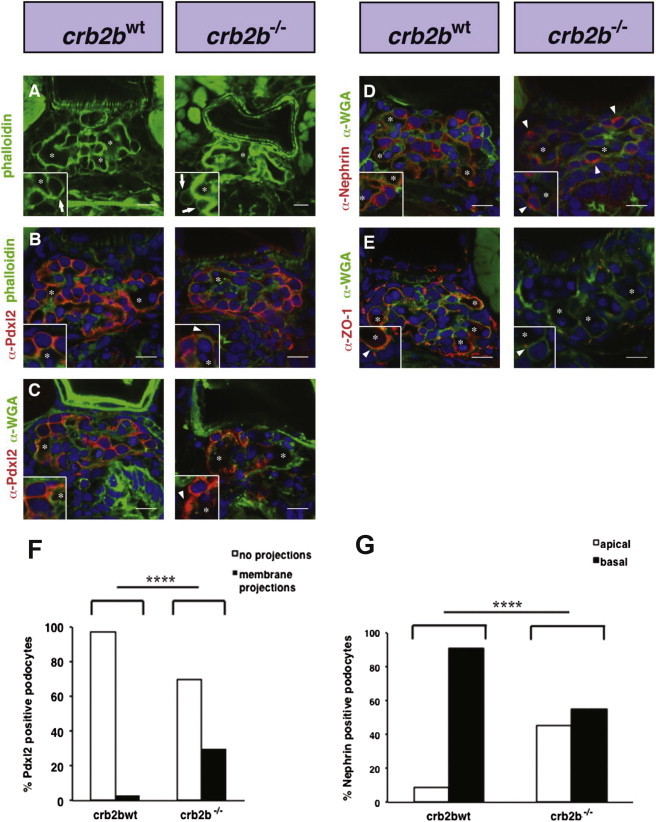
Apical Basal Polarity Is Affected in Zebrafish crb2b−/− Glomerular Podocytes
(A) Phalloidin staining outlines the dense podocyte actin foot process network surrounding capillary lumens. In crb2b−/− mutants, capillary lumens are not compartmentalized but fused into larger vessels. Insets show enlarged images of podocytes. Asterisks mark glomerular capillary lumens.
(B) In crb2bwt, α-Pdxl2 localizes to apical podocyte membranes. In crb2b−/− mutant podocytes, α-Pdxl2 staining is found in ectopic apical projections (arrowheads).
(C) Glomerular basement membranes are visualized by α-wheat germ agglutinin (α-WGA) staining. Asterisks mark glomerular capillary lumens. Pdxl2 staining is again found in ectopic apical projections in crb2b−/− mutant podocytes (arrowheads).
(D) α-WGA shows glomerular basement membranes. α-Nephrin staining is basally localized in control podocytes but apically mislocalized in crb2b−/− podocytes (arrowheads).
(E) α-ZO-1 (Zymed) podocyte staining lines the GBM in crb2bwt but is diminished in crb2b−/− mutants. Scale bars represent 10 μm.
(F) Quantification of α-Pdxl2-positive membrane projections in crb2bwt and crb2b−/− podocytes. n = 37 (tallied from 6 embryos) crb2bwt control and n = 30 (tallied from 4 embryos) crb2b−/− podocytes.
(G) Quantification of α-Nephrin localization in crb2bwt and crb2b−/− podocytes. n = 106 (tallied from 9 embryos) control and n = 67 (tallied from 5 embryos) crb2b−/− podocytes. p < 0.0001.
We employed the zebrafish crb2b−/− mutant to test the functional consequences of CRB2 mutations identified in the human families. The human CRB2 open reading frame (RefSeq NM_173689 [MIM 609720]) was synthesized and cloned into pcDNA 3.1 by Genescript. Mutations were introduced into the human CRB2 open reading frame by site-directed mutagenesis. In crb2b+/− ♂ × ♀ incrosses, crb2b−/− embryos were generated in Mendelian ratios (Figure 5). However, when an in vitro synthesized mRNA encoding the human wild-type CRB2 was injected, only 9% of the resulting embyros were phenotypically crb2b mutant, demonstrating rescue and functional conservation of the human and zebrafish CRB2 genes. Injection of mRNA harboring the human CRB2 mutation c.1859G>C (p.Cys620Ser); CRB2C620S into crb2b+/−, ♂ × ♀ incrosses resulted in 19% crb2b−/−, suggesting that mutation c.1859G>C (p.Cys620Ser) disrupts CRB2 ability to rescue and represents a loss-of-function mutation (Figure 5). The CRB2 protein harboring p.Cys629Ser showed an intermediate level of rescue compared to wild-type CRB2, suggesting a milder loss of protein function compared to p.Cys620Ser.
Figure 5.
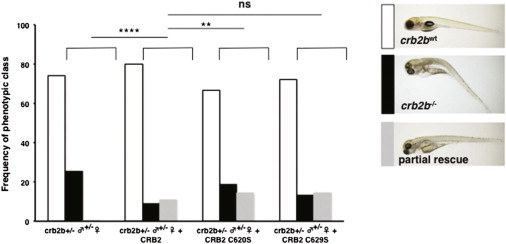
Functional Assay of Human CRB2 Mutations in Zebrafish crb2b+/− ♂ × ♀ Incrosses
Phenotypic frequencies of crb2bwt and crb2b−/− mutant embryos after injection of human CRB2 control mRNA and the mRNAs harboring the mutations c.1859G>C; CRB2C620S and c.1882C>T; CRB2C629S (crb2b+/− ♂ × ♀, n = 363; crb2b+/− ♂ × ♀ + CRB2, n = 167, p = 0.02; crb2b+/− ♂ × ♀ + CRB2C620S, n = 117, p < 0.0001; crb2b+/− ♂ × ♀ + CRB2C629S, n = 283, p = 0.06; ns, no significant difference). Phenotypic classes of embryos recovered from rescue experiments. Partially rescued embryos have a straight body axis, phenotypically wild-type eyes, and lack pronephric cysts but still show some pericardial effusion. No rescue (black bars) and partial rescue (gray bars) frequencies are shown. Phenotypes were scored at 4.5 dpf.
In this report, we show that heritable mutations in the gene encoding human polarity complex protein CRB2 cause monogenic SRNS in humans. In addition, by testing for phenotypic complementation in the zebrafish crb2b−/− mutant, we were able to demonstrate that these mutations resulted in loss of function and were probably pathogenic alterations in human CRB2. The discovery that CRB2 mutations cause a recessive Mendelian form of SRNS suggests that the misregulation of podocyte apical basal polarity is an important causative factor in primary FSGS. Foot process arborization, cytoskeletal architecture, trafficking, and membrane biogenesis take part in the regulation of apical basal polarity. Our findings raise the possiblity that genes encoding other polarity complex members could also be mutated in heritable and sporadic forms of NS.
Acknowledgments
We would like to acknowledge the late Michelle Winn (Duke University) for initial attempts at finding human CRB2 mutations. We thank Markus Affolter and Hans Georg Belting for the α-Pdxl2 antibody. Special thanks to Lars Holmgren (Karolinska Institute) and Lena Claesson-Welsh (Uppsala University) for their overall support and encouragement. We thank Leslie Steed for clinical samples, Giselbert Hauptmann and Iris Sol for zebrafish care, and Katarina Garpenstrand and Johan Ledin at the SciLife zebrafish, Uppsala University. The authors also thank the families who contributed to this study. This research was supported by grants from the NIH to F.H. (DK076683, DK086542) and by the Nephcure Foundation to F.H. H.Y.G. is supported by the NephCure-ASN Foundation Kidney Research grant. F.H. is an Investigator of the Howard Hughes Medical Institute and the Warren E. Grupe Professor. A.K. is supported by a HEFC Senior Clinical Lectureship. The research was also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, Kids Kidney Research, Kidney Research UK, and the British Kidney Patients Association. We would like to acknowledge RADAR, the UK SRNS study group, especially Dr. Larissa Kerecuk for participation and support. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Contributor Information
Friedhelm Hildebrandt, Email: friedhelm.hildebrandt@childrens.harvard.edu.
Arindam Majumdar, Email: arindam.majumdar@igp.uu.se.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/index.html
HomozygosityMapper software, http://www.homozygositymapper.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
Renal Genes, http://www.renalgenes.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Reiser J., Kriz W., Kretzler M., Mundel P. The glomerular slit diaphragm is a modified adherens junction. J. Am. Soc. Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 2.Wiggins R.C. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 3.Mekahli D., Liutkus A., Ranchin B., Yu A., Bessenay L., Girardin E., Van Damme-Lombaerts R., Palcoux J.B., Cachat F., Lavocat M.P. Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: a multicenter study. Pediatr. Nephrol. 2009;24:1525–1532. doi: 10.1007/s00467-009-1138-5. [DOI] [PubMed] [Google Scholar]
- 4.ISKDC Primary nephrotic syndrome in children: clinical significance of histopathologic variants of minimal change and of diffuse mesangial hypercellularity. A Report of the International Study of Kidney Disease in Children. Kidney Int. 1981;20:765–771. doi: 10.1038/ki.1981.209. [DOI] [PubMed] [Google Scholar]
- 5.Benoit G., Machuca E., Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr. Nephrol. 2010;25:1621–1632. doi: 10.1007/s00467-010-1495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lovric S., Fang H., Vega-Warner V., Sadowski C.E., Gee H.Y., Halbritter J., Ashraf S., Saisawat P., Soliman N.A., Kari J.A., Nephrotic Syndrome Study Group Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2014;9:1109–1116. doi: 10.2215/CJN.09010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 9.Katoh M., Katoh M. Identification and characterization of Crumbs homolog 2 gene at human chromosome 9q33.3. Int. J. Oncol. 2004;24:743–749. [PubMed] [Google Scholar]
- 10.den Hollander A.I., Davis J., van der Velde-Visser S.D., Zonneveld M.N., Pierrottet C.O., Koenekoop R.K., Kellner U., van den Born L.I., Heckenlively J.R., Hoyng C.B. CRB1 mutation spectrum in inherited retinal dystrophies. Hum. Mutat. 2004;24:355–369. doi: 10.1002/humu.20093. [DOI] [PubMed] [Google Scholar]
- 11.Bujakowska K., Audo I., Mohand-Saïd S., Lancelot M.E., Antonio A., Germain A., Léveillard T., Letexier M., Saraiva J.P., Lonjou C. CRB1 mutations in inherited retinal dystrophies. Hum. Mutat. 2012;33:306–315. doi: 10.1002/humu.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebarasi L., He L., Hultenby K., Takemoto M., Betsholtz C., Tryggvason K., Majumdar A. A reverse genetic screen in the zebrafish identifies crb2b as a regulator of the glomerular filtration barrier. Dev. Biol. 2009;334:1–9. doi: 10.1016/j.ydbio.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Amsterdam A., Hopkins N. Mutagenesis strategies in zebrafish for identifying genes involved in development and disease. Trends Genet. 2006;22:473–478. doi: 10.1016/j.tig.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Jao L.E., Maddison L., Chen W., Burgess S.M. Using retroviruses as a mutagenesis tool to explore the zebrafish genome. Brief. Funct. Genomics Proteomics. 2008;7:427–443. doi: 10.1093/bfgp/eln038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alves C.H., Sanz A.S., Park B., Pellissier L.P., Tanimoto N., Beck S.C., Huber G., Murtaza M., Richard F., Sridevi Gurubaran I. Loss of CRB2 in the mouse retina mimics human retinitis pigmentosa due to mutations in the CRB1 gene. Hum. Mol. Genet. 2013;22:35–50. doi: 10.1093/hmg/dds398. [DOI] [PubMed] [Google Scholar]
- 16.Zou J., Wang X., Wei X. Crb apical polarity proteins maintain zebrafish retinal cone mosaics via intercellular binding of their extracellular domains. Dev. Cell. 2012;22:1261–1274. doi: 10.1016/j.devcel.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerjaschki D., Sharkey D.J., Farquhar M.G. Identification and characterization of podocalyxin—the major sialoprotein of the renal glomerular epithelial cell. J. Cell Biol. 1984;98:1591–1596. doi: 10.1083/jcb.98.4.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herwig L., Blum Y., Krudewig A., Ellertsdottir E., Lenard A., Belting H.G., Affolter M. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr. Biol. 2011;21:1942–1948. doi: 10.1016/j.cub.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 19.Babayeva S., Rocque B., Aoudjit L., Zilber Y., Li J., Baldwin C., Kawachi H., Takano T., Torban E. Planar cell polarity pathway regulates nephrin endocytosis in developing podocytes. J. Biol. Chem. 2013;288:24035–24048. doi: 10.1074/jbc.M113.452904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartleben B., Schweizer H., Lübben P., Bartram M.P., Möller C.C., Herr R., Wei C., Neumann-Haefelin E., Schermer B., Zentgraf H. Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J. Biol. Chem. 2008;283:23033–23038. doi: 10.1074/jbc.M803143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satoh D., Hirose T., Harita Y., Daimon C., Harada T., Kurihara H., Yamashita A., Ohno S. aPKCλ maintains the integrity of the glomerular slit diaphragm through trafficking of nephrin to the cell surface. J. Biochem. 2014;156:115–128. doi: 10.1093/jb/mvu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnabel E., Anderson J.M., Farquhar M.G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell Biol. 1990;111:1255–1263. doi: 10.1083/jcb.111.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukasawa H., Bornheimer S., Kudlicka K., Farquhar M.G. Slit diaphragms contain tight junction proteins. J. Am. Soc. Nephrol. 2009;20:1491–1503. doi: 10.1681/ASN.2008101117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.