

Abstract
Genome-wide association studies (GWASs) have revealed SNP rs889312 on 5q11.2 to be associated with breast cancer risk in women of European ancestry. In an attempt to identify the biologically relevant variants, we analyzed 909 genetic variants across 5q11.2 in 103,991 breast cancer individuals and control individuals from 52 studies in the Breast Cancer Association Consortium. Multiple logistic regression analyses identified three independent risk signals: the strongest associations were with 15 correlated variants (iCHAV1), where the minor allele of the best candidate, rs62355902, associated with significantly increased risks of both estrogen-receptor-positive (ER+: odds ratio [OR] = 1.24, 95% confidence interval [CI] = 1.21–1.27, ptrend = 5.7 × 10−44) and estrogen-receptor-negative (ER−: OR = 1.10, 95% CI = 1.05–1.15, ptrend = 3.0 × 10−4) tumors. After adjustment for rs62355902, we found evidence of association of a further 173 variants (iCHAV2) containing three subsets with a range of effects (the strongest was rs113317823 [pcond = 1.61 × 10−5]) and five variants composing iCHAV3 (lead rs11949391; ER+: OR = 0.90, 95% CI = 0.87–0.93, pcond = 1.4 × 10−4). Twenty-six percent of the prioritized candidate variants coincided with four putative regulatory elements that interact with the MAP3K1 promoter through chromatin looping and affect MAP3K1 promoter activity. Functional analysis indicated that the cancer risk alleles of four candidates (rs74345699 and rs62355900 [iCHAV1], rs16886397 [iCHAV2a], and rs17432750 [iCHAV3]) increased MAP3K1 transcriptional activity. Chromatin immunoprecipitation analysis revealed diminished GATA3 binding to the minor (cancer-protective) allele of rs17432750, indicating a mechanism for its action. We propose that the cancer risk alleles act to increase MAP3K1 expression in vivo and might promote breast cancer cell survival.
Introduction
One of the first genome-wide association studies (GWASs) for breast cancer (MIM 114480) susceptibility identified a 5q11.2 SNP (rs889312) associated with risk of breast cancer in women of European ancestry.1 In the most recent analyses by the Breast Cancer Association Consortium (BCAC), the minor allele of rs889312 was associated with a per-allele odds ratio (OR) = 1.12 (95% confidence interval [CI] = 1.10–1.15; ptrend = 1.8 × 10−26).2 The association was stronger for estrogen-receptor-positive (ER+) disease (OR = 1.14, 95% CI = 1.11–1.17, p = 1.1 × 10−26 in the most recent BCAC analysis) but was also seen for estrogen-receptor-negative (ER−) disease (OR = 1.06, 95% CI = 1.03–1.10, p = 0.0024) and triple negative disease (OR = 1.11, 95% CI = 1.02–1.20, p = 0.016).3 SNP rs889312 was also reported to be associated with an increased breast cancer risk in carriers of BRCA2 (MIM 600185) mutations.4
The GWAS SNP rs889312 lies approximately 80 kb centromeric to MAP3K1 (MIM 600982), the gene encoding mitogen-activated protein kinase kinase kinase 1, also known as MEK kinase 1 (MEKK1), a stress-induced serine/threonine kinase with apparent dual functions: MEKK1 induces cell proliferation through a RAS-RAF-MEK-ERK signaling pathway,5 but upon caspase cleavage, it generates a fragment with a proapoptotic function.6,7 Furthermore, MEKK1 regulates transcription of key cancer-related genes, such as MYC8 (MIM 190080), TP539 (MIM 191170), and JUN10 (MIM 165160), through its signal-transduction pathway. There is already evidence of a role for MAP3K1 in breast cancer pathogenesis: MAP3K1 driver mutations have been observed in luminal A and B type breast tumors,11 and MAP3K1 expression has been associated with specific breast tumor subtypes.12
In this study, we performed genetic epidemiological analyses on all common variants at 5q11.2, together with in silico and in vitro analyses of candidate causal variants, and identified strong candidates that we propose are functionally related to breast cancer risk. Specifically, we provide evidence that these associations are mediated through MAP3K1.
Material and Methods
SNP Selection and Genotyping
Using the March 2010 release of the 1000 Genomes Project,13 we searched a 305 kb interval on 5q11.2 (GRCh37 positions 55,983,657–56,288,810) and identified all SNPs with a minor allele frequency (MAF) > 0.02 in Europeans. SNPs with an Illumina designability score > 0.8 (and r2 > 0.1 with rs889312), together with a tagging set (r2 > 0.9) for all other known SNPs in the interval, were selected for inclusion on the iCOGS custom array.2 A total of 352 SNPs, of which 300 passed postgenotyping quality-control criteria, were selected.2 For improvement of SNP coverage across the locus, 16 further SNPs selected from the October 2010 release of the 1000 Genomes Project were genotyped in a subset of two BCAC studies (SEARCH and the combined Copenhagen studies CPGS [Copenhagen General Population Study] and CCHS [Copenhagen City Heart Study]) by using a Fluidigm array according to the manufacturer’s instructions. These two data sets, as well as IMPUTE2 and the January 2012 release of the 1000 Genomes Project as references,14 were used for imputing all genotypes of other common variants in this interval. All participants provided written informed consent, and all BCAC studies had local human ethical approvals.2 Study characteristics and iCOGS methodology have been previously reported.2
Statistical Analyses
Associations with breast cancer risk in BCAC were evaluated by a comparison of SNP genotype frequencies in case and control individuals by unconditional logistic regression. Analyses were adjusted by study and seven principal components.2 The primary analysis fit each SNP as an allelic dose and tested for association with a 1-degree-of-freedom trend test (ptrend) with associated OR and 95% CI. To identify independent risk signals, we performed stepwise conditional analysis in R with the function “step,” which included any variant with p value < 10−4 in the single-SNP analysis to calculate the most parsimonious model with a penalty value of k = 10. The null model included study and principal components. Haplotype analysis was performed in R with the package “haplo.stats,” and the analyses were adjusted for study and principal components.
Cell Lines
The normal breast epithelial cell line Bre-80 was cultured as described previously.15 The breast cancer cell lines MCF7, T-47D, and MDA-MB231 were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, antibiotics, sodium pyruvate, and in the case of MCF7 and T-47D cells, 10 μg/ml insulin.
Chromosome Conformation Capture
Chromosome conformation capture (3C) libraries were generated with EcoRI from the cell lines above as described previously.16 3C interactions were quantitated by quantitative PCR (qPCR) with primers designed within the EcoRI restriction fragments spanning the 5q11.2 risk locus (Table S1, available online). qPCR was performed as described previously17 with at least two independent 3C libraries from each cell line; each experiment was quantified in duplicate. We used two bacterial artificial clones (RP11-378G4 and RP11-1146C6) covering the 5q11.2 region to create an artificial library of ligation products in order to normalize for PCR efficiency. As an internal control, interaction frequencies were normalized to that of the EcoRI fragment immediately upstream of the promoter fragment.
Plasmid Construction
A MAP3K1-promoter-driven luciferase reporter construct was generated by the insertion of a 1,928 bp fragment containing the MAP3K1 promoter and the transcription start site (chr5: 56,109,070–56,110,997, GRch37) into the MluI and HindIII sites of pGL3-Basic. To assist cloning, AgeI and SbfI sites were inserted into the BamHI and SalI sites downstream of the luciferase gene. A 1,575 bp putative regulatory element (PRE)-A fragment, a 1,765 bp PRE-B2 fragment, a 2,357 bp PRE-B3 fragment, a 2,203 bp PRE-C fragment, and a 1,519 bp PRE-D fragment were generated by PCR using primers designed with AgeI and SbfI sites and cloned into the modified pGL3-MAP3K1 promoter construct. PRE-B was too large (∼7 kb) to be cloned in its entirety, so three subregions termed PRE-B1, PRE-B2, and PRE-B3 were cloned separately. The minor alleles of individual SNPs were introduced into promoter and PRE sequences, containing the major alleles of any other causal candidate variants, by overlap extension PCR. Sequencing of all constructs confirmed variant incorporation (AGRF, Brisbane). PCR primers are listed in Table S2. For the PRE-B1 construct, a 2,129 bp region spanning chr5: 56,028,968–56,031,097 (GRCh37) was synthesized with AgeI and SbfI sites incorporated at the 5′ and 3′ ends (GenScript, Piscataway) to assist cloning into the MAP3K1 promoter construct. The cloned regions are highlighted in Figure 2B.
Figure 2.
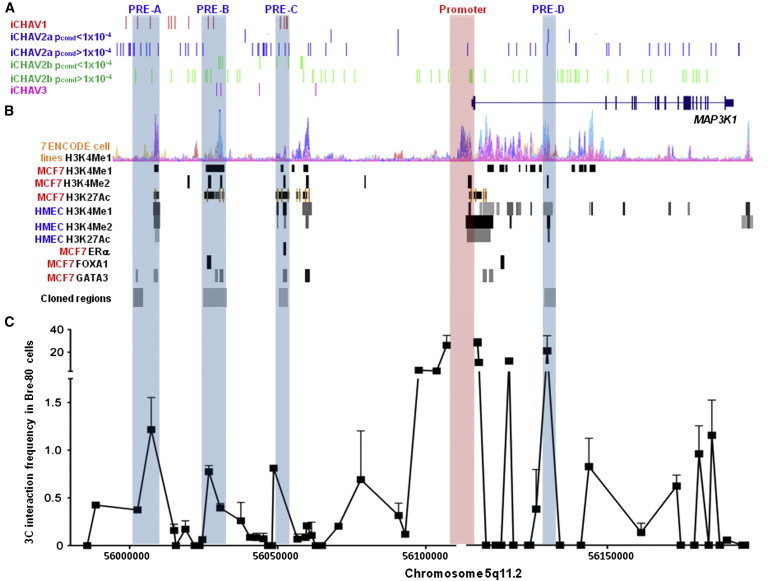
Candidate Causal Variants Are Located in PREs that Interact with the MAP3K1 Promoter
(A) The candidate causal variants associated with breast cancer risk from iCHAV1, iCHAV2a, iCHAV2b, and iCHAV3 were mapped to PREs at the 5q11.2 locus.
(B) PREs (highlighted) were identified with ChIP-seq data (H3K4Me1 studies in seven ENCODE cell lines [GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK, and NHLF]; H3K4Me1 and H3K4Me2 in MCF7 and human mammary epithelial cells [HMECs]; and transcription factors ER-α, FOXA1, and GATA3 in MCF7 cells) accessed from the UCSC Genome Browser. Regions cloned into reporter gene constructs are also shown.
(C) 3C analysis of interactions between EcoRI fragments at the 5q11.2 locus, encompassing the PREs coinciding with candidate causal variants, and the MAP3K1 promoter in Bre-80 cells (error bars represent SD, and a representative graph is shown).
Reporter Assays and Estrogen Induction
Bre-80 and MCF7 cells were transfected with equimolar amounts of luciferase reporter plasmids and 50 ng of pRLTK transfection control plasmid with Lipofectamine 2000. The total amount of transfected DNA was kept constant at 600 ng for each construct by the addition of pUC19 as a carrier plasmid. Luciferase activity was measured 24 hr posttransfection by the Dual-Glo Luciferase Assay System. Normalizing firefly luciferase activity to Renilla luciferase corrected for differences in transfection efficiency or cell-lysate preparation. For the assays under basal conditions, the activity of each construct was calculated in relation to the activity (defined as 1) of the construct containing the MAP3K1 promoter alone.
For estrogen-induction assays, we treated cells as described.18 In brief, 24 hr after plating MCF7 cells into wells, we replaced medium with that containing 10 nM fulvestrant for 48 hr to inhibit estrogen-induced gene expression and thereby create a baseline of expression for reporter assays. Cells were then incubated with fresh medium containing either 10 nM estrogen (17β-estradiol) or DMSO (vehicle control) and transfected with reporter plasmids. Luciferase assays were performed as above after 24 hr. Statistical significance was tested by repeated-measures ANOVA with the Greenhouse-Geisser correction for nonsphericity and by a subsequent Dunnett’s multiple-comparisons test in GraphPad Prism.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation (ChIP) assays were carried out as previously described.19 GATA3-specific mouse monoclonal antibodies (sc268) were obtained from Santa Cruz Biotech. Precipitate and input were used in qPCR using SYBR green master mix as previously described.19 Primer sequences are listed in Table S3. Both rs17432750 primer sets gave identical enrichment, and the identity of the larger fragment was confirmed by Sanger sequencing. All values obtained were normalized to input, and enrichment was given in relation to the negative CCND1 (MIM 168461) control. Allele-specific ChIP was carried out with a TaqMan SNP genotyping assay (Applied Biosystems) on the ChIP material. In the TaqMan assay, two different fluorophores were each linked to a probe detecting the two different alleles. Each allele was subsequently amplified with an Applied Biosystems Real Time PCR machine (7900HT), and the data were analyzed with the SDS software. The SDS software converts raw data to fluorescence intensity for each allele and then plots the results as a scatter graph of allele X versus allele Y. We tested the accuracy of this assay by genotyping known mixtures of homozygous ZR751 and T47D (C/C) and MDA-MB0-468 (A/A) cell-line DNA (Figure S1). For the allelic discrimination, three independent experiments were carried out and gave similar results; a representative example is shown.
GATA3 siRNA Knockdown for Reporter Assay
GATA3 (MIM 131320; L-003781-00) and nontargeting (D-001810-10-20) ON-TARGETplus SMARTpool siRNAs were purchased from Thermo Scientific. For knockdown, Bre-80 cells were cotransfected with the relevant luciferase reporter plasmids and 100 nM of either GATA3 or nontargeting siRNAs with Lipofectamine 2000. Luciferase assays were performed as described above after 24 hr. qPCR was performed as described previously17 to validate GATA3 knockdown (Figure S9).
Analysis of Expression Quantitative Trait Loci
Analysis of expression quantitative trait loci (eQTLs) was undertaken in two sample sets of adjacent normal breast samples from women of European decent: the first set contained 135 samples collected for the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) study,20 and the second set contained 56 samples extracted from The Cancer Genome Atlas (TCGA) breast cancer study.21 Matched gene expression (Illumina HT-12 v3 microarray for the METABRIC data; Agilent G4502A-07-3 microarray for the TCGA data) and germline SNP data that were either genotyped (Affymetrix SNP 6.0) or imputed (1000 Genomes Project March 2012 data, IMPUTE 2.0) were used. Correlations between all imputed and genotyped variants at the 5q11.2 locus and expression levels of eight (METABRIC) or four (TCGA) genes present in the fine-mapped region were assessed with a linear regression model in which an additive effect on expression level was assumed for each copy of the rare allele. Calculations were carried out with the eMap library in R on the METABRIC data and with SNPTEST software22 on the TCGA data.
Results
Genotyping of Case-Control Studies
Three hundred SNPs at the 5q11.2 locus (GRch37 positions 55,983,657–56,288,810) were successfully genotyped with the iCOGs chip in 103,991 breast cancer case and control individuals from 52 BCAC studies, of which 41 included individuals of European ancestry (46,451 case and 42,599 control subjects), nine included individuals of Asian ancestry (6,269 case and 6,624 control subjects), and two included individuals of African American ancestry (1,116 case and 932 control subjects). Using these data, together with data on a further 16 SNPs genotyped in two BCAC studies (SEARCH and the combined Copenhagen studies CPGS and CCHS), we imputed genotypes for 909 (out of a possible 911) variants with MAF > 0.02 and imputation r2 > 0.3 by using the January 2012 release of the 1000 Genomes Project as a reference.
Potential Breast Cancer Risk Signals in European Studies
Figure 1A shows the Manhattan plot of the 909 genotyped and imputed SNPs (r2 > 0.3) for overall breast cancer risk in European studies. Genotype and association results for all 909 SNPs are presented in Table S4. Five hundred and forty-one variants display association with overall breast cancer risk at ptrend < 10−4 (Table S5). All associations are consistent with a log-additive model.
Figure 1.
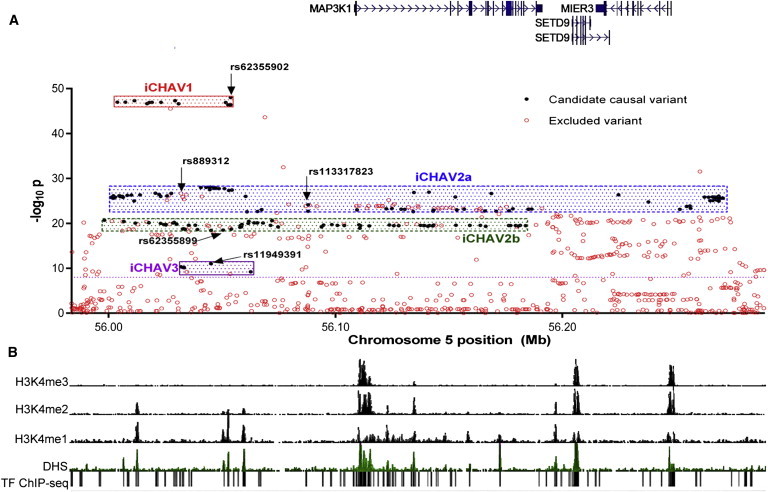
Genetic Mapping and Chromatin State of the 5q11.2 Locus
(A) Manhattan plot of overall breast cancer risk in Europeans at the 5q11.2 locus. SNPs are plotted on the basis of their chromosomal position on the x axis and p values (log10 values) for association. The span of the iCHAVs in terms of chromosomal location and p value is displayed with shading, and candidate causal variants from the iCHAV are colored black. The lead SNPs from each iCHAV, the original GWAS tag SNP (rs889312), and the three genes present in the region are shown. The dotted line intersects the y axis at p = 10−8 and indicates conventional genome-wide significance.
(B) The chromatin state of the 5q11.2 locus in human mammary epithelial cells is shown with ENCODE ChIP-seq data from H3K4me3, H3K4me2, H3k4me1, and DNaseI studies accessed from the UCSC Genome Browser. Transcription factor (TF) binding from ENCODE ChIP-seq studies of 161 TFs in 91 cell lines is also displayed.
In a forward stepwise conditional analysis using the SNPs listed in Table S5, the best model included three SNPs: (1) rs62355902 (conditional p value [pcond] = 8.6 × 10−26), (2) rs113317823 (pcond = 2.8 × 10−5), and (3) rs11949391 (pcond = 9.7 × 10−5). No significant evidence of heterogeneity was observed among ORs for these SNPs among studies of European ancestry (minimum observed phet = 0.14 and maximum l2 = 19.3% for SNP rs11949391; Figure S2). Each SNP remaining in the conditional-analysis model indicates the existence of a separate genetic risk signal (previously defined23 as an independent set of correlated highly associated variants [iCHAV]), each of which will contain at least one directly causal variant.
The most significantly associated variant overall was rs62355902 (OR per minor allele = 1.21, 95% CI = 1.19–1.24, ptrend = 9.5 × 10−49). This was the most significant of 15 strongly correlated SNPs (r2 > 0.93) lying within a 50 kb interval (GRCh37 positions 56,003,831–56,053,745, marked in red in Table S5); we designated these SNPs as iCHAV1. These iCHAV1 SNPs all had likelihood ratios of <100:1 in relation to the best candidate SNP (rs62355902) and thus could not be excluded from further analysis, given that they remained strong candidate causal variants on the basis of epidemiological evidence. After conditioning on iCHAV1 top SNP rs62355902, the most strongly associated variant was SNP rs113317823 (OR per minor allele = 1.22, 95% CI = 1.18–1.26, ptrend = 7.0 × 10−25; conditional OR = 1.12, 95% CI = 1.05–1.20, pcond = 2.8 × 10−5). One hundred and seventy-two variants had likelihood ratios of <100:1 in relation to rs113317823, and these constituted iCHAV2 (highlighted in shades of blue and yellow in Table S5). SNP rs113317823 was partially correlated with iCHAV1 top candidate rs62355902 (r2 = 0.19), and this added complexity to the subsequent analysis—this is explored in more detail by the haplotype analysis described below.
After adjustment for the top iCHAV1 (rs62355902) and iCHAV2 (rs113317823) SNPs, the best remaining significantly associated SNP was rs11949391 in iCHAV3 (OR per minor allele = 0.91, 95% CI = 0.89–0.94, ptrend = 9.4 × 10−12; conditional OR = 0.95, 95% CI = 0.92–0.98, pcond = 9.7 × 10−5). Four other SNPs had likelihood ratios of <100:1 in relation to rs11949391 and were highly correlated with rs11949391 (r2 ≥ 0.95), but not with either of the top iCHAV1 or iCHAV2 SNPs (r2 < 0.04, marked in yellow in Table S5), and these were thus candidate causal variants for iCHAV3.
Effects on ER+ and ER− Tumor Subtypes
Candidate causal SNPs in iCHAV1 were associated with risks of both ER+ and ER− disease (Table S5). However, the OR was greater for ER+ disease (rs62355902, OR = 1.24, 95% CI = 1.21–1.27) than for ER− disease (OR = 1.10, 95% CI = 1.05–1.15, p difference [pdiff] = 1.5 × 10−5; Table 1). By contrast, the minor alleles of candidate causal SNPs in iCHAV3 were protective against ER+ tumor development (rs11949391, OR = 0.90, 95% CI = 0.87–0.93, p = 1.0 × 10−10) but had no apparent effect on ER− tumor risk (OR = 1.01, 95% CI = 0.96–1.06, p = 0.81, pdiff = 1.3 × 10−4; Table 1). The lead SNP in iCHAV2, rs113317823, remained significantly associated with ER+ tumor risk (pcond = 9.7 × 10−5 after adjustment for rs62355902), but not with ER− tumor risk (pcond = 0.099), but the difference in the OR by ER subtype was only borderline significant (pdiff = 0.02; Table 1).
Table 1.
The Best Candidate Variants Representing the Four iCHAVs with the Strongest Effects on Breast Cancer Risk in European Studies
| iCHAV | SNP | Chr Position (GRch37) | Alleles (Major/Minor) | MAF | r2with Lead iCHAV1 SNP | Imputation r2 |
Overall Breast Cancer Risk |
ER+Breast Cancer Risk |
ER−Breast Cancer Risk |
ER Status |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | (95% CI) | ptrend | pcond | OR | (95% CI) | ptrend | pcond | OR | (95% CI) | ptrend | pcond | pdiff | |||||||
| 1 | rs62355902 | 56,053,723 | A/T | 0.18 | – | 0.95 | 1.21 | (1.19–1.24) | 9.50 × 10−49 | – | 1.24 | (1.21–1.27) | 5.71 × 10−44 | – | 1.10 | (1.05–1.15) | 3.02 × 10−4 | – | 1.47 × 10−5 |
| 2a | rs113317823 | 56,087,883 | C/T | 0.08 | 0.19 | 0.72 | 1.22 | (1.18–1.26) | 7.00 × 10−25 | 1.61 × 10−5 | 1.24 | (1.20–1.29) | 2.02 × 10−21 | 9.74 × 10−5 | 1.12 | (1.05–1.20) | 2.56 × 10−3 | 9.90 × 10−2 | 1.93 × 10−2 |
| 2b | rs62355899 | 56,050,465 | A/G | 0.13 | 0.62 | 1.00 | 1.15 | (1.12–1.18) | 2.98 × 10−19 | 3.04 × 10−5 | 1.17 | (1.13–1.20) | 5.52 × 10−18 | 2.04 × 10−3 | 1.08 | (1.02–1.14) | 1.11 × 10−2 | 5.42 × 10−1 | 1.86 × 10−2 |
| 3 | rs11949391 | 56,045,081 | T/C | 0.16 | 0.04 | 1 | 0.91 | (0.89–0.94) | 9.36 × 10−12 | 5.57 × 10−5 | 0.90 | (0.87–0.93) | 1.00 × 10−10 | 1.44 × 10−4 | 1.01 | (0.96–1.06) | 8.14 × 10−1 | 3.50 × 10−1 | 1.27 × 10−4 |
Single-SNP risk estimates for the top candidates in each iCHAV as well as overall breast cancer risk and subtypes by estrogen receptor status. Results are given as ORs with 95% CI (with the minor variant alleles as the reference), per-allele ptrend, and pcond (pcond is conditional on the iCHAV1 lead SNP rs62355902). The pdiff for ER status is from a case-only analysis comparing the effect sizes between the ER+ and ER− subtypes. The complete list of variants for these iCHAVs can be found in Table S7).
Effects of Haplotypes on Breast Cancer Risk
Whereas iCHAV1 and iCHAV3 represent sets of highly correlated SNPs (r2 > 0.93 with the lead SNP), the set of 173 SNPs, labeled iCHAV2, includes three subsets defined according to their correlations with rs113317823 (iCHAV2) and rs62355902 (iCHAV1). These subsets are (1) iCHAV2a (lead SNP rs113317823), which includes 90 SNPs correlated with rs113317823 (r2 > 0.53) and iCHAV1 SNP rs62355902 (r2 = 0.19–0.29) and is marked in dark blue in Table S5; (2) iCHAV2b (lead SNP rs62355899), which includes 66 SNPs independent of rs113317823 (r2 ≤ 0.01) but correlated with rs62355902 (r2 = 0.59–0.62) (conditional OR = 0.90, 95% CI = 0.86–0.95, pcond = 3.0 × 10−5) and is marked in teal in Table S5; and (3) iCHAV2c (lead SNP rs7721581), which includes 17 SNPs that are modestly correlated with rs113317823 (r2 = 0.14–0.16) but independent of rs62355902 (r2 ≤ 0.01) (conditional OR = 0.96, 95% CI = 0.93–0.98, pcond = 4.8 × 10−4) and is marked in pale blue Table S5.
To further clarify these association signals, we performed haplotype analyses based on the lead SNPs from iCHAV1, iCHAV2a, iCHAV2b, iCHAV2c, and iCHAV3. These five SNPs define seven common haplotypes (Table 2). Two of these—h5 (which carries the risk alleles of iCHAV1, iCHAV2a, and iCHAV2b) and h6 (which carries the risk alleles of iCHAV1 and iCHAV2b)—are strongly associated with risk, although the risk is higher for haplotype h6 (p = 1.66 × 10−29). These results are consistent with the observation that SNPs in iCHAV1 are the most strongly associated with risk. They are also consistent with a model in which either a SNP in iCHAV2a or iCHAV2c (in combination with iCHAV1) increases risk or a SNP in iCHAV2b reduces risk. These hypotheses are difficult to distinguish given that the iCHAV1 risk allele never occurs alone—it occurs in combination with either iCHAV2a or iCHAV2c or with iCHAV2b, but not both. Some support for the iCHAV1+iCHAV2a hypothesis is provided by the fact that although rare, haplotypes h3 and h4, which carry the risk alleles for iCHAV2a, but not iCHAV1, are associated with increased risk. Evidence against the iCHAV1+iCHAV2c hypothesis is provided by the fact that h1, which carries the risk allele for iCHAV2c alone, is not associated with increased risk. These observations are consistent with the regression analyses, in which iCHAV2c was less likely than iCHAV2a or iCHAV2b SNPs to harbor a causal variant (likelihood ratio ∼ 30:1 after adjustment for iCHAV1). Haplotype h2, which carries the minor allele of iCHAV3 SNP rs11949391, was associated with a reduced ER+ (but not ER−) breast cancer risk, consistent with the effect of the iCHAV3 SNP in the regression analysis.
Table 2.
Breast Cancer Risk in Europeans by Haplotypes of Five iCHAV Representative SNPs
|
Haplotypea |
|||||||
|---|---|---|---|---|---|---|---|
| h0 | h1 | h2 | h3 | h4 | h5 | h6 | |
| iCHAV1b | 1 | 1 | 1 | 1 | 1 | 2 | 2 |
| iCHAV2ac | 1 | 1 | 1 | 2 | 2 | 1 | 2 |
| iCHAV2bd | 1 | 1 | 1 | 1 | 1 | 2 | 1 |
| iCHAV2ce | 1 | 2 | 1 | 1 | 2 | 1 | 2 |
| iCHAV3f | 1 | 1 | 2 | 1 | 1 | 1 | 1 |
| Frequency | 0.52 | 0.12 | 0.15 | 0.02 | 0.01 | 0.11 | 0.05 |
| Overall Breast Cancer Risk | |||||||
| OR | – | 1.01 | 0.95 | 1.08 | 1.05 | 1.16 | 1.31 |
| 95% CI | – | 0.99–1.05 | 0.92–0.97 | 0.99–1.17 | 0.99–1.17 | 1.12–1.20 | 1.25–1.37 |
| p value | – | 4.50 × 10−1 | 2.08 × 10−4 | 7.57 × 10−2 | 3.81 × 10−1 | 2.00 × 10−20 | 1.66 × 10−29 |
| ER+Breast Cancer Risk | |||||||
| OR | – | 1.03 | 0.95 | 1.13 | 0.99 | 1.19 | 1.34 |
| 95% CI | – | 0.99–1.07 | 0.92–0.99 | 1.02–1.24 | 0.87–1.13 | 1.14–1.23 | 1.27–1.42 |
| p value | – | 1.12 × 10−1 | 5.23 × 10−3 | 1.68 × 10−2 | 8.75 × 10−1 | 9.37 × 10−20 | 3.74 × 10−27 |
| ER−Breast Cancer Risk | |||||||
| OR | – | 1.04 | 1.04 | 1.08 | 1.17 | 1.1 | 1.15 |
| 95% CI | – | 0.98–1.11 | 0.98–1.10 | 0.92–1.27 | 0.96–1.42 | 1.04–1.18 | 1.05–1.25 |
| p value | – | 2.08 × 10−1 | 1.67 × 10−1 | 3.45 × 10−1 | 1.12 × 10−1 | 1.82 × 10−3 | 3.64 × 10−3 |
1 represents major alleles, and 2 represents minor alleles in each SNP.
h1–h6 are compared to h0 (the reference haplotype carrying the major alleles of all five SNPs).
iCHAV1 represents 15 SNPs, of which rs62355902 is the best candidate.
iCHAV2a represents 90 SNPs that have r2 ≥ 0.53 with rs113317823 and r2 = 0.19–0.29 with rs62355902. rs113317823 is the best candidate.
iCHAV2b represents 66 SNPs that have r2 ≤ 0.01 with rs113317823 and r2 = 0.59–0.62 with rs62355902; rs62355899 is the best candidate.
iCHAV2c represents 17 SNPs that have r2 = 0.14–0.16 with rs113317823 and r2 ≤ 0.01 with rs6235590; rs7721581 is the best candidate.
iCHAV3 represents five SNPs, of which rs11949391 is the best candidate.
We conclude that at least one of the 90 SNPs in iCHAV2a (positions 56,001,002–56,270,717) or one of the 66 SNPs in iCHAV2b (positions 55,998,085–56,183,743) is causally related to risk together with a variant in iCHAV1 and a variant in iCHAV3.
Risk Associations in Asian and African American Studies
We tested all genotyped and imputed SNPs with MAF > 0.02 and imputation r2 > 0.3 in the nine Asian studies (6,269 case and 6,624 control individuals; 1,045 SNPs) and the two African American studies (1,116 case and 932 control individuals; 1,601 SNPs) for association with overall breast cancer risk. None reached genome-wide levels of significance (p < 5 × 10−8), but the lead SNPs of each iCHAV displayed compatible effects in all three ethnic groups. This was most apparent for iCHAV2a SNP rs113317823 (European unadjusted OR = 1.22, 95% CI = 1.18–1.26, ptrend = 7.0 × 10−25; Asian OR = 1.19, 95% CI = 1.11–1.27, ptrend = 1.4 × 10−5; African American OR = 1.04, 95% CI = 0.77–1.31, ptrend = 0.78; Table S6).
Identification of PREs that Interact with the MAP3K1 Promoter
After the epidemiological analyses, 15 iCHAV1, 90 iCHAV2a, 66 iCHAV2b, and 5 iCHAV3 variants remained as strong causal candidates (Figure 1A; Table S7), whereas the data provide weaker support that iCHAV2c is causal. Because iCHAV2a and iCHAV2b were composed of a large number of variants, we prioritized these for functional analysis by using a threshold of pcond < 1 × 10−4 and focused on the remaining 30 iCHAV2a and 10 iCHAV2b variants, in addition to the iCHAV1 and iCHAV3 candidates. Next, we examined whether these 62 iCHAV variants coincide with PREs that might affect gene expression.
Using publicly available ENCODE ChIP sequencing (ChIP-seq) data from MCF7 and human mammary epithelial cells, we identified regions marked by histone modifications associated with transcriptional enhancers (mono- or dimethylation of H3 lysine 4 [H3K4Me1 or H3K4Me2, respectively] and acetylation of H3 lysine 27 [H3K27Ac]) or bound by transcription factors ER-α, FOXA1, or GATA3, known to play a role in breast cancer. Next, we mined RNA polymerase II ChIA-PET (chromatin-interaction analysis with paired-end tag sequencing) data, previously generated in MCF7 cells,24,25 and identified multiple long-range chromatin interactions between discrete regions of the iCHAV loci and the promoter of MAP3K1 (Figure S3). Consequently, we performed 3C experiments to analyze interactions between the MAP3K1 promoter and these regions within 5q11.2. Using 3C in a normal mammary epithelial cell line, Bre-80, we found several regions that interacted with the MAP3K1 promoter (Figure 2C). Similar 3C profiles were observed in two ER+ cell lines (MCF7 and T-47D) and in ER− MDA-MB231 breast cancer cells. (Figure S4).
PREs were defined as the loci encompassing functional elements identified from the ENCODE data within a region interacting with the MAP3K1 promoter. This analysis revealed four PREs (PRE-A, PRE-B, PRE-C, and PRE-D) that coincided with the iCHAV candidates prioritized for functional analyses (Figure 2). Consistently, ENCODE ChIA-PET data demonstrated that all four PREs interacted with the MAP3K1 promoter (Figure S3A), but not with the promoters of other nearby genes in MCF7 cells (Figure S3B). It should be noted that all four PREs contained iCHAV2a or iCHAV2b variants (pcond > 1 × 10−4) that were not prioritized for functional analyses but could not be excluded as causal candidates after the log-likelihood testing (Figure 2A). Furthermore, additional PREs, some containing such iCHAV2a or iCHAV2b variants, were apparent at this locus (Figure 2). Twenty-six percent of the 62 iCHAV variants prioritized for functional analyses coincided with a PRE. In contrast, only 16% of SNPs with a MAF in the range of the MAF of the prioritized iCHAV variants (dbSNP 138, MAF = 0.04–0.18, accessed through the UCSC Genome Browser) at this locus were located in a PRE.
The Risk Alleles of iCHAV1 Candidates rs74345699 and rs62355900 Further Induce PRE-C Enhancer Activity after Estrogen Stimulation
For functional analysis of iCHAV1, we focused on 7 of the 15 candidate causal variants coinciding with a PRE (PRE-A, PRE-B, and PRE-C; Figure 2). We first examined the effects of these PREs on MAP3K1 promoter activity by cloning the relevant genomic regions into luciferase reporter gene constructs containing the MAP3K1 promoter. All three of the PREs had effects on MAP3K1 promoter activity: PRE-A acted as a silencer and reduced promoter activity by 62% (p = 0.006) in Bre-80 cells (Figure 3A) and had a similar but nonsignificant trend (p = 0.056) in MCF7 cells (Figure S5A). PRE-B1, a PRE-B subregion containing two candidate variants, acted as an enhancer in Bre-80 cells and increased promoter activity by 42% (p = 0.047; Figure 3B) but had no significant effect on MCF7 cells, indicating that PRE-B1 might function in specific breast cell types (Figure S5B). PRE-C acted as an enhancer and increased MAP3K1 promoter activity by 90% (p = 0.034) in MCF7 cells (Figure 3C) and by 77% (p = 0.034) in Bre-80 cells (Figure S5D). Introduction of the iCHAV1 minor alleles into the PRE-A, PRE-B1, and PRE-C reference constructs did not detectably alter MAP3K1 promoter activity (Figures 3A–3C; Figures S5A, S5B, and S5D).
Figure 3.
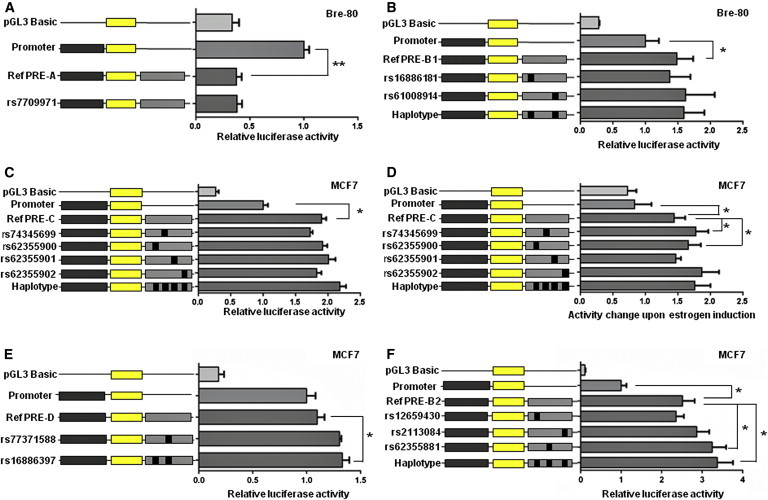
Risk Alleles of iCHAV1 and iCHAV2a SNPs Enhance MAP3K1 Promoter Activity in Luciferase Reporter Assays
PRE-A, PRE-B, PRE-C, and PRE-D regions containing the major allelic variants of iCHAV1, iCHAV2a, and iCHAV2b SNPs were cloned downstream of a MAP3K1-promoter-driven luciferase construct (promoter) for the creation of reference (ref) PRE constructs. Minor allelic variants of the iCHAV1, iCHAV2a, and iCHAV2b SNPs were engineered into the constructs and are designated by the rs ID of the corresponding SNP. Constructs containing minor allelic haplotypes (haplotype) were also generated. Cells were transiently transfected with each of these constructs and assayed for luciferase activity after 24 hr.
(A and B) Results from assays of PRE-A (A) and PRE-B1 (B) in Bre-80 cells.
(C, E, and F) Results from assays of PRE-C (C), PRE-D (E), and PRE-B2 (F) in MCF7 cells under basal conditions.
(D) Results after estrogen induction of MCF7 cells. For each reporter construct in this assay, the luciferase activity of estrogen-treated cells was normalized to the activity of the corresponding vehicle-treated cells.
Error bars denote the SEM from three experiments performed in triplicate. p values were determined by repeated-measures ANOVA followed by Dunnett’s multiple comparisons test (∗p < 0.05 and ∗∗p < 0.01).
ChIP-seq data from the ENCODE project indicate that at least two different transcription factors implicated in estrogen signaling, FOXA1 and ER-α, bind within PRE-B1 and PRE-C, respectively (Figure 2B). We thus tested whether the iCHAV1 candidates within PRE-B1 and PRE-C confer estrogen-dependent effects on MAP3K1 promoter activity. We first confirmed that MAP3K1 expression was upregulated by estrogen stimulation (Figure S6). Then, using reporter assays, we examined the effects of estrogen induction on PRE-B1 and PRE-C by measuring the changes in MAP3K1 promoter activity between estrogen-stimulated and -unstimulated cells. We showed that compared with the promoter construct, the PRE-C enhancer containing the protective (major) alleles (Figure 3D), but not PRE-B1 (Figure S5C), had a significant induction in activity (72%, p = 0.012) after estrogen stimulation. Induction was 23% (p = 0.032) and 15% (p = 0.011) greater in PRE-C enhancers containing the risk (minor) alleles of iCHAV1 candidate SNPs rs74345699 and rs62355900, respectively, than in this reference PRE-C enhancer (Figure 3D). Of note, neither of these candidates significantly affected MAP3K1 promoter activity in the absence of estrogen.
The Risk Allele of iCHAV2a Candidate rs16886397 in PRE-D Enhances MAP3K1 Promoter Activity
Of the 30 candidate causal variants for iCHAV2a at pcond < 1 × 10−4, one variant (rs77371588) coincided with a PRE (Figure 2), and we thus prioritized this SNP for functional analysis. Using luciferase reporter assays, we demonstrated that the reference PRE-D acted as an enhancer of the MAP3K1 promoter in Bre-80 cells and increased MAP3K1 promoter activity by 69% (p = 0.013; Figure S7A). The PRE-D enhancer containing the risk (minor) allele of rs77371588 had 23% greater enhancer activity than the reference PRE-D, but this effect did not reach statistical significance (p = 0.103; Figure S7A). By contrast, the same PRE-D reference construct did not affect MAP3K1 promoter activity in MCF7 cells, nor did the introduction of the risk allele of rs77371588 into PRE-D significantly alter its activity (Figure 3E). Because we had generated the PRE-D construct, it was straightforward to test rs16886397, an additional iCHAV2a causal candidate located in PRE-D. It did not reach the threshold for the functional prioritization (pcond < 1 × 10−4) but did pass the likelihood-ratio threshold of <100:1 for defining the causal iCHAV candidates. In MCF7 cells, the construct containing the minor (risk) allele of rs16886397 had 21% (p = 0.049) greater MAP3K1 promoter activity than the reference PRE-D (Figure 3E), and thus rs16886397 appears to confer enhancer activity on PRE-D. In Bre-80 cells, in contrast, the minor allele of rs16886397 had no effect on PRE-D activity (Figure S7A).
The Minor Allele of iCHAV2b Candidate rs62355881 Increases PRE-B2 Enhancer Activity
Of the ten iCHAV2b candidates at pcond < 1 × 10−4, three variants coincided with PRE-B, and two flanked the boundaries of PRE-C (Figure 2). We prioritized the three variants in PRE-B because these were the most compelling functional candidates given their central location in several functional elements (Figure 2B). Using reporter assays, we demonstrated that the reference PRE-B2 construct (a PRE-B subregion containing the iCHAV2b variants) acted as an enhancer and increased the activity of the MAP3K1 promoter by 152% (p = 0.032; Figure 3F) and 143% (p = 0.048; Figure S7B) in MCF7 and Bre-80 cells, respectively. The introduction of the minor (potentially protective) allele of the iCHAV2b candidate rs62355881 into the reference PRE-B2 construct led to a 29% (p = 0.017) increase in the enhancer activity of PRE-B2 in MCF7 cells, and the haplotype construct containing the minor alleles of all three iCHAV2b candidates demonstrated a similar effect (p = 0.030; Figure 3F). However, these effects were not seen in Bre-80 cells (Figure S7B), indicating another cell-type-specific effect, and the other iCHAV2b candidates did not have any effect on PRE-B2 activity in either cell line (Figure 3F; Figure S7B).
The Protective Allele of iCHAV3 Candidate rs17432750 Reduces PRE-B3 Enhancer Activity and GATA3 Binding
Of the five iCHAV3 candidates, we focused on two variants coinciding with PRE-B3 (Figure 2B) for functional analysis. Using reporter assays, we demonstrated that the reference PRE-B3 construct (a third subregion of PRE-B), containing the risk (major) iCHAV3 alleles, increased MAP3K1 promoter activity by 166% (p = 0.041; Figure 4A) and 110% (p = 0.035; Figure S8A) in Bre-80 and MCF7 cells, respectively. Reversing the orientation of PRE-B3 in the reporter gene construct had no effect on its activity in either cell line (Figures S8B and S8C), indicating that it acts as a typical enhancer. Next, we introduced the protective (minor) alleles of the iCHAV3 candidates into the reference PRE-B3 construct. The protective A allele of rs17432750 had a repressive effect and reduced PRE-B3 enhancer activity by 43% (p = 0.024; Figure 4A) in Bre-80 cells. The same allele had a similar but nonsignificant effect (p = 0.150) in MCF7 cells (Figure S8A). By contrast, the protective allele of the second iCHAV3 candidate, rs11956804, had no significant effect on enhancer activity in either cell line (Figure 4A; Figure S8A). The haplotype construct containing both iCHAV3 variants also had no significant effect on enhancer activity (Figure 4A; Figure S8A), suggesting the possibility of an interaction between the two iCHAV3 minor alleles in this construct.
Figure 4.
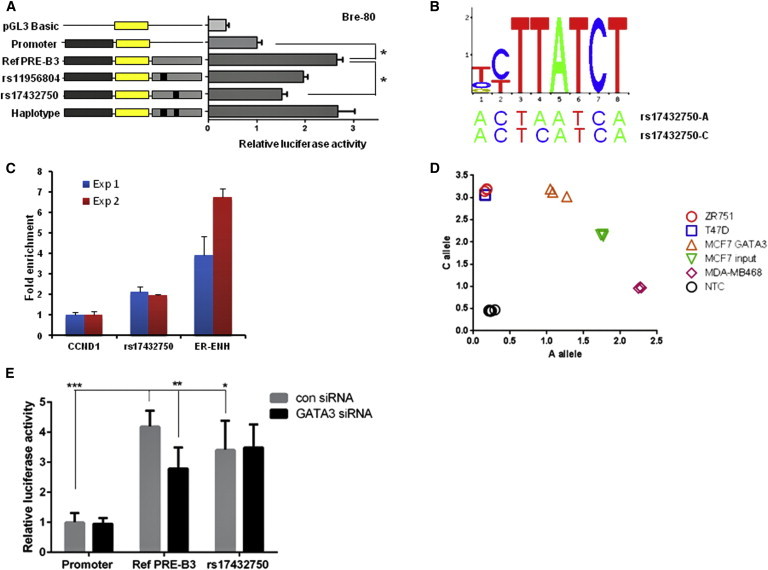
The Protective Allele of iCHAV3 SNP rs17432750 Demonstrates Diminished PRE-B3 Enhancer Activity in Luciferase Reporter Assays and Reduced GATA3 Binding in ChIP Analysis
(A) The PRE-B3 region containing the major allelic variants of iCHAV3 SNPs was cloned downstream of a MAP3K1-promoter-driven luciferase construct (promoter) for the creation of reference (ref) PRE constructs. Minor allelic variants of iCHAV3 SNPs were engineered into the constructs and are designated by the rs ID of the corresponding SNP. A construct containing the minor allelic haplotypes (haplotype) was also generated. Bre-80 cells were transiently transfected with each of these constructs and assayed for luciferase activity after 24 hr. Error bars denote SEM from three experiments performed in triplicate. p values were determined by repeated-measures ANOVA followed by Dunnett’s multiple comparisons test (∗p < 0.05).
(B) Position weight matrix of GATA3 is shown in relation to the negative strand of the sequences surrounding rs17432750.
(C) GATA3 ChIP assays demonstrate enrichment of rs17432750 in relation to the CCND1 negative control. A GATA3 site from the ER-α enhancer was included as a positive control. Results from two biological repeats are shown, and error bars denote SD of three technical repeats.
(D) Genotyping of rs17432750 in MCF7 genomic DNA versus MCF7 GATA3-ChIP DNA. Homozygous cell lines ZR751 (C/C), T47D (C/C), and MDA-MB-468 (A/A) and no template controls (NTCs) were included as references for the assay. The risk (major) C allele was preferentially precipitated in the ChIP experiment.
(E) Luciferase assay in Bre-80 cells shows the effect of GATA3 siRNA silencing on the activity of the MAP3K1 promoter alone (promoter) and with PRE-B3 constructs containing the C allele (ref PRE-B3) and protective A (minor) allele rs17432750 (rs17432750). Error bars denote SEM from three experiments performed in triplicate. p values were determined by two-way repeated-measures ANOVA followed by either Sidak’s multiple-comparisons test (to analyze the effect of GATA3 knockdown within constructs) or Dunnett’s multiple-comparisons test (to analyze differences in activity between constructs) (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). The level of GATA3 knockdown is shown in Figure S9.
We observed from MCF7 ChIP-seq data that a region containing rs17432750 bound the transcription factor GATA3 (Figure 2B) and that the sequence around rs17432750 showed homology to the GATA3 position weight matrix (Figure 4B). Using GATA3 ChIP assays, followed by qPCR detection, we confirmed that compared to the CCND1 negative control, the sequence surrounding this SNP showed consistent 2-fold enrichment in precipitated DNA (Figure 4C). We also tested the allele specificity of GATA3 binding by using a TaqMan genotyping assay for rs17432750 on ChIP samples from MCF7 cells. The allelic-discrimination plot of these data showed an enrichment of the risk (major) C allele in the GATA3 ChIP samples (Figure 4D). The ratio of the two alleles in three independent ChIP experiments indicated 3.7-fold greater GATA3 binding to the risk C allele than to the protective A allele in MCF7 cells.
To determine whether differential GATA3 transcription factor binding might explain the effects of rs17432750 in the reporter assays, we used siRNA to knock down GATA3 and found the enhancer activity of the reference PRE-B3 construct, containing the risk C allele, to be reduced by 33% in Bre-80 cells (p = 0.001; Figure 4E). GATA3 knockdown had no effect on the construct containing the protective A allele of rs17432750 or the MAP3K1 promoter alone (Figure 4E). Diminished GATA3 binding to the protective A allele thus appears to be responsible for the observed decrease in PRE-B3 enhancer activity under basal conditions (Figure 4A).
eQTL Analyses
Given the findings of these functional studies, an obvious hypothesis is that the candidate causal variants in the iCHAVs are associated with differences in expression of MAP3K1 and possibly other local genes in normal breast cells. We therefore explored potential eQTL associations between all locus SNPs and genes lying within ∼1 Mb of the locus in 135 normal breast tissue samples from the METABRIC study and 56 further normal breast samples from the TCGA study. Summary results for representative SNPs from the three iCHAVs are presented in Table S8. None of the iCHAV representative SNPs showed detectable differences in MAP3K1 expression in this data set. This locus had a positive control eQTL: SNP rs832402 was the most strongly associated SNP in that it showed SETD9 expression in both METABRIC (p = 5.93 × 10−9) and TCGA (p = 1.96 × 10−7) studies, but it was not a strong candidate SNP for breast cancer risk (pcond = 1.46 × 10−3). The positive control SNP was in iCHAV2c (correlated with lead SNP rs7721581 at r2 = 0.74), and SNP rs7721581 was consequently also associated, although less significantly, with SETD9 expression (METABRIC p = 4.38 × 10−8; TCGA p = 2.78 × 10−4). However, because none of the other representative iCHAV SNPs were associated with SETD9 expression, it appears unlikely that detectable SETD9 expression differences in normal breast cells are the underlying cause of breast cancer generated by the candidate functional variants we have identified.
Discussion
In this fine-scale mapping study, we found clear evidence of at least three independent breast cancer risk variants in European women: SNPs in iCHAV1 and iCHAV2 each had the greatest effects on breast cancer in the unadjusted analysis in that their minor alleles conferred increased risks of 25%–30% for ER+ and ∼10% for ER− tumor development, whereas the minor alleles of SNPs in iCHAV3 had a protective effect of ∼10% against ER+ breast cancer but no apparent effect on ER− tumor risk (Table 1). The originally detected GWAS tag SNP, rs889312, was most correlated with iCHAV1 (r2 = 0.5) and could be excluded from causality within iCHAV1 with a likelihood ratio of >1021:1. Within iCHAV2, we additionally found evidence of three subsets of variants (iCHAV2a, iCHAV2b, and iCHAV2c) with a range of effects. The epidemiological analyses suggest that iCHAV2c is least likely to be causally related to risk, and we conclude that at least one of the variants in iCHAV2a or iCHAV2b is functional. It should be noted that the correlations between candidate causal SNPs in iCHAV1 and iCHAV2 have added to this analysis and its interpretation a level of complexity that we have not recognized in previous fine-scale mapping studies of breast cancer risk loci.
We separately identified at least four 5q11.2 PREs that contain iCHAV candidate variants and interact with the MAP3K1 promoter in normal and cancer mammary epithelial cells. 3C analysis indicates that there are long-range chromatin interactions between these PREs and the MAP3K1 promoter, whereas interactions between the PREs and the promoters of other nearby genes are not evident from available ChIA-PET studies.24,25 Although we cannot rule out interactions between the iCHAVs and the promoters of other genes in the region, such as SETD9 or MIER3 (a proposed candidate gene for this risk locus26), we propose that MAP3K1 is the likely target gene of the 5q11.2 breast cancer susceptibility locus. Consistent with this proposition and our analyses, Corradin et al. identified an enhancer (chr5: 56,052,477–56,053,943) that coincides with the element we have termed PRE-C and that is predicted to regulate MAP3K1 on the basis of correlation between cell-type-specific H3K4me1 modification and MAP3K1 expression data.27
Our reporter assays indicate that PRE-B, PRE-C, and PRE-D act as enhancers, whereas PRE-A is a silencer of the MAP3K1 promoter. Having identified these regulatory elements, we investigated whether the iCHAV candidate causal variants within these PREs detectably modify their regulatory activity. We found that (1) the risk alleles of two iCHAV1 candidates, rs74345699 and rs62355900, in the PRE-C enhancer acted to further induce MAP3K1 promoter activity in breast cancer cells under estrogen stimulation; (2) the risk allele of iCHAV2a candidate rs16886397 in PRE-D conferred enhancer activity on this PRE for MAP3K1 promoter activity in breast cancer cells; (3) the potentially protective allele of iCHAV2b candidate rs62355881 in the PRE-B2 enhancer increased MAP3K1 promoter activity in breast cancer cells; and (4) the protective allele of iCHAV3 SNP rs17432750 diminished the enhancer activity of PRE-B3 for the MAP3K1 promoter in these cells.
Because of experimental constraints, we were only able to examine the functions of a minority of the iCHAV causal candidates, and we thus cannot exclude the presence of more functional variants across the recognizable iCHAVs. Of the candidates we examined in reporter gene assays, four support a hypothesis that alleles that increase MAP3K1 expression also increase breast cancer risk. Currently, the allelic effects of a fifth candidate, iCHAV2b SNP rs62355881, might be inconsistent with this hypothesis given that the conditional analysis suggests that iCHAV2b has a protective effect. However, we do not have clear epidemiological evidence that iCHAV2b has an individual effect on risk: iCHAV2b was only observed in the presence of iCHAV1, which acted in combination on the same haplotype to increase breast cancer risk, and we could not test such iCHAV haplotype effects in reporter gene assays given the number of candidates and the size of the region they encompass.
Consistent with our hypothesis that candidate causal risk alleles act by increasing transcriptional activation of MAP3K1, Godde et al. have recently demonstrated that upregulation of MAP kinase activity in mouse mammary basal progenitor cells is associated with ductal hyperplasia and accelerated tumor progression.28 This hypothesis is also supported by one known function of MEKK1: knockdown of MAP3K1 in human breast cancer cells reduces tumor invasiveness and progression in a mouse model.29 Furthermore, studies have shown that MEKK1 has an antiapoptotic effect and enhances cancer cell survival,30,31 although upon stress stimulus, caspase cleavage of the protein generates a fragment that plays a proapoptotic role.6,7 These dual functions of MEKK1 suggest that it has a complex role in cell-fate decisions. In this context, it is intriguing that somatic MAP3K1 driver mutations, found in tumor sequencing studies, are mostly truncating and are predicted to disrupt MEKK1 signaling,11 given that inactivation of the kinase domain at the protein terminus reduces apoptotic responses in cells exposed to stress.7 Thus, it appears that germline cancer risk alleles act by increasing MAP3K1 expression, but once a tumor has developed, somatic mutations drive cancer progression by disrupting MEKK1 signaling within the tumor. Dysregulation of MAP3K1 expression or MEKK1 function might thus promote tumorigenesis by perturbing a balance between cell apoptosis and survival. Similar conflicting effects of germline risk variants and somatic mutations have been observed at other breast cancer risk loci. For example, CCND1 is frequently amplified in breast tumors, and here, overexpression appears to play an important role in breast cancer pathogenesis,32 even though germline breast cancer risk alleles at 11q13 reduce CCND1 transcriptional activity.17 Amplification of TERT (MIM 187270) is also common within breast and other tumors,33 but again, germline breast cancer risk alleles reduce TERT transcriptional activity.34 These observations therefore challenge the notion that variants at loci such as 5q11.2 act in the same manner as somatic tumor driver mutations to confer germline risk of tumor development.
iCHAV1 spans multiple PREs (PRE-A, PRE-B, and PRE-C) and is consistent with a recent proposal that genetic susceptibility to common diseases can be explained by multiple enhancer variants that are in linkage disequilibrium, that each have modest effects on gene expression, and that cooperatively act to alter gene expression.27 There is also some suggestion from the haplotype data that iCHAV2a, which is in linkage disequilibrium (r2 = 0.19–0.29) with iCHAV1, might have a cooperative effect on risk in combination with iCHAV1. The modest effect sizes observed in the reporter assays for iCHAV1 and iCHAV2a variants might be a consequence of the fact that, as a result of size limitations of reporter gene constructs, we could not examine these variants in combination.
Some SNP effects in our reporter assays were suggestive of cell-line and stimulus dependence, highlighting the importance of cellular and environmental context in the assessment of SNP functionality.23 Similarly, we defined our PREs on the basis of chromatin modification and conformation states, both of which can change during development or in response to stimulus. Additionally, the effects of some SNPs might not have been observed because plasmid reporter gene constructs do not reflect the native genomic context or the chromatin or methylation state of genomic DNA. The chromatin state of transiently transfected DNA, for example, is considered to be more open and disorganized than the corresponding chromatin in the native genomic context and might not have the repressive chromatin structures, found in genomic DNA, that inhibit binding of ubiquitous transcription factors.35 A disorganized chromatin state might explain the inconsistent effect of rs17432750 in the PRE-B3 constructs (Figure 4A). The abrogation of the effect of rs17432750 in the haplotype construct suggests some interaction or cooperative effect of the minor alleles of rs17432750 and rs11956804 to enhance promoter activity. This effect might be possible as a result of the more open and permissive chromatin structures associated with transfected plasmid DNA.
It is noteworthy that, in available normal breast tissue, the top candidate causal variants showed no association with MAP3K1 expression, although iCHAV2c variants were associated with significant differences in SETD9 mRNA levels. Similarly, normal-breast-tissue-eQTL studies with strong candidate causal variants at the 11q13,1710q26,15 2q35,36 and 5p15.3334 breast cancer risk loci have indicated that available normal breast samples might be inappropriate for these studies. Sample sizes are large enough for detecting significant eQTLs at these loci, but those detected do not appear to drive breast cancer risk. It is possible that tissue-heterogeneity-, developmental-stage-, or stimulus-dependent effects prevent the detection of risk-driving eQTLs in currently available normal breast samples. Indeed, the finding that upregulation of MAP kinase activity in mammary progenitor cells is associated with mammary tumorigenesis28 suggests that increased MAP3K1 expression in specific breast cell populations, possibly at a specific point in time, could drive breast cancer risk.
Studies on transcription factor binding indicate that the C (risk) allele of rs17432750 preferentially binds GATA3 over the A (protective) allele. Increased binding of the C allele by GATA3 appears to explain the activity of the PRE-B3 enhancer in which it is located (Figure 4E). These findings suggest that SNP rs17432750 is a strong causal candidate for the protective effect of iCHAV3. The transcription factor GATA3 has multiple regulatory roles and can affect histone modifications associated with enhancers and the binding of other breast-cancer-related transcription factors, such as ER-α and FOXA1.37 We have previously identified GATA3 to be a mediator of breast cancer risk across multiple loci,38 specifically at the 11q13 locus.17
In conclusion, we have found evidence of the existence of at least three breast cancer risk iCHAVs that partially coincide with four MAP3K1 regulatory elements at 5q11.2. Genetic epidemiological studies within BCAC reduced the catalog of potentially causal variants from 909 to 193 candidates within five iCHAVs, of which at least three must be functional. Functional studies on candidates that lie within the identified regulatory elements have shown that the effects of strong candidate cancer risk alleles in iCHAV1, iCHAV2a, and iCHAV3 are compatible with the hypothesis that they act via increased expression of MAP3K1. Moreover, the function of MEKK1 suggests that increased expression might alter the balance between apoptosis and cell survival in breast cancer cells, thus explaining the risks conferred by the candidate alleles.
Published: December 18, 2014
Contributor Information
Alison M. Dunning, Email: amd24@medschl.cam.ac.uk.
Juliet D. French, Email: juliet.french@qimrberghofer.edu.au.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Easton D.F., Pooley K.A., Dunning A.M., Pharoah P.D., Thompson D., Ballinger D.G., Struewing J.P., Morrison J., Field H., Luben R., SEARCH collaborators. kConFab. AOCS Management Group Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J., Milne R.L., Schmidt M.K., Chang-Claude J., Bojesen S.E., Bolla M.K., Breast and Ovarian Cancer Susceptibility Collaboration. Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) kConFab Investigators. Australian Ovarian Cancer Study Group. GENICA (Gene Environment Interaction and Breast Cancer in Germany) Network Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013;45:353–361. doi: 10.1038/ng.2563. e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broeks A., Schmidt M.K., Sherman M.E., Couch F.J., Hopper J.L., Dite G.S., Apicella C., Smith L.D., Hammet F., Southey M.C., Genica Network. kConFab. AOCS Low penetrance breast cancer susceptibility loci are associated with specific breast tumor subtypes: findings from the Breast Cancer Association Consortium. Hum. Mol. Genet. 2011;20:3289–3303. doi: 10.1093/hmg/ddr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antoniou A.C., Spurdle A.B., Sinilnikova O.M., Healey S., Pooley K.A., Schmutzler R.K., Versmold B., Engel C., Meindl A., Arnold N., Kathleen Cuningham Consortium for Research into Familial Breast Cancer. OCGN. Swedish BRCA1 and BRCA2 study collaborators. DNA-HEBON collaborators. EMBRACE. GEMO. CIMBA Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am. J. Hum. Genet. 2008;82:937–948. doi: 10.1016/j.ajhg.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keshet Y., Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 6.Schlesinger T.K., Bonvin C., Jarpe M.B., Fanger G.R., Cardinaux J.R., Johnson G.L., Widmann C. Apoptosis stimulated by the 91-kDa caspase cleavage MEKK1 fragment requires translocation to soluble cellular compartments. J. Biol. Chem. 2002;277:10283–10291. doi: 10.1074/jbc.M106885200. [DOI] [PubMed] [Google Scholar]
- 7.Widmann C., Gerwins P., Johnson N.L., Jarpe M.B., Johnson G.L. MEK kinase 1, a substrate for DEVD-directed caspases, is involved in genotoxin-induced apoptosis. Mol. Cell. Biol. 1998;18:2416–2429. doi: 10.1128/mcb.18.4.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alarcon-Vargas D., Tansey W.P., Ronai Z. Regulation of c-myc stability by selective stress conditions and by MEKK1 requires aa 127-189 of c-myc. Oncogene. 2002;21:4384–4391. doi: 10.1038/sj.onc.1205543. [DOI] [PubMed] [Google Scholar]
- 9.Fuchs S.Y., Adler V., Pincus M.R., Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. USA. 1998;95:10541–10546. doi: 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke N., Arenzana N., Hai T., Minden A., Prywes R. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol. Cell. Biol. 1998;18:1065–1073. doi: 10.1128/mcb.18.2.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephens P.J., Tarpey P.S., Davies H., Van Loo P., Greenman C., Wedge D.C., Nik-Zainal S., Martin S., Varela I., Bignell G.R., Oslo Breast Cancer Consortium (OSBREAC) The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nordgard S.H., Johansen F.E., Alnaes G.I., Naume B., Børresen-Dale A.L., Kristensen V.N. Genes harbouring susceptibility SNPs are differentially expressed in the breast cancer subtypes. Breast Cancer Res. 2007;9:113. doi: 10.1186/bcr1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer K.B., O’Reilly M., Michailidou K., Carlebur S., Edwards S.L., French J.D., Prathalingham R., Dennis J., Bolla M.K., Wang Q., GENICA Network. kConFab Investigators. Australian Ovarian Cancer Study Group Fine-scale mapping of the FGFR2 breast cancer risk locus: putative functional variants differentially bind FOXA1 and E2F1. Am. J. Hum. Genet. 2013;93:1046–1060. doi: 10.1016/j.ajhg.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan-Wong S.M., French J.D., Proudfoot N.J., Brown M.A. Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc. Natl. Acad. Sci. USA. 2008;105:5160–5165. doi: 10.1073/pnas.0801048105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.French J.D., Ghoussaini M., Edwards S.L., Meyer K.B., Michailidou K., Ahmed S., Khan S., Maranian M.J., O’Reilly M., Hillman K.M., GENICA Network. kConFab Investigators Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am. J. Hum. Genet. 2013;92:489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prall O.W., Sarcevic B., Musgrove E.A., Watts C.K., Sutherland R.L. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 1997;272:10882–10894. doi: 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt D., Wilson M.D., Spyrou C., Brown G.D., Hadfield J., Odom D.T. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48:240–248. doi: 10.1016/j.ymeth.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 23.Edwards S.L., Beesley J., French J.D., Dunning A.M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.denDekker A.D., Xu X., Vaughn M.D., Puckett A.H., Gardner L.L., Lambring C.J., Deschenes L., Samuelson D.J. Rat Mcs1b is concordant to the genome-wide association-identified breast cancer risk locus at human 5q11.2 and MIER3 is a candidate cancer susceptibility gene. Cancer Res. 2012;72:6002–6012. doi: 10.1158/0008-5472.CAN-12-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corradin O., Saiakhova A., Akhtar-Zaidi B., Myeroff L., Willis J., Cowper-Sal lari R., Lupien M., Markowitz S., Scacheri P.C. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014;24:1–13. doi: 10.1101/gr.164079.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godde N.J., Sheridan J.M., Smith L.K., Pearson H.B., Britt K.L., Galea R.C., Yates L.L., Visvader J.E., Humbert P.O. Scribble modulates the MAPK/Fra1 pathway to disrupt luminal and ductal integrity and suppress tumour formation in the mammary gland. PLoS Genet. 2014;10:e1004323. doi: 10.1371/journal.pgen.1004323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuevas B.D., Winter-Vann A.M., Johnson N.L., Johnson G.L. MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene. 2006;25:4998–5010. doi: 10.1038/sj.onc.1209507. [DOI] [PubMed] [Google Scholar]
- 30.Hirano T., Shino Y., Saito T., Komoda F., Okutomi Y., Takeda A., Ishihara T., Yamaguchi T., Saisho H., Shirasawa H. Dominant negative MEKK1 inhibits survival of pancreatic cancer cells. Oncogene. 2002;21:5923–5928. doi: 10.1038/sj.onc.1205643. [DOI] [PubMed] [Google Scholar]
- 31.Nawata R., Yujiri T., Nakamura Y., Ariyoshi K., Takahashi T., Sato Y., Oka Y., Tanizawa Y. MEK kinase 1 mediates the antiapoptotic effect of the Bcr-Abl oncogene through NF-kappaB activation. Oncogene. 2003;22:7774–7780. doi: 10.1038/sj.onc.1206901. [DOI] [PubMed] [Google Scholar]
- 32.Arnold A., Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. 2005;23:4215–4224. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]
- 33.Cao Y., Bryan T.M., Reddel R.R. Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci. 2008;99:1092–1099. doi: 10.1111/j.1349-7006.2008.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bojesen S.E., Pooley K.A., Johnatty S.E., Beesley J., Michailidou K., Tyrer J.P., Edwards S.L., Pickett H.A., Shen H.C., Smart C.E., Australian Cancer Study. Australian Ovarian Cancer Study. Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Gene Environment Interaction and Breast Cancer (GENICA) Swedish Breast Cancer Study (SWE-BRCA) Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) Epidemiological study of BRCA1 & BRCA2 Mutation Carriers (EMBRACE) Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO) Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 2013;45:371–384. doi: 10.1038/ng.2566. e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith C.L., Hager G.L. Transcriptional regulation of mammalian genes in vivo. A tale of two templates. J. Biol. Chem. 1997;272:27493–27496. doi: 10.1074/jbc.272.44.27493. [DOI] [PubMed] [Google Scholar]
- 36.Ghoussaini M., Edwards S.L., Michailidou K., Nord S., Cowper-Sal Lari R., Desai K., Kar S., Hillman K.M., Kaufmann S., Glubb D.M., Australian Ovarian Cancer Management Group. Australian Ovarian Cancer Management Group Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat. Commun. 2014;4:4999. doi: 10.1038/ncomms5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theodorou V., Stark R., Menon S., Carroll J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12–22. doi: 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fletcher M.N.C., Castro M.A.A., Wang X., de Santiago I., O’Reilly M., Chin S.-F., Rueda O.M., Caldas C., Ponder B.A.J., Markowetz F., Meyer K.B. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013;4:2464. doi: 10.1038/ncomms3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.