

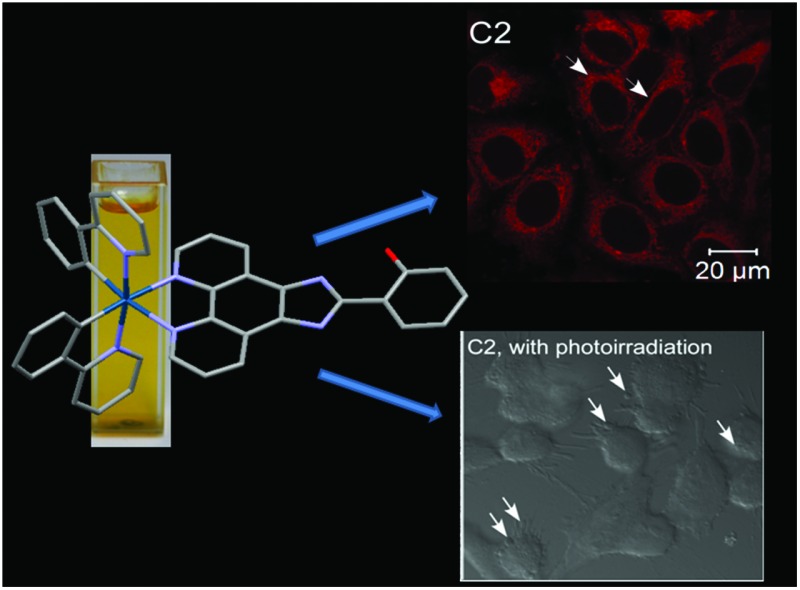
 A phosphorescent iridium(iii) complex shows selective localization and high fluorescence in the endoplasmic reticulum along with photoinduced cell death of breast carcinoma cells.
A phosphorescent iridium(iii) complex shows selective localization and high fluorescence in the endoplasmic reticulum along with photoinduced cell death of breast carcinoma cells.
Abstract
Cyclometalated iridium complexes have important applications as phosphorescent probes for cellular imaging due to their photophysical properties. Moreover, these properties also make them potential candidates as photosensitizers for photodynamic therapy (PDT) of tumors and skin diseases. Treatment of MCF7 breast carcinoma cells with a heteroleptic phosphorescent cyclometalated iridium(iii) complex C2 followed by confocal imaging indicates that the complex selectively localizes and exhibits high fluorescence in the endoplasmic reticulum. In an unprecedented approach, systematic alteration of functional groups or the metal core in C2 to synthesize a series of iridium(iii) complexes (C1–C10) and an organometallic rhenium complex C11 with an imidazolyl modified phenanthroline ligand has indicated the functional groups and their interactions that are responsible for this selective localization. Remarkably, the exposure of the cells treated with C2 to irradiation at 405 nm for one hour led to membrane blebbing and cell death, demonstrating a photosensitizing property of the compound.
Introduction
The cyclometalated heteroleptic iridium(iii) complexes of polypyridyl ligands are highly efficient in imaging of cells and cellular organelles, and are important tools for studying cellular function. The efficacy of the cyclometalated iridium(iii) complexes in this field has been demonstrated with a range of substituted phenyl pyridine and polypyridyl units.1,2 Presently, small-molecular dyes, fluorophore-conjugated antibodies or fluorescent proteins are used commercially as imaging agents for cellular organelles such as nuclei, mitochondria, endoplasmic reticulum (ER), Golgi complex and lysosomes.3 The cyclometalated iridium(iii) complexes provide a number of important advantages over these currently available imaging agents. These include enhanced chemical and photochemical stability within the cellular environment, permeability, minimization of self-quenching due to the significant Stokes’ shift, long-lived triplet excited state (τ ∼ μs) compared to organic dyes (τ ∼ ns) that helps to eliminate the short-lived autofluorescence of biological samples, and microenvironment-sensitive emission characteristics which are less damaging to cellular components.4,5 Kinetic inertness of the low-spin octahedral d6 complexes contributes to both reducing the cytotoxicity of the heavy metal ions and enhancing the stability of the complexes. The high photostability of these complexes allows the continuous monitoring of biological events by fluorescence spectroscopy and microscopy. High fluorescence lifetimes and tunable emission properties have resulted in their exploitation in sensors,6 OLEDs,7 OLECs,8 catalysts for water splitting9 and dye-sensitized solar cells (DSSCs),10 together with successful usage as cellular imaging agents.11
A major challenge in the field of cellular imaging is the selective staining of specific cell organelles or sub-cellular compartments. The factors that influence the cellular uptake and distribution within the microenvironment include permeability through the cell membrane, diffusion in the cellular cytoplasm, the charge-interaction associated with the organellar membranes during the uptake and interaction with organellar components through non-covalent forces, e.g. hydrogen bonds, ionic bonds, van der Waals forces and hydrophobic interactions, which results in specific localization. Within the micro- or nano-environment of the cell, specific localization is mostly regulated by pH,12 dielectric constant, hydrophobic and hydrophilic interactions and redox potential.13–16 Cyclometalated iridium complexes have been reported as cellular imaging probes for visualization of different organelles like nucleoli,17,18 Golgi complex,19 cytoplasm,1,2,20 mitochondria,21 lysosomes and endosomes22 and endoplasmic reticulum23 as well as intracellular metal-containing molecules21,24 and they have also been reported to act as efficient photosensitizing agents in cells.25 Therefore they have the potential of being used for photodynamic therapy (PDT) which involves the destruction of tumor cells by photoirradiation.25,26 PDT uses photosensitizing agents in a less invasive and more targeted form of cancer treatment. In many cases, the photosensitizing agents such as porphyrins, phthalocyanines or methylene blue27 have been shown to generate reactive singlet oxygen which causes cell death.25,28 The iridium(iii) complexes also have great potential in the field due to their high phosphorescence lifetime and cell permeability.29
In order to rationally design cyclometalated iridium(iii) complexes with specific subcellular localization properties, we have initially synthesized three simple imidazolyl-phenanthroline ligands and treated them with a cyclometalated iridium(iii) precursor to form phosphorescent iridium(iii) complexes. The complexes exhibit significant fluorescence intensity between pH 3 and 8 in a pH-dependent manner in vitro. Cellular imaging with these complexes showed differential fluorescence intensity in cells, and remarkable selectivity for the ER by one of the complexes. Alteration of the ancillary ligand in a systematic manner and synthesis of eight more complexes enabled us to locate the precise functional moieties and their interactions responsible for the ER localization and fluorescence emission. A detailed analysis of the complexes revealed that presence of strong intramolecular hydrogen bonding interactions and cationic nature of the metal complex will contribute significantly to the specific localization and fluorescence intensity. Finally, the ER localized complex was also found to have significant photosensitizing property, efficiently causing cell death induced by photoirradiation at 405 nm.
Results
Synthesis and characterization of complexes C1–C11
The primary criteria for designing a cell-organelle specific imaging agent involve the provision of interactions between the sub-cellular microenvironment and the potential imaging agent, resulting in specific localization20 and efficient fluorescence emission characteristics. Taking these criteria into consideration we selected the 2-phenyl-(1H-imidazo[4,5-f][1,10]phenanthroline backbone as the polypyridyl ligand with substitutions at different positions of the appended phenyl ring (Scheme 1). We initially synthesized three complexes (C1–C3) followed by eight other complexes (C4–C11). Complexes C1–C10 have been synthesized by refluxing [Ir(C^N)2Cl]2 in dichloromethane–acetonitrile solution at 60 °C with ligand 1–9, where systematic substitution of various functional groups have been done. C11 was obtained by the reaction of [Re(CO)5Cl] with ligand 2 in toluene at 110 °C (Chart 1). The synthesized complexes were purified and characterized by 1H NMR (Fig. S1 and S2†), high-resolution mass spectrometry (Fig. S3 and S4†), IR and elemental analysis. The photophysical studies showed that the complexes have absorption maxima at around 466–469 nm, 390–410 nm, 344–348 nm, 321–334 nm and 272–276 nm in organic solvents. The absorption bands with very high extinction coefficient at ca. 270–350 nm have been assigned to spin-allowed ligand-centered 1IL transitions (1π → π*), from both the ppy and phen ligands. The low-energy absorption shoulders at ca. 380–410 nm have been assigned to an admixture of spin-allowed metal-to-ligand charge-transfer (metal-to-ligand (1MLCT)(dπ–π* phen) and ligand-to-ligand (1LLCT)(π(ppy)–π*phen)) processes. The very low intensity band around 466–469 nm is assigned to spin-forbidden 3MLCT and 3LLCT transitions.30 C1–C11, upon excitation at 390 nm in acetonitrile at 298 K, phosphoresces with λ max ∼ 570–600 nm with phosphorescence quantum yield (Φ PL) and emission lifetime (τ e) in the range of 5.40–41.21% and 8.60–17.72 μs respectively (Table S1 and Fig. S5†). These systems possess a large ligand-field stabilization of iridium owing to its high oxidation state, the presence of 5d orbitals and, most importantly, a high ligand-field splitting energy due to the presence of two C∧N ligands. These enable the iridium to allow intersystem crossing from the first singlet excited state to the triplet state efficiently. Thus a long-lived triplet excited state (τ ∼ μs) and strong spin–orbit coupling (ζIr = 3909 cm–1) are characteristics of these complexes.31–33 The photophysical parameters of the complexes were quite satisfactory to explore their application as cellular imaging agents.
Scheme 1. Details of the synthesis of ligands 1–9 and complexes C1–C11, with procedures starting from 1,10-phenanthroline as the ligand and IrCl3·3H2O as the metal precursor.

Chart 1. Complexes at a glance. Molecular structures of C1–C11 showing the cyclometalated core and the ancillary ligands. C1–C9 have [Ir(ppy)2]+ as the cyclometalated core whereas C11 contains [Re(CO)3Cl] as the metal core. C1–C9 contain various substitutions in the phenolic ring of the ancillary ligand. C10 has benzo(h)quinoline instead of 2-phenylpyridine attached to Ir(iii).

C2 specifically localizes in the endoplasmic reticulum of cells
Initially, MCF7 breast carcinoma cells were treated with C1–C3 for 30 minutes following which fixed cells were imaged under a confocal laser scanning microscope at 405 nm excitation wavelength and 550–610 nm emission wavelength. Cells treated with C1 and C3, which contained –OH groups in position 4′ and at positions 2′ and 3′ of the appended phenolic ring respectively, showed diffused fluorescence throughout the cytoplasm of the cells and lower fluorescence in the nuclei (Fig. 1). In contrast, C2, with a –OH group at position 2′ of the phenolic ring (Scheme 1), showed specific and intense fluorescence only in the perinuclear region of the cells, suggesting specific localization in the ER. The cells were also transduced with a baculovirus-mammalian expression vector encoding Green Fluorescent Protein (GFP) fused with the calreticulin ER insertion sequence and the KDEL tetrapeptide retention sequence (CellLight ER-GFP from Life Technologies) which specifically caused the localization of the GFP in the ER. The fluorescence from C2 was found to completely and specifically colocalize with the GFP fluorescence in the ER whereas C1 and C3 did not show any colocalization with the GFP fluorescence. This demonstrated that C2 acted as a cell permeable, fluorescent imaging agent specifically marking the ER. The CellLight ER-GFP exhibited fluorescence nonhomogeneously, and in only a subset of cells, whereas C2 showed fluorescence in a majority of cells, suggesting the higher staining efficiency of C2 compared to the expression vector-based system.
Fig. 1. Confocal fluorescence images of MCF7 cells treated with C1–C4. The same cells were transduced with CellLight ER-GFP. The cells were imaged at λ ex = 405 nm and λ em = 550–610 nm for the complexes and at λ ex = 488 nm and λ em = 490–540 nm for CellLight ER-GFP. Green (left panels) represents CellLight ER-GFP, red (central panels) represents the complexes and the right panels represent merged images of both.

The 2′-OH group in the phenyl ring of C2 and its possible interaction with the imidazolyl N plays a crucial role in ER localization and fluorescence
Cyclometalated iridium(iii) complexes with different ligand systems have shown localization in specific cellular organelles but the involvement of the functional groups of the ligands, and their chemical interactions, in this process has not been elucidated.1,2,12,18–24 We therefore attempted to systematically determine the precise functional group(s) and their interactions responsible for the specific localization by following a “mutational approach” of systematically altering the various functional groups on the phenyl ring.
Firstly, in order to investigate the role of the 2′-OH group, cells were treated with cyclometalated iridium(iii) complex C4 containing the 2-phenyl-(1H-imidazo[4,5-f][1,10]phenanthroline) as an ancillary ligand, i.e. without any –OH groups. This complex (C4) showed high intensity fluorescence (quantum yield = 41.21%, τ = 14.57 μs) and considerable ER localization, but the localization was less specific compared to C2, with fluorescence exhibited in the nucleus and to some extent in the cytoplasm (Fig. 1). This suggests that the specific localization in the ER requires the presence of the –OH group at 2′ position of the appended phenyl ring. However, the other groups in the complex also play a role in conferring the fluorescence property. High fluorescence of C4 might be due to the absence of fluorescence quenching by the –OH moiety.
We then further investigated the role of the 2′-OH group, and its possible interaction with other groups in the complex. In order to investigate whether the charged nature of the –OH group and a possible intramolecular hydrogen bonding interaction (O–H···N) with the cyclic N of the imidazolyl group is responsible for the ER localization and fluorescence, we synthesized two more complexes (C5 and C6) by replacing –OH at positions 2′ and 4′ by –OCH3. C5 and C6 were found to have fluorescence lifetimes of 10.34 and 10.52 μs respectively. They also showed a very intense fluorescence with a quantum yield of 23.73% (C5) and 16.31% (C6). The only difference between C6 and C2 is the absence of O–H···N interaction. On treating the cells with these complexes and imaging, C5 diffused nearly uniformly over the entire cell, including the cytoplasm (Fig. 2). In the case of C6 although the complex did not localize in the nucleus, it diffused more in the cytoplasm than C2. This suggested that the O–H···N interaction is important for the very specific localization observed in the case of C2.
Fig. 2. Confocal fluorescence images of MCF7 cells treated with C5–C8. The same cells were transduced with CellLight ER-GFP. The cells were imaged at λ ex = 405 nm and λ em = 550–610 nm for the complexes and at λ ex = 488 nm and λ em = 490–540 nm for CellLight ER-GFP. Green (left panels) represents CellLight ER-GFP, red (central panels) represents the complexes and the right panels represent merged images of both.

To further study how substitutions at other positions in the phenolic ring might influence the ER localization and fluorescence emission of C2, a bulky tertiary butyl group was incorporated at positions 3′ and 5′ of the phenolic ring (C7) and an –OCH3 group was incorporated at position 3′ (C8). These complexes were also found to have phosphorescence lifetimes in the microsecond range (C7 = 11.66 μs, C8 = 8.60 μs). The quantum yield of the C7 (18.99%) was quite comparable to complexes C1–C6 whereas C8 had a reduced quantum yield of 6.74%. On treating the cells with these complexes, and subsequent imaging, it was found that the fluorescence emission was strongly reduced in both the cases, being nearly absent in the case of C8 (Fig. 3). Also, there was no specific localization in the case of C7. This suggested that substitution of other groups in the phenolic ring possibly caused structural distortions in the molecule which prevented the O–H···N interaction between the 2′-OH and the cyclic N of the imidazolyl group in the case of C8. On the other hand, in C7, the introduction of a new O–H···O(–CH3) type intramolecular hydrogen bonding interaction might disturb the existing O–H···N interaction and thus result in loss of fluorescence intensity and specific localization.
Fig. 3. Confocal fluorescence images of MCF7 cells treated with C9–C11. The same cells were transduced with CellLight ER-GFP. The cells were imaged at λ ex = 405 nm and λ em = 550–610 nm for the complexes and at λ ex = 488 nm and λ em = 490–540 nm for CellLight ER-GFP. Green (left panels) represents CellLight ER-GFP, red (central panels) represents the complexes and the right panels represent merged images of both.

Alteration of the cationic metal core, but not of the ligand systems, abrogates the fluorescence of C2
Finally, we altered the crucial functional groups in the imidazolyl ring of C2 to estimate their contribution to the localization specificity and fluorescence properties of the compound. Therefore we have replaced the hydrogen atom of the –NH group in the imidazole ring with 4-methoxyphenyl group (C9). We also replaced the 2-phenylpyridine ligand with benzo(h)quinoline (C10) and the [Ir(ppy)2]+ core with a [Re(CO)3Cl] core (C11) to investigate the role of the ligand and the metal ion in the properties of the molecule. Interestingly, the replacement of the hydrogen in the –NH group did not abrogate the fluorescence and localization of C9, but resulted in a decreased fluorescence intensity (Fig. 3). This suggested that the O–H···N interaction in C9 is weaker in comparison with C2, due to the loss in planarity of the imidazolyl and the phenyl rings. This is due to the steric hindrance between the 4-methoxyphenyl group attached to the imidazolyl nitrogen and the ortho-hydrogen of the appended phenyl group. Replacement of the 2-phenylpyridine ligand with benzo(h)quinoline did not diminish the fluorescence emission or change the localization of C10 suggesting that the nature of the spectator ligand is not significant for ER localization, provided it does not add any specific functional moieties outside the C–H framework (Fig. 3). On the other hand, replacement of the [Ir(ppy)2]+ core with a [Re(CO)3Cl] core completely abrogated all fluorescence, indicating the importance of iridium(iii) in determining the properties of these cyclometalated complexes and also the possible involvement of cationic [Ir(ppy)2]+ in the cellular uptake process (Fig. 3).34–37
Fluorescence and localization of C2 in cells are not pH-dependent but are dependent on intermediate lipophilicity
As the fluorescence, uptake and specific localization of a compound would be influenced by the pH of the medium and the lipophilicity of the compound, the pH dependence of the fluorescence emission (Fig. S5 and S6†) as well as the lipophilicity of the complexes (Table S1†) were estimated. The pH dependent fluorescence spectra (pH = 1–9) showed that all the complexes exhibited pH-dependent fluorescence in vitro, with highest fluorescence at acidic pH (Fig. S5 and S6†). C4 showed the maximum fluorescence intensity at pH 4, as well as in the organic solvent. Only C9 showed maximum fluorescence intensity at basic pH (pH = 9). C2 also fluoresced with the highest intensity at acidic pH, and the fluorescence intensity decreased with an increase in the pH, although there was considerable fluorescence in the range of pH 4–8. (Table S2†). The maximum fluorescence of C2 at acidic pH was surprising, considering that the pH of the ER lumen is thought to be nearly neutral.38 Therefore we tested whether the fluorescence of C2 in cells was dependent on pH by treating the cells with Bafilomycin A (BA), a macrolide antibiotic that specifically inhibits the vacuolar type ATPase (V-ATPase) in cells and thereby increases the pH of subcellular organelles.29 It was found that BA did not affect the fluorescence intensity or ER localization of C2, thereby suggesting that the fluorescence of C2 in cells is not pH-dependent (Fig. 4a). We also tested whether C2 marked other organelles with acidic pH (Golgi complex and lysosomes, pH ∼ 6.5 and 5.5 respectively) or alkaline pH (mitochondria pH ∼ 8) by treating the cells with a Golgi marker (CellLight Golgi-GFP), a mitochondrial marker (MitoTracker) and a lysosome marker (LysoTracker) (Fig. 4b). The localization of C2 was distinct from any of these organelles, suggesting that it specifically localizes to the ER rather than entering multiple cellular organelles and exhibiting pH-dependent fluorescence. This provides a better handle to design organelle-specific fluorescent markers than the previously reported iridium(iii) complexes which stain all acidic organelles in an indiscriminate manner.25
Fig. 4. (a) Confocal fluorescence images of MCF7 cells treated with C2 and Bafilomycin A. The same cells were transduced with CellLight ER-GFP. The cells were imaged at λ ex = 405 nm and λ em = 550–610 nm for the complexes and at λ ex = 488 nm and λ em = 490–540 nm for CellLight ER-GFP. (b) Confocal fluorescence images of MCF7 cells transduced with CellLight Golgi-GFP or treated with MitoTracker, LysoTracker or C2.

We also tested the lipophilicity of the compounds to estimate their cellular uptake and distribution (Table 1). Highly lipophilic compounds may be taken up easily through the cell membrane, but will have poor diffusion in the aqueous cell cytoplasm, and also low solubility in the cell culture medium, making it less available for cellular uptake. Measurements of partition coefficients showed that the increase in hydrophobic interaction increased the log P value, and interestingly, C8 and C11, which were found to have the highest log P values, showed the least cellular fluorescence. This might suggest a low cellular uptake as a possible reason for the low fluorescence exhibited by C8 and C11, although many other factors, such as interaction with the cellular microenvironment, might contribute to the loss of fluorescence. Conversely, C5, with the lowest log P values, show the maximum fluorescence within the cellular environment, but poor specific localization. C2, with an intermediate lipophilicity, showed good uptake, together with specific sub-cellular localization.
Table 1. Lipophilicity of complexes C1–C11 .
| Complex | Log Pa |
| C1 | 1.21 |
| C2 | 1.12 |
| C3 | 1.08 |
| C4 | 1.08 |
| C5 | 0.94 |
| C6 | 1.56 |
| C7 | 1.78 |
| C8 | 1.86 |
| C9 | 1.55 |
| C10 | 1.41 |
| C11 | 1.83 |
aPartition coefficient P = Co/Cw (Co and Cw are the complex concentrations in n-octanol and water, respectively); absorbance at 371 nm.
Molecular structure of C2 shows the presence of hydrogen bonding between 2′-OH of the phenyl ring and cyclic N of imidazole
To understand the specific intramolecular hydrogen bonding interactions in C2, the molecular structure of C2 was elucidated by single crystal X-ray crystallography (Fig. 5a, Tables 2 and S3†). The iridium(iii) center is coordinated in a slightly distorted octahedral fashion with the two N-donor groups of ligand 2 and the metalated C atoms of the 2-phenylpyridine ligands in a mutually cis arrangement. All bond lengths and bond angles are within normal ranges. The average values of the Ir–C(ppy), Ir–N(ppy) and Ir–N(ligand 2) distances are 2.068 (3) Å, 2.059 (3) Å and 2.129 (10) Å respectively.39 The structure consists of an intramolecular hydrogen-bonding interaction between imidazolyl nitrogen N5 (as a heterocyclic N-donor) and O1–H1A (as an acceptor). It has already been established that the probability of intramolecular ring formation correlates with the acceptor strength, and heterocyclic N is an efficient acceptor for this type of hydrogen bonding interaction.40 Standard geometric parameters for ‘hydrogen bond’ formation (distance between H and the acceptor atom = 2.30 Å) are well satisfied with 1.740 Å for O–H···N distance,41 with the formation of a 6-membered conjugated ring (Fig. 5b). The structure suggests that C2 is unique among these complexes as it contains strong intramolecular hydrogen bonding interaction.
Fig. 5. (a) ORTEP diagram of C2 with 50% thermal ellipsoids and all atoms numbered with hydrogen bonding interaction. Counter anions and hydrogen atoms are omitted for clarity except for O–H and N–H hydrogens. (b) Functionalized phenanthroline ligands showing bond lengths and bond angles around the hydrogen bond.

Table 2. Crystallographic parameters of C2.
| Empirical formula | C41H28ClIrN6O |
| F.w. | 848.36 |
| Cryst syst | Orthorhombic |
| Space group | P212121 |
| a (Å) | 9.650(4) |
| b (Å) | 15.353(6) |
| c (Å) | 25.967(11) |
| V (Å3) | 3847(3) |
| Z | 4 |
| λ (Å) | 0.71073 |
| Crystal size (mm3) | 0.22 × 0.10 × 0.05 |
| T (K) | 100 |
| ρ calcd (g cm–3) | 1.465 |
| μ (mm–1) | 3.579 |
| θ (°) | 1.54–25.00 |
| F(000) | 1672 |
| Index ranges | –7 ≤ h ≤ 11 |
| –16 ≤ k ≤ 18 | |
| –27 ≤ l ≤ 30 | |
| Data/restraints/params | 6787/1/152 |
| GOF (F 2) | 0.964 |
| R 1, wR2 (I > 2σ(I)) | 0.0658, 0.1354 |
| R 1, wR2 (all data) | 0.0986, 0.1489 |
| W | 1/[σ2(Fo2) + (0.0566P)2 + 0.0000P], where P = (Fo2 + 2Fc2)/3 |
C2 acts as a photosensitizing agent to cause cell death by limited photoirradiation
Cyclometalated iridium complexes have also been reported to act as efficient photosensitizing agents and induce cell death by photoirradiation.23 Therefore, we investigated whether C2 can also act as a photosensitizing agent and cause photoinduced cell death. Cells treated with C2 were exposed to laser irradiation at 405 nm for 1 h (2% AOTF laser power, 0.6 mW intensity), and live cells were imaged during the time of exposure. Cells treated with C2 and exposed to light at 405 nm showed progressive membrane blebbing, contraction of cells and generation of cellular processes, leading to nearly complete cell death after 1 h of exposure (Fig. 6a and b and ESI videos 1 and 2†). On the other hand, cells treated with C2, but not exposed to photoirradiation, did not show cell death (Fig. 6a). These observations suggested that C2 is an efficient photosensitizing agent which can cause cell death with a limited exposure to photoirradiation. We also tested whether C2 could cause cellular toxicity by treating cells for prolonged times (10 h, 22 h and 34 h) with 1 μM, 10 μM and 50 μM of C2, and measuring cell viability by performing an MTT assay. 10 μM is the concentration that has been used for all the imaging studies. We found that 10 μM of C2 does not cause any reduction of cell viability till 22 h of treatment, and shows a 50% decrease in cell viability only at 34 h of treatment (Fig. 6c). On the other hand, treatment with C2, followed by photoirradiation for only 1 h, was sufficient to cause cell death. This further reinforced the role of C2 as an efficient imaging and photosensitizing agent with minimal autonomous cytotoxicity.
Fig. 6. (a) DIC and confocal fluorescence images of live MCF7 cells treated with C2 and exposed to photoirradiation at 405 nm for 1 hour. The upper panel shows the cells prior to photoirradiation whereas the lower panel shows cells at the end of the photoirradiation period. The central tile represents the field which was exposed to the laser whereas the surrounding tiles represent the fields not exposed. (b) Frames captured from the video of the live imaging of the cells exposed to photoirradiation. Frames for both photoirradiated and non-photoirradiated cells are at 30 minutes of exposure to photoirradiation. (c) MTT assay of MCF7 cells exposed to no or three increasing concentrations of C2 and incubated for 10, 22 and 34 hours.

C2 and photoirradiation cause apoptotic cell death by generation of reactive oxygen species
To understand the mechanism of photoinduced cell death caused by C2 we subjected the cells to four conditions: (1) C2 in the presence of photoirradiation, (2) C2 in the absence of photoirradiation, (3) photoirradiation in the absence of C2 and (4) the absence of both C2 and photoirradiation (Fig. 7). After the experimental treatment, the cells were stained with Annexin V-FITC, which binds to externalized phosphatidyl serine of cell membranes which is a marker of the early stage of apoptosis, and propidium iodide (PI) which stains the DNA in membrane permeabilized cells which is a marker of the late stage of apoptosis. Only cells that were treated with both C2 and photoirradiation at 405 nm were found to be positive for both Annexin V and PI staining, which suggests apoptotic cell death. Also, the cells showed membrane blebbing and characteristic changes in morphology, as observed in the DIC image. Cells treated with C2 alone in the absence of photoirradiation showed faint Annexin V staining whereas cells which were either only photoirradiated for 1 hour or not treated at all did not show any cell death. These observations demonstrated that only photoirradiation of cells after uptake of C2 can cause photoinduced cell death by apoptosis.
Fig. 7. DIC and fluorescence images of MCF7 cells treated with (1) C2 and exposed to photoirradiation at 405 nm for 1 hour, (2) treated with C2 but not exposed to photoirradiation, (3) not treated with C2 but exposed to photoirradiation and (4) not treated with C2 or photoirradiated. Following treatment the cells were stained with Annexin V-FITC (right panels) and propidium iodide (central panel).

We then investigated whether the photosensitizing property of C2 was mediated by the generation of reactive oxygen species (ROS). Singlet oxygen has been reported to be generated on photoirradiation of Ir(iii) complexes and suggested to cause cell death although in vivo singlet oxygen generation has not been demonstrated.25 To understand the photosensitizing property of C2, we initially checked the generation of singlet oxygen (1O2) by the complex (5 μM) in organic solvents upon photoirradiation at 366 nm measured by the change in UV/vis spectra of 1,3-diphenylisobenzofuran (DPBF, 25 μM). DPBF is possibly the most well known 1O2 acceptor, and the absorbance maximum of DPBF at 415 nm decreases by the reaction with 1O2.42 We observed a decline in DPBF absorbance on photoirradiation in the presence of C2 in an organic solvent (methanol) (Fig. 8a). The 1O2 quantum yield of the C2 (Φ = 0.21; Φ methylene blue = 0.5) was determined by comparing with a methylene blue standard. However, the generation of singlet oxygen was not detected in DMSO–H2O, as DPBF dimerizes in aqueous media and thus becomes unreactive toward singlet oxygen in the H2O-rich mixture.43 We then investigated the generation of ROS in photoirradiated cells treated with C2. Cells were treated with C2 and photoirradiated at 405 nm for 30 min, following which they were treated with 2′,7′-dichlorofluorescein diacetate (DCFDA) which fluoresces at 529 nm on generation of ROS. It was observed that among the cells treated with C2, only those cells that were photoirradiated and showed the characteristic morphology of photoinduced cell death demonstrated the DCFDA fluorescence, suggesting that the cell death was induced by the generation of ROS (Fig. 8b). Cells that were irradiated in the absence of C2 treatment did not show any fluorescence with DCFDA (Fig. S7†).
Fig. 8. (a) Comparative photooxidation of only 1,3-diphenylisobenzofuran (DPBF) (25 μM) in methanol (red dot), C2 (5 μM) + DPBF (black square) and methylene blue (5 μM) + DPBF (blue triangle) in methanol degassed by O2. Excitation at 366 nm for C2 and DPBF. (b) DIC and confocal fluorescence images of live MCF7 cells treated with C2 and exposed to photoirradiation at 405 nm for 30 min. The cells were treated with DCFDA and fluorescence images were obtained at 529 nm after excitation at 495 nm. The central tile represents the field which was exposed to the laser whereas the surrounding tiles represent the fields not exposed.

Discussion and conclusion
Rational designing of a cell organelle-specific fluorescent marker would involve the provision of interactions between the sub-cellular microenvironment and the potential imaging agent, which would result in specific localization18 and efficient fluorescence emission properties. These interactions may be influenced both by the properties of the imaging agent and the cellular microenvironment such as pH of the cellular compartment, intramolecular and intermolecular hydrogen bonding interactions, sensitivity towards redox potential and charge interactions during cellular uptake, and diffusion and localization within the cell. In our attempt to design cyclometalated iridium complexes with imidazolyl modified phenanthroline ligands based on these criteria, one particular complex, C2, showed specific localization and high fluorescence emission in the endoplasmic reticulum of cells. Using an unprecedented “mutational” approach, systematic substitution of functional groups and elucidation of the crystal structure of C2 have provided insights into the intramolecular chemical interactions responsible for its photophysical properties and specific subcellular localization. A specific hydrogen bonding interaction between the 2′-OH group in the appended phenyl ring and the –NH group attached to the imidazole ring was found to confer localization specificity to the complex. Also, the highly stable resonance-assisted hydrogen bonding resulted in enhanced π-delocalization within the complex C2, and resulted in improved fluorescence due to excited state intramolecular proton transfer (ESIPT).44 On the other hand, replacement of the –NH group with N-anisidine in C9, which is expected to maintain the hydrogen bonding interaction, did not alter the ER specific localization, but reduced the fluorescence intensity substantially. This is possibly due to the distortion of planarity between the imidazolyl group and the appended phenyl group, increasing the proportion of the open form of the molecule relative to the closed form contributed by the intramolecular hydrogen bonding.
While changes in the functional groups in the phenyl and imidazolyl rings only altered subcellular localization or reduced fluorescence emission, the replacement of the [Ir(ppy)2]+ with [Re(CO)3Cl] in C11 completely abrogated all fluorescence, suggesting that the cationic nature of Ir(iii) is playing a crucial role in cellular uptake. On the other hand, replacement of 2-phenylpyridine ligand with benzo(h)quinoline did not substantially alter ER localization or fluorescence emission suggesting that a change in the C–H framework of the spectator ligand does not have a profound effect on subcellular localization.
The fluorescence emission of cyclometalated iridium(iii) complexes is known to be pH dependent based on the ancillary ligand system.45 In our study, in vitro fluorescence of the complexes was found to be pH-dependent, with maximum fluorescence of most of the complexes being in the acidic pH range. However, the high fluorescence of C2 in the ER, which is reported to have neutral pH, and the absence of a decrease in fluorescence intensity under Bafilomycin A treatment showed that the fluorescence of C2 in cells was not pH dependent. This is unlike the properties of cyclometalated Ir(iii) complexes reported previously, which specifically stained acidic organelles.25 This provides a better tool to design organelle-specific fluorescent markers which might be targeted to specific sub-cellular compartments or organelles by substituting different functional groups. Also, the intermediate lipophilicity of the complex appears to have contributed to the efficient uptake and subcellular localization, suggesting that efficient diffusion through the cytoplasm might be beneficial for targeting to specific organelles.
Furthermore, the complex has been found to be an efficient photosensitizing agent, causing extensive photoinduced cell death upon exposure to light at 405 nm for one hour. Ir(iii) complex functionalized nanoparticles have been reported to have applications in PDT and a recent study has shown an iridium(iii) complex with the N,N-diethylamino group in the ancillary ligand21,23–25 to cause photoinduced necrosis-like cell death. However, C2 was found to cause photoinduced apoptotic cell death and very little cytotoxicity in the absence of photoirradiation. These results suggest that the complex has enormous potential for application as a cellular imaging agent in cell biology as well as a photosensitizer for PDT in medicine. Further studies need to be directed at elucidating the mechanism by which C2 causes apoptotic cell death as a result of photoirradiation.
Experimental section
The starting materials IrCl3·3H2O, 2-phenylpyridine, 1,10-phenanthroline, aldehydes, and n-octanol were purchased from Sigma-Aldrich and used without further purification. All the solvents were dried by usual methods prior to use. The cyclometalated iridium(iii) chloro bridged dimer [Ir(ppy)2Cl]2 was prepared according to the methods described in the literature.46
Synthesis of ligands
The ligands 1–8 47 and 9 (–N-Ph has been replaced by N-p-anisidine)48 were synthesized with a similar procedure to that reported earlier. Ligands 3, 5, 6, and 8 were not reported earlier. See the ESI† for details of the synthesis and Fig. S1 and S3† for NMR and ESI-MS data.
Synthesis of complexes
Complexes C2 and C4 were synthesized with a similar procedure to that reported earlier.40 Complexes C1, C3, C5, C6, C7, C8, C9, C10 and C11 were not reported earlier. See the ESI for details of the synthesis and Fig. S2 and S4† for NMR and ESI-MS data.
Physical measurements
IR spectra were obtained on a Perkin-Elmer Spectrum RXI spectrophotometer with samples prepared as KBr pellets. Elemental analyses were performed on a Perkin-Elmer 2400 series II CHN series. Electronic spectra were recorded on a U-4100, HITACHI spectrometer. 1H NMR spectra were obtained on a Jeol 400 MHz NMR spectrometer using TMS as the internal standard. Electrochemical measurements were made using a PAR model 273 potentiostat. A platinum disk working electrode, a platinum wire auxiliary electrode and an aqueous Ag/Ag+ reference electrode were used in a three electrode configuration. Electrochemical measurements were made under a dinitrogen atmosphere. All electrochemical data were collected at 298 K and are uncorrected for junction potentials. Mass spectra were recorded on a Q-Tof Micromass spectrometer by positive-ion mode electrospray ionization. Fluorescence spectra were taken on a HORIBA JOBINYVON spectrofluorometer Fluoromax 4. Quantum yield data reported here were measured relative to quinine sulfate in 0.1 M H2SO4 (λ ex = 350 nm, Φ = 0.577). The integration of the emission spectra was obtained from the Fluoromax-4 instrument directly. The photoluminescence spectra were measured on a Horiba Jobin Yvon spectrometer equipped with a 150 W Xe lamp. The lifetime measurements were done by exciting the sample using a pulsed Xe lamp. Lifetime measurements were performed with dry deaerated dichloromethane solution (conc. 10–5 M). The first order exponential decay curve was fitted to obtain the phosphorescence lifetime.
X-ray crystallography
Crystal data were collected on a Bruker SMART APEXII CCD area-detector diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). For the crystal, X-ray data reduction was carried out using the Bruker SAINT program. The structures were solved by direct methods using the SHELXS-97 program and refinement was done using the SHELXL-97 program. Selected crystal data and data collection parameters for all the complexes are given in Table 1. X-ray data reduction, structure solution and refinement were done using the SHELXL-97 program package.49
Partition coefficient determination50
The lipophilicity of complexes C1–C11 was determined by the “shake flask” method using a pH 7.4 phosphate buffer (0.129 M NaCl) and n-octanol as solvents. Each compound was dissolved in the phase in which it is most soluble, resulting in typical concentrations of 50–350 μM. Duplicate determinations using three different solvent ratios were performed for each complex. Following the mixing and phase separation, each phase was analyzed for solute content, and the concentration was determined using spectrophotometric methods. All the n-octanol–water partition coefficients were determined by UV/vis spectroscopy. Octanol and buffer solutions were presaturated. Fifty rotations were performed by hand, followed by 1 h of settling time. Equilibration and absorption measurements were made at 20 °C.
Study of pH-dependent fluorescence
Tris buffer solution (500 ml, 0.1 M) was prepared and a series of buffer solutions having pH in the range 2–9 was prepared by adding 1 M HCl/NaOH aqueous solution to the Tris solution. The pH of the resulting solutions was recorded using a pH meter. 31 × 10–5 M stock solutions of all the complexes were prepared in dry DMSO, and in the fluorescence cuvette, 3.0 ml buffer solution and 0.1 ml complex solution in DMSO were mixed and fluorescence emission was recorded at 25 °C.
Singlet oxygen (1O2) quantum yield
Quantum yields for singlet oxygen generation in CH3OH were determined by monitoring the photooxidation of 1,3-diphenylisobenzofuran (DPBF) sensitized by the Ir complexes. Values of the triplet photosensitizers were calculated according to a modified literature method.51 The DPBF and samples with DPBF were irradiated at 405 nm and methylene blue was irradiated at 575 nm.25 The light source of a spectrofluorometer was used as an irradiation light source (150 W Xe source) to obtain constant monochromatic light. The absorbance of DPBF was adjusted to around 1.0 at 415 nm in oxygen saturated CH3OH, and the absorbance of the sensitizers was adjusted to 0.2–0.3 at the irradiation wavelength. The photooxidation of DPBF was monitored at intervals of 5 s. The quantum yields of singlet oxygen generation (Φ Δ) were calculated by a relative method using methylene blue (in methanol) (Φ Δ = 0.50)52 as the reference. The following equation was used:Φ Δ(C2) = Φ Δ(std)(m C2·F std)/(m std·F C2)where subscripts ‘C2’ and ‘std’ designate the iridium complex and methylene blue, respectively, m is the slope of a plot of differences in changes in absorbance of DPBF (at 415 nm) with the irradiation time and F is the absorption correction factor, which is given by F = 1–10–A (absorbance at the irradiation wavelength).
Cellular imaging
MCF7 human breast carcinoma cells were maintained in DMEM with 10% FBS. Cells were treated with 10 μM of the complexes in 0.1% DMSO in DMEM medium. Cells were treated for 30 minutes, following which they were washed with PBS (pH 7.4) and fixed with 4% paraformaldehyde. Fixed cells were imaged in a Zeiss LSM710 confocal laser scanning microscope at 405 nm excitation wavelength and 550 nm–610 nm emission wavelengths. For identifying the ER, cells were also transduced with a baculovirus-mammalian expression vector encoding Green Fluorescent Protein (GFP) fused with the calreticulin ER insertion sequence and the KDEL tetrapeptide retention sequence (CellLight ER-GFP from Life Technologies) according to the manufacturer's instructions. A similar treatment was also performed for identifying the Golgi complex (CellLight ER-GFP from Life Technologies). Mitochondria and lysosomes were stained with MitoTracker and LysoTracker dyes (Life Technologies) respectively according to the manufacturer's instructions. For the inhibition of V-type ATPases, cells were treated with 0.5 μM Bafilomycin A1 (Sigma Life Sciences) for 1 hour prior to treatment with the complex. For live cell imaging, cells were treated with the complex 10 minutes prior to the initiation of live cell imaging. Live imaging at 405 nm was done for 1 hour. For checking apoptotic cell death, cells were stained with Annexin-V FITC and propidium iodide (PI) after treatment with C2 and photoirradiation. For checking ROS generation, cells were treated with 2′,7′-dichlorofluorescein diacetate (DCFDA), a fluorogenic dye that fluoresces in the presence of ROS with excitation and emission maxima at 495 nm and 529 nm respectively.
Cell viability assay
MCF7 cells were treated with 1 μM, 10 μM and 50 μM of the complex and kept for 10 hours, 22 hours and 34 hours. At the end of these incubation times, cells were washed with PBS and 0.5 mg ml–1 of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT from Sigma Life Sciences) was added to the medium. Cells were incubated for 3 hours following which cells were lysed with 0.1 N HCl in isopropanol and absorption readings were taken at 595 nm.
Acknowledgments
Soumik Mandal and Dipak K. Poria are thankful to CSIR, New Delhi for Senior Research Fellowship. This work is supported by DST, New Delhi through research grants ST/FT/CS-057/2009 awarded to Parna Gupta and Wellcome Trust-DBT India Alliance Intermediate Fellowship WT500139/Z/09/Z awarded to Partho Sarothi Ray. We thank Dr. G. Subba Rao, MCB, IISc. Bangalore for helpful discussions.
Footnotes
References
- Lo K. K.-W., Choi A. W.-T., Law W. H.-T. Dalton Trans. 2012;41:6021. doi: 10.1039/c2dt11892k. [DOI] [PubMed] [Google Scholar]
- Zhao Q., Huanga C., Li F. Chem. Soc. Rev. 2011;40:2508. doi: 10.1039/c0cs00114g. [DOI] [PubMed] [Google Scholar]
- Hilderbrand S. A. Methods Mol. Biol. 2010;591:17. doi: 10.1007/978-1-60761-404-3_2. [DOI] [PubMed] [Google Scholar]
- Ho C. L., Wong K.-L., Kong H.-K., Ho Y.-M., Chan C. T.-L., Kwok W.-M., Leung K. S.-Y., Tam H.-L., Lam M. H.-W., Ren X.-F., Ren A.-M., Feng J.-K., Wong W.-Y. Chem. Commun. 2012;48:2525. doi: 10.1039/c2cc16993b. [DOI] [PubMed] [Google Scholar]
- Sun H., Yang L., Yang H., Liu S., Xu W., Liu X., Tu Z., Su H., Zhao Q., Huang W. RSC Adv. 2013;3:8766. [Google Scholar]
- Yam V. W.-W., Wo K. M.-C. Chem. Commun. 2011;47:11579. doi: 10.1039/c1cc13767k. [DOI] [PubMed] [Google Scholar]
- Chang C.-H., Wu Z.-J., Chiu C.-H., Liang Y.-H., Tsai Y.-S., Liao J.-L., Chi Y., Hsieh H.-Y., Kuo T.-Y., Lee G.-H., Pan H.-A., Chou P.-T., Lin J.-S., Tseng M.-R. ACS Appl. Mater. Interfaces. 2013;5:7341. doi: 10.1021/am401694s. [DOI] [PubMed] [Google Scholar]
- Brooks A. C., Basore K., Bernhard S. Inorg. Chem. 2013;52:5794. doi: 10.1021/ic302629q. [DOI] [PubMed] [Google Scholar]
- Curtin P. N., Tinker L. L., Burgess C. M., Cline E. D., Bernhard S. Inorg. Chem. 2009;48:10498. doi: 10.1021/ic9007763. [DOI] [PubMed] [Google Scholar]
- Baranoff E., Yum J.-H., Jung I., Vulcano R., Grtzel M., Nazeeruddin M. K. Chem. – Asian J. 2010;5:496. doi: 10.1002/asia.200900429. [DOI] [PubMed] [Google Scholar]
- You Y., Nam W. Chem. Soc. Rev. 2012;41:7061. doi: 10.1039/c2cs35171d. [DOI] [PubMed] [Google Scholar]
- Han J., Burgess K. Chem. Rev. 2010;110:2709. doi: 10.1021/cr900249z. [DOI] [PubMed] [Google Scholar]
- Patra M., Gasser G., Metzler-Nolte N. Dalton Trans. 2012;41:6350. doi: 10.1039/c2dt12460b. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G., Metzler-Nolte N., Dyson P. J. Organometallics. 2012;31:5677. [Google Scholar]
- Romero-Canelón I., Sadler P. J. Inorg. Chem. 2013;52:12276. doi: 10.1021/ic400835n. [DOI] [PubMed] [Google Scholar]
- Liu Z., Sadler P. J. Acc. Chem. Res. 2014;47:1174. doi: 10.1021/ar400266c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Yu M., Sun Y., Wu Y., Huang C., Li F. A. J. Am. Chem. Soc. 2011;133:11231. doi: 10.1021/ja202344c. [DOI] [PubMed] [Google Scholar]
- Zhang K. Y., Po-Yam Li S., Zhu N., Wai-Shan Or I., Shau-Ha Cheung M., Lam Y.-W., Lo K. K.-W. Inorg. Chem. 2010;49:2530. doi: 10.1021/ic902465b. [DOI] [PubMed] [Google Scholar]
- Zhang K. Y., Liu H.-W., Fong T. T.-H., Chen X.-G., Lo K. K.-W. Inorg. Chem. 2010;49:5432. doi: 10.1021/ic902443e. [DOI] [PubMed] [Google Scholar]
- Wu H., Yang T., Zhao Q., Zhou J., Li C., Li F. A. Dalton Trans. 2011;40:1969. doi: 10.1039/c0dt01366h. [DOI] [PubMed] [Google Scholar]
- Li S. P.-Y., Lau C. T.-S., Louie M.-W., Lam Y.-W., Cheng S. H., Lo K. K.-W. Biomaterials. 2013;34:7519. doi: 10.1016/j.biomaterials.2013.06.028. [DOI] [PubMed] [Google Scholar]
- Steunenberg P., Ruggi A., van den Berg N. S., Buckle T., Kuil J., van Leeuwen F. W. B., Velders A. H. Inorg. Chem. 2012;51:2105. doi: 10.1021/ic201860s. [DOI] [PubMed] [Google Scholar]
- Cao R., Jia J., Ma X., Zhou M., Fei H. J. Med. Chem. 2013;56:3636. doi: 10.1021/jm4001665. [DOI] [PubMed] [Google Scholar]
- You Y., Cho S., Nam W. Inorg. Chem. 2013;53:1804. doi: 10.1021/ic4013872. [DOI] [PubMed] [Google Scholar]
- Moromizato S., Hisamatsu Y., Suzuki T., Matsuo Y., Abe R., Aoki S. Inorg. Chem. 2012;51:12697. doi: 10.1021/ic301310q. [DOI] [PubMed] [Google Scholar]
- Celli J. P., Spring B. Q., Rizvi I., Evans C. L., Samkoe K. S., Verma S., Pogue B. W., Hasan T. Chem. Rev. 2010;110:2795. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H., van Lier J. E. Chem. Rev. 1999;99:2379. doi: 10.1021/cr980439y. [DOI] [PubMed] [Google Scholar]
- Yogo T., Urano Y., Mizushima A., Sunahara H., Inoue T., Hirose K., Iino M., Kikuchi K., Nagano T. Proc. Natl. Acad. Sci. U. S. A. 2008;105:28. doi: 10.1073/pnas.0611717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.-W., Wang Y.-H., Lai C.-H., Yang M.-J., Chen C.-Y., Chou P.-T., Chan C.-S., Chi Y., Chen Y.-C., Hsiao J.-K. Small. 2008;4:218. doi: 10.1002/smll.200700283. [DOI] [PubMed] [Google Scholar]
- Ladouceur S., Zysman-Colman E. Eur. J. Inorg. Chem. 2013:2985. [Google Scholar]
- Lowry M. S., Bernhard S. Chem. – Eur. J. 2006;12:7970. doi: 10.1002/chem.200600618. [DOI] [PubMed] [Google Scholar]
- Campagna S., Cavazzini M., Cusumano M., Di Pietro M. L., Giannetto A., Puntoriero F., Quici S. Inorg. Chem. 2011;50:10667. doi: 10.1021/ic2010437. [DOI] [PubMed] [Google Scholar]
- Lee P-K., Law W. H.-T., Liu H.-W., Lo K. K.-W. Inorg. Chem. 2011;50:8570. doi: 10.1021/ic201153d. [DOI] [PubMed] [Google Scholar]
- Zheng Z.-B., Wu Y.-Q., Wang K.-Z., Li F. Dalton Trans. 2014;53:3273. doi: 10.1039/c3dt52568f. [DOI] [PubMed] [Google Scholar]
- Langdon-Jones E. E., Symonds N. O., Yates S. E., Hayes A. J., Lloyd D., Williams R., Coles S. J., Horton P. N., Pope S. J. A. Inorg. Chem. 2014;53:3788. doi: 10.1021/ic500142z. [DOI] [PubMed] [Google Scholar]
- Balasingham R. G., Coogan M. P., Thorp-Greenwood F. L. Dalton Trans. 2011;40:11663. doi: 10.1039/c1dt11219h. [DOI] [PubMed] [Google Scholar]
- Raszeja L., Maghnouj A., Hahn S., Metzler-Nolte N. ChemBioChem. 2011;12:371. doi: 10.1002/cbic.201000576. [DOI] [PubMed] [Google Scholar]
- Kim J. H., Johannes L., Goud B., Antony C., Lingwood C. A., Daneman R., Grinstein S. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2997. doi: 10.1073/pnas.95.6.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Liu S., Shi M., Li F., Jing H., Yi T., Huang C. Organometallics. 2007;26:5922. [Google Scholar]
- Laurence C., Brameld K. A., Graton J., Le Questel J.-Y., Renault E. J. Med. Chem. 2009;52:4073. doi: 10.1021/jm801331y. [DOI] [PubMed] [Google Scholar]
- Bilton C., Allen F. H., Shields G. P., Howard J. A. K. Acta Crystallogr., Sect. B: Struct. Sci. 2000;56:849. doi: 10.1107/S0108768100003694. [DOI] [PubMed] [Google Scholar]
- Wilkinson F., Helman W. P., Ross A. B. J. Phys. Chem. Ref. Data. 1993;22:113. [Google Scholar]
- Kraljic I. Biochimie. 1986;68:807. doi: 10.1016/s0300-9084(86)80096-7. [DOI] [PubMed] [Google Scholar]
- Ciuciu A. I., Skonieczny K., Koszelewski D., Gryko D. T., Flamigni L. J. Phys. Chem. C. 2013;117:791. doi: 10.1039/c3cp52291a. [DOI] [PubMed] [Google Scholar]
- Himeda Y., Onozawa-Komatsuzaki N., Miyazawa S., Sugihara H., Hirose T., Kasuga K. Chem. – Eur. J. 2008;14:11076. doi: 10.1002/chem.200801568. [DOI] [PubMed] [Google Scholar]
- Nonoyama M. Bull. Chem. Soc. Jpn. 1974;47:767. [Google Scholar]
- Coban B., Yildiz U., Sengul A. J. Biol. Inorg. Chem. 2013;18:461. doi: 10.1007/s00775-013-0991-7. [DOI] [PubMed] [Google Scholar]
- Skonieczny K., Ciuciu A. I., Nichols E. M., Hugues V., Blanchard-Desce M., Flamigni L., Gryko D. T. J. Mater. Chem. 2012;22:20649. [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Fundam. Crystallogr. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Angeles-Boza A. M., Chifotides H. T., Aguirre J. D., Chouai A., Fu P. K. L., Dunbar K. R., Turro C. J. Med. Chem. 2006;49:6841. doi: 10.1021/jm060592h. [DOI] [PubMed] [Google Scholar]
- Adarsh N., Avirah R. R., Ramaiah D. Org. Lett. 2010;12:5720. doi: 10.1021/ol102562k. [DOI] [PubMed] [Google Scholar]
- DeRosa M. C., Crutchley R. J. Coord. Chem. Rev. 2002;233–234:351. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.