

Abstract
Background
Non-small cell lung cancer (NSCLC) remains lethal despite the development of numerous drug therapy technologies. About 85% to 90% of lung cancers are NSCLC and the 5-year survival rate is at best still below 50%. Thus, it is important to find drugable target genes for NSCLC to develop an effective therapy for NSCLC.
Results
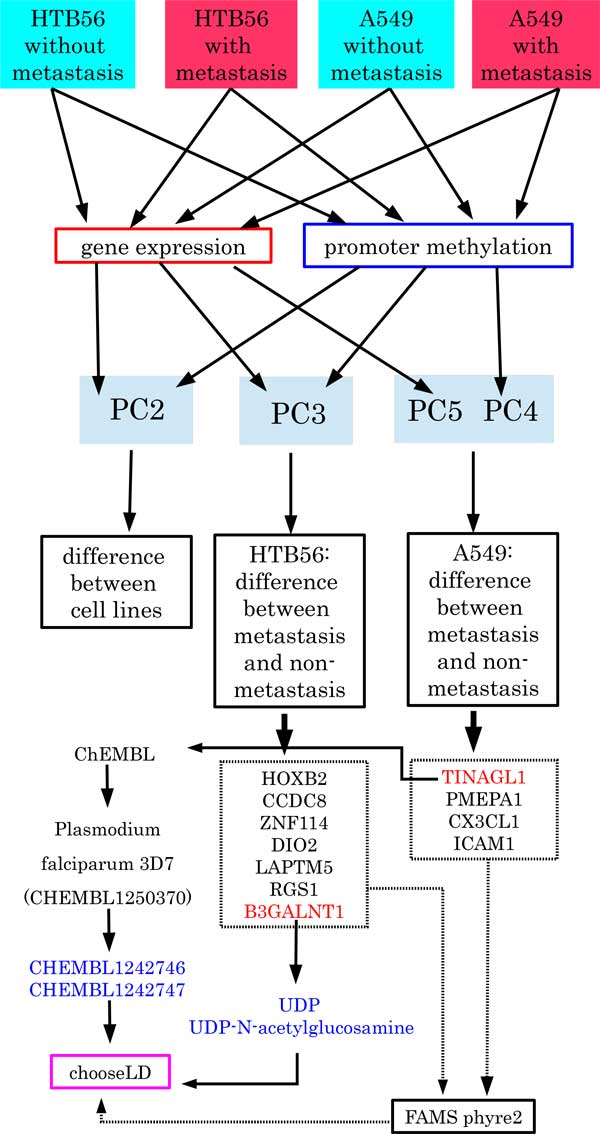
Integrated analysis of publically available gene expression and promoter methylation patterns of two highly aggressive NSCLC cell lines generated by in vivo selection was performed. We selected eleven critical genes that may mediate metastasis using recently proposed principal component analysis based unsupervised feature extraction. The eleven selected genes were significantly related to cancer diagnosis. The tertiary protein structure of the selected genes was inferred by Full Automatic Modeling System, a profile-based protein structure inference software, to determine protein functions and to specify genes that could be potential drug targets.
Conclusions
We identified eleven potentially critical genes that may mediate NSCLC metastasis using bioinformatic analysis of publically available data sets. These genes are potential target genes for the therapy of NSCLC. Among the eleven genes, TINAGL1 and B3GALNT1 are possible candidates for drug compounds that inhibit their gene expression.
Keywords: Gene expression, promoter methylation, integrated analysis, unsupervised feature selection, non-small cell lung cancer, metastasis, principal component analysis, in silico drug discovery, protein structure prediction
Background
Currently, there is no effective therapy for non-small cell lung cancer (NSCLC), thus NSCLC remains lethal [1]. The 5-year survival rate is at best still below 50%. In addition, NSCLC consists of several subtypes that require distinct therapies. Thus, from both a diagnosis and therapy point of view, the identification of genes critical to NSCLC is urgent. Few studies have identified NSCLC critical genes. Fawdar et al [2] recently found that mutations in FGFR4, MAO3K and PAK5 have critical roles in lung cancer progression. Li et al [3] also recently identified EML4-ALK fusion gene and EGFR and KRAS gene mutations were associated with NSCLC. Takeuchi et al [4] also reported that RET, ROS1 and ALK gene fusions were observed in lung cancer. However, it is likely that other critical gene candidates for NSCLC exist.
In this study, we attempted to identify new critical candidate genes important for NSCLC using recently proposed principal component (PCA) based unsupervised feature extraction (FE) mediated integrated analysis [5-8] of publically available promoter methylation and gene expression patterns of two NSCLC cell lines with and without enhanced metastasis ability.
In contrast to the standard usage of PCA, PCA based unsupervised FE does not embed samples but features (that is, probes in this study) into a low dimensional space. Then, features identified as outliers are extracted (for details, see method). Empirically this methodology was successful and identified biologically significant features [5-8] even when other conventional methods tested in the current study failed.
Most of the genes identified in the present study by this methodology were also previously reported as significant cancer-related genes. To understand the functionality of the selected genes, we predicted the tertiary structures of selected genes by Full Automatic Modeling System (FAMS) [9] and phyre2 [10] profile-based protein structure prediction software. This system also allowed the identification of drug target candidate genes.
Results
The first principal components show no significant difference between samples
Since conventional methodology was not useful for the identification of differences between samples, we decided to employ PCA based unsupervised FE to extract biologically relevant genes (probes) when cell lines with and without metastasis were compared. Since it was not our main purpose to emphasize novelty and superiority of the present method compared with the conventional methods, but to identify critical genes for NSCLC metastasis, how the conventional methods failed to identify critical genes in NSCLC metastasis will be discussed in Discussion sections below (the section "Superiority and novelty of the proposed method"). Figure 1 shows two-dimensional embeddings of probes using PCA for gene expression and promoter methylation. To apply PCA based unsupervised FE, first we identified PCs to be used for FE. To determine what each principal component (PC) represented, the contributions of samples to the first PC (PC1) are shown (Figure 2). As previously observed [6,8], the first PC did not identify distinct features among the samples, although they had major contributions (97% for gene expression and 87% for promoter methylation). Contributions of samples to PC1 are almost constantly independent of samples for gene expression and promoter methylation. Thus, we concluded that PC1 did not exhibit any significant differences among samples. It should be noted that this does not always mean that PC1s are biologically meaningless, but rather that most gene expression and promoter methylation is sample independent; thus, the cell lines are very similar to each other independent of their ability for metastasis. This is not surprising, as they are similar NSCLC cell lines; therefore, significantly different outcomes caused by sample dependence and/or metastasis presence would be unusual.
Figure 1.
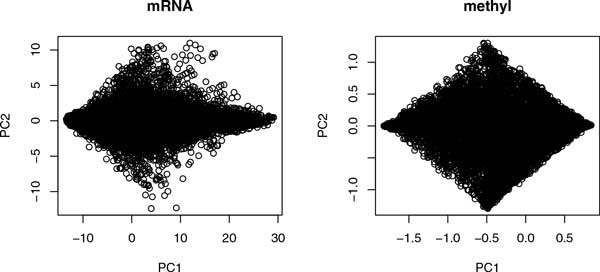
Two-dimensional embeddings of probes using PCA. Two-dimensional embeddings of probes (left: gene expression, right: promoter methylation) spanned by the first (horizontal axes) and the second (vertical) axes.
Figure 2.
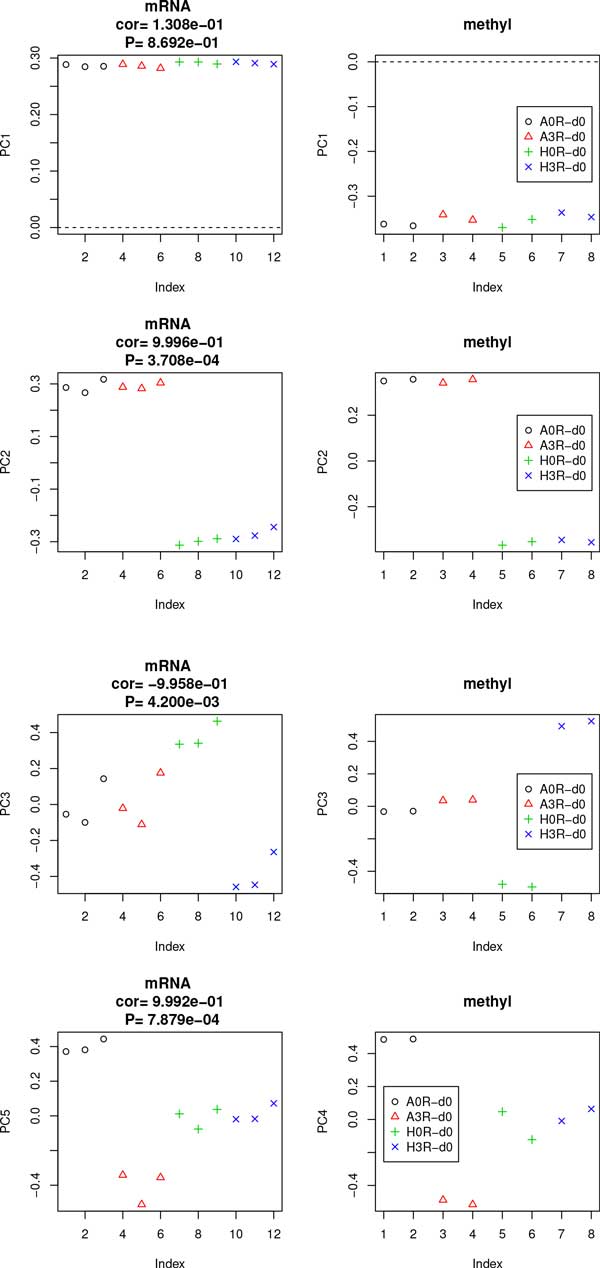
Contributions of samples to PCs. Contributions of samples (black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis) to PCs. Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients of PCs between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
The second PCs demonstrates distinction between cell lines
Because the first PCs did not distinguish between samples, we next considered second PCs (PC2s). As can be seen by two-dimensional embeddings of probes (Figure 1), the second PCs had a relatively smaller contribution. However, this does not always mean that PC2s are biologically meaningless, since PC2 having common values for all samples simply means that all samples behave similarly which does not always contradict with biological significance if all samples are equally biologically significant in the very similar fashions. The second PC of gene expression has only a 1.7% contribution, while for promoter methylation it is 9.6%. These values for contributions, especially for gene expression, can usually be ignored. However, in this case, since the samples were similar NSCLC cell lines, differences between samples were expected to be small. Thus, PCs with small contributions may represent biologically critical differences between samples, as shown in Figure 2, where the contributions of samples to PC2s are demonstrated. PC2s did not distinguish between samples with and without metastasis ability, but did distinguish between A549 and HTB56 cell lines. Because we are interested in metastasis-causing genes in HSCLC, what PC2 expresses is out of the scope of the present study. However, it is useful to identify genes associated with PC2 to determine which genes are different between the two cell lines, A549 and HTB56. PC2s showed good correlation between gene expression and promoter methylation (Figure 3). Thus, integrated analysis using PCA based unsupervised FE is applicable. Selected genes are shown in Table 1. Figure S1 (Additional file 1) shows the gene expression and promoter methylation of selected genes. If analysis is successful, genes selected based on mRNA expression and those based on promoter methylation should overlap significantly. Indeed, P-values attributed to selected genes common between gene expression and promoter methylation were 4.1×10-9 and 5.1×10-12, respectively. Thus, integrated analysis using PCA based unsupervised FE was successful. In contrast to expectations, the selected genes were frequently and significantly related to cancers by the Gendoo server [11] (Table 1), which was used because it attributed P-values to each association between genes and disease. Thus, the reliability of associations can be more easily identified. The successful identification of associations between genes and disease by the Gendoo server suggests that HTB56 and A549 cell lines are potentially distinct to each other and should be considered separately. This is coincident with findings that when distinct genes are present between samples with and without metastasis, they can also reflect differences between the HTB56 and A549 cell lines. Conversely, in contrast to the high correlation of PC2 for gene expression and promoter methylation, correlations between gene expression and promoter methylation of individual genes were not significant (Fig S1). This might be because of the too small contribution of PC2s.
Figure 3.
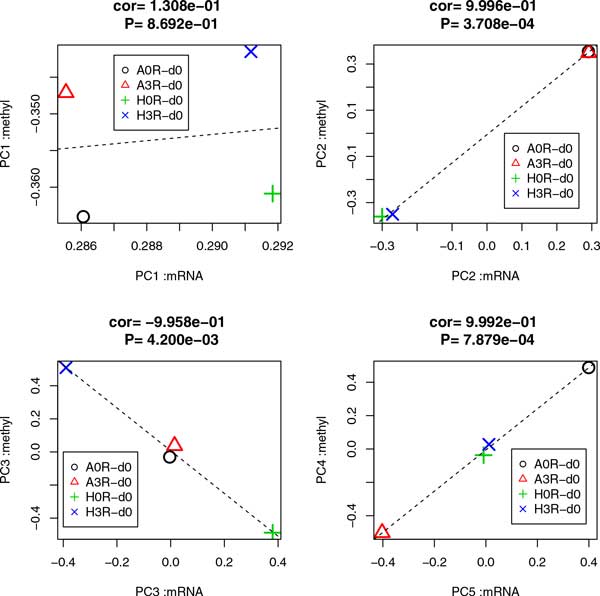
Scatter plots of contributions of samples to PCs between gene expression and promoter methylation. Scatter plots of PCs averaged within each of four categories. Contributions of samples (black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis) to PCs. Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients of PCs between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Table 1.
Cancer disease association with genes selected in the present study based on Gendoo server.
| Gene Symbol | RefSeq mRNA | Cancer associations (P-value) |
|---|---|---|
| PC2 vs PC2 | ||
| SLC22A3 | NM_021977 | Gonadoblastoma (0.0002), Dysgerminoma (0.00075), Testicular Neoplasms (0.00456), Ovarian Neoplasms (0.0297), Cell Transformation, Neoplastic (0.0384) |
| DFNA5 | NM_004403 | Melanoma (0.006), |
| SPG20 | NM_015087 | Hepatoblastoma (0.0033), Liver Neoplasms (0.00496) |
| CYP1B1 | NM_000104 | Breast Neoplasms (1.13 × 10-45), Endometrial Neoplasms (2.44 × 10-12), Lung Neoplasms (1.56 × 10-9), Prostatic Neoplasms (4.65e-9), Adenocarcinoma (6.03 × 10-6), Ovarian Neoplasms (1.35 × 10-5) Carcinoma, Squamous Cell (0.00018), Colorectal Neoplasms (0.000337), Head and Neck Neoplasms (0.00052), Adenoma, Liver Cell (0.0072), Urinary Bladder Neoplasms (0.012), Neoplasms (0.019), Carcinoma, Small Cell (0.028), Carcinoma, Non-Small-Cell Lung (0.0326) |
| ALX1 | NM_006982 | Carcinoma (0.000305), Chondrosarcoma (0.00129), Bone Neoplasms (0.0106), Uterine Cervical Neoplasms (0.011) |
| TFPI2 | NM006528 | Uterine Neoplasms (2.6 × 10-21), Neoplasm Invasiveness (1.18 × 10-14), Choriocarcinoma (2.33 × 10-13), Fibrosarcoma (7.98 × 10-9), Glioma (2.50 × 10-8), Cystadenocarcinoma (1.68 × 10-5), Lung Neoplasms (6.74 × 10-5), Carcinoma, Non-Small-Cell Lung (0.00559) |
| HOXA9 | NM_152739 | Leukemia, Myeloid (2.0 × 10-48), Leukemia, Myeloid, Acute (9.24 × 10-30), Cell Transformation, Neoplastic (4.64 × 10-29), Leukemia (9.46 × 10-19), Leukemia, Myelogenous, Chronic, BCR-ABL Positive (2.64 × 10-14), Precursor Cell Lymphoblastic Leukemia-Lymphoma (2.46 × 10-8), Precursor B-Cell Lymphoblastic Leukemia-Lymphoma (1.65 × 10-6), Myoma (0.00046), Leukemia, T-Cell (0.0012), Endodermal Sinus Tumor (0.0079), Seminoma (0.0157), |
| HOXA11 | NM_005523 | Uterine Neoplasms (8.23 × 10-7), Choriocarcinoma (3.97 × 10-5), Carcinoma, Endometrioid (0.0065), Adenocarcinoma, Clear Cell (0.00662), Wilms Tumor (0.0076), |
| PCSK1 | NM000439 | Bronchial Neoplasms (0.0022), Adenoma (0.0030), Adenoma, Islet Cell (0.0035), Bile Duct Neoplasms (0.011) |
| SPARC | NM_003118 | Neoplasm Invasiveness (8.42 × 10-14), Glioma (1.35 × 10-8), Brain Neoplasms (1.01 × 10-7), Melanoma (2.99 × 10-7), Lung Neoplasms (1.43 × 10-5), Carcinoma (0.00013), Carcinoma, Non-Small-Cell Lung (0.0009) |
| PC3 vs PC3 | ||
| HOXB2 | NM_002145 | Lung Neoplasms (0.000159), Leukemia, Myeloid (0.000326), Pulmonary Emphysema (0.00139), Carcinoma, Embryonal (0.0025), Adenocarcinoma (0.0054), Leukemia, Erythroblastic, Acute (0.0096), Leukemia, Promyelocytic, Acute (0.0124), Carcinoma, Small Cell (0.0148), Carcinoma, Non-Small-Cell Lung (0.0387) |
| CCDC8 | NM_032040 | |
| ZNF114 | NM_153608 | |
| DIO2 | NM_000793 | Choriocarcinoma (0.000616), Carcinoma, Papillary (0.00366), Hemangioma (0.0099), Adenoma (0.019), Neuroblastoma (0.025) |
| LAPTM5 | NM_006762 | Carcinoma, Hepatocellular (0.000396), Liver Neoplasms (0.000495), Multiple Myeloma (0.00947), Neoplasm Recurrence (0.010), Cell Transformation, Neoplastic (0.032) |
| RGS1 | NM_002922 | Burkitt Lymphoma (3.55 × 10-5), Lymphoma, B-Cell (9.14 × 10-5), Leukemia-Lymphoma, Adult T-Cell (0.0076), Lymphatic Metastasis (0.0329), Skin Neoplasms (0.0364), Stomach Neoplasms (0.0454), Melanoma (0.0455) |
| B3GALNT1 | NM_003781 | Neuroblastoma (0.0034) |
| PC5 vs PC4 | ||
| TINAGL1 | NM_022164 | Carcinoma, Hepatocellular (0.000119), Neoplasms (0.0295) |
| PMEPA1 | NM_020182 | Prostatic Neoplasms (2.30e-12), Carcinoma, Renal Cell (0.0233), Kidney Neoplasms (0.032) |
| CX3CL1 | NM_002996 | Neuroblastoma (0.0014) |
| ICAM1 | NM_000201 | Melanoma (0.00305), astrocytoma (0.00644), Granular Cell Tumor (0.0166), Colonic Neoplasms (0.0233), Lymphoma, AIDS-Related (0.023), Adenoma, Oxyphilic (0.0433) |
The third PCs distinguish differences between samples with and without metastasis for HTB56 but not for A549
Because no PCs reflected differences between samples with and without metastasis, we considered additional PCs. Figure 2 shows the contributions of samples to the third PC (PC3). Although PC3s have even smaller contributions (0.2% for gene expression and 1.5% for promoter methylation) than PC1s or PC2s (Figure 3) their correlation is high. Thus, genes associated with PC3 represent differences between samples with and without metastasis and we finally identified a useful PC. Interestingly, PC3 exhibited differences between samples with and without metastasis only for the HTB56 cell line. However, since the two cell lines are distinct in terms of their oncogenic potential, it is not surprising that genes that exhibit differences between samples with and without metastasis for HTB56 did not exhibit differences between samples with and without metastasis for A549. Thus, we further applied integrated analysis using PCA based unsupervised FE. Selected genes are shown in Table 1. Figure S2 (Additional file 2) shows gene expression and promoter methylation of the selected genes. Again, whether genes selected based on mRNA expression and those based on promoter methylation were significantly overlapped was analyzed and P-values attributed to selected genes common between gene expression and promoter methylation were 3.5×10-5 and 5.1×10-4. Thus, integrated analysis using PCA based unsupervised FE successfully identified genes with both aberrant mRNA expression and promoter methylation. The association of cancer disease and the selected genes were investigated by the Gendoo server, and the results are shown in Table 1. As expected, most of the selected genes were significantly associated with cancer disease. Correlations between gene expression and promoter methylation of individual genes were not significant (Fig. S2).
The fourth PC of promoter methylation and the fifth PC of gene expression represent differences between samples with and without metastasis for A549 but not for HTB56
We further sought PCs that exhibited differences between samples with and without metastasis for A549. The fourth PC (PC4) of promoter methylation and the fifth PC (PC5) of gene expression demonstrated differences between samples with and without metastasis for the A549 cell line (Figures 2 and 3). Because the correlation between PC4 and PC5 were very high despite their small contributions (0.6% for PC4 of promoter methylation and 0.09% for PC5 of gene expression), integrated analysis using PCA based unsupervised FE could still be used. Selected genes are shown in Table 1. Figure S3 (Additional file 3) shows gene expression and promoter methylation of the individual genes.
P-values attributed to selected genes common between gene expression and promoter methylation were 9.8×10-8. Thus, integrated analysis using PCA based unsupervised FE was successful. Cancer diseases associated with the selected genes are listed in Table 1, and more than 50% were reported to be associated with cancer-related diseases. However, correlations between gene expression and promoter methylation of individual genes were not significant (Fig. S3).
Discussion
Although the association of cancer-related disease and the selected genes were annotated by the Gendoo server, more detailed information, regarding whether genes are expressed or repressed in cancers, will be useful. In addition, since the Gendoo server was last updated in April 2012, recent information might be missing. To fill these gaps, we will discuss the selected genes individually citing actual studies.
HOXB2
HOXB2 has a Homeobox domain in the central region. Figure S4 (Additional file 4) shows the tertiary structure of the homeobox domain in HOXB2 predicted by FAMS. The homeodomain fold is a protein structural domain that binds to DNA or RNA and is commonly found in transcription factors. HOXB2 was upregulated in pancreatic cancer [12] as a part of the retinoic acid (RA) signaling pathway, which is generally regarded to be a potential anti-tumor agent [13]. HOXB2 also promotes the invasion of lung cancer cells by regulating metastasis-related genes [14]. Considering these studies, it was not surprising that HOXB2 might also have a critical role in NSCLC.
CCDC8
Neither FAMS nor phyre2 predicted a significant tertiary structure for CCDC8, which is reported to be a cofactor required for p53-mediated apoptosis through interactions with OBSL1 [15]. Thus, because p53 protein is a typical tumor suppressor, it is likely that CCDC8 has a critical role in NSCLC.
ZNF114
ZNF114 has one KRAB box and four Zinc-finger double domains. Figure S5 (Additional file 4) shows the Zinc-finger domains as predicted by FAMS. Since the Zinc-finger double domain functions in DNA binding, ZNF114 might also be a DNA binding protein. KRAB is a transcription repression domain, thus ZNF114 might be a transcription suppressor. Unfortunately, very few studies of ZNF114 have been published. However, mutation of CTCF that has seven Zinc-finger double domains was reported to be associated with tumors [16]. GC79 that has multiple Zinc-finger double domains was reported to be associated with apoptosis of prostate cancer cells [17]. Studies related to proteins with Zinc-finger double domains indicate ZNF114 might also have a role in NSCLC.
DIO2
Figure S6 (Additional file 4) shows the tertiary structure of DIO2 as predicted by FAMS. DIO2 belongs to the iodothyronine deiodinase family and is underexpressed in benign and malignant thyroid tumors [18]. DIO2 expression was also shown to be higher in most brain tumors [19]. Thus, although it is unclear whether DIO2 is generally oncogenic or tumor suppressive, it appears to be related to cancer. Therefore, DIO2 is likely to be related to NSCLC.
LAPTM5
Neither FAMS nor phyre2 predicted a significant tertiary structure for LAPTM5, a transmembrane protein that was reported to be associated with spontaneous regression of neuroblastomas [20]. Inactivation of the E3/LAPTM5 gene by chromosomal rearrangement and DNA methylation in human multiple myeloma was observed [21]. Expression of LAPTM5 was also elevated in human B lymphomas [22]. Although there have been no reports indicating a relationship between LAPTM5 and NSCLC, LAPTM5 might have a critical role in NSCLC.
RGS1
RGS1 contains a regulator of G protein signaling domain. The tertiary structure of RGS1 is available in the Protein Data Bank (PDB) (Fig. S7 in Additional file 4). Regulator of G-protein signaling (RGS) proteins are related to cancer biology [23] and genetic variations in these genes are associated with survival in late-stage NSCLC [24]. RGS1 was overexpressed in a gene expression-profiling study of melanoma [25]. RGS is thought to be related to the functionality of G protein-coupled receptors [26] that are often drug targets. Thus, RGS might be a promising drug target candidate for therapy of NSCLC.
B3GALNT1
B3GALNT1 is a galactosyl transferase that catalyzes the transfer of galactose. Fig. S8 (Additional file 4) shows the tertiary structure of B3GALNT1 as predicted by FAMS. Numerous studies have suggested a relationship between galactosyl transferase and cancer, including the use of galactosyl transferase as a tumor biomarker for ovarian clear cell carcinoma [27,28]. Alternatively, cancer-associated isoenzymes of serum galactosyl transferase were reported in various cancers [29]. Thus, B3GALNT1 might have a role in NSCLC progression, although no reports have demonstrated a specific relationship between B3GALNT1 and cancer.
TINAGL1
TINAGL1 is papain family cysteine protease that degrades proteins. Figure S9 (Additional file 4) shows the tertiary structure as predicted by FAMS. TINAGL1 is a Sec23a-dependent metastasis suppressor [30] and was reported to be upregulated in highly metastatic tumors [31]. Thus, it was reasonable that it was selected as a cancer-related gene candidate by our methodology.
PMEPA1
Neither FAMS nor phyre2 predicted a significant tertiary structure for PMEPA1, a transmembrane prostate androgen-induced protein that enhances tumorigenic activity in lung cancer cells [32]. It was also reported to be upregulated in ovarian cancer [33], colon cancer [34] and renal cell carcinoma [35]. Considering these studies, PMEPA1 was reasonably selected as a NSCLC-related gene by PCA based unsupervised feature extraction (FE) mediated integrated analysis.
CX3CL1
CX3CL1 contains a small cytokine (intecrine/chemokine) interleukin-8 (IL-8)-like domain. Figure S10 (Additional file 4) shows the tertiary structure of the IL-8 domain as predicted by FAMS. The IL-8 pathway was reported to be important in cancer [36] and CX3CL1 expression was associated with a poor outcome in breast cancer patients [37] as it promotes breast cancer via transactivation of the epidermal growth factor pathway [38]. A complex role for CX3CL1 in cancer has been reported [39]. Thus, it was reasonable that CX3CL1 was selected by our methodology.
ICAM1
Intercellular adhesion molecule (ICAM) contains an N-terminal domain and three immunoglobulin domains. The tertiary structure of ICAM1 was available in PDB (Fig. S11 in Additional file 4). Many studies have reported a relationship between ICAM1 and cancer. ICAM1 expression was reported to determine the malignant potential of cancer [40], to have a role in the invasion of human breast cancer cells [41], and upregulated endogenous ICAM-1 reduced ovarian cancer cell growth in the absence of immune cells [42]. Thus, it is reasonable that ICAM1 was selected as a potential NSCLC therapy target by our methodology.
TINAGL1 as a drug target gene candidate
In this study, we selected multiple genes that might be involved in the progression of NSCLC metastasis. Most of selected genes are potential cancer-related genes. Thus, it is reasonable to regard these genes as therapy targets. Among those selected, we investigated TINAGL1 as a potential drug target gene, because although TINAGL1 is regarded to be a tumor suppressor, in this study it was upregulated in a metastasis-enhanced cell line. Naba et al [31] also reported that TINAGL1 was up-regulated in highly metastatic tumors. Thus, inhibition of TINAGL1 might be a potential therapeutic target for the treatment of metastatic NCSLC. Furthermore, although we used a profile based drug discovery software, chooseLD (Insilico Science Co., Tokyo, Japan) [43], for in silico drug screening, it required the tertiary structure of the target protein and multiple ligand compounds whose binding structure to the protein were known. TINAGL1 satisfied these requirements as follows. To infer the tertiary structure of TINAGL1, we uploaded the amino acid sequence NP_071447.1 retrieved from RefSeq to FAMS and phyre2. Because there was no significant difference between tertiary structures inferred by FAMS and phyre2, hereafter we used the structure inferred by FAMS.
Based on FAMS, TINAGL1 has many tertiary structures registered in PDB that can be used for tertiary structure predictions. Among them, the "A" chain of PDB ID: 2DCC (2DCC_A) has a 32% sequence similarity with TINAGL1 and is accompanied by multiple highly similar (> 95% sequence similarity) tertiary structures registered in PDB (PDB ID: 1IT0_A, 1QDQ_A, 2DC6_A, 2DC7_a, 2DC8_A, 2DC9_A, 2DCA_A, and 2DCD_A). Because all of these structures have more than one ligand that binds to protein, we had a large number of ligand-protein binding structures that could be used for in silico drug screening using chooseLD. We selected 2DCC_A, a protein structure of TINAGL1 from aa 204 to 455 for modeling by FAMS. Because 2DCC_A is cathepsin, hereafter we called this structure the cathepsin domain. To confirm that chooseLD could predict ligand binding to the cathepsin domain, we attempted to identify a known ligand that binds to the cathepsin domain. ChEMBL [44] was identified by a BLAST search using the cathepsin domain amino acid sequence. Thus, Plasmodium falciparum 3D7 (CHEMBL1250370), a putative protease, was found to have 47.22% sequence similarity with the cathepsin domain. There were five assay experiments for this protein. Among them, CHEMBL1244076 was employed to list candidate binding ligands. Three ligands were reported to inhibit Plasmodium falciparum 3D7. Among them, CHEMBL1242746 and CHEMBL1242747 (Fig. S12 in Additional file 4) were employed as potential binding ligands to TINAGL1. Then chooseLD was used to test the two ligands using 15 template ligand proteins 3S3Q_C1P, 3S3R_0IW_00, 3AI8_HNQ_01, 1GMY_hld_00, 2DCC_77B, 1ITO_E6C, 1QDQ_074, 2DC6_73V, 2DC7_042, 2DC8_59A, 2DC9_74M, 2DCA_75V, 2DCB_76V, 2DCD_78A, and 3PDF_LXV. Fig. S13 (Additional file 4) shows the binding of CHEMBL1242565 and CHEMBL514348 to TINAGL1 (Additional file 5 for template ligand binding to TINAGL1).
Binding affinity of ligands to TINAGL1 was evaluated by Cyscore [45] (Table 2). All template ligands had negative, thus better, Cyscores. Although CHEMBL1242746 or CHEMBL1242747 did not achieve a negative Cyscore, CHEMBL1242747 had a low positive Cyscore that was lower than the lowest template ligands. If we consider these two were only reported to bind to proteins with a TINAGL1 sequence similarity of 45%, the Cyscore attributed to CHEMBL1242746 or CHEMBL1242747 was good and demonstrated the ability of chooseLD to infer proper binding configurations of potential ligands. Since chooseLD inferred suitable binding modes for two potential ligands, we concluded that in silico drug discovery is possible for TINAGL1, which might be a promising candidate drug target.
Table 2.
Inference of ligand binding affinity to TINAGL1.
| Hydrophobic | Vdw | HBond | Ent | Cyscore | |
|---|---|---|---|---|---|
| 1GMY_hld | -0.4156 | -1.77 | 0 | 0.378 | -1.8086 |
| 1ITO_E6C | -0.3024 | -0.56 | 0 | 0.42 | -0.4461 |
| 1QDQ_074 | -1.3611 | -1.88 | 0 | 0.42 | -2.8243 |
| 2DC6_73V | -0.552 | -1.79 | 0 | 0.63 | -1.7081 |
| 2DC7_042 | -0.5374 | -1.56 | 0 | 0.588 | -1.514 |
| 2DC8_59A | -0.5766 | -1.6 | 0 | 0.462 | -1.717 |
| 2DC9_74M | -1.3254 | -1.24 | 0 | 0.462 | -2.1026 |
| 2DCA_75V | -1.1413 | -1.52 | 0 | 0.546 | -2.1103 |
| 2DCB_76V | -1.6095 | -1.76 | 0 | 0.63 | -2.7419 |
| 2DCC_77B | -0.5932 | -2.07 | 0 | 0.63 | -2.0373 |
| 2DCD_78A | -1.5344 | -1.65 | 0 | 0.42 | -2.7627 |
| 3AI8_HNQ | -0.7476 | 0.635 | 0 | 0.042 | -0.0704 |
| 3PDF_LXV | -0.6498 | -0.96 | 0 | 0.168 | -1.4434 |
| 3S3Q_C1P | -1.2913 | -1.61 | 0 | 0.504 | -2.4019 |
| 3S3R_0IW | -1.0358 | -0.97 | 0 | 0.504 | -1.5028 |
| CHEMBL1242746 | -1.3699 | 2.768 | 0 | 0.42 | 1.8177 |
| CHEMBL1242747 | -1.561 | 1.431 | 0 | 0.336 | 0.206 |
Cyscore and Hydrophobic, Vdw (van der Waals), Hbond (hydrogen bond), and Ent (entropy) computed by Cyscore programs of 15 template ligands and two drug candidate compounds.
B3GALNT1 as a candidate drug target gene
Another potential drug target gene is B3GALNT1. Substrates such as UDP-galactose and UDP-N-acetylglucosamine bind to B3GALNT1 and after various catalytic reactions, UDP remains bound to B3GALNT1. Thus, if compounds that inhibit UDP binding to B3GALNT1 that compete with UDP can be identified, the function of B3GALNT1 can be inhibited. B3GALNT1 is a galactosyl transferase, which is often reported to be upregulated in various cancers. Thus, inhibition of B3CALNT1 might be a potential therapeutic target for NSCLC. Fig. S14 (Additional file 4) shows the UDP and UDP-N-acetylglucosamine binding to B3GALNT1 predicted by chooseLD. Because both bind to the same pockets of B3GALNT1, chooseLD can be used to identify other compounds that bind to and inhibit B3GALNT1.
Inconsistency between gene expression and promoter methylation of individual genes
Although Figs. S1, S2 and S3 show the gene expression and promoter methylation of individual genes associated with selected PCs, coincidence between gene expression and promoter methylation is relatively poor. Gene selection was reliable because P-values attributed to the simultaneous selection of genes for gene expression and promoter methylation PCs were significant and the selected genes were associated with cancer-related genes (Table 1). To resolve the discrepancy between the significant selection of genes and poor coincidence of individual genes between gene expression and promoter methylation, we considered promoter methylation by sequencing technology, which was performed simultaneously with microarray measurements. Figure S15 (Additional file 6) shows the promoter methylation profile of selected genes by sequencing technology. Although measurements were unfortunately not available for all observations, promoter methylation measured by sequencing technology was more coincident (negatively correlated) with gene expression than by microarray. Since sequencing technology is more reliable than microarray, poor consistency between gene expression and promoter methylation might be explained by the poor ability of microarray to measure promoter methylation. Thus, discrepancies are expected to be resolved when promoter methylation is measured with high accuracy.
Superiority and novelty of the proposed method
The novel method employed in the current study has a number of advantages compared with existing conventional methodologies. To demonstrate failure of the conventional approaches, first we tried detect genes that have a negative correlation between mRNA expression and promoter methylation. Pairs of mRNA expression microarray probes and promoter methylation probes to which common mRNA RefSeq IDs are attributed were collected. Then Pearson correlation coefficients were computed for all pairs as in Figure 3. The obtained P-values were adjusted by Benjamin-Hochberg criterion since there were more than twenty thousand pairs. Only one gene had an adjusted P-value <0.05. This clearly demonstrates the usefulness of applying PCA, without which almost no significant correlations would be detected.
Next, we used the t-test to detect significant differences between samples with and without metastasis. The t-test was applied to all probes and obtained P-values were again adjusted by Benjamin-Hochberg criterion to suppress failed positives because of high numbers of observations. For comparison between A549 cell lines with and without metastasis, no probes had adjusted P-values <0.05 for either mRNA expression or promoter methylation. For comparison between HTB56 cell lines with and without metastasis, although as many as 434 probes had adjusted P-values <0.05 for mRNA expression, there were no probes for promoter methylation. This also suggests the usefulness of applying PCA without which no significant aberrant promoter methylation or mRNA expression would be detected.
These difficulties for the detections of significant correlations and aberrant mRNA expression/promoter methylation were caused by the small number of replicates in the study (three replicates for each mRNA expression and two replicates for each promoter methylation). Since PCA can detect the behavior of a group of probes, this difficulty can be compensated for and explains why only applying PCA can detect significant outcomes.
Finally, we would like to emphasize some of the novelties of the PCA based methodology. Although PCA itself is a frequently used method, the current study applied PCA differently from conventional methodology. First, PCA is usually used to embed samples into low dimensional space to demonstrate the groupings of samples, while this study used embedded probes. This enabled the identification of what each PC discriminates as in Figure 2. To our knowledge, PCA has rarely been used this way. Second, we did not ignore PCs to which only tiny contributions were attributed (PC2, PC3, PC4 and PC5 investigated in this study had at most 10% contributions), while standard procedures recommend ignoring such PCs since it is impossible to distinguish them from background noise. The reason why such small a contribution can have meaning is because of the huge number of probes used. Since as many as twenty to thirty thousand probes are analyzed, contributions as little as 0.1% can correspond to several tens of probes. This is a new concept, and thus it is not generally recognized that small contributions can have meaning. Therefore, although the usage of PCA itself is not novel, the method used in this study is new.
Comparison with tissue samples associated with metastasis
Although we detected many significant results in this study, the original observation of the analysis was cell lines. Since cell lines have a greater tendency to exhibit clear outcomes than tissues, it is unclear whether the study results could be observed using more relevant samples, such as tissues. To confirm this, we consulted a database that stores the expression of tissues with and without metastasis. Unfortunately, we could not find a data set where NSCLC tissues were treated. Thus, we compared melanoma with and without metastasis (GEO ID: GDS3966 [46]). Because metastasis is commonly caused by similar factors, e.g, the loss of cell adhesion, independent of the type of cancer, comparison of melanoma with and without metastasis was expected to share aberrant mRNA expression with NSCLC with and without metastasis (Table 3). Although not all eleven genes were observed, among seven of the genes included, six had a P-vales <0.05 by Kolmogorov-Smirnov (KS) test. The KS test was used because it is more robust (non-metric) than the t-test, thus was more suitable for samples with a high background noise such as tissues. The results shown in Table 3 suggest that the findings in this cell line might also be observed in tissue samples.
Table 3.
P-values that represent significant differences of melanoma tissue samples between those with and without metastasis.
| Gene | Metastasis > no metastasis | No metastasis >metastasis |
|---|---|---|
| HOXB2 | 3 × 10-3 | 1 |
| LAPTM5 | 5 × 10-2 | 8 × 10-1 |
| RGS1 | 1 × 10-1 | 4 × 10-2 |
| TINAGL1 | 9 × 10-1 | 3 × 10-2 |
| PMEPA1 | 6 × 10-1 | 5 × 10-3 |
| CX3CL1 | 1 | 1 × 10-1 |
| ICAM1 | 3 × 10-3 | 1 |
Values were computed using the Kolmogorov-Smirnov test. P-values <0.05 highlighted in bold.
Transcription factor aryl hydrocarbon receptor targets selected eleven genes
Although the biological significance of individual genes was confirmed, it would be more useful if biological reasons for the commonality between the genes could be identified. We uploaded mRNA RefSeq IDs for eleven genes to DAVID [47] and found that all eleven genes were targets of the transcription factor aryl hydrocarbon receptor (AHR), reported to be primary factor that causes lung cancer [48]. AHR was also suggested to promote metastasis [48]. Given that promoter methylation is primarily related to transcription factors binding to promoters, it is reasonable that AHR was identified by the integrated analysis of mRNA expression and promoter methylation. This also suggests that our methodology and analysis are suitable for identification of potential cancer causing genes for NSCLC.
Conclusions
This study performed the integrated analysis of promoter methylation and gene expression using PCA based unsupervised FE. It selected eleven genes that were differently expressed and which had different promoter methylation patterns between cell lines with and without metastasis ability. P-values attributed to the simultaneous selection between gene expression and promoter methylation were significant and many cancer-related diseases were associated with the eleven genes selected. Two of the eleven genes selected, B3GALNT1 and TINAGL1, were identified as drug target candidates that might suppress metastasis in NSCLC. Further detailed and advanced studies are required to confirm these findings.
Methods
Promoter methylation and gene expression profiles
Promoter methylation profiles were downloaded from Gene Expression Omnibus (GEO) with GEO ID: GSE52144 that included two replicates of HTB56 cell lines with (H3R_d0) and without (H0R_d0) metastasis ability and A549 cell lines with (A3R_d0) and without (A0R_d0) metastasis ability. Gene expression profiles were downloaded from GEO with GEO ID: GSE52143 that included three replicates of the samples in GSE52144. For these two cell lines, data sets deposited in the "Series Matrix Files" were retrieved. Promoter methylation measured by sequencing was obtained from GEO with GEO ID: GSE52140. Within GSE52140_RAW.tar, eight files corresponding to those in GSE52144, (two replicates of H0R_d0. H3R_d0, A0R_d0 and A3R_d3) were used.
Retrieval of promoter methylation from sequencing data
Information of genes annotated by RefSeq mRNA, (transcription start and end sites, strand, chromosome name) in hg19 human genome were retrieved using the Table browser of the Genome Browser [49]. Using this information, methylation sites between 1500 bps upstream and 500 bps of transcription start sites were collected. Mean β-values, (the ratio of methylated sites among the total number of methylation sites), were employed as promoter methylation for each RefSeq gene.
Integrated analysis of gene expression and promoter methylation using PCA based unsupervised FE
First, PCA was applied to gene expression and promoter methylation and each probe was embedded into a two dimensional space spanned with the first and the second PC scores. Then contributions of each probe to each PC were investigated and biologically meaningful PCs were selected. The 100 top outlier probes with larger (positively larger) or smaller (negatively larger) PC scores were extracted for each PC. The coincidence between selected probes for gene expression and promoter methylation was estimated as follows. If contributions of each probe to PCs were positively correlated between gene expression and promoter methylation, then intersections between gene expression outlier probes having larger (smaller) PC scores and promoter methylation outlier probes having smaller (larger) PC scores were sought, since gene expression and promoter methylation were expected to be negatively correlated with each other. Conversely, if contributions of each probe to PCs were negatively correlated between gene expression and promoter methylation, intersections between gene expression outlier probes having larger (smaller) PC scores and promoter methylation outlier probes having larger (smaller) PC scores were sought. P-values attributed to simultaneous selection of probes between gene expression and promoter methylation were computed by distribution that obeyed binomial distribution as follows:
where × is the number of commonly selected probes between the top 100 outliers of gene expression and promoter methylation, y is total number of probes on the microarray, and P is the cumulative frequency of binomial distribution.
Cancer-related disease association of selected genes by Gendoo server
Cancer-related disease association was identified using the Gendoo server. RefSeq mRNA was transformed to a gene symbol, which was uploaded to the Gendoo server with "human" as the target species. Among the associated diseases, those related to cancer and with P-values <0.05 are listed in Table 1.
Tertiary protein structure prediction using profile based inference servers
Amino acid sequences retrieved from Uniprot (Additional file 7) were uploaded to two profile based tertiary structure databases, FAMS and phyre2. Because no significant differences were observed between the two databases, inferences by FAMS were used for all further analyses.
Domain annotation by Pfam
To annotate domains included in each gene, we uploaded amino acid sequences to pfam [50], and determined the domains contained in each amino acid sequence.
Ligand-protein docking inference by chooseLD
ChooseLD is a profile-based ligand-protein docking affinity evaluation software. ChooseLD requires well-predicted or observed tertiary structures of target genes and known binding configurations of multiple compounds to which drug candidate compounds can be aligned. For the TINAGL1 gene, there are 15 known ligands that bind TINAGL1 or highly similar proteins (> 95% sequence similarity). Thus, in silico drug discovery was performed for TINAGL1. However, UDP is the only ligand with a known configuration that can bind to B3GALNT1. Fortunately, B3GALNT1 can bind substrates in contrast to other proteins that only bind to other proteins, thus in silico drug discovery is easier since compounds that bind to B3GALNT1 by substituting UDP can be determined. Therefore, TINAGL1 and B3GALNT1 might be potential drug candidate genes.
Ligand-protein docking affinity evaluation by Cyscore
Cyscore was used to evaluate binding affinity to TINAGL1, [45]. Hydrogen was added to PDB files of TINAGL1 by pymol [51] using the "h_add" command and was added to mol2 files of ligands by babel [52] using the "-h" option. Then, Cyscore was applied to the PDB file of TINAGL1 and mol2 files of ligands. Although pdb2pqr [53] (default settings) was used to add hydrogen to TINAGL1, the resulting Cyscore was not improved. Thus, we decided to use pymol. These processes are shown in Figure 4.
Figure 4.
Schematic figure of data processing. Schematic of data processing.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HU performed in silico ligand-protein binding inference by chooseLD. MI predicted tertiary structures of proteins using FAMS. YHT designed and supervised the whole research project, performed integrated analysis, predicted tertiary structures of proteins by phyre2, obtained Cyscores and wrote the paper.
Supplementary Material
Fig. S1 Gene expression and promoter methylation associated with PC2. Gene expression and promoter methylation associated with PC2. Left column: gene expression, right column: promoter methylation. NM_021977 (SLC22A3), NM_004403 (DFNA5), NM_015087 (SPG20), NM_000104 (CYP1B1), NM_006982 (ALX1), NM_006528 (TFPI2), NM_152739 (HOXA9), NM_005523 (HOXA11 ), NM_000439 (PCSK1), NM_003118 (SPARC). (Black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis). Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Fig S2 Gene expression and promoter methylation associated with PC3. Gene expression and promoter methylation associated with PC3. Left column: gene expression, right column: promoter methylation. NM_002145 (HOXB2), NM_032040 (CCDC8), NM_153608 (ZNF114), NM_000793 (DIO2), NM_006762 (LAPTM5), NM_002922 (RGS1), NM_003781 (B3GALNT1). (Black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis). Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Fig. S3 Gene expression and promoter methylation associated with PC4 (promoter methylation) and PC5 (gene expression). Gene expression and promoter methylation associated with PC4 (promoter methylation) and PC5 (gene expression). Left column: gene expression, right column: promoter methylation. NM_022164 (TINAGL1), NM_020182 (PMEPA1), NM_002996 (CX3CL1), NM_000201 (ICAM1). Contributions of samples (black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis) to PCs. Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Supplementary figures. Supplementary figures from Fig. S4 to Fig. S14.
Ligand binding configuration to TINAGL1. Full list of ligands that bind to TINAGL1.
Fig. S15 Promoter methylation profile of selected genes measured by sequencing technology. Promoter methylation measured by sequencing, NM_002145 (HOXB2), NM_032040 (CCDC8), NM153608 (ZNF114), NM_006762 (LAPTM5), NM_003781 (B3GALNT1), NM_022164 (TINAGL1), NM_020182 (PMEPA1), NM_002996 (CX3CL1), and NM_000201 (ICAM1).
Amino acid sequences used for tertiary protein structure prediction. Amino acid sequences retrieved from Uniprot of selected genes (in fasta format).
Contributor Information
Hideaki Umeyama, Email: umeyama@bio.chuo-u.ac.jp.
Mitsuo Iwadate, Email: iwadate@bio.chuo-u.ac.jp.
Y-h Taguchi, Email: tag@granular.com.
Acknowledgements
This study was supported by KAKENHI 23300357 and 26120528 from the National Institute of Informatics, Japan and a Chuo University Joint Research Grant.
Declarations
Publication costs for this article were funded by grant KAKENHI 26120528 from National Institute of Informatics, Japan.
This article has been published as part of BMC Genomics Volume 15 Supplement 9, 2014: Thirteenth International Conference on Bioinformatics (InCoB2014): Computational Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/15/S9.
References
- Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, Shepherd FA. Non-small-cell lung cancer. Lancet. 2011;378:1727–1740. doi: 10.1016/S0140-6736(10)62101-0. [DOI] [PubMed] [Google Scholar]
- Fawdar S, Trotter EW, Li Y, Stephenson NL, Hanke F, Marusiak AA, Edwards ZC, Ientile S, Waszkowycz B, Miller CJ, Brognard J. Targeted genetic dependency screen facilitates identification of actionable mutations in FGFR4, MAP3K9, and PAK5 in lung cancer. Proc Natl Acad Sci USA. 2013;110:12426–12431. doi: 10.1073/pnas.1305207110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li Y, Yang T, Wei S, Wang J, Wang M, Wang Y, Zhou Q, Liu H, Chen J. Clinical significance of EML4-ALK fusion gene and association with EGFR and KRAS gene mutations in 208 Chinese patients with non-small cell lung cancer. PLoS One. 2013;8:e52093. doi: 10.1371/journal.pone.0052093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, Nakagawa K, Mano H, Ishikawa Y. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Toyoda H, Tanahashi T, Tanaka J, Kumada T, Yoshioka Y, Kosaka N, Ochiya T, Taguchi YH. Comprehensive miRNA expression analysis in peripheral blood can diagnose liver disease. PLoS One. 2012;7:e48366. doi: 10.1371/journal.pone.0048366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida S, Umeyama H, Iwadate M, Taguchi YH. Bioinformatic Screening of Autoimmune Disease Genes and Protein Structure Prediction with FAMS for Drug Discovery. Protein Pept Lett. 2013;8:823–829. doi: 10.2174/09298665113209990052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi YH, Murakami Y. Principal component analysis based feature extraction approach to identify circulating microRNA biomarkers. PLoS One. 2013;8:e66714. doi: 10.1371/journal.pone.0066714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita R, Iwadate M, Umeyama H, Taguchi YH. Genes associated with genotype-specific DNA methylation in squamous cell carcinoma as candidate drug targets. BMC Syst Biol. 2014;8:S4. doi: 10.1186/1752-0509-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeyama H, Iwadate M. FAMS and FAMSBASE for protein structure. Curr Protoc Bioinformatics. 2004;4:5.2.1–5.2.16. doi: 10.1002/0471250953.bi0502s04. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Nakazato T, Bono H, Matsuda H, Takagi T. Gendoo: functional profiling of gene and disease features using MeSH vocabulary. Nucleic Acids Res. 2009;37:W166–169. doi: 10.1093/nar/gkp483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segara D, Biankin AV, Kench JG, Langusch CC, Dawson AC, Skalicky DA, Gotley DC, Coleman MJ, Sutherland RL, Henshall SM. Expression of HOXB2, a retinoic acid signaling target in pancreatic cancer and pancreatic intraepithelial neoplasia. Clin Cancer Res. 2005;11:3587–3596. doi: 10.1158/1078-0432.CCR-04-1813. [DOI] [PubMed] [Google Scholar]
- Theodosiou M, Laudet V, Schubert M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell Mol Life Sci. 2010;67:1423–1445. doi: 10.1007/s00018-010-0268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura K, Togashi Y, Ninomiya H, Shimoji T, Noda T, Ishikawa Y. HOXB2, an adverse prognostic indicator for stage I lung adenocarcinomas, promotes invasion by transcriptional regulation of metastasis-related genes in HOP-62 non-small cell lung cancer cells. Anticancer Res. 2008;28:2121–2127. [PubMed] [Google Scholar]
- Hanson D, Murray PG, O'Sullivan J, Urquhart J, Daly S, Bhaskar SS, Biesecker LG, Skae M, Smith C, Cole T, Kirk J, Chandler K, Kingston H, Donnai D, Clayton PE, Black GC. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet. 2011;89:148–153. doi: 10.1016/j.ajhg.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova GN, Qi CF, Ulmer JE, Moore JM, Ward MD, Hu YJ, Loukinov DI, Pugacheva EM, Klenova EM, Grundy PE, Feinberg AP, Cleton-Jansen AM, Moerland EW, Cornelisse CJ, Suzuki H, Komiya A, Lindblom A, Dorion-Bonnet F, Neiman PE, Morse HC 3rd, Collins SJ, Lobanenkov VV. Tumor-associated zinc finger mutations in the CTCF transcription factor selectively alter tts DNA-binding specificity. Cancer Res. 2002;62:48–52. [PubMed] [Google Scholar]
- Chang GT, Steenbeek M, Schippers E, Blok LJ, van Weerden WM, van Alewijk DC, Eussen BH, van Steenbrugge GJ, Brinkmann AO. Characterization of a zinc-finger protein and its association with apoptosis in prostate cancer cells. J Natl Cancer Inst. 2000;92:1414–1421. doi: 10.1093/jnci/92.17.1414. [DOI] [PubMed] [Google Scholar]
- Arnaldi LA, Borra RC, Maciel RM, Cerutti JM. Gene expression profiles reveal that DCN, DIO1, and DIO2 are underexpressed in benign and malignant thyroid tumors. Thyroid, 2005. pp. 210–221. [DOI] [PubMed]
- Casula S, Bianco AC. Thyroid hormone deiodinases and cancer. Front Endocrinol (Lausanne) 2012;3:74. doi: 10.3389/fendo.2012.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue J, Misawa A, Tanaka Y, Ichinose S, Sugino Y, Hosoi H, Sugimoto T, Imoto I, Inazawa J. Lysosomal-associated protein multispanning transmembrane 5 gene (LAPTM5) is associated with spontaneous regression of neuroblastomas. PLoS One. 2009;4:e7099. doi: 10.1371/journal.pone.0007099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayami Y, Iida S, Nakazawa N, Hanamura I, Kato M, Komatsu H, Miura I, Dave BJ, Sanger WG, Lim B, Taniwaki M, Ueda R. Inactivation of the E3/LAPTm5 gene by chromosomal rearrangement and DNA methylation in human multiple myeloma. Leukemia. 2003;17:1650–1657. doi: 10.1038/sj.leu.2403026. [DOI] [PubMed] [Google Scholar]
- Seimiya M, O-Wang J, Bahar R, Kawamura K, Wang Y, Saisho H, Tagawa M. Stage-specific expression of Clast6/E3/LAPTM5 during B cell differentiation: elevated expression in human B lymphomas. Int J Oncol. 2003;22:301–304. [PubMed] [Google Scholar]
- Hurst JH, Hooks SB. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochem Pharmacol. 2009;78:1289–1297. doi: 10.1016/j.bcp.2009.06.028. [DOI] [PubMed] [Google Scholar]
- Dai J, Gu J, Lu C, Lin J, Stewart D, Chang D, Roth JA, Wu X. Genetic variations in the regulator of G-protein signaling genes are associated with survival in late-stage non-small cell lung cancer. PLoS One. 2011;6:e21120. doi: 10.1371/journal.pone.0021120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel J, Nosrati M, Leong SP, Haqq C, Miller JR III, Sagebiel RW, Kashani-Sabet M. Novel role for RGS1 in melanoma progression. Am J Surg Pathol. 2008;32:1207–1212. doi: 10.1097/PAS.0b013e31816fd53c. [DOI] [PubMed] [Google Scholar]
- Croft W, Hill C, McCann E, Bond M, Esparza-Franco M, Bennett J, Rand D, Davey J, Ladds G. A physiologically required G protein-coupled receptor (GPCR)-regulator of G protein signaling (RGS) interaction that compartmentalizes RGS activity. J Biol Chem. 2013;288:27327–27342. doi: 10.1074/jbc.M113.497826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa S, Yajima M, Sakuma T, Udagawa Y, Kiguchi K, Sakayori M, Narisawa S, Iizuka R, Uemura M. Cancer-associated galactosyltransferase as a new tumor marker for ovarian clear cell carcinoma. Cancer Res. 1990;50:754–759. [PubMed] [Google Scholar]
- Saitoh E, Aoki D, Susumu N, Udagawa Y, Nozawa S. Galactosyltransferase associated with tumor in patients with ovarian cancer: factors involved in elevation of serum galactosyltransferase. Int J Oncol. 2003;23:303–310. [PubMed] [Google Scholar]
- Weiser MM, Podolsky DK, IselbacherProc KJ. Cancer-associated isoenzyme of serum galactosyltransferase. Natl Acad Sci USA. 1976;73:1319–1322. doi: 10.1073/pnas.73.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M1, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife. 2014;3:e01308. doi: 10.7554/eLife.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo Nguyen TT, Watanabe Y, Shiba A, Noguchi M, Itoh S, Kato M. TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci. 2014;105:334–341. doi: 10.1111/cas.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini G, Ambrosini MI, Di Marcotullio L, Cerignoli F, Zani M, MacKay AR, Screpanti I, Frati L, Gulino A. EGF- and cell-cycle-regulated STAG1/PMEPA1/ERG1.2 belongs to a conserved gene family and is overexpressed and amplified in breast and ovarian cancer. Mol Carcinog. 2003;38:188–200. doi: 10.1002/mc.10162. [DOI] [PubMed] [Google Scholar]
- Brunschwig EB, Wilson K, Mack D, Dawson D, Lawrence E, Willson JK, Lu S, Nosrati A, Rerko RM, Swinler S, Beard L, Lutterbaugh JD, Willis J, Platzer P, Markowitz S. PMEPA1, a transforming growth factor-beta-induced marker of terminal colonocyte differentiation whose expression is maintained in primary and metastatic colon cancer. Cancer Res. 2003;63:1568–1575. [PubMed] [Google Scholar]
- Rae FK, Hooper JD, Nicol DL, Clements JA. Characterization of a novel gene, STAG1/PMEPA1, upregulated in renal cell carcinoma and other solid tumors. Mol Carcinog. 2001;32:44–53. doi: 10.1002/mc.1063. [DOI] [PubMed] [Google Scholar]
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- Tsang JY, Ni YB, Chan SK, Shao MM, Kwok YK, Chan KW, Tan PH, Tse GM. CX3CL1 expression is associated with poor outcome in breast cancer patients. Breast Cancer Res Treat. 2013;140:495–504. doi: 10.1007/s10549-013-2653-4. [DOI] [PubMed] [Google Scholar]
- Tardáguila M, Mira E, García-Cabezas MA, Feijoo AM, Quintela-Fandino M, Azcoitia I, Lira SA, Mañes S. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013;73:4461–4473. doi: 10.1158/0008-5472.CAN-12-3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardáguila M, Mañes S. In: Oncology Theory & Practice. 1. iConcept Press Ltd, editor. The complex role of chemokines in cancer: the case of the CX3CL1/CX3CR1 axis. in press . [Google Scholar]
- Roland CL, Harken AH, Sarr MG, Barnett CC Jr. ICAM-1 expression determines malignant potential of cancer. Surgery. 2007;141:705–707. doi: 10.1016/j.surg.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Rosette C, Roth RB, Oeth P, Braun A, Kammerer S, Ekblom J, Denissenko MF. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis. 2005;26:943–950. doi: 10.1093/carcin/bgi070. [DOI] [PubMed] [Google Scholar]
- de Groote ML, Kazemier HG, Huisman C, van der Gun BT, Faas MM, Rots MG. Upregulation of endogenous ICAM-1 reduces ovarian cancer cell growth in the absence of immune cells. Int J Cancer. 2014;134:280–290. doi: 10.1002/ijc.28375. [DOI] [PubMed] [Google Scholar]
- Takaya D, Takeda-Shitaka M, Terashi G, Kanou K, Iwadate M, Umeyama H. Bioinformatics based Ligand-Docking and in-silico screening. Chem Pharm Bull. 2008;56:742–744. doi: 10.1248/cpb.56.742. [DOI] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Li L. Improved protein-ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics. 2014. [DOI] [PubMed]
- Xu L1, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet JP, Wagner SN, Ramaswamy S, Mesirov JP, Hynes RO. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res. 2008;6:760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Tsay JJ, Tchou-Wong KM, Greenberg AK, Pass H, Rom WN. Aryl hydrocarbon receptor and lung cancer. Anticancer Res. 2013;33:1247–1256. [PMC free article] [PubMed] [Google Scholar]
- Genome Browser. http://genome.ucsc.edu/
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System. Version 1.7.0.0 Schrödinger, LLC.
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- pdb2pqr. http://nbcr-222.ucsd.edu/pdb2pqr_1.9.0/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Gene expression and promoter methylation associated with PC2. Gene expression and promoter methylation associated with PC2. Left column: gene expression, right column: promoter methylation. NM_021977 (SLC22A3), NM_004403 (DFNA5), NM_015087 (SPG20), NM_000104 (CYP1B1), NM_006982 (ALX1), NM_006528 (TFPI2), NM_152739 (HOXA9), NM_005523 (HOXA11 ), NM_000439 (PCSK1), NM_003118 (SPARC). (Black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis). Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Fig S2 Gene expression and promoter methylation associated with PC3. Gene expression and promoter methylation associated with PC3. Left column: gene expression, right column: promoter methylation. NM_002145 (HOXB2), NM_032040 (CCDC8), NM_153608 (ZNF114), NM_000793 (DIO2), NM_006762 (LAPTM5), NM_002922 (RGS1), NM_003781 (B3GALNT1). (Black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis). Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Fig. S3 Gene expression and promoter methylation associated with PC4 (promoter methylation) and PC5 (gene expression). Gene expression and promoter methylation associated with PC4 (promoter methylation) and PC5 (gene expression). Left column: gene expression, right column: promoter methylation. NM_022164 (TINAGL1), NM_020182 (PMEPA1), NM_002996 (CX3CL1), NM_000201 (ICAM1). Contributions of samples (black open circles: A549 without metastasis, red triangles: A549 with metastasis, green crosses: HTB56 without metastasis, blue crosses: HTB56 with metastasis) to PCs. Left column: gene expression, right column: promoter methylation. "cor" indicates Pearson correlation coefficients between gene expression and promoter methylation averaged within each of four categories and "P" is attributed to "cor".
Supplementary figures. Supplementary figures from Fig. S4 to Fig. S14.
Ligand binding configuration to TINAGL1. Full list of ligands that bind to TINAGL1.
Fig. S15 Promoter methylation profile of selected genes measured by sequencing technology. Promoter methylation measured by sequencing, NM_002145 (HOXB2), NM_032040 (CCDC8), NM153608 (ZNF114), NM_006762 (LAPTM5), NM_003781 (B3GALNT1), NM_022164 (TINAGL1), NM_020182 (PMEPA1), NM_002996 (CX3CL1), and NM_000201 (ICAM1).
Amino acid sequences used for tertiary protein structure prediction. Amino acid sequences retrieved from Uniprot of selected genes (in fasta format).