

Abstract
The universal second messenger cAMP is generated upon stimulation of Gs protein-coupled receptors, such as the β2-adreneoceptor, and leads to the activation of PKA, the major cAMP effector protein. PKA oscillates between an on and off state and thereby regulates a plethora of distinct biological responses. The broad activation pattern of PKA and its contribution to several distinct cellular functions lead to the introduction of the concept of compartmentalization of cAMP. A-kinase anchoring proteins (AKAPs) are of central importance due to their unique ability to directly and/or indirectly interact with proteins that either determine the cellular content of cAMP, such as β2-adrenoceptors, ACs and PDEs, or are regulated by cAMP such as the exchange protein directly activated by cAMP. We report on lessons learned from neurons indicating that maintenance of cAMP compartmentalization by AKAP5 is linked to neurotransmission, learning and memory. Disturbance of cAMP compartments seem to be linked to neurodegenerative disease including Alzheimer's disease. We translate this knowledge to compartmentalized cAMP signalling in the lung. Next to AKAP5, we focus here on AKAP12 and Ezrin (AKAP78). These topics will be highlighted in the context of the development of novel pharmacological interventions to tackle AKAP-dependent compartmentalization.
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| A2B receptor |
| β2-adrenoceptor |
| M3 muscarinic receptor |
| Ligand gated ion channelsb |
| AMPA (GluA) receptors |
| Ionotropic glutamate receptors |
| NMDA (GluN) receptor |
| Ion channelsc |
| Cav1.2 |
| IP3 receptor |
| Potassium channels |
| Enzymesd |
| AC (adenylyl cyclases) |
| Epac |
| ERK1/2 |
| GRK2 |
| GSK3β |
| LKB1 |
| PDE4B |
| PDE7 |
| PDE8 |
| PKA |
| PKB (Akt) |
| PKC |
| PLCε1 |
| Other protein targets |
| CREB binding protein |
| LIGANDS |
|---|
| ACh |
| Amyloid β |
| Calmodulin |
| cAMP |
| Fenoterol |
| H-89 |
| IL-8 (CXCL8) |
| Isoprenaline |
| LPS |
| Roflumilast |
| Rolipram |
| Salbutamol |
| Tiotropium |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d,,,).
Introduction
GPCRs, such as the Gs-coupled β2-adrenoceptor, currently represent one of the largest groups of drug targets (Rask-Andersen et al., 2011). After receptor binding of β2-agonists, such as isoprenaline and fenoterol, elevation in the cellular content of cAMP is catalyzed by membrane-bound ACs (Hanoune and Defer, 2001; Beavo and Brunton, 2002; Dessauer, 2009), a process known to be shaped by cAMP-degrading PDEs (Conti and Beavo, 2007; McCahill et al., 2008; Houslay, 2010; Keravis and Lugnier, 2009; Cheepala et al., 2013). Among the PDE superfamily members, PDE4, PDE7 and PDE8 exhibit substrate specificity towards cAMP (Houslay, 2010; Keravis and Lugnier, 2009).
The best known effector of cAMP is PKA. The PKA holoenzyme consists of two catalytic (C) subunits, which exist in three isoforms (Cα, Cβ and Cγ), and two regulatory (R) subunits. There are two major isoforms of PKA, designated as PKA(I) and PKA(II), which differ exclusively due to the RI and RII subunits, each again subdivided in an α and β isoform (RIα, RIβ, RIIα, RIIβ). Upon binding of the two cAMP molecules to each R subunit, the dimer releases the C subunits and thereby initiates target protein phosphorylation. PKA is known to oscillate between an on and off state and thereby regulates a plethora of cellular responses (Taylor et al., 2013). With the discovery of the exchange factor directly activated by cAMP (Epac) (Kawasaki et al., 1998; de Rooij et al., 1998), the subset of biological functions driven by cAMP started to become even more diverse (Cheng et al., 2008; Oldenburger et al., 2012a; Dekkers et al., 2013; Schmidt et al., 2013), and thereby further supported the concept of compartmentalization of cAMP. Tough cyclic nucleotide-gated ion channels represent another cAMP-targeted group, a detailed description of this is beyond the scope of our current review and we would like to refer the reader to a recent review (Biel, 2009).
Concept of compartmentalization of cAMP
The localization of the different PKA isoforms and of the Epac proteins as well as of cAMP generating and degrading enzymes is strictly regulated. Indeed, PKA has already, some time ago found to be activated in either particulate or soluble cellular fractions (Corbin et al., 1977; Hayes et al., 1980). Clustering of PKA to lipid rafts and caveolae further support the existence of subcellular regions specialized in cAMP signalling that are characterized by a rather dynamic composition of a specific subset of signalling molecules, which include Gs-coupled receptors, ACs, PDEs and Epac (Insel and Ostrom, 2003; Hanzal-Bayer and Hancock, 2007; Gosens et al., 2008; Patel et al., 2008; Parton and del Pozo, 2013).
About 40 years ago, studies primarily performed in heart tissue reported that the two prototypical Gs-coupled receptor agonists isoprenaline and PGE1 elevated both the cellular content of cAMP, while only isoprenaline increased cardiac contractility (Corbin et al., 1977; Hayes et al., 1979; 1980,; Buxton and Brunton, 1983). Based on these early studies, the concept of compartmentalization of cAMP signalling was introduced, which ignited a new surge of cAMP-related research. Since then, several studies have provided further insights into the diversity of cellular strategies to compartmentalize intracellular signalling, a concept currently believed to enable a tightly and fine-tuned control of biological functions. Of particular interest is a recent study from Feinstein et al. (2012). Combining mathematical modelling and experimental measurements, the authors demonstrated that the microvascular endothelial barrier strictly relies on subtle local changes in cellular cAMP. Cytosolic-produced cAMP disrupted the microvascular endothelial barrier integrity, whereas cAMP produced at the plasma membrane increased pulmonary microvascular endothelial barrier integrity (Feinstein et al., 2012). Thus, studies on compartmentalization of cellular cAMP emerged as a theme of central importance to unravel the multiple facets of cAMP signalling and its effect in physiological and pathophysiological situatons. Such cAMP gradients may display high spatial resolution, as cAMP signalling often occurs within one protein complex orchestrated by a scaffold protein; the most studied family of scaffold proteins coordinating cAMP signalling is the A-kinase anchoring protein (AKAP) family, outlined in the next paragraph.
AKAPs
Microtubule-associated protein 2 (MAP2) was found to be the AKAP that tethers PKA together with microtubules (Theurkauf and Vallee, 1982). Members of the AKAP family represent important scaffolding proteins, which determine the specificity of cellular cAMP signalling. AKAPs control the spatio-temporal activity of the main cAMP effector PKA and some AKAPs have been shown to bind Epac (Dodge-Kafka et al., 2005; Nijholt et al., 2008; Sehrawat et al., 2011).
Through their association with cAMP-elevating receptors, ACs and/or cAMP-degrading PDEs, AKAPs are able to create and maintain local cAMP pools (Dodge et al., 2001; Smith et al., 2006a; Dessauer, 2009; Willoughby et al., 2010). To date, over 50 members and splice variants of the AKAP family have been identified (Tasken and Aandahl, 2004; Pidoux and Tasken, 2010; Skroblin et al., 2010; Welch et al., 2010; Scott et al., 2012; Troger et al., 2012).
AKAPs: PKA-RI and PKA-C
Differentiation between AKAPs is based on their ability to bind exclusively to PKA-RI, PKA-RII subunits or in the case of dual-specific AKAP members both PKA-R subtypes. Most of the AKAP superfamily members bind the PKA-RII subunit (Skroblin et al., 2010). In 2010, however, sphingosine kinase interacting protein (SKIP) was identified as the first mammalian AKAP specific for the binding of PKA-RI (Kovanich et al., 2010; Means et al., 2011; Burgers et al., 2012). In RIα −/− mouse embryonic fibroblasts, SKIP was unable to bind any PKA thereby strongly supporting the notion that SKIP specifically binds PKA-RI (Means et al., 2011). SKIP is also one of the few AKAPs shown to sequester two PKA holoenzymes thereby leading to their sequestration at the inner mitochondrial membrane (Means et al., 2011). Most AKAPs bind with the R subunits and thereby interact also indirectly with the C subunit of PKA. This is distinctly different from the scaffolding proteins A-kinase interacting protein (AKIP1) (Sastri et al., 2005) and caveolin-1 (Razani et al., 1999), which directly interact with the C subunit. Upon binding to both the C subunit of PKA and the p65 subunit of NF-κB, AKIP1 seems to act as a molecular switch for PKA-driven NF-κB signalling (Gao et al., 2010; King et al., 2011). In cardiomyocytes, AKIP1 protected against ischaemia/reperfusion damage by decreasing reactive oxygen species generation, a process requiring the mitochondrial localization of AKIP1 (Sastri et al., 2013). As both SKIP and AKIP1 seem to exert their primary biological functions in close proximity to mitochondria, it is tempting to speculate that AKAP scaffolding mechanisms via the PKA-RI subunit and/or PKA-C subunit most likely represent novel molecular mechanisms to unravel yet undefined cellular roles of AKAP-dependent compartmentalization of cAMP.
AKAPs: functional diversity and oligomerization
Utilization of distinct combinations of broad-spectrum signalling proteins, such as PKA, PKC and protein phosphatase 2B/calcineurin (PP2B/CaN), on the same AKAP, namely AKAP5, modulated the activity of the two distinct neuronal ion channels: AMPA-type glutamate receptor and M-type potassium channels, thereby triggering precise localized cellular responses (Hoshi et al., 2005). With this notion, it is meanwhile generally accepted that AKAPs act as a Swiss army knife that seem to execute differential cellular tasks upon subtle changes in their interacting proteins. Together with the huge number of different members of the AKAP family, the multitude of cellular tasks being performed in different cellular compartments is largely increased.
Even further complexity is added with the finding that AKAPs form homodimers (Baisamy et al., 2005; Gao et al., 2011b; Gold et al., 2011) and heterodimers (Gao et al., 2011a), a process initially described for AKAP-Lbc (Baisamy et al., 2005). For example, overexpression of AKAP12 in cells that endogenously express AKAP5, such as HEK293 or A431 cells, potentiates AKAP5-mediated phosphorylation of ERK1/2 in response to the β2-agonist isoprenaline (Gao et al., 2011a). Interestingly, however, AKAP12-mediated recycling of the β2-adrenoceptor was unaffected upon AKAP5 overexpression (Gao et al., 2011a) (Figure 1). Thus, oligomerization of AKAP family members may regulate a distinct subset of signalling properties. However, mechanisms involved in AKAP oligomerization, and how such dimer formation is triggered by molecular cues still remain obscure.
Figure 1.
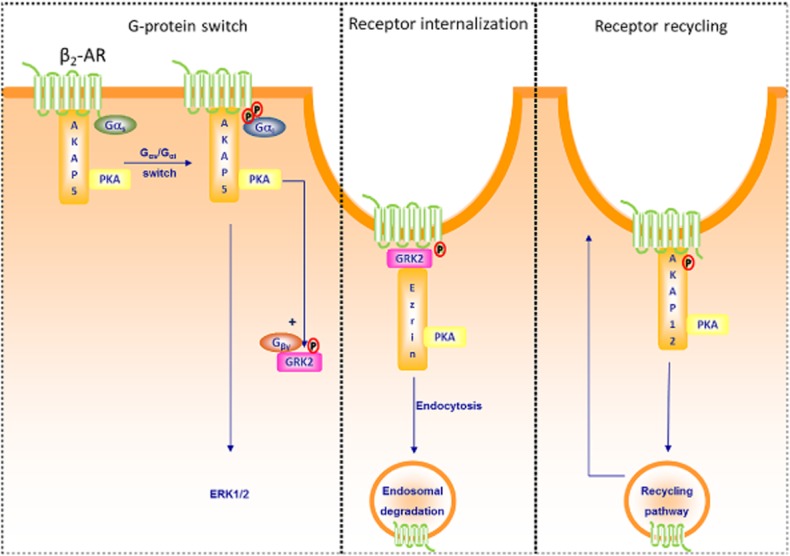
Members of the AKAP family and the function of the β2-adrenoceptor (β2–AR). Left: AKAP5 has been shown to constitutively associate with the β2-adrenoceptor receptor (Fraser et al., 2000; Lynch et al., 2005; Chen and Malbon, 2013). Upon β2-adrenoceptor activation, AKAP5-bound PKA phosphorylates the receptor, facilitates the switch of Gs to Gi and thereby permits signalling to ERK (Fraser et al., 2000; Lynch et al., 2005). In addition, AKAP5-bound PKA phosphorylates GRK2, enhances the affinity of GRK2 for Gβγ subunits and subsequent interaction with the β2-adrenoceptor (Cong et al., 2001). Middle: Receptor-bound GRK2 has the ability to interact with Ezrin (AKAP78), the latter known to be required for the internalization of the β2-adrenoceptor (Cant and Pitcher, 2005). Right: β2-adrenoceptor activation leads also to phosphorylation of AKAP12 via bound PKA and increases the association of AKAP12 with the β2-adrenoceptor receptor, a process known to be essential for the recycling of the β2-adrenoceptor (Shih et al., 1999; Tao et al., 2003).
For the purpose of this review, we will summarize the most important features of AKAP5, AKAP12 and Ezrin (AKAP78) (Table 1). Neuronal key discoveries will be recapitulated to introduce paradigm shifts that illustrate the general spatio-temporal nature of the compartmentalized cAMP signalling. Our goal is to translate the lessons learned from neurons to the lung as our current knowledge about cAMP compartmentalization in the airways is rather limited. Before that, we will focus in the next section on cAMP compartmentalization via AKAPs acting alone with PKA or in concert with Epac, starting in the following section with the different tools currently available or under development.
Table 1.
Subset of AKAP family members known to regulate biological functions in the lung and brain
| AKAP | Interactions | Processes involved |
|---|---|---|
| AKAP5 (HUGO) AKAP79 (Human) AKAP150 (Murine) AKAP75 (Bovine) H21 |
AKAP12, AKAP5 β1/β2-adrenoceptor AC5, Epac2 PKB/Akt, PKC PP2A/B, Calcineurin, Calmodulin PSD-95, MAGUK, SAP97 PIP2, F-actin E-/N-Cadherin AMPA/NMDA receptor Cav1.2 |
β2-AR switching to ERK β2-AR desensitization Cell cycle progression Synaptic plasticity |
| AKAP12 (HUGO) AKAP250 Gravin (Human) SSeCKS (Murine) Tsga12 Srcs5 AI317366 |
AKAP5, AKAP12 β2-adrenoceptor PKC |
β2-AR resensitization Cell cycle progression Synaptic plasticity |
| Ezrin AKAP78 Cytovillin p81 Villin-2 |
EBP50 (NHERF1) GRK2 RhoGDI Rho Rac Epac |
β2-AR internalization Actin-binding linker protein |
The most important AKAP interactions are highlighted, except of their primary binding partner PKA. Text between parentheses, AKAP synonym using the HUGO gene nomenclature or name of a certain orthologue. For further details and references, see text. β2-AR, β2-adrenoceptor.
Tools to study compartmentalization of cAMP
In the following section, we will highlight novel tools used to study the effect of AKAP-bearing multiprotein complexes on a diverse subset of biological functions. As some AKAPs bind to Epac in addition to PKA, we will briefly discuss some tools that are used to interfere at the level of PKA or Epac. For further details about Epac, we would like to refer the reader to recent reviews on this topic (Oldenburger et al., 2012a; Dekkers et al., 2013; Schmidt et al., 2013). Our main focus is the tools that interfere with AKAP-bearing multiprotein complexes.
Epac and PKA
To distinguish between PKA and Epac, cell membrane-permeable cyclic nucleotide analogues have been developed, such as N6-benzyladenosine-3′,5′-cyclic monophosphate for PKA or 8-(4-chlorophenylthio)-2′-O-methyl-cAMP for Epac (Holz et al., 2008; Grandoch et al., 2010; Schmidt et al., 2013). In addition, inhibitors of PKA have also been synthesized, such as Rp-8-CPT-cAMPS, and have been shown to abolish the dissociation of PKA-C subunits from the PKA-R subunits (Grandoch et al., 2010). These compounds seem to provide more specificity compared with PKA inhibitors known to act on the ATP binding site, such as H-89 (Bain et al., 2007). Inhibition of PKA can also be achieved with the PKA inhibitor (PKI) (Yan et al., 2011). Very recently, pharmacological inhibitors of Epac have been identified, which seem to act primarily on Epac1 (CE3F4) or Epac2 (ESI-05) (Courilleau et al., 2012; Tsalkova et al., 2012a,b,). Even though researchers worldwide use the novel compounds to gain insight into the contribution of Epac1 and/or Epac2 to biological functions (Chen et al., 2013), their mode of action and specificity warrants further studies (Rehmann, 2013). Specific activators for Epac1 and Epac2 are still lacking.
AKAPs: genetically modified mice
To address the physiological importance of specific AKAPs in vivo, mice deficient for a specific AKAP gene (e.g. AKAP5−/−) or for a specific AKAP-protein interaction, for example by introducing a truncation for AKAP5-PKA interactions have been developed (AKAP5Δ36). Ablation of AKAP members has led to several phenotypes such as decreased fertility (e.g. AKAP1, AKAP4), cardiac arrhythmias [e.g. AKAP10 (D-AKAP2)], developmental [e.g. mAKAP (AKAP6), WAVE-1] and neuronal defects (e.g. AKAP5, MAP2) (Hundsrucker and Klussmann, 2008; Skroblin et al., 2010). Based on these findings, it has been suggested that drug targets interfering at the level of AKAPs might have the ability to disturb signalling driven by cAMP and might, therefore, represent a novel layer of pharmacological interventions (Dodge-Kafka et al., 2008; Hundsrucker and Klussmann, 2008; Troger et al., 2012).
AKAPs: dynamics of PKA and AKAP
To assess the dynamics of the primary AKAP interaction partner PKA in vivo, several FRET tools have been developed, taking advantage of the genetically encoded A-kinase activity reporters (Nagai et al., 2000; Zhang et al., 2001; 2005,; Allen and Zhang, 2006; Depry et al., 2011; Komatsu et al., 2011). The addition of cellular localization signals permits the recruitment of these tools to subcellular compartments, including the cytosol, the nucleus, the sarcoplasmic reticulum, the mitochondria (using an AKAP-based localization), the plasma membrane (Allen and Zhang, 2006; Liu et al., 2011) and even the raft or non-raft domains of the cell membrane (Depry et al., 2011). Interestingly, the PKA-based biosensors have been transferred to AKAP research by combining them with AKAP12 (Tao et al., 2010) and AKAP5 (Kocer et al., 2012). Using this novel approach, distinct dynamics of PKA bound to either AKAP12 or AKAP5 at the membrane compared with cytosolic/perinuclear regions were identified (Tao et al., 2010; Kocer et al., 2012). Currently, several novel insights into the subcellular dynamics of AKAP-bound PKA are based on cell transduction with PKA-defined AKAP reporters and studies in genetically modified mice.
AKAPs: pharmacological tools
Novel pharmacological tools have been developed to overcome the technical limitations and to study the biological effect of AKAP-based multiprotein complexes in vivo. A conserved amphipathic helix represents a well-defined domain structure present in all AKAP superfamily members, which is required for the interaction with the primary AKAP-binding partner PKA (Malbon et al., 2004; Wang et al., 2006). The amphipathic helix is inserted into the hydrophobic pockets formed by the dimer of the PKA-R subunits (Gold et al., 2006; Kinderman et al., 2006). It is this amphipathic helix that provided the first basis for the design of dominant interfering peptides able to disrupt the interaction between PKA and AKAP, such as Ht31 (Figure 2A). The stearated form of Ht31, st-Ht31, exhibits an improved membrane permeability (Skroblin et al., 2010). It is important to note that the generation of such PKA-AKAP interfering peptides has enabled the research community to gain insights into the contribution of AKAP–PKA interactions to a diverse subset of cellular functions in physiology and pathophysiology (Tasken and Aandahl, 2004; Hundsrucker and Klussmann, 2008; Skroblin et al., 2010; Troger et al., 2012).
Figure 2.
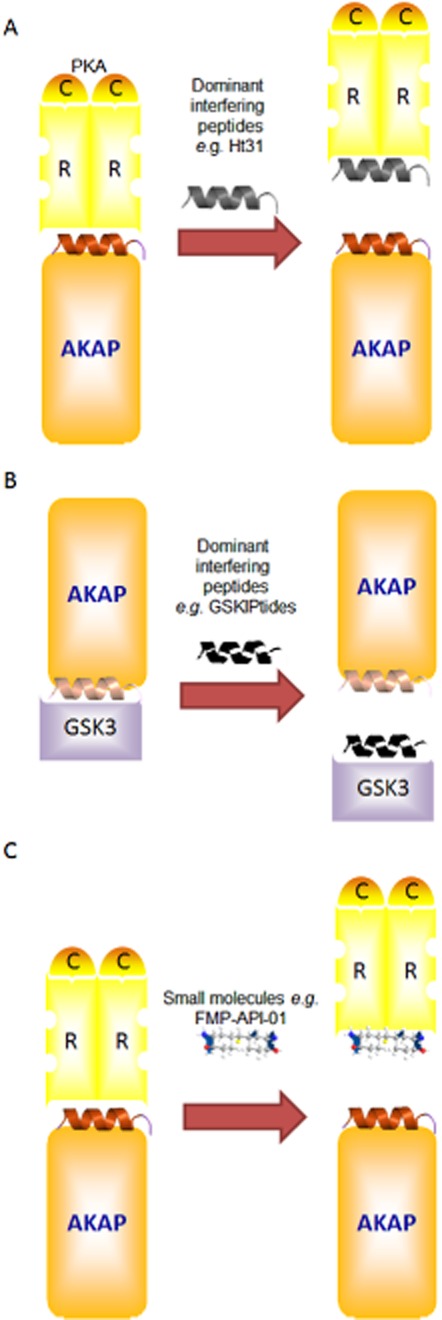
Strategies for disrupting AKAP complexes. Schematic illustration of the different ways to disrupt AKAP complexes. (A) Using PKA-AKAP dominant interfering peptides, such as Ht31, to displace PKA as the archetypical AKAP interaction partner. (B) Using dominant interfering peptides to disrupt interactions between proteins and AKAPs, such as GSKIPtide, to remove GSK3 from AKAP complexes. (C) Similar strategies are now applied using small molecules such as FMP-API-1. For further details, see text.
The original peptides, however, provided little, if any, distinction between PKA-RI and PKA-RII subtypes and members of the AKAP family. Through bioinformatics RI [A-kinase binding (AKB), RI anchoring disruptor] (Burns-Hamuro et al., 2003; Carlson et al., 2006) and RII-specific [AKB-RII, (Super)-AKAP-IS] (Alto et al., 2003; Burns-Hamuro et al., 2003; Gold et al., 2006) compounds were designed to discriminate between different type of PKA–AKAP interactions, PKA-RI or PKA-RII subunits. In attempts to overcome the central limitation in the current AKAP research field, a recent study from Scott and colleagues reported on the design of Rselect peptides, based on the RII subunits of PKA that seem to exhibit selective affinity for certain members of the AKAP family (Gold et al., 2013). Intriguingly, using a phage selection procedure combined with high-resolution structural bioinformatics AKAP2 (AKAP-kidney/lung) and AKAP7 (AKAP18) selective Rselect peptides were validated by biochemical and cell-based experiments (Gold et al., 2013). The AKAP5 (AKAP79, AKAP150) Rselect peptide, however, not only interfered with the binding of PKA to AKAP5, but also its binding to AKAP7 and AKAP11 (Gold et al., 2013). Functional data for these new tools have yet to come; however, the importance of this development is evident as for the first time it is possible to distinguish between the individual PKA compartmentalizers without genetic modifications.
In addition, recent studies intend to facilitate a distinction between different AKAPs based on their ability to interact with a discrete interaction partner and/or on mechanisms distinct from the AKAP–PKA interaction outlined earlier. The dominant interfering peptide, GSKIPtide, structurally based on the glycogen synthase kinase 3β (GSK3β) binding site of GSK3β interaction protein (GSKIP), competes with AKAP members known to bind to GSK3β, including GSKIP, AKAP11 and MAP2D (in rat) and thereby to disrupt the compartmentalization of GSK3β (Chou et al., 2006) (Figure 2B). Meanwhile, similar peptides were designed, such as a phospholamban peptide, which is able to prevent the interaction with AKAP7δ (Lygren et al., 2007), and EBP50 (also known as NHERF1, SLC9A3R1) peptide, which prevents the interaction with Ezrin (AKAP78) (Stokka et al., 2009) (Table 1). Also of particular interest are peptides that specifically inhibit the interaction between mAKAP and the AC isoform 5 (AC5), leaving the interaction between AKAP5-AC5 unaltered (Kapiloff et al., 2009). Recently, a disruptor for the Hsp20-PDE4 interaction has been described that liberates PDE4 from the AKAP-Lbc based complex (Sin et al., 2011).
Most tools being developed thus far, however, are still peptide based and might therefore exert some unknown interactions. For example, it has been reported that st-Ht31P, generated from st-Ht31 by two proline substitutions believed to render the molecule incapable of disrupting the AKAP–PKA interaction (Skroblin et al., 2010), seems to inhibit PKA (Klussmann et al., 1999). The aim of current research is to design small-molecule inhibitors for PKA–AKAP interactions (Christian et al., 2011; Schafer et al., 2013). Intriguingly, it has been reported that the small molecule 3,3′-diamino-4,4′-dihydroxydiphenylmethane (FMP-API-1) and its derivatives inhibit AKAP–PKA interactions in vitro and in cultured cardiomyocytes (Christian et al., 2011) (Figure 2C). As FMP-API-1, however, also activates PKA (Christian et al., 2011), synthesis of additional small molecules is still warranted. Indeed, new terpyridine scaffolds has been recently synthesized (Schafer et al., 2013), representing the non-peptidic compounds which might exert less unwanted biological side effects.
Relation to disease
Disturbance of AKAPs either at the level of their expression profile or biological functions has been associated with a variety of diseases (Tasken and Aandahl, 2004; Hundsrucker and Klussmann, 2008; Skroblin et al., 2010; Troger et al., 2012). For example, AKAP12, also known as AKAP250 or Gravin, was first identified as an auto-antigen in myasthenia gravis (Nauert et al., 1997). Down-regulation of AKAP12 is associated with prostate hyperplasia (Akakura et al., 2008) and several types of cancer (Gelman, 2010), including gastric cancer (Choi et al., 2004). It is tempting to speculate that down-regulation of AKAP12 might be mediated by promoter hypermethylation, a mechanism described before in the context of oesophageal and colon cancer (Mori et al., 2006; Jin et al., 2008; Paintlia et al., 2009). Such a mechanism is important for the promotion of cancer cell invasiveness by AKAP12 (Su et al., 2010). In line with this, AKAP12 inhibits cell proliferation (Gelman, 2010; Akakura and Gelman, 2012). In addition to AKAP12, other members of the AKAP family, such as AKAP4 and AKAP9, are discussed as cancer markers (Hasegawa et al., 2004; Sharma et al., 2005; Ferrari et al., 2007; Chiriva-Internati et al., 2008; Frank et al., 2008).
In the following sections, we will first focus on the compartmentalization of cAMP maintained by AKAPs in the context of neuronal learning and memory processes related to neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, Huntington's disease, multiple sclerosis and Wallerian degeneration. Then, we will highlight our current knowledge about compartmentalized cAMP signalling networks in the context of obstructive pulmonary diseases, such as chronic obstructive pulmonary disease and asthma, and whenever appropriate we will emphasize the effect of AKAP-based multiprotein complexes.
Lessons from neurons and neurodegenerative diseases
In the following sections, we will discuss the most recent findings on compartmentalized cAMP signalling to maintain proper neuron functions and to alleviate symptoms of neurodegenerative disease. In particular, we will highlight studies that focus on members of the AKAP family.
Concept of neuronal cAMP compartmentalization: PKA and Epac
Neurons represent highly polarized structures, displaying short, tapered dendrites and long, thin axons (Andersen and Bi, 2000; Hutchins, 2010; Shelly et al., 2010). In primary rat hippocampal neurons, Poo and colleagues demonstrated that liver kinase B1 phosphorylation by PKA represents an early event in axonal differentiation, whereas Smurf1 phosphorylation by PKA directs selective neuronal degradation of Par6 or RhoA (Shelly et al., 2010; Cheng et al., 2011). In rat dorsal root ganglion neurons, local cAMP levels regulate axonal guidance through the attraction and repulsion of axons, a process involving netrin-1 and myelin-associated glycoprotein (Murray and Shewan, 2008; Murray et al., 2009). High cAMP levels during the embryonic stage regulate axonal guidance by Epac, whereas low cAMP levels during the postnatal stage result in growth cone repulsion by PKA (Murray et al., 2009).
Local changes in cAMP determine hippocampus-dependent learning and memory stages such as acquisition, consolidation, retrieval, reconsolidation and extinction (Abel and Lattal, 2001; Abel and Nguyen, 2008). Since the pioneering work in Aplysia 30 years ago (Castellucci et al., 1980; Abrams et al., 1984), several studies link cAMP and PKA to locally defined synaptic plasticity, learning and different memory stages (Arnsten et al., 2005; Abel and Nguyen, 2008; Gelinas et al., 2008; Nijholt et al., 2008; Nijholt et al., 2007). In addition, several recent genetic and pharmacological studies report on the role of Epac in a context-dependent fear-conditioning paradigm (Morozov et al., 2003; Ouyang et al., 2008; Kelly et al., 2009; Ma et al., 2009; Ostroveanu et al., 2010; Schutsky et al., 2011; Srivastava et al., 2012; Yang et al., 2012).
Concept of compartmentalization of cAMP: AKAP5
As outlined earlier, both PKA and Epac seem to sense local changes in cAMP to control neuronal development and differentiation, learning and memory. Compartmentalization of cAMP in the brain seems to be maintained primarily by AKAP5 (Moita et al., 2002). AKAP5 is regulated during neuronal development (Robertson et al., 2009) and provides a platform to integrate neuronal cAMP signalling networks (Nijholt et al., 2008). Thus, AKAP5 most likely coordinates the fine tuning of cAMP by regulating the temporal and spatial events controlling cAMP levels. Indeed, a neuronal cAMP-sensing multiprotein complex maintained by AKAP5, PKA, Epac2 and PKB(Akt)-controlled the survival PKB (Akt) pathway (Nijholt et al., 2008).
AKAP5: neurotransmission, learning and memory
Binding of PKA to AKAPs alters synaptic protein phosphorylation and thereby controls synaptic plasticity and memory consolidation (Moita et al., 2002). In hippocampal neurons, AKAP5 acts as a postsynaptic scaffold protein that also binds to PP3 in addition to PKA protein PP2B/CaN (Bauman et al., 2004) and PKC (Smith et al., 2006c; Tunquist et al., 2008) (Figure 3). The postsynaptic AKAP5 localization is dependent on its association with the actin cytoskeleton, acidic phospholipids and cadherins (Gomez et al., 2002; Gorski et al., 2005b). Binding of AKAP5 with membrane-associated guanylate kinase (MAGUK) is required for maturation of dendritic protrusions into large, dendritic spines with an increased density of synaptic AMPA receptors (Robertson et al., 2009). The functional relation between AKAP5 and AMPA receptors may also be linked to the binding of AKAP5 to the MAGUK family member SAP-97 (Colledge et al., 2000). AKAP5 can also bind the postsynaptic density protein (PSD)-95 to regulate NMDA receptors (Smith et al., 2006b; Bhattacharyya et al., 2009) (Figure 3). Next to the interaction of AKAP5 with several members of the scaffold protein PSD family, binding of AKAP5 to cadherins may also influence synaptic plasticity mechanisms, a process implicated in the regulation of NMDA receptors (Gorski et al., 2005a) (Figure 3).
Figure 3.
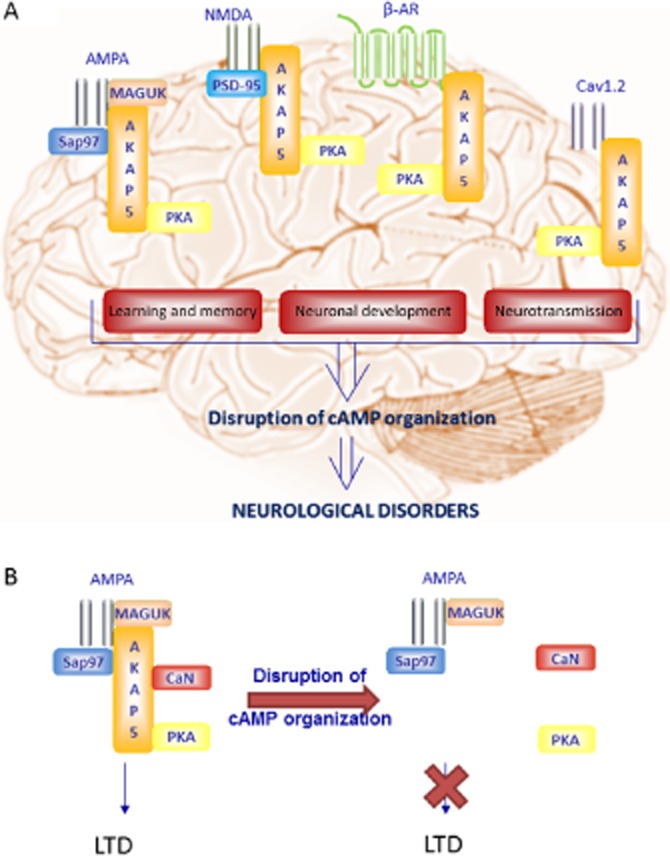
Compartmentalization of cAMP in neurons in relation to neurodegenerative diseases. (A) Illustration of cAMP compartmentalization with emphasis on AKAP5 and selected adaptor proteins in neurons and their alterations under pathological conditions including Alzheimer's disease, Parkinson's disease, Huntington's disease, multiple sclerosis and Wallerian degeneration. (B) An example of disrupted cAMP compartmentalization, as it was shown that AKAP5 coordinated calcineurin (CaN) was required for AMPA receptor internalization and LTD, removal of the AKAP5 caused an impairment of this LTD. For further details, see text.
AKAP5 directly interacts with the neuronal L-type calcium channel subunit Cav1.2 (Oliveria et al., 2007), and thereby forms a complex with AC, PKA and PP2A, and is, therefore, able to modulate Ca2+ signalling downstream of the β2-adrenoceptor (Davare et al., 2001). Anchoring of PP2B/CaN to AKAP5 regulates internalization and rapid dephosphorylation of the AMPA receptor, and most likely reflects a form of molecular and cellular memory associated with long-term depression (LTD) (Figure 3B) (Smith et al., 2006b). Indeed, brain slices derived from adult AKAP5 knockout mice display normal basal hippocampal spine density and synaptic transmission, but exhibit a deficiency in LTD, learning and memory (Robertson et al., 2009). Malenka and colleagues (Jurado et al., 2010) reported that AKAP5 modulates LTD, most likely through binding of AKAP5 to PSD-95, causing the release of PP2B/CaN, and subsequently enhances endocytosis of synaptic AMPA receptors. As a consequence, AKAP5 may leave the spine and thereby contribute to the shrinkage of spines that accompanies LTD (Jurado et al., 2010). Currently, the best genetic models for studying AKAP5 function are the Δ36 mice, which lack the PKA binding site at the C-terminus of AKAP5 (Lu et al., 2007), and AKAP5-deficient mice (Weisenhaus et al., 2010). Δ36 mice display both long-term potentiation and LTD defects. In contrast, the AKAP5-deficient mice exhibit only LTD defects. Such differences suggest that the most critical function of AKAP5 is most likely related to its interaction with PKA, to control the formation and/or maintenance of dendritic spines (Lu et al., 2011). It is clear that regulation of PKA signalling by AKAP5 is necessary to facilitate neurotransmission, learning and defined stages of the memory.
Throughout the mouse brain, AKAP5 is widely distributed in regions linked to learning and memory in rodents, such as the cortex, the hippocampus and the amygdala (Glantz et al., 1992; Ulfig and Setzer, 1999; Moita et al., 2002; Ostroveanu et al., 2007). Using contextual fear conditioning in mice, the expression of AKAP5 protein was increased in the hippocampus in a late phase of memory consolidation of associative memory (Nijholt et al., 2007). Disruption of hippocampal AKAP–PKA interactions by st-Ht31 or st-superAKAP-IS facilitates the extinction and impairs the consolidation of contextual fear memories, whereas acquisition and retrieval remain unchanged (Nijholt et al., 2008) (Figure 3). Disruption of AKAP–PKA interactions by st-Ht31 in the rat lateral amygdala impaired memory consolidation in auditory fear conditioning (Moita et al., 2002). Using the Morris water maze to study learning and spatial memory, AKAP5-deficient mice exhibit deficits in spatial memory retention most likely caused by delocalization of PKA and subsequent alterations in the local environment of cAMP signalling in the hippocampus (Tunquist et al., 2008). Taken together, the results from several recent studies illustrate the importance of AKAP5 for maintaining neuronal compartmentalized cAMP signalling to coordinate learning and memory.
AKAP5: lessons from Alzheimer's disease
As discussed earlier, cAMP in neurons is crucial for learning, memory and physiological events but it is not known how this system is altered under pathological neurodegenerative circumstances. Elucidating this is likely to provide mechanistic insights that may give some clues for the development of novel pharmacological tools. Ample evidence suggests that perturbation of local cAMP signalling contributes to the development and progression of neurodegenerative diseases. Here, we focus on the role of the players discussed previously in the context of Alzheimer's disease.
Alzheimer's disease is a neurodegenerative disease characterized by the progressive decline of cognitive function and memory, and is the fourth largest cause of death for people over 65 years of age (Sonkusare et al., 2005). The disease is characterized by extracellular β-amyloid plaques, intracellular neurofibrillary tangles, cholinergic transmission defects and neuronal loss preferentially in the entorhinal cortex and hippocampus (Sonkusare et al., 2005). As several inflammatory markers are up-regulated in Alzheimer's disease, it is generally assumed that inflammation is linked to the pathogenesis of Alzheimer's disease. Indeed, amyloid plaques seem to trigger inflammatory processes (McGeer and McGeer, 1995; Martinez et al., 1999; Halliday et al., 2000; Rogers, 2008).
Chronic infusion of lipopolysaccharide (LPS) has been used as an experimental model to mimic certain aspects of Alzheimer's disease (Jaeger et al., 2009). Chronic lipopolysaccharide infusion into the fourth ventricle of young rats induces brain inflammation and subsequently activation of microglial, a process accompanied by a reduction in the expression of adenosine A2B receptors and cAMP (Rosi et al., 2003). Mengod and colleagues (Perez-Torres et al., 2003), using in situ hybridization, showed that at early stages of Alzheimer's disease, PDE4, in particular PDE4B, and PDE7 are up-regulated, while at later stages of Alzheimer's disease, PDE8 is up-regulated (Perez-Torres et al., 2003). Both studies imply that progression of Alzheimer's disease is associated with a limitation in cellular cAMP.
Accumulating evidence suggests that Aβ-induced neurotoxicity alters NMDA receptor signalling through the cAMP response element-binding protein (CREB), a transcription involved in learning and memory processes (Snyder et al., 2005). Moreover, CREB phosphorylation was reduced in the hippocampus of Alzheimer's post-mortem brains (Yamamoto-Sasaki et al., 1999). Intriguingly, Shelanski and colleagues showed that treatment of rat hippocampal neurons with Aβ peptides decreases the dissociation of PKA C and R subunits and thereby phosphorylation of downstream targets such as CREB (Vitolo et al., 2002). The PDE4 inhibitor rolipram promotes the dissociation of PKA's C and R subunits and reverses the inhibitory effects of Aβ peptides on CREB phosphorylation (Vitolo et al., 2002; Cheng et al., 2010; Wang et al., 2012). As PKA-dependent signalling studied by CREB phosphorylation in the hippocampus of Alzheimer's post-mortem brains was reduced (Yamamoto-Sasaki et al., 1999), Arima and colleagues proposed that CREB phosphorylation may serve as a molecular biomarker of ageing-related pathological processes (Satoh et al., 2009), in particular of Alzheimer's disease.
In addition to PKA, recent studies indicate that Epac may also be linked to Alzheimer's disease. Lezloualc'h and colleagues show that the Epac effector Rap1 promotes the activation of Rac, and subsequently leads to the cleavage of the amyloid precursor protein (APP) and production of secreted APPα (sAPPα) (Maillet et al., 2003). Rap1 can directly interact with Sif- and Tiam1-like exchange factor, a specific guanine exchanging factor (GEF) for Rac1, and this association is involved in the secretion of the sAPPα (Zaldua et al., 2007). Moreover, activation of the serotonin receptor of the subtype 4 increases sAPPα through Epac1/Rap1/Rac (Robert et al., 2005). It has been postulated that sAPPα acts as a memory enhancer and neuroprotector (Maillet et al., 2003; Robert et al., 2005). Thus, production of sAPPα by Epac may reduce the symptoms of Alzheimer's disease. Indeed, in human brain regions associated with Alzheimer's disease, Epac1 mRNA is up-regulated, which is accompanied by a down-regulation of Epac2 mRNA (McPhee et al., 2005).
Next to Alzheimer's disease, cAMP and its players are associated with others neurodegenerative disease such as Parkinson's disease, Huntington's disease, multiple sclerosis and Wallerian degeneration (Table 2). Several lines of evidence indicate that alterations in local cAMP dynamics might be caused by inhibition of PKA, up-regulation of a specific PDE subset, up-/down-regulation of Epacs or a combination of these events. Persistent limitations in the cellular cAMP level, due to either defects in the cAMP-producing receptors and/or elevations of the cAMP-degrading PDEs, such as PDE4, seem to underpin the development and progression of neurodegenerative diseases (Table 2). Even though not yet being studied in detail in the context of neurodegenerative diseases, a central role for the AKAP family member AKAP5 might be envisaged due to its ability to interact with the β2-adrenoceptor and/or PDE4 (Lynch et al., 2005), and due to its ability to maintain neuronal cAMP compartmentalization.
Table 2.
cAMP compartmentalization in neurodegenerative diseases
| Pathology | Modulator involved | cAMP-dependent effects | References |
|---|---|---|---|
| Alzheimer's disease | PKA | Reduced phosphorylation of CREB | Cheng et al., 2010 |
| Inactivation of PKA | Vitolo et al., 2002 | ||
| τ phosphorylation at Ser214 and Ser409 | Jicha et al., 1999 | ||
| Down-regulation of A2B receptor/PKA signalling | Rosi et al., 2003 | ||
| Epac | sAPPα production via Epac1/Rap1/Rac | Zaldua et al., 2007 | |
| AKAPs | AKAP79, associated with neurofibrillary pathology | Jicha et al., 1999 | |
| PDEs | PDE4, PDE4B and PDE7 up-regulation at early stage of Alzheimer's disease | Perez-Torres et al., 2003 | |
| PDE8 up-regulation at later stage of Alzheimer's disease | |||
| Parkinson's disease | PKA | Down-regulation of A2A receptor/PKA signalling | Hara et al., 2010 |
| α-synuclein stimulates τ phosphorylation by PKA | Qureshi et al., 2011 | ||
| PDEs | PDE7 and PDE4 inhibition enhances neuroprotection | Morales-Garcia et al., 2011 | |
| Yang et al., 2008 | |||
| Huntington's disease | PKA | Decreased levels and CREB activation | Gines et al., 2003 |
| Sugars et al., 2004 | |||
| PDEs | Inhibition of PDE4 or PDE10A promotes neuroprotective effects | DeMarch et al., 2007; Giampa et al., 2009; 2010, | |
| Multiple sclerosis | PKA | β2-AR deficient astrocytes produce less cAMP | Chesik et al., 2008 |
| Lipoic acid treatment increased PKA activity | Salinthone et al., 2010 | ||
| PDEs | Lovastatin treatment and inhibition of PDE4 promote neuroprotection and neurorepair | Paintlia et al., 2009 |
For further details, see text.
Airway smooth muscle and obstructive pulmonary diseases
Chronic obstructive pulmonary disease (COPD) and asthma are both obstructive inflammatory airway diseases characterized by chronic inflammation, airway obstruction and airway remodelling, albeit with different aetiology and specific pathological features (Barnes, 2008; Hogg and Timens, 2009). COPD is predicted to be the third leading cause of death by disease worldwide in 2020 (Rycroft et al., 2012). Airflow limitation in asthma is reversible with bronchodilators and associated with airway hyperresponsiveness, whereas airway obstruction in COPD is largely irreversible and lung function decline is progressive (Meurs et al., 2008; Guerra, 2009; Hogg and Timens, 2009; Barnes, 2011). Airway smooth muscle cells contribute to disease symptoms in both asthma and COPD due to their multifunctional behaviour that supports airway remodelling and airway obstruction, causing the limitation of airflow (Halayko et al., 2008; Damera and Panettieri, 2011; Billington et al., 2013).
Different classes of bronchodilators are used in practice: β2-adrenoceptor agonists (β2-agonists), muscarinic receptor antagonists (anticholinergics), individually or in combinations, with or without the addition of anti-inflammatory glucocorticosteroids (Peters et al., 2010; Vogelmeier et al., 2011; Sethi et al., 2012; Kandeel et al., 2013; Meurs et al., 2013). The main targets for the therapeutic treatment of obstructive pulmonary diseases have a direct or indirect link to GPCR signalling, mainly to the β2-adrenoceptor and the M3 muscarinic receptor. In obstructive airway diseases, increase in smooth muscle mass and hypercontractility cause severe limitations in the airflow. Airway smooth muscle cell growth is inhibited by several β2-agonists such as fenoterol and salbutamol (Ibe et al., 2006; Yan et al., 2011). Increased smooth muscle mass is believed to reduce the lumen size of the airways, a process associated with aberrant β2-adrenoceptor signalling (Deshpande and Penn, 2006). Despite the fact that β2-agonists are generally well tolerated (Donohue et al., 2008; Hanania et al., 2010), long-term use of β2-agonists caused variations in the treatment outcome in asthma and COPD patients, being either less efficacious in COPD patients or even leading to an increased incidence of asthma exacerbations and other markers of morbidity and mortality (Liesker et al., 2002; Giembycz and Newton, 2006; Aguilaniu, 2010; Kliber et al., 2010).
Another treatment option in obstructive airway diseases is represented by PDE inhibition, for instance the selective PDE4 inhibitors rolipram and roflumilast (Calverley et al., 2009; Global Initiative for Chronic Obstructive Lung Disease, 2006; Rabe, 2011). PDE inhibitors increase the cellular level of cAMP by preventing its degradation. Although both β2-agonists and PDE inhibitors show anti-inflammatory properties in vitro (Hallsworth et al., 2001; Kaur et al., 2008; Spina, 2008), a notable difference is seen in vivo. PDE4 inhibitors show anti-inflammatory properties in vivo, but largely lack airway smooth muscle relaxing properties. In contrast, β2-agonists show bronchorelaxing properties in vivo, but lack anti-inflammatory properties (Calverley et al., 2009; Hurst et al., 2010). Possible explanations for this discrepancy are most likely β2-adrenoceptor desensitization and/or biased signalling of the β2-adrenoceptor towards ERK signalling (Dickey et al., 2010; Walker et al., 2011), features largely absent with PDE4 inhibitors due to their post-receptor mode of action. Both the process of β2-adrenoceptor desensitization and biased signalling seem to be facilitated by scaffolding proteins such as AKAP5 and AKAP12 (Lefkowitz et al., 2006; Tao and Malbon, 2008) (Figure 1). Subcellular localized cAMP pools seem to cause differential biological effects upon scaffolding protein-mediated targeting of either the β2-adrenoceptor or PDEs.
An innovative alternative is, therefore, urgently required to safeguard long-term treatment of obstructive lung disorders. Compartmentalized cAMP signalling may provide a novel opportunity for pharmacological interventions. For example, targeting downstream of the β2-adrenoceptor will most likely circumvent receptor desensitization. One might also expect that such strategies will increase treatment specificity, and thereby minimize unwanted side effects, by targeting only the desired cAMP pool. In the following section, the potential effect of compartmentalized cAMP signalling in the lung for further improvement of obstructive airway diseases will be discussed.
AKAPs: signalling in the airway smooth muscle
In the airway smooth muscle, the main signalling pathways that determine its functionality are receptors coupling to Gq or Gs proteins. The Gq protein-coupled receptor family is the M3 muscarinic receptor known to be activated by ACh and to be inhibited by anticholinergics such as tiotropium (Meurs et al., 2013). After agonist binding, the Gαq subunit activates PLC, thereby leading to the elevation in cellular calcium and activation of calcium/calmodulin-dependent myosin light chain (MLC), a process known to result in airway smooth muscle contraction (Billington and Penn, 2003; Mizuno and Itoh, 2009). Activation of PKC by DAG also alters the (de)phosphorylation of the MLC through several pathways and thereby contributes to the airway smooth muscle tone (Billington and Penn, 2003). Activation of the Gs protein-coupled receptors by drugs targeting the β2-adrenoceptor causes elevation of cAMP production via Gs and subsequent activation of ACs (Figure 4).
Figure 4.

Compartmentalization of cAMP in relation to airway smooth muscle functioning. Schematic illustration of central biological ASM functions, namely contraction, cytokine secretion and proliferation. Endogenous expression of AKAP5, AKAP12 and Ezrin (AKAP78) in ASM seems to maintain defined subcellular signalling compartments. Abbreviations not mentioned in the text; CaM, calmodulin; ROCK, Rho-kinase.
Two members of the AKAP superfamily are known to interact with the β2-adrenoceptor, AKAP5 and AKAP12. Whereas the association of AKAP5 with the β2-adrenoceptor is constitutive (Fraser et al., 2000; Lynch et al., 2005), agonist binding to the β2-adrenoceptor increases the interaction of the receptor with AKAP12 (Tao et al., 2003). Despite the fact that AKAP5 and AKAP12 share many common features, no redundancy is seen between them with regard to this cellular response (Tao and Malbon, 2008). AKAP5 has been reported to switch the coupling of the β2-adrenoceptor from Gs to Gi, a process most likely facilitated by a PKA-mediated phosphorylation of the receptor (Daaka et al., 1997; Fraser et al., 2000; Hill and Baker, 2003; Lynch et al., 2005) (Figure 1). It has been reported that coupling of the β2-adrenoceptor to Gi leads to activation of ERK signalling (Chen and Malbon, 2013). The ERK pathway is known to be linked to both proliferative and cytokine production pathways in airway smooth muscle (Roscioni et al., 2011;a,c Dekkers et al., 2013; Schmidt et al., 2013). In the context of obstructive pulmonary diseases, it is worthwhile to emphasize reports indicating that AKAP5 seems to determine the cell surface expression of the β2-adrenoceptor by increasing the affinity of GPCR kinase 2 (GRK2) for βγ subunits of the G-proteins, causing their translocation to the membrane, leading to the desensitization and internalization of the β2-adrenoceptor (Cong et al., 2001) (Figure 1). In contrast, after desensitization, AKAP12 is essential for the dephosphorylation, resensitization and recycling of the β2-adrenoceptor back to the cell membrane (Tao et al., 2003; Tao and Malbon, 2008; Chen and Malbon, 2013). In addition, interaction of GRK2 with Ezrin (AKAP78) determines the β2-adrenoceptor internalization (Cant and Pitcher, 2005) (Figure 1).
Based on these findings, it is reasonable to assume that β2-adrenoceptor functions are determined by the balance between AKAP5, AKAP12 and Ezrin (AKAP78) (Figure 1). Indeed, a recent study from Penn and colleagues reported on the expression of AKAP5, AKAP12 and Ezrin (AKAP78) in human airway smooth muscle cells (Horvat et al., 2012). Penn and colleagues did not observe effects of Ht31 or AKAP-IS studying whole-cell cAMP after stimulation with isoprenaline or the direct AC activator forskolin. However, using a cyclic nucleotide-gated ion channel reporter, the authors showed that local cAMP concentrations close to the near-membrane compartment were significantly and transiently increased (Horvat et al., 2012). Using a combination of st-Ht31 and a PDE inhibitor cocktail, the authors demonstrated that disruption of PKA–AKAP interactions resulted in sustained AC activity (Horvat et al., 2012). Mathematical models predicted that tethering of PKA to AKAP should cause a threefold increase in PKA at the β2-adrenoceptor compartment, thereby decreasing input of the β2-adrenoceptor acting as a negative feedback for AC and PDE activity (Horvat et al., 2012). Indeed, direct inhibition of PKA with the PKI completely blunted the rapid decay of the cAMP signal over time (Horvat et al., 2012). With multiple AKAPs possibly involved to create such PKA pool, utilization of tools recently described by Gold et al. (2013) would be necessary to assess the individual contribution of each AKAP.
In the following sections, we will discuss the role of cAMP compartmentalization in some of the important features of COPD: contraction, inflammation and remodelling. Herein, we will keep the focus on studies performed in airway smooth muscle.
AKAPs: airway smooth muscle contraction
Elevation of cAMP leads to the activation of both PKA and Epac and thereby modulates airway smooth muscle responses (Dekkers et al., 2013; Schmidt et al., 2013). It is well established that PKA on its own deactivates MLC kinase and desensitizes the inositol 1,4,5-triphosphate (IP3) receptor, thereby functionally counteracting the PLC-PKC pathway. In our research group and by others, Epac has been identified as a novel factor being involved in the regulation of airway smooth muscle relaxation. Epac, acting most likely via its main effector Rap1, deactivates RhoA and up-regulates Rac1 activation, causing the balance to shift from phosphorylated MLC to non-phosphorylated MLC and thus to airway smooth muscle relaxation (Roscioni et al., 2011b; Zieba et al., 2011) (Figure 4). Interestingly, Ezrin (AKAP78) is phosphorylated by Rho-regulated Rho-kinase and binds via its ezrin-radixin-moesin domain, the Rho inhibitor Rho guanine-nucleotide-dissociation inhibitor (RhoGDI) (Bretscher et al., 2002). Airway smooth muscle cells express both Epac and Ezrin (AKAP78) (Roscioni et al., 2009; Horvat et al., 2012). Thus deactivation of Rho by Epac might involve mechanisms driven by Ezrin (AKAP78) and RhoGDI.
In a Madin-Darby canine kidney cell line, activated Ezrin (AKAP78) binds in a calcium-dependent manner to Rac and thereby delayed membrane localization of E-cadherin (Pujuguet et al., 2003). Calcium also underlies cellular compartmentalization and cross-talk with cAMP, a process being facilitated by members of the AKAP family. For example, AKAP5, known to be involved in β2-adrenoceptor desensitization as outlined earlier (Figure 1), interacts with calcineurin (Coghlan et al., 1995; Oliveria et al., 2003) and calmodulin (Sarkar et al., 1984). Calmodulin competes with PKC in a Ca2+-dependent manner for binding to AKAP5 (Faux and Scott, 1997). More recently, AKAP12, known to be involved in β2-adrenoceptor sensitivity (Figure 1), rapidly redistributes from the plasma membrane to the cytosol upon stimulation with calcium-elevating agents such as ionomycin or thapsigargin (Schott and Grove, 2013). Moreover, it has been reported that AKAP12 displace PKA-RII from the membrane (Schott and Grove, 2013).
A striking example of cooperativity between cAMP and calcium facilitated by AKAPs is shown for AKAP11 upon assembly of a complex that includes IQGAP1, GSK3β and PKA. It has been shown that binding of AKAP11 and IQGAP2 requires high intracellular calcium levels (Logue et al., 2011a,c,). At lower intracellular calcium, AKAP11-anchored PKA phosphorylates IQGAP2 and thereby leads to an increase in Rac binding. In the presence of inactive GSK3β, however, AKAP11 serves as a platform for the assembly of a complex between IQGAP and cytoplasmic linker proteins-associating proteins 2 (CLASP2), a plus-end microtubule tracking protein involved in microtubule polymerization. PKA phosphorylation of GSK3β and elevations in calcium cooperatively drive the formation of an IQGAP1-CLASP2. Both the IQGAP1-Rac and IQGAP1-CLASP2 complexes have been suggested to be involved in microtubule dynamics and cell motility (Logue et al., 2011a,c,). AKAP11 was found to be expressed in airway smooth muscle (Horvat et al., 2012). In addition, Epac not only interacts with AKAP5, but also with the microtubule network and with the calcium-elevating PLCε1 (Schmidt et al., 2013). Future studies should point out if similar mechanisms contribute to airway smooth muscle contraction.
AKAPs: airway smooth muscle inflammation
Recently, we reported in human airway smooth muscle cells that direct pharmacological activation of PKA and Epac synergistically enhances Gq protein-coupled receptor-induced release of the neutrophil chemoattractant CXCL8 (IL-8) (Roscioni et al., 2009). Silencing of Epac expression decreased not only IL-8 release in response to Epac activation but also in response to PKA activation, and vice versa PKA inhibition by Rp-8-CPT-cAMPS reduced CXCL8 release induced by both PKA and Epac (Roscioni et al., 2009). Using st-Ht31 to disrupt PKA–AKAP interactions (Figure 2A), preliminary results of our group suggest that PKA and Epac regulate the CXCL8 release in an AKAP-dependent manner.
Results from our research groups and others implicate that such close interconnectivity requires the presence of spatial regulation. AKAP5 was shown to be present in the same AKAP-PKA-Epac complex described before in neuronal cells (Nijholt et al., 2008). In a related study, we showed that induction of CXCL8 release by cigarette smoke extract (CSE) was attenuated by the β2-agonist fenoterol, seemingly via Epac and PKA (Oldenburger et al., 2012b). Disturbance of AKAP-based multiprotein complexes might be expected due to the down-regulation of Epac1 and members of the AKAP family by CSE (Oldenburger et al., 2012b; 2014,). Indeed, AKAP12 is down-regulated in lung cancer (Wikman et al., 2002). With AKAP5 and AKAP12 known to determine β2-adrenoceptor functions (Figure 1), an important role for PKA and Epac localization close to GPCRs in asthma and COPD could be imagined. This could explain the varying treatment outcomes seen for these bronchodilators in COPD (Liesker et al., 2002; Aguilaniu, 2010; Kliber et al., 2010).
The underlying molecular mechanisms of the attenuation of CXCL8 release by cAMP seem to be coordinated via parallel routes. Epac was shown to inhibit the NF-κB translocation to the nucleus caused by CSE, and PKA counteracts CSE-induced ERK phosphorylation, both known to underlie CXCL8 production (Oldenburger et al., 2012b; Saito et al., 2012). Although limited knowledge is currently available on Epac compartmentalization, both NF-κB and ERK are known to interact with proteins that anchor C and/or R PKA subunits respectively (Gao et al., 2008; 2010,; King et al., 2011; Smith et al., 2011). Thus, it is tempting to speculate that a distinct subset of AKAP members mediate the anti-inflammatory properties of both PKA and Epac, a research topic open for future investigation.
Our current knowledge implicates AKAPs as important factors of both inflammation and contraction. The question that remains: what role AKAPs play in airway remodelling?
AKAPs: airway smooth muscle remodelling
Another important functional feature of airway smooth muscle cells encompasses the existence of multiple phenotypes, a process reported to involve both PKA and Epac. Upon chronic exposure to stimuli, such as growth factors, airway smooth muscle cells switch between a contractile and proliferative (synthetic) phenotype (Halayko et al., 2008). Some researchers have suggested that ASM proliferation is primarily inhibited by Epac, but not by PKA (Kassel et al., 2008), while others state a more prominent role for PKA (Ibe et al., 2006; Yan et al., 2011). Recently, our research group demonstrated that pharmacological activation of either Epac or PKA prevented PDGF-induced hypocontractility of airway smooth muscle strips and airway smooth muscle proliferation, a process being accompanied by the inhibition of ERK1/2 (Roscioni et al., 2011a,c,), suggesting a possible synergism between PKA and Epac. Our findings were strengthened by other studies in vascular smooth muscle cells (Hewer et al., 2011). Here, a concerted action of PKA and Epac inhibited serum-induced bromodeoxyuridine incorporation, Rb phosphorylation and the expression of cell cycle progression proteins, in a Rap1a-independent fashion (Hewer et al., 2011).
Several signalling pathways have been shown to be involved airway smooth muscle cell proliferation, including ERK1/2 (Lee et al., 2001) and phosphoinositide 3-kinase/PKB (Akt) (Ibe et al., 2006; Ma et al., 2011). Until now, molecular interactions between the cAMP effectors PKA and Epac have been studied in great detail in non-pulmonary systems pointing to compartmentalization of both cAMP effectors via muscle-specific mAKAP (Dodge-Kafka et al., 2005), via β2-adrenoceptor-associated AKAP5 (Nijholt et al., 2008) and via the cytoskeletal scaffolding AKAP11 complex (Logue et al., 2011b). Interestingly, AKAP11 was found to be expressed in ASM using real-time PCR (Horvat et al., 2012). AKAP11 is not only able to bind PKA, but also GSK3, a kinase shown to be involved in expression of contractile proteins in airway smooth muscle (Oenema et al., 2012), their proliferation (Gosens et al., 2007; Nunes et al., 2008) and profibrotic signalling (Baarsma et al., 2011). Thus, AKAP11-driven cAMP compartmentalization may regulate airway smooth muscle remodelling.
In summary, several lines of evidence point towards the logical conclusion that AKAP family members are most likely of key importance for cAMP compartmentalization and thereby signalling to maintain a fine-tuned control over structural lung cell responses. Future studies will surely add additional insights into our current knowledge of signal compartmentalization and perhaps cross-talk between calcium and cAMP in the lung.
Outlook and future perspectives
Compartmentalization of cAMP by AKAP family members represents a highly specialized and dynamic process to fine-tune intracellular signalling. Disturbance of cAMP compartmentalization, either due to alterations in AKAP expression or complex composition with a variety of tools outlined herein, seems to profoundly regulate biological functions and thereby to contribute to neurodegenerative and obstructive lung diseases.
Ageing of the worldwide population will require further improvement of the management of chronic diseases. Notably, cAMP and its effectors seem to be critical in regulating several processes both in chronic brain and lung diseases. Next to PKA, Epac seems to act as a novel pharmacological target in both groups of diseases; however, the impact of Epac compared with PKA might be diverse and sometimes even conflicting. Members of the AKAP superfamily maintain cellular compartmentalization of cAMP primarily via direct interaction with PKA, a process now also linked to Epac. As AKAP-bearing multiprotein complexes regulate receptor desensitization and are able to target simultaneously cAMP and calcium, the AKAP superfamily most likely represent an interesting novel pharmacological concept. In COPD, targeting calcium-mediated bronchoconstriction and cAMP-mediated bronchorelaxation by one AKAP-related drug might give an additional benefit above the current combination therapy with anticholinergics and β2-agonists (Karner and Cates, 2012).
As outlined herein, the design of small molecule inhibitors seems to represent one of the most recent key findings in the field of AKAP research (Christian et al., 2011; Schafer et al., 2013). The AKAP–PKA interaction has also been used as a template for drug design based on the ‘Dock-and-Lock method’ (Rossi et al., 2012a,b,). Here, a trivalent drug is created upon conjugation of two identical (pro-) drugs [e.g. IFN-α 2b (Rossi et al., 2013)] to the PKA-RII dimer and another drug-(targeting) antibody to an AKAP peptide derived from the amphipathic helix (such as AKAP-IS) (Rossi et al., 2012a,b; 2013,,), a process being stabilized by cysteine residues allowing covalent ‘locking’ of the subunits via disulphide bridges. In theory, it should be possible to combine any RII module with any AKAP module (Rossi et al., 2012a,b,), the benefit of this method most likely should be envisaged for the creation of a diverse set of potential pharmacological drugs.
Within the AKAP research field, pharmacological tools focus on PKA–AKAP interactions and disruption of other interaction partners from the AKAP complexes. Until now, however, no reports focus on the disruption of AKAP-Epac complexes. Even though an increasing amount of evidence indicates that Epac interacts with AKAPs, and other scaffolds independently of PKA (Schmidt et al., 2013). These Rap-GEF interacting proteins might add another dimension to the concept of subcellular compartmentalization of cAMP, in particular in the context of the physiology and pathophysiology of biological functions.
Acknowledgments
W. J. P. was supported by a grant from the Dutch Lung Foundation (3.2.11.015). P. M.-L. was a recipient of an Abel Tasman Talent Program Fellowship from the University of Groningen. C. G.-B. was supported by FONDECYT International, Chile. M. S. was supported by a Rosalind Franklin Fellowship from the University of Groningen and a grant from the Deutsche Forschungsgemeinschaft (IRTG1874/1).
Glossary
- AKAP
A-kinase anchoring protein
- AKIP
A-kinase interacting protein
- APP
amyloid precursor protein
- CREB
cAMP response element-binding protein
- CSE
cigarette smoke extract
- Epac
exchange factor directly activated by cAMP
- GSKIP
GSK3β interaction protein
- LTD
long-term depression
- MAGUK
membrane-associated guanylate kinase
- MAP2
microtubule-associated protein 2
- MLC
myosin light chain
- PKI
PK inhibitor
- PP2B/CaN
phosphatase 2B/calcineurin (also PPP3)
- PSD
postsynaptic density
- RhoGDI
Rho guanine-nucleotide-dissociation inhibitor
- RI
regulatory subunit I of PKA
- RII
regulatory subunit II of PKA
- SKIP
sphingosine kinase interacting protein
Author contributions
W. J. P., P. M.-L., C. G.-B. and M.S. were responsible for the conception and design of the review. W. J. P. prepared the figures and tables. W. J. P. and M.S. edited and revised the manuscript. M. S. approved the final version of the manuscript.
Conflict of interest
None.
References
- Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;11:180–187. doi: 10.1016/s0959-4388(00)00194-x. [DOI] [PubMed] [Google Scholar]
- Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams TW, Castellucci VF, Camardo JS, Kandel ER, Lloyd PE. Two endogenous neuropeptides modulate the gill and siphon withdrawal reflex in Aplysia by presynaptic facilitation involving cAMP-dependent closure of a serotonin-sensitive potassium channel. Proc Natl Acad Sci U S A. 1984;81:7956–7960. doi: 10.1073/pnas.81.24.7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilaniu B. Impact of bronchodilator therapy on exercise tolerance in COPD. Int J Chron Obstruct Pulmon Dis. 2010;5:57–71. doi: 10.2147/copd.s7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akakura S, Gelman IH. Pivotal role of AKAP12 in the regulation of cellular adhesion dynamics: control of cytoskeletal architecture, cell migration, and mitogenic signaling. J Signal Transduct. 2012;2012:529179. doi: 10.1155/2012/529179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akakura S, Huang C, Nelson PJ, Foster B, Gelman IH. Loss of the SSeCKS/Gravin/AKAP12 gene results in prostatic hyperplasia. Cancer Res. 2008;68:5096–5103. doi: 10.1158/0008-5472.CAN-07-5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand –gated ion channels. Br J Pharmacol. 2013b;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013c;170:1607–1675. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–721. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, et al. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci U S A. 2003;100:4445–4450. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SS, Bi GQ. Axon formation: a molecular model for the generation of neuronal polarity. Bioessays. 2000;22:172–179. doi: 10.1002/(SICI)1521-1878(200002)22:2<172::AID-BIES8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Ramos BP, Birnbaum SG, Taylor JR. Protein kinase A as a therapeutic target for memory disorders: rationale and challenges. Trends Mol Med. 2005;11:121–128. doi: 10.1016/j.molmed.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Baarsma HA, Menzen MH, Halayko AJ, Meurs H, Kerstjens HA, Gosens R. beta-Catenin signaling is required for TGF-beta1-induced extracellular matrix production by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011;301:L956–L965. doi: 10.1152/ajplung.00123.2011. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baisamy L, Jurisch N, Diviani D. Leucine zipper-mediated homo-oligomerization regulates the Rho-GEF activity of AKAP-Lbc. J Biol Chem. 2005;280:15405–15412. doi: 10.1074/jbc.M414440200. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Biochemical basis of asthma therapy. J Biol Chem. 2011;286:32899–32905. doi: 10.1074/jbc.R110.206466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman AL, Goehring AS, Scott JD. Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology. 2004;46:299–310. doi: 10.1016/j.neuropharm.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schluter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M. Cyclic nucleotide-regulated cation channels. J Biol Chem. 2009;284:9017–9021. doi: 10.1074/jbc.R800075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington CK, Penn RB. Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res. 2003;4:2. [PMC free article] [PubMed] [Google Scholar]
- Billington CK, Ojo OO, Penn RB, Ito S. cAMP regulation of airway smooth muscle function. Pulm Pharmacol Ther. 2013;26:112–120. doi: 10.1016/j.pupt.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Burgers PP, Ma Y, Margarucci L, Mackey M, van der Heyden MA, Ellisman M, et al. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J Biol Chem. 2012;287:43789–43797. doi: 10.1074/jbc.M112.395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, et al. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci U S A. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton IL, Brunton LL. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J Biol Chem. 1983;258:10233–10239. [PubMed] [Google Scholar]
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374:685–694. doi: 10.1016/S0140-6736(09)61255-1. [DOI] [PubMed] [Google Scholar]
- Cant SH, Pitcher JA. G protein-coupled receptor kinase 2-mediated phosphorylation of ezrin is required for G protein-coupled receptor-dependent reorganization of the actin cytoskeleton. Mol Biol Cell. 2005;16:3088–3099. doi: 10.1091/mbc.E04-10-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CR, Lygren B, Berge T, Hoshi N, Wong W, Tasken K, et al. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J Biol Chem. 2006;281:21535–21545. doi: 10.1074/jbc.M603223200. [DOI] [PubMed] [Google Scholar]
- Castellucci VF, Kandel ER, Schwartz JH, Wilson FD, Nairn AC, Greengard P. Intracellular injection of the catalytic subunit of cyclic AMP-dependent protein kinase simulates facilitation of transmitter release underlying behavioral sensitization in Aplysia. Proc Natl Acad Sci U S A. 1980;77:7492–7496. doi: 10.1073/pnas.77.12.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheepala S, Hulot JS, Morgan JA, Sassi Y, Zhang W, Naren AP, et al. Cyclic nucleotide compartmentalization: contributions of phosphodiesterases and ATP-binding cassette transporters. Annu Rev Pharmacol Toxicol. 2013;53:231–253. doi: 10.1146/annurev-pharmtox-010611-134609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wild C, Zhou X, Ye N, Cheng X, Zhou J. Recent advances in the discovery of small molecules targeting exchange proteins directly activated by cAMP (EPAC) J Med Chem. 2013;57:3651–3665. doi: 10.1021/jm401425e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Malbon CC. G-protein-coupled receptor-associated A-kinase anchoring proteins AKAP5 and AKAP12: differential trafficking and distribution. Cell Signal. 2009;21:136–142. doi: 10.1016/j.cellsig.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011;69:231–243. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YF, Wang C, Lin HB, Li YF, Huang Y, Xu JP, et al. Inhibition of phosphodiesterase-4 reverses memory deficits produced by Aβ25–35 or Aβ1–40 peptide in rats. Psychopharmacology (Berl) 2010;212:181–191. doi: 10.1007/s00213-010-1943-3. [DOI] [PubMed] [Google Scholar]
- Chesik D, Wilczak N, De Keyser J. IGF-1 regulates cAMP levels in astrocytes through a beta2-adrenergic receptor-dependant mechanism. Int J Med Sci. 2008;5:240–243. doi: 10.7150/ijms.5.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriva-Internati M, Ferrari R, Yu Y, Hamrick C, Gagliano N, Grizzi F, et al. AKAP-4: a novel cancer testis antigen for multiple myeloma. Br J Haematol. 2008;140:465–468. doi: 10.1111/j.1365-2141.2007.06940.x. [DOI] [PubMed] [Google Scholar]
- Choi MC, Jong HS, Kim TY, Song SH, Lee DS, Lee JW, et al. AKAP12/Gravin is inactivated by epigenetic mechanism in human gastric carcinoma and shows growth suppressor activity. Oncogene. 2004;23:7095–7103. doi: 10.1038/sj.onc.1207932. [DOI] [PubMed] [Google Scholar]
- Chou HY, Howng SL, Cheng TS, Hsiao YL, Lieu AS, Loh JK, et al. GSKIP is homologous to the Axin GSK3beta interaction domain and functions as a negative regulator of GSK3beta. Biochemistry. 2006;45:11379–11389. doi: 10.1021/bi061147r. [DOI] [PubMed] [Google Scholar]
- Christian F, Szaszak M, Friedl S, Drewianka S, Lorenz D, Goncalves A, et al. Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J Biol Chem. 2011;286:9079–9096. doi: 10.1074/jbc.M110.160614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, et al. Association of protein-kinase-a and protein-phosphatase-2b with a common anchoring protein. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Cong M, Perry SJ, Lin FT, Fraser ID, Hu LA, Chen W, et al. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J Biol Chem. 2001;276:15192–15199. doi: 10.1074/jbc.M009130200. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Sugden PH, Lincoln TM, Keely SL. Compartmentalization of adenosine 3′:5′-monophosphate and adenosine 3′:5′-monophosphate-dependent protein kinase in heart tissue. J Biol Chem. 1977;252:3854–3861. [PubMed] [Google Scholar]
- Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, et al. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J Biol Chem. 2012;287:44192–44202. doi: 10.1074/jbc.M112.422956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Damera G, Panettieri RA., Jr Does airway smooth muscle express an inflammatory phenotype in asthma? Br J Pharmacol. 2011;163:68–80. doi: 10.1111/j.1476-5381.2010.01165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, et al. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- Dekkers BG, Racke K, Schmidt M. Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacol Ther. 2013;137:248–265. doi: 10.1016/j.pharmthera.2012.10.006. [DOI] [PubMed] [Google Scholar]
- DeMarch Z, Giampa C, Patassini S, Martorana A, Bernardi G, Fusco FR. Beneficial effects of rolipram in a quinolinic acid model of striatal excitotoxicity. Neurobiol Dis. 2007;25:266–273. doi: 10.1016/j.nbd.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Depry C, Allen MD, Zhang J. Visualization of PKA activity in plasma membrane microdomains. Mol Biosyst. 2011;7:52–58. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Penn RB. Targeting G protein-coupled receptor signaling in asthma. Cell Signal. 2006;18:2105–2120. doi: 10.1016/j.cellsig.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Dessauer CW. Adenylyl cyclase-A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey BF, Walker JK, Hanania NA, Bond RA. beta-Adrenoceptor inverse agonists in asthma. Curr Opin Pharmacol. 2010;10:254–259. doi: 10.1016/j.coph.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, et al. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, et al. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Bauman A, Kapiloff MS. A-kinase anchoring proteins as the basis for cAMP signaling. Handb Exp Pharmacol. 2008;186:3–14. doi: 10.1007/978-3-540-72843-6_1. [DOI] [PubMed] [Google Scholar]
- Donohue JF, Hanania NA, Sciarappa KA, Goodwin E, Grogan DR, Baumgartner RA, et al. Arformoterol and salmeterol in the treatment of chronic obstructive pulmonary disease: a one year evaluation of safety and tolerance. Ther Adv Respir Dis. 2008;2:37–48. doi: 10.1177/1753465808089455. [DOI] [PubMed] [Google Scholar]
- Faux MC, Scott JD. Regulation of the AKAP79-protein kinase C interaction by Ca2+/calmodulin. J Biol Chem. 1997;272:17038–17044. doi: 10.1074/jbc.272.27.17038. [DOI] [PubMed] [Google Scholar]
- Feinstein WP, Zhu B, Leavesley SJ, Sayner SL, Rich TC. Assessment of cellular mechanisms contributing to cAMP compartmentalization in pulmonary microvascular endothelial cells. Am J Physiol Cell Physiol. 2012;302:C839–C852. doi: 10.1152/ajpcell.00361.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Kelly A, Grizzi F, Yuefei Y, Cobos E, Chiriva-Internati M. Is AKAP-4 a novel cancer testis antigen for multiple myeloma? J Invest Med. 2007;55:S266. doi: 10.1111/j.1365-2141.2007.06940.x. [DOI] [PubMed] [Google Scholar]
- Frank B, Wiestler M, Kropp S, Hemminki K, Spurdle AB, Sutter C, et al. Association of a common AKAP9 variant with breast cancer risk: a collaborative analysis. J Natl Cancer Inst. 2008;100:437–442. doi: 10.1093/jnci/djn037. [DOI] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, et al. Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol. 2000;10:409–412. doi: 10.1016/s0960-9822(00)00419-x. [DOI] [PubMed] [Google Scholar]
- Gao N, Asamitsu K, Hibi Y, Ueno T, Okamoto T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J Biol Chem. 2008;283:7834–7843. doi: 10.1074/jbc.M710285200. [DOI] [PubMed] [Google Scholar]
- Gao N, Hibi Y, Cueno M, Asamitsu K, Okamoto T. A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J Biol Chem. 2010;285:28097–28104. doi: 10.1074/jbc.M110.116566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Wang HY, Malbon CC. AKAP12 and AKAP5 form higher-order hetero-oligomers. J Mol Signal. 2011a;6:8. doi: 10.1186/1750-2187-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Wang HY, Malbon CC. AKAP5 and AKAP12 form homo-oligomers. J Mol Signal. 2011b;6:3. doi: 10.1186/1750-2187-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Banko JL, Peters MM, Klann E, Weeber EJ, Nguyen PV. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learn Mem. 2008;15:403–411. doi: 10.1101/lm.830008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman IH. Emerging roles for SSeCKS/Gravin/AKAP12 in the control of cell proliferation, cancer malignancy, and barriergenesis. Genes Cancer. 2010;1:1147–1156. doi: 10.1177/1947601910392984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampa C, Patassini S, Borreca A, Laurenti D, Marullo F, Bernardi G, et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington's disease. Neurobiol Dis. 2009;34:450–456. doi: 10.1016/j.nbd.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS ONE. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giembycz MA, Newton R. Beyond the dogma: novel beta2-adrenoceptor signalling in the airways. Eur Respir J. 2006;27:1286–1306. doi: 10.1183/09031936.06.00112605. [DOI] [PubMed] [Google Scholar]
- Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum Mol Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- Glantz SB, Amat JA, Rubin CS. cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell. 1992;3:1215–1228. doi: 10.1091/mbc.3.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for diagnosis, management, and prevention of COPD (December 2010). Available at: http://www.goldcopd.org/uploads/users/files/GOLDReport_April112011.pdf (accessed 07/14/2014)
- Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, et al. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2010;24:383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Gold MG, Stengel F, Nygren PJ, Weisbrod CR, Bruce JE, Robinson CV, et al. Architecture and dynamics of an A-kinase anchoring protein 79 (AKAP79) signaling complex. Proc Natl Acad Sci U S A. 2011;108:6426–6431. doi: 10.1073/pnas.1014400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Fowler DM, Means CK, Pawson CT, Stephany JJ, Langeberg LK, et al. Engineering AKAP-selective regulatory subunits of PKA through structure-based phage selection. J Biol Chem. 2013;288:17111–17121. doi: 10.1074/jbc.M112.447326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML. Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell. 2005a;16:3574–3590. doi: 10.1091/mbc.E05-02-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML. Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell. 2005b;16:3574–3590. doi: 10.1091/mbc.E05-02-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens R, Dueck G, Rector E, Nunes RO, Gerthoffer WT, Unruh H, et al. Cooperative regulation of GSK-3 by muscarinic and PDGF receptors is associated with airway myocyte proliferation. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1348–L1358. doi: 10.1152/ajplung.00346.2007. [DOI] [PubMed] [Google Scholar]
- Gosens R, Mutawe M, Martin S, Basu S, Bos ST, Tran T, et al. Caveolae and caveolins in the respiratory system. Curr Mol Med. 2008;8:741–753. doi: 10.2174/156652408786733720. [DOI] [PubMed] [Google Scholar]
- Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra S. Asthma and chronic obstructive pulmonary disease. Curr Opin Allergy Clin Immunol. 2009;9:409–416. doi: 10.1097/ACI.0b013e3283300baf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halayko AJ, Tran T, Gosens R. Phenotype and functional plasticity of airway smooth muscle: role of caveolae and caveolins. Proc Am Thorac Soc. 2008;5:80–88. doi: 10.1513/pats.200705-057VS. [DOI] [PubMed] [Google Scholar]
- Halliday G, Robinson SR, Shepherd C, Kril J. Alzheimer's disease and inflammation: a review of cellular and therapeutic mechanisms. Clin Exp Pharmacol Physiol. 2000;27:1–8. doi: 10.1046/j.1440-1681.2000.03200.x. [DOI] [PubMed] [Google Scholar]
- Hallsworth MP, Twort CH, Lee TH, Hirst SJ. Beta(2)-adrenoceptor agonists inhibit release of eosinophil-activating cytokines from human airway smooth muscle cells. Br J Pharmacol. 2001;132:729–741. doi: 10.1038/sj.bjp.0703866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanania NA, Donohue JF, Nelson H, Sciarappa K, Goodwin E, Baumgartner RA, et al. The safety and efficacy of arformoterol and formoterol in COPD. COPD. 2010;7:17–31. doi: 10.3109/15412550903499498. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Hanzal-Bayer MF, Hancock JF. Lipid rafts and membrane traffic. FEBS Lett. 2007;581:2098–2104. doi: 10.1016/j.febslet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Hara M, Fukui R, Hieda E, Kuroiwa M, Bateup HS, Kano T, et al. Role of adrenoceptors in the regulation of dopamine/DARPP-32 signaling in neostriatal neurons. J Neurochem. 2010;113:1046–1059. doi: 10.1111/j.1471-4159.2010.06668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Ono T, Matsushita H, Shimono M, Noguchi Y, Mizutani Y, et al. A-kinase anchoring protein 3 messenger RNA expression in ovarian cancer and its implication on prognosis. Int J Cancer. 2004;108:86–90. doi: 10.1002/ijc.11565. [DOI] [PubMed] [Google Scholar]
- Hayes JS, Brunton LL, Brown JH, Reese JB, Mayer SE. Hormonally specific expression of cardiac protein kinase activity. Proc Natl Acad Sci U S A. 1979;76:1570–1574. doi: 10.1073/pnas.76.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JS, Brunton LL, Mayer SE. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J Biol Chem. 1980;255:5113–5119. [PubMed] [Google Scholar]
- Hewer RC, Sala-Newby GB, Wu YJ, Newby AC, Bond M. PKA and Epac synergistically inhibit smooth muscle cell proliferation. J Mol Cell Cardiol. 2011;50:87–98. doi: 10.1016/j.yjmcc.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Baker JG. The ups and downs of Gs- to Gi-protein switching. Br J Pharmacol. 2003;138:1188–1189. doi: 10.1038/sj.bjp.0705192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvat SJ, Deshpande DA, Yan H, Panettieri RA, Codina J, DuBose TD, Jr, et al. A-kinase anchoring proteins regulate compartmentalized cAMP signaling in airway smooth muscle. FASEB J. 2012;26:3670–3679. doi: 10.1096/fj.11-201020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Hundsrucker C, Klussmann E. Direct AKAP-mediated protein-protein interactions as potential drug targets. Handb Exp Pharmacol. 2008;186:483–503. doi: 10.1007/978-3-540-72843-6_20. [DOI] [PubMed] [Google Scholar]
- Hurst JR, Vestbo J, Anzueto A, Locantore N, Mullerova H, Tal-Singer R, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138. doi: 10.1056/NEJMoa0909883. [DOI] [PubMed] [Google Scholar]
- Hutchins BI. Competitive outgrowth of neural processes arising from long-distance cAMP signaling. Sci Signal. 2010;3:jc1. doi: 10.1126/scisignal.3118jc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibe BO, Portugal AM, Raj JU. Levalbuterol inhibits human airway smooth muscle cell proliferation: therapeutic implications in the management of asthma. Int Arch Allergy Immunol. 2006;139:225–236. doi: 10.1159/000091168. [DOI] [PubMed] [Google Scholar]
- Insel PA, Ostrom RS. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol. 2003;23:305–314. doi: 10.1023/A:1023684503883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer's disease. Brain Behav Immun. 2009;23:507–517. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, et al. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer's disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Hamilton JP, Yang J, Mori Y, Olaru A, Sato F, et al. Hypermethylation of the AKAP12 promoter is a biomarker of Barrett's-associated esophageal neoplastic progression. Cancer Epidemiol Biomarkers Prev. 2008;17:111–117. doi: 10.1158/1055-9965.EPI-07-0407. [DOI] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–1055. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M, Balaha M, Inagaki N, Kitade Y. Current and future asthma therapies. Drugs Today (Barc) 2013;49:325–339. doi: 10.1358/dot.2013.49.5.1950149. [DOI] [PubMed] [Google Scholar]
- Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, et al. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem. 2009;284:23540–23546. doi: 10.1074/jbc.M109.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner C, Cates CJ. Long-acting beta(2)-agonist in addition to tiotropium versus either tiotropium or long-acting beta(2)-agonist alone for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012;(4) doi: 10.1002/14651858.CD008989.pub2. CD008989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel KM, Wyatt TA, Panettieri RA, Jr, Toews ML. Inhibition of human airway smooth muscle cell proliferation by beta 2-adrenergic receptors and cAMP is PKA independent: evidence for EPAC involvement. Am J Physiol Lung Cell Mol Physiol. 2008;294:L131–L138. doi: 10.1152/ajplung.00381.2007. [DOI] [PubMed] [Google Scholar]
- Kaur M, Holden NS, Wilson SM, Sukkar MB, Chung KF, Barnes PJ, et al. Effect of beta2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. Am J Physiol Lung Cell Mol Physiol. 2008;295:L505–L514. doi: 10.1152/ajplung.00046.2008. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kelly MP, Stein JM, Vecsey CG, Favilla C, Yang X, Bizily SF, et al. Developmental etiology for neuroanatomical and cognitive deficits in mice overexpressing Galphas, a G-protein subunit genetically linked to schizophrenia. Mol Psychiatry. 2009;14:398–415. doi: 10.1038/mp.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol. 2012;165:1288–1305. doi: 10.1111/j.1476-5381.2011.01729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, et al. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CC, Sastri M, Chang P, Pennypacker J, Taylor SS. The rate of NF-kappaB nuclear translocation is regulated by PKA and A kinase interacting protein 1. PLoS ONE. 2011;6:e18713. doi: 10.1371/journal.pone.0018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliber A, Lynd LD, Sin DD. The effects of long-acting bronchodilators on total mortality in patients with stable chronic obstructive pulmonary disease. Respir Res. 2010;11:56. doi: 10.1186/1465-9921-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klussmann E, Maric K, Wiesner B, Beyermann M, Rosenthal W. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J Biol Chem. 1999;274:4934–4938. doi: 10.1074/jbc.274.8.4934. [DOI] [PubMed] [Google Scholar]
- Kocer SS, Wang HY, Malbon CC. ‘Shaping’ of cell signaling via AKAP-tethered PDE4D: probing with AKAR2-AKAP5 biosensor. J Mol Signal. 2012;7:4. doi: 10.1186/1750-2187-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovanich D, van der Heyden MA, Aye TT, van Veen TA, Heck AJ, Scholten A. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. Chembiochem. 2010;11:963–971. doi: 10.1002/cbic.201000058. [DOI] [PubMed] [Google Scholar]
- Lee JH, Johnson PR, Roth M, Hunt NH, Black JL. ERK activation and mitogenesis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1019–L1029. doi: 10.1152/ajplung.2001.280.5.L1019. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Liesker JJ, Wijkstra PJ, Hacken NHT, Koeter GH, Postma DS, Kerstjens HA. A systematic review of the effects of bronchodilators on exercise capacity in patients with COPD. Chest. 2002;121:597–608. doi: 10.1378/chest.121.2.597. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhang J, Xiang YK. FRET-based direct detection of dynamic protein kinase A activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem Biophys Res Commun. 2011;404:581–586. doi: 10.1016/j.bbrc.2010.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JS, Whiting JL, Scott JD. Sequestering Rac with PKA confers cAMP control of cytoskeletal remodeling. Small GTPases. 2011a;2:173–176. doi: 10.4161/sgtp.2.3.16487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JS, Whiting JL, Tunquist B, Langeberg LK, Scott JD. Anchored protein kinase A recruitment of active Rac GTPase. J Biol Chem. 2011b;286:22113–22121. doi: 10.1074/jbc.M111.232660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JS, Whiting JL, Tunquist B, Sacks DB, Langeberg LK, Wordeman L, et al. AKAP220 protein organizes signaling elements that impact cell migration. J Biol Chem. 2011c;286:39269–39281. doi: 10.1074/jbc.M111.277756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Lu YS, Shuai Y, Feng C, Tully T, Xie Z, et al. The AKAP Yu is required for olfactory long-term memory formation in Drosophila. Proc Natl Acad Sci U S A. 2007;104:13792–13797. doi: 10.1073/pnas.0700439104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zha XM, Kim EY, Schachtele S, Dailey ME, Hall DD, et al. A kinase anchor protein 150 (AKAP150)-associated protein kinase a limits dendritic spine density. J Biol Chem. 2011;286:26496–26506. doi: 10.1074/jbc.M111.254912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygren B, Carlson CR, Santamaria K, Lissandron V, McSorley T, Litzenberg J, et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007;8:1061–1067. doi: 10.1038/sj.embor.7401081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MJ, Baillie GS, Mohamed A, Li X, Maisonneuve C, Klussmann E, et al. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta(2)-adrenergic receptor to activation of ERK in HEK293B2 cells. J Biol Chem. 2005;280:33178–33189. doi: 10.1074/jbc.M414316200. [DOI] [PubMed] [Google Scholar]
- Ma L, Brown M, Kogut P, Serban K, Li X, McConville J, et al. Akt activation induces hypertrophy without contractile phenotypic maturation in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2011;300:L701–L709. doi: 10.1152/ajplung.00119.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N, Abel T, Hernandez PJ. Exchange protein activated by cAMP enhances long-term memory formation independent of protein kinase A. Learn Mem. 2009;16:367–370. doi: 10.1101/lm.1231009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet M, Robert SJ, Cacquevel M, Gastineau M, Vivien D, Bertoglio J, et al. Crosstalk between Rap1 and Rac regulates secretion of sAPPalpha. Nat Cell Biol. 2003;5:633–639. doi: 10.1038/ncb1007. [DOI] [PubMed] [Google Scholar]
- Malbon CC, Tao J, Shumay E, Wang HY. AKAP (A-kinase anchoring protein) domains: beads of structure-function on the necklace of G-protein signalling. Biochem Soc Trans. 2004;32:861–864. doi: 10.1042/BST0320861. [DOI] [PubMed] [Google Scholar]
- Martinez M, Fernandez E, Frank A, Guaza C, de la Fuente M, Hernanz A. Increased cerebrospinal fluid cAMP levels in Alzheimer's disease. Brain Res. 1999;846:265–267. doi: 10.1016/s0006-8993(99)01981-2. [DOI] [PubMed] [Google Scholar]
- McCahill AC, Huston E, Li X, Houslay MD. PDE4 associates with different scaffolding proteins: modulating interactions as treatment for certain diseases. Handb Exp Pharmacol. 2008;186:125–166. doi: 10.1007/978-3-540-72843-6_6. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- McPhee I, Gibson LC, Kewney J, Darroch C, Stevens PA, Spinks D, et al. Cyclic nucleotide signalling: a molecular approach to drug discovery for Alzheimer's disease. Biochem Soc Trans. 2005;33:1330–1332. doi: 10.1042/BST0331330. [DOI] [PubMed] [Google Scholar]
- Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc Natl Acad Sci U S A. 2011;108:E1227–E1235. doi: 10.1073/pnas.1107182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs H, Gosens R, Zaagsma J. Airway hyperresponsiveness in asthma: lessons from in vitro model systems and animal models. Eur Respir J. 2008;32:487–502. doi: 10.1183/09031936.00023608. [DOI] [PubMed] [Google Scholar]
- Meurs H, Oenema TA, Kistemaker LE, Gosens R. A new perspective on muscarinic receptor antagonism in obstructive airways diseases. Curr Opin Pharmacol. 2013;13:316–323. doi: 10.1016/j.coph.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Mizuno N, Itoh H. Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals. 2009;17:42–54. doi: 10.1159/000186689. [DOI] [PubMed] [Google Scholar]
- Moita MA, Lamprecht R, Nader K, LeDoux JE. A-kinase anchoring proteins in amygdala are involved in auditory fear memory. Nat Neurosci. 2002;5:837–838. doi: 10.1038/nn901. [DOI] [PubMed] [Google Scholar]
- Morales-Garcia JA, Redondo M, Alonso-Gil S, Gil C, Perez C, Martinez A, et al. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE. 2011;6:e17240. doi: 10.1371/journal.pone.0017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Cai K, Cheng Y, Wang S, Paun B, Hamilton JP, et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology. 2006;131:797–808. doi: 10.1053/j.gastro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio I, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, et al. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cell Neurosci. 2008;38:578–588. doi: 10.1016/j.mcn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Tucker SJ, Shewan DA. cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci. 2009;29:15434–15444. doi: 10.1523/JNEUROSCI.3071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Miyazaki M, Aoki R, Zama T, Inouye S, Hirose K, et al. A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nat Biotechnol. 2000;18:313–316. doi: 10.1038/73767. [DOI] [PubMed] [Google Scholar]
- Nauert JB, Klauck TM, Langeberg LK, Scott JD. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr Biol. 1997;7:52–62. doi: 10.1016/s0960-9822(06)00027-3. [DOI] [PubMed] [Google Scholar]
- Nijholt I, Ostroveanu A, de Bruyn M, Luiten P, Eisel U, Van der Zee E. Both exposure to a novel context and associative learning induce an upregulation of AKAP150 protein in mouse hippocampus. Neurobiol Learn Mem. 2007;87:693–696. doi: 10.1016/j.nlm.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Nijholt IM, Dolga AM, Ostroveanu A, Luiten PG, Schmidt M, Eisel UL. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell Signal. 2008;20:1715–1724. doi: 10.1016/j.cellsig.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Nunes RO, Schmidt M, Dueck G, Baarsma H, Halayko AJ, Kerstjens HA, et al. GSK-3/beta-catenin signaling axis in airway smooth muscle: role in mitogenic signaling. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1110–L1118. doi: 10.1152/ajplung.00500.2007. [DOI] [PubMed] [Google Scholar]
- Oenema TA, Smit M, Smedinga L, Racke K, Halayko AJ, Meurs H, et al. Muscarinic receptor stimulation augments TGF-beta1-induced contractile protein expression by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2012;303:L589–L597. doi: 10.1152/ajplung.00400.2011. [DOI] [PubMed] [Google Scholar]
- Oldenburger A, Maarsingh H, Schmidt M. Multiple facets of cAMP signalling and physiological impact: cAMP compartmentalization in the lung. Pharmaceuticals (Basel) 2012a;5:1291–1331. doi: 10.3390/ph5121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburger A, Roscioni SS, Jansen E, Menzen MH, Halayko AJ, Timens W, et al. Anti-inflammatory role of the cAMP effectors Epac and PKA: implications in chronic obstructive pulmonary disease. PLoS ONE. 2012b;7:e31574. doi: 10.1371/journal.pone.0031574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburger A, Poppinga WJ, Kos F, de Bruin HG, Rijks WF, Heijink IH, et al. A-kinase anchoring proteins contribute to loss of E-cadherin and bronchial epithelial barrier by cigarette smoke. Am J Physiol Cell Physiol. 2014;306:C585–C597. doi: 10.1152/ajpcell.00183.2013. [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML. Imaging kinase-AKAP79-phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroveanu A, Van der Zee EA, Dolga AM, Luiten PGM, Eisel ULM, Nijholt IM. A-kinase anchoring protein 150 in the mouse brain is concentrated in areas involved in learning and memory. Brain Res. 2007;1145:97–107. doi: 10.1016/j.brainres.2007.01.117. [DOI] [PubMed] [Google Scholar]
- Ostroveanu A, van der Zee E, Eisel U, Schmidt M, Nijholt I. Exchange protein activated by cyclic AMP 2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus. 2010;20:1018–1026. doi: 10.1002/hipo.20700. [DOI] [PubMed] [Google Scholar]
- Ouyang M, Zhang L, Zhu JJ, Schwede F, Thomas SA. Epac signaling is required for hippocampus-dependent memory retrieval. Proc Natl Acad Sci U S A. 2008;105:11993–11997. doi: 10.1073/pnas.0804172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Skoff RB, Singh AK. Combination therapy of lovastatin and rolipram provides neuroprotection and promotes neurorepair in inflammatory demyelination model of multiple sclerosis. Glia. 2009;57:182–193. doi: 10.1002/glia.20745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- Patel HH, Murray F, Insel PA. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb Exp Pharmacol. 2008;186:167–184. doi: 10.1007/978-3-540-72843-6_7. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Torres S, Cortes R, Tolnay M, Probst A, Palacios JM, Mengod G. Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer's disease brains examined by in situ hybridization. Exp Neurol. 2003;182:322–334. doi: 10.1016/s0014-4886(03)00042-6. [DOI] [PubMed] [Google Scholar]
- Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, Ameredes BT, et al. Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med. 2010;363:1715–1726. doi: 10.1056/NEJMoa1008770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux G, Tasken K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J Mol Endocrinol. 2010;44:271–284. doi: 10.1677/JME-10-0010. [DOI] [PubMed] [Google Scholar]
- Pujuguet P, Del Maestro L, Gautreau A, Louvard D, Arpin M. Ezrin regulates E-cadherin-dependent adherens junction assembly through Rac1 activation. Mol Biol Cell. 2003;14:2181–2191. doi: 10.1091/mbc.E02-07-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi HY, Paudel HK. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J Biol Chem. 2011;286:5055–5068. doi: 10.1074/jbc.M110.178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol. 2011;163:53–67. doi: 10.1111/j.1476-5381.2011.01218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Razani B, Rubin CS, Lisanti MP. Regulation of cAMP-mediated signal transduction via interaction of caveolins with the catalytic subunit of protein kinase A. J Biol Chem. 1999;274:26353–26360. doi: 10.1074/jbc.274.37.26353. [DOI] [PubMed] [Google Scholar]
- Rehmann H. Epac-inhibitors: facts and artefacts. Sci Rep. 2013;3:3032. doi: 10.1038/srep03032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert S, Maillet M, Morel E, Launay JM, Fischmeister R, Mercken L, et al. Regulation of the amyloid precursor protein ectodomain shedding by the 5-HT4 receptor and Epac. FEBS Lett. 2005;579:1136–1142. doi: 10.1016/j.febslet.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML. Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J Neurosci. 2009;29:7929–7943. doi: 10.1523/JNEUROSCI.6093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J. The inflammatory response in Alzheimer's disease. J Periodontol. 2008;79:1535–1543. doi: 10.1902/jop.2008.080171. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Roscioni SS, Kistemaker LE, Menzen MH, Elzinga CR, Gosens R, Halayko AJ, et al. PKA and Epac cooperate to augment bradykinin-induced interleukin-8 release from human airway smooth muscle cells. Respir Res. 2009;10:88. doi: 10.1186/1465-9921-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioni SS, Dekkers BG, Prins AG, Menzen MH, Meurs H, Schmidt M, et al. cAMP inhibits modulation of airway smooth muscle phenotype via the exchange protein activated by cAMP (Epac) and protein kinase A. Br J Pharmacol. 2011a;162:193–209. doi: 10.1111/j.1476-5381.2010.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioni SS, Maarsingh H, Elzinga CR, Schuur J, Menzen M, Halayko AJ, et al. Epac as a novel effector of airway smooth muscle relaxation. J Cell Mol Med. 2011b;15:1551–1563. doi: 10.1111/j.1582-4934.2010.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioni SS, Prins AG, Elzinga CR, Menzen MH, Dekkers BG, Halayko AJ, et al. Protein kinase A and the exchange protein directly activated by cAMP (Epac) modulate phenotype plasticity in human airway smooth muscle. Br J Pharmacol. 2011;164:958–969. doi: 10.1111/j.1476-5381.2011.01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S, McGann K, Hauss-Wegrzyniak B, Wenk GL. The influence of brain inflammation upon neuronal adenosine A2B receptors. J Neurochem. 2003;86:220–227. doi: 10.1046/j.1471-4159.2003.01825.x. [DOI] [PubMed] [Google Scholar]
- Rossi EA, Goldenberg DM, Chang CH. Complex and defined biostructures with the dock-and-lock method. Trends Pharmacol Sci. 2012a;33:474–481. doi: 10.1016/j.tips.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Rossi EA, Goldenberg DM, Chang CH. The dock-and-lock method combines recombinant engineering with site-specific covalent conjugation to generate multifunctional structures. Bioconjug Chem. 2012b;23:309–323. doi: 10.1021/bc2004999. [DOI] [PubMed] [Google Scholar]
- Rossi EA, Chang CH, Cardillo TM, Goldenberg DM. Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties. Bioconjug Chem. 2013;24:63–71. doi: 10.1021/bc300488f. [DOI] [PubMed] [Google Scholar]
- Rycroft CE, Heyes A, Lanza L, Becker K. Epidemiology of chronic obstructive pulmonary disease: a literature review. Int J Chron Obstruct Pulmon Dis. 2012;7:457–494. doi: 10.2147/COPD.S32330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Sugimoto N, Ohta K, Shimizu T, Ohtani K, Nakayama Y, et al. Phosphodiesterase inhibitors suppress Lactobacillus casei cell-wall-induced NF-kappaB and MAPK activations and cell proliferation through protein kinase A–or exchange protein activated by cAMP-dependent signal pathway. ScientificWorldJournal. 2012;2012:748572. doi: 10.1100/2012/748572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinthone S, Yadav V, Schillace RV, Bourdette DN, Carr DW. Lipoic acid attenuates inflammation via cAMP and protein kinase A signaling. PLoS ONE. 2010;5:e13058. doi: 10.1371/journal.pone.0013058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, Erlichman J, Rubin CS. Identification of a calmodulin-binding protein that co-purifies with the regulatory subunit of brain protein kinase II. J Biol Chem. 1984;259:9840–9846. [PubMed] [Google Scholar]
- Sastri M, Barraclough DM, Carmichael PT, Taylor SS. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc Natl Acad Sci U S A. 2005;102:349–354. doi: 10.1073/pnas.0408608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastri M, Haushalter KJ, Panneerselvam M, Chang P, Fridolfsson H, Finley JC, et al. A kinase interacting protein (AKIP1) is a key regulator of cardiac stress. Proc Natl Acad Sci U S A. 2013;110:E387–E396. doi: 10.1073/pnas.1221670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Tabunoki H, Arima K. Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis Markers. 2009;27:239–252. doi: 10.3233/DMA-2009-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer G, Milic J, Eldahshan A, Gotz F, Zuhlke K, Schillinger C, et al. Highly functionalized terpyridines as competitive inhibitors of AKAP-PKA interactions. Angew Chem Int Ed Engl. 2013;52:12187–12191. doi: 10.1002/anie.201304686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65:670–709. doi: 10.1124/pr.110.003707. [DOI] [PubMed] [Google Scholar]
- Schott M, Grove B. Receptor-mediated Ca and PKC signaling triggers the loss of cortical PKA compartmentalization through the redistribution of gravin. Cell Signal. 2013;25:2125–2135. doi: 10.1016/j.cellsig.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutsky K, Ouyang M, Thomas S. Xamoterol impairs hippocampus-dependent emotional memory retrieval via Gi/o-coupled β2-adrenergic signaling. Learn Mem. 2011;18:598–604. doi: 10.1101/lm.2302811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JD, Dessauer CW, Tasken K. Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol. 2012;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehrawat S, Ernandez T, Cullere X, Takahashi M, Ono Y, Komarova Y, et al. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties. Blood. 2011;117:708–718. doi: 10.1182/blood-2010-02-268870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Mahler DA, Marcus P, Owen CA, Yawn B, Rennard S. Inflammation in COPD: implications for management. Am J Med. 2012;125:1162–1170. doi: 10.1016/j.amjmed.2012.06.024. [DOI] [PubMed] [Google Scholar]
- Sharma S, Qian F, Keitz B, Driscoll D, Scanlan MJ, Skipper J, et al. A-kinase anchoring protein 3 messenger RNA expression correlates with poor prognosis in epithelial ovarian cancer. Gynecol Oncol. 2005;99:183–188. doi: 10.1016/j.ygyno.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science. 2010;327:547–552. doi: 10.1126/science.1179735. [DOI] [PubMed] [Google Scholar]
- Shih M, Lin F, Scott JD, Wang HY, Malbon CC. Dynamic complexes of beta2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J Biol Chem. 1999;274:1588–1595. doi: 10.1074/jbc.274.3.1588. [DOI] [PubMed] [Google Scholar]
- Sin YY, Edwards HV, Li X, Day JP, Christian F, Dunlop AJ, et al. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the beta-agonist induced hypertrophic response in cardiac myocytes. J Mol Cell Cardiol. 2011;50:872–883. doi: 10.1016/j.yjmcc.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E. Mechanisms of protein kinase a anchoring. Int Rev Cell Mol Biol. 2010;283:235–330. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- Smith FD, Langeberg LK, Scott JD. The where's and when's of kinase anchoring. Trends Biochem Sci. 2006a;31:316–323. doi: 10.1016/j.tibs.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Smith FD, Langeberg LK, Scott JD. Plugging PKA into ERK scaffolds. Cell Cycle. 2011;10:731–732. doi: 10.4161/cc.10.5.14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006b;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006c;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sonkusare SK, Kaul CL, Ramarao P. Dementia of Alzheimer's disease and other neurodegenerative disorders–memantine, a new hope. Pharmacol Res. 2005;51:1–17. doi: 10.1016/j.phrs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D, Jones K, Woolfrey K, Burgdorf J, Russell T, Kalmbach A, et al. Social, communication, and cortical structural impairments in Epac2-deficient mice. J Neurosci. 2012;32:11864–11878. doi: 10.1523/JNEUROSCI.1349-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokka AJ, Mosenden R, Ruppelt A, Lygren B, Tasken K. The adaptor protein EBP50 is important for localization of the protein kinase A-Ezrin complex in T-cells and the immunomodulating effect of cAMP. Biochem J. 2009;425:381–388. doi: 10.1042/BJ20091136. [DOI] [PubMed] [Google Scholar]
- Su B, Bu Y, Engelberg D, Gelman IH. SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a protein kinase C- Raf/MEK/ERK pathway. J Biol Chem. 2010;285:4578–4586. doi: 10.1074/jbc.M109.073494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J Biol Chem. 2004;279:4988–4999. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- Tao J, Malbon CC. G-protein-coupled receptor-associated A-kinase anchoring proteins AKAP5 and AKAP12: differential signaling to MAPK and GPCR recycling. J Mol Signal. 2008;3:19. doi: 10.1186/1750-2187-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wang HY, Malbon CC. Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2-adrenergic receptor. EMBO J. 2003;22:6419–6429. doi: 10.1093/emboj/cdg628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wang HY, Malbon CC. AKAR2-AKAP12 fusion protein ‘biosenses’ dynamic phosphorylation and localization of a GPCR-based scaffold. J Mol Signal. 2010;5:3. doi: 10.1186/1750-2187-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84:137–167. doi: 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: lessons learned after twenty years. Biochim Biophys Acta. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurkauf WE, Vallee RB. Molecular characterization of the cAMP-dependent protein kinase bound to microtubule-associated protein 2. J Biol Chem. 1982;257:3284–3290. [PubMed] [Google Scholar]
- Troger J, Moutty MC, Skroblin P, Klussmann E. A-kinase anchoring proteins as potential drug targets. Br J Pharmacol. 2012;166:420–433. doi: 10.1111/j.1476-5381.2011.01796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Cheng X. A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic amp (epac) antagonists. PLoS ONE. 2012a;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, et al. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A. 2012b;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, et al. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfig N, Setzer M. Expression of a kinase anchoring protein 79 in the human fetal amygdala. Microsc Res Tech. 1999;46:48–52. doi: 10.1002/(SICI)1097-0029(19990701)46:1<48::AID-JEMT4>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmeier C, Hederer B, Glaab T, Schmidt H, Rutten-van Molken MP, Beeh KM, et al. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med. 2011;364:1093–1103. doi: 10.1056/NEJMoa1008378. [DOI] [PubMed] [Google Scholar]
- Walker JK, Penn RB, Hanania NA, Dickey BF, Bond RA. New perspectives regarding beta(2)-adrenoceptor ligands in the treatment of asthma. Br J Pharmacol. 2011;163:18–28. doi: 10.1111/j.1476-5381.2010.01178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yang XM, Zhuo YY, Zhou H, Lin HB, Cheng YF, et al. The phosphodiesterase-4 inhibitor rolipram reverses Aβ-induced cognitive impairment and neuroinflammatory and apoptotic responses in rats. Int J Neuropsychopharmacol. 2012;15:749–766. doi: 10.1017/S1461145711000836. [DOI] [PubMed] [Google Scholar]
- Wang HY, Tao J, Shumay E, Malbon CC. G-protein-coupled receptor-associated A-kinase anchoring proteins: AKAP79 and AKAP250 (gravin) Eur J Cell Biol. 2006;85:643–650. doi: 10.1016/j.ejcb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, et al. Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS ONE. 2010;5:e10325. doi: 10.1371/journal.pone.0010325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol Interv. 2010;10:86–97. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikman H, Kettunen E, Seppanen JK, Karjalainen A, Hollmen J, Anttila S, et al. Identification of differentially expressed genes in pulmonary adenocarcinoma by using cDNA array. Oncogene. 2002;21:5804–5813. doi: 10.1038/sj.onc.1205726. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, Everett KL, et al. A-kinase anchoring protein 79/150 interacts with adenylyl cyclase type 8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J Biol Chem. 2010;285:20328–20342. doi: 10.1074/jbc.M110.120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Sasaki M, Ozawa H, Saito T, Rosler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- Yan H, Deshpande DA, Misior AM, Miles MC, Saxena H, Riemer EC, et al. Anti-mitogenic effects of beta-agonists and PGE2 on airway smooth muscle are PKA dependent. FASEB J. 2011;25:389–397. doi: 10.1096/fj.10-164798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calingasan NY, Lorenzo BJ, Beal MF. Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp Neurol. 2008;211:311–314. doi: 10.1016/j.expneurol.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shu X, Liu D, Shang Y, Wu Y, Pei L, et al. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron. 2012;73:774–788. doi: 10.1016/j.neuron.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaldua N, Gastineau M, Hoshino M, Lezoualc'h F, Zugaza JL. Epac signaling pathway involves STEF, a guanine nucleotide exchange factor for Rac, to regulate APP processing. FEBS Lett. 2007;581:5814–5818. doi: 10.1016/j.febslet.2007.11.053. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A. 2001;98:14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437:569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- Zieba BJ, Artamonov MV, Jin L, Momotani K, Ho R, Franke AS, et al. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem. 2011;286:16681–16692. doi: 10.1074/jbc.M110.205062. [DOI] [PMC free article] [PubMed] [Google Scholar]