

Abstract
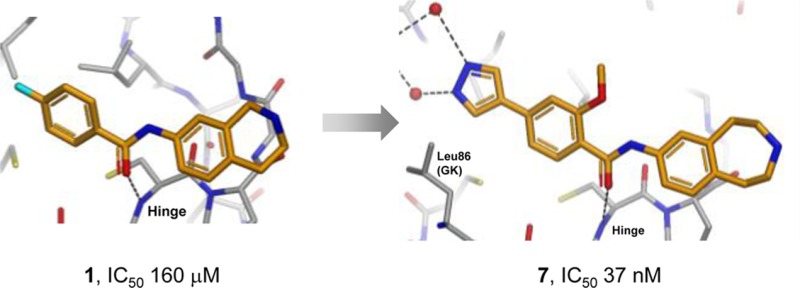
Fragment-based drug design was successfully applied to maternal embryonic leucine zipper kinase (MELK). A low affinity (160 μM) fragment hit was identified, which bound to the hinge region with an atypical binding mode, and this was optimized using structure-based design into a low-nanomolar and cell-penetrant inhibitor, with a good selectivity profile, suitable for use as a chemical probe for elucidation of MELK biology.
Keywords: Maternal embryonic leucine zipper kinase, fragment-based drug design, structure-based optimization
Maternal embryonic leucine zipper kinase (MELK) is a serine/threonine kinase belonging to the CAMK family of kinases.1 MELK is structurally related to AMPK, SNARK, and NUAK, which are implicated in metabolism2 but, in contrast to these, is not regulated by LKB1.3 Early studies suggested that MELK might be a promising target for the treatment of several cancers.4 Increased MELK expression has been reported in brain, breast, prostate, melanoma, and colorectal carcinomas, and is correlated with poor prognosis.5−10 Normal adult cells have low expression of MELK, offering promise for selective treatment, rendering the assessment of the role of MELK in carcinogenesis, tumor growth, and metastasis even more urgent. In addition, it has been shown recently that MELK plays an important role in the proliferation of glioma stem-like cells in glioblastoma multiforme11 through phosphorylation of the transcription factor FOXM1, which can be disrupted by the antibiotic Siomycin A. Similarly, depletion of MELK by shRNA decreases tumor growth and malignancy and increases survival in a GSC-derived tumor mouse model.12 MELK has also been shown to be upregulated in mammary tumor-initiating cells, and its function is required for mammary tumorigenesis in vivo.13 Consequently, inhibition of MELK was shown to prevent mammosphere formation in breast cancer cells.14
Elucidation of the role of MELK in cancer15,16 is hampered by the lack of well-characterized, selective inhibitors of the kinase activity. At the time of the execution of this project, only nonselective inhibitors of MELK were described.17 Most potent was dorsomorphin, developed as a KDR (VEGFR) inhibitor18 and subsequently described as an AMPK inhibitor.19,20 More recently, a number of publications on MELK inhibitors have appeared,21−25 for example, OTSSP167,14 but none discloses both ligand structure and data showing a broad selectivity profile.
This letter describes the discovery of a dual-selective MELK inhibitor using fragment-based drug design (FBDD). FBDD has been successfully applied across a number of target classes, and fragment-derived kinase inhibitors have progressed to clinical trials.26,27 An important factor in the success of FBDD campaigns is access to high quality structural information in the form of multiple protein–ligand X-ray crystal structures. For this work, soakable crystal forms of MELK were developed that were suitable for high throughput structure determination of protein–ligand complexes (see Supporting Information). Notably, crystal structures for MELK have recently been published.25,28
Approximately 1500 compounds constituting an Astex fragment library were screened using ligand observed nuclear magnetic resonance (NMR) spectroscopy and protein thermal shift (Tm). Hits from both biophysical assays were progressed to X-ray crystallography, from which a large number of structurally validated hinge-binding compounds were obtained. Multiple factors are taken into account when selecting fragment hits for progression to hit-to-lead chemistry, for example, high ligand efficiency (LE)29 and the presence of suitable growth vectors. However, in this case, it was also important to obtain high selectivity over other kinases in order to obtain a good quality chemical probe. Furthermore, selectivity versus closely related AMPK (60% identity in kinase domain) was highly desirable since off-target inhibition of AMPK has been reported as the cause of cardiotoxicity induced by sunitinib.30 We hypothesized that a hinge-binding fragment with an atypical binding motif might provide an inherent bias toward selective inhibition during the optimization process compared with a typical “frequent-hitter” kinase hinge binding motif. An atypical hinge binder was also thought to be desirable in terms of exploring highly novel chemical space for kinase inhibition.
Here we describe the efficient optimization of a secondary amide hinge binder 1 leading to a potent and selective MELK inhibitor 7, designated MELK-T1. The binding mode of 1 from the X-ray crystal structure is shown in Figure 1A. Notably, the amide binds in the E- or trans-form (a rarely precedented binding mode), and this key feature distinguishes it from cyclic amide- and pyridone-containing hinge binders previously reported. One exception is a series of p38 inhibitors,31 though these molecules are structurally distinct from the series derived from 1.
Figure 1.

X-ray crystal structures of trans-amide ligands bound to MELK: (A) compound 1; (B) compound 2; (C) compound 3; (D) compound 4 (see Table 1); with hydrogen bond and salt bridge interactions shown as dotted lines.

Another key feature of the binding mode is the movement of the side chain position of Tyr88 in the hinge region, which was not seen in structures of more typical hinge binding fragments identified in the MELK fragment screen, or in the published crystal structures.25,28 Examination of Protein Data Bank kinase structures showed that this residue movement was rare and hence might be an important contributor to obtaining a beneficial selectivity profile. The structure shows a DFG-in conformation of the kinase activation loop; hence, 1 binds as a Type I inhibitor. The benzo ring of the tetrahydroisoquinoline (THIQ) moiety forms an edge-to-face interaction with the displaced Tyr88, while the unsaturated ring is oriented toward the solvent exposed region of the ATP binding cleft, and the potentially charged cyclic amine therein provides a means of conferring favorable aqueous solubility for the chemotype. Although 1 (IC50 160 μM, LE 0.26) was by no means the most ligand efficient hit obtained from the fragment screen, the novel features described above made it an attractive starting point for testing our selectivity hypothesis.
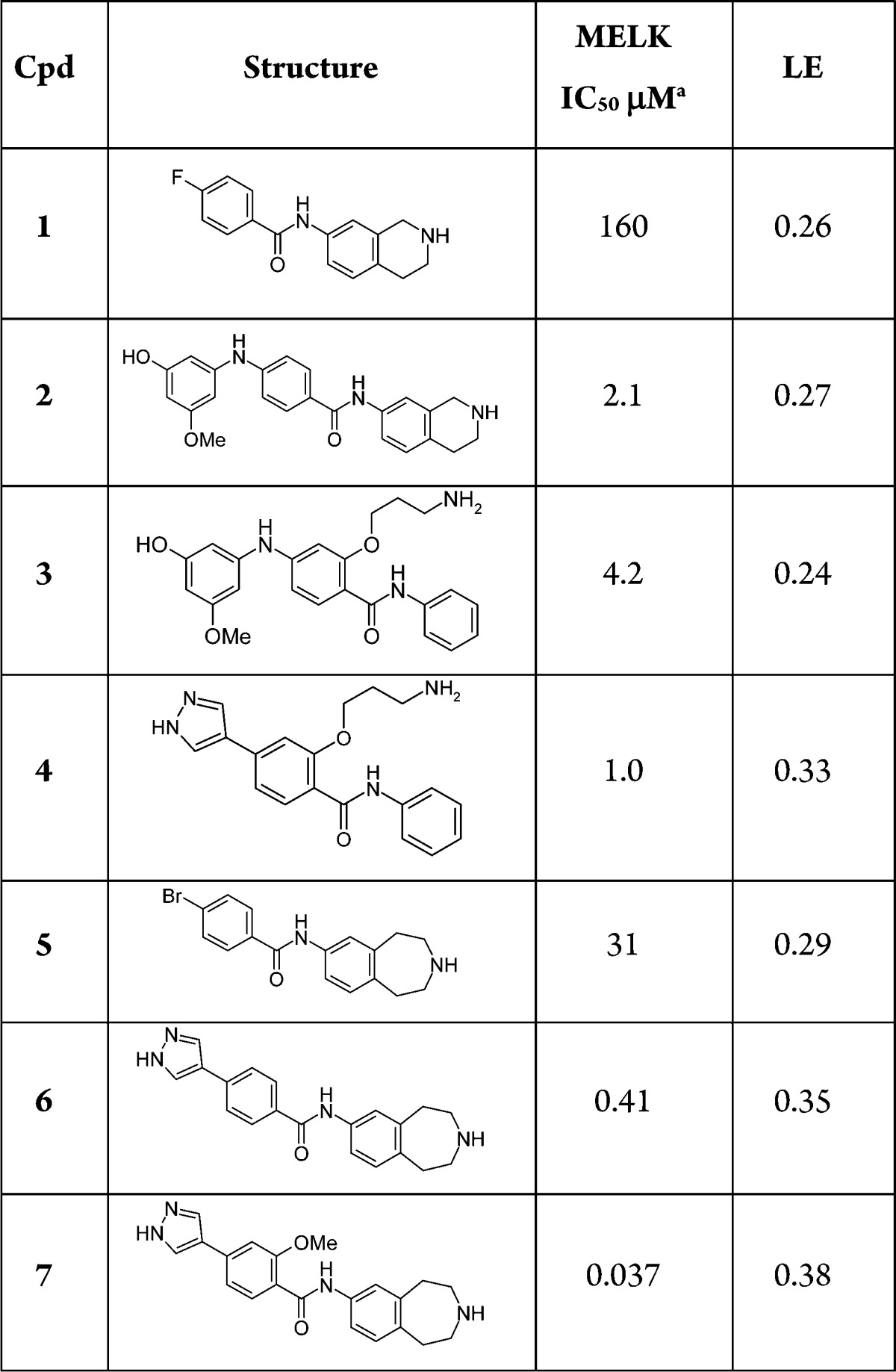
Compounds 3–7 (Table 1) were prepared as illustrated in Scheme 1 (for experimental details and compound characterization, see Supporting Information). Nitration of benzazepine 8 gave 9, which was Boc-protected and then reduced by catalytic hydrogenation to give the protected bicyclic aniline 10. Acylation gave amides 12, which were then subjected to palladium-mediated cross-coupling with an optionally protected pyrazole boronate ester, followed by acid mediated deprotection to give final compounds 6 and 7. Alternatively 12 (R = H) could be deprotected directly to give bromobenzamide 5. Synthesis of aminopropoxy derivatives 3 and 4 started from 2-fluoro-4-bromobenzoyl chloride 13. Amide coupling gave anilide 14, then nucleophilic substitution of the 2-fluoro substituent with the appropriately protected amino-alcohol under basic conditions gave the required aryl alkyl ether 15. Introduction of the pyrazole and final deprotection proceeded as previously described to afford compound 4. The N-linked phenol derivative 3 was also synthesized from 15 using a palladium catalyzed coupling with 3-amino-5-methoxyphenol and subsequent deprotection. An analogous two step sequence starting from N-1,2,3,4-tetrahydroisoquinolin-7-yl-4-bromobenzamide afforded compound 2. Compounds were assessed for inhibition of MELK kinase activity using a radioactive filter binding assay (see Supporting Information).
Table 1. Structure-Based Optimization of 1.
Assay details described in Supporting Information.
Scheme 1. Preparation of Compounds 3–7.

Reagents and conditions: (i) 1. H2SO4 (aq), 1,4-dioxane, RT. 2. HNO3, H2SO4, 0 °C; (ii) 1. (Boc)2O, CH2Cl2, RT. 2. H2, Pd/C, MeOH, RT; (iii) 11, EDC, HOBT, Et3N, CH2Cl2, RT; or 4-bromobenzoyl chloride, NEt3, CH2Cl2, RT; (iv) 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyrazole or 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyrazole-1-carboxylic acid tert-butyl ester, Pd(PPh3)4, Na2CO3, H2O, DME, reflux; (v) HCl, 1,4-dioxane, EtOAc, RT; (vi) PhNH2, NEt3, CH2Cl2, RT; (vii) N-Boc-3-hydroxy-1-propylamine, Cs2CO3, DMF, 60 °C; (viii) 3-amino-5-methoxyphenol, Pd2dba3, XPhos, K3PO4, DME, 140 °C. For yields, see experimental procedures in Supporting Information.
Compound 1 provided good growth vectors toward the back pocket via conserved catalytic Lys40 and the DFG motif, a region that has been targeted successfully in kinase optimizations. Using C-4 of the benzamide moiety as a growth point, a range of groups bearing H-bond donors and/or acceptors was designed, with a view to forming interactions with polar residues and backbone amides in this area. Encouragingly, substituted phenol 2 (IC50 2.1 μM, LE 0.27; Figure 1B) gave an 80-fold improvement in potency compared to 1 while maintaining ligand efficiency. The phenol OH functions as both donor and acceptor, engaging both the catalytic lysine and Glu57 situated on the α-C helix. The methoxy group fills the lipophilic selectivity pocket adjacent to the gate-keeper residue, while the NH spacer is important in allowing the bulky disubstituted phenyl group to bypass the relatively large Leu86 gate-keeper.
An alternative growth strategy was explored with the goal of forming a salt bridge interaction with Glu93. A targeted set of basic side chains was designed, attached via an oxygen atom to the ortho position of the benzamide phenyl ring. The oxygen linker was deemed important in ensuring a coplanar arrangement of the aryl benzamide core as seen in the crystal structures of 1 and 2, by virtue of forming an intramolecular H-bond to the adjacent amide NH, an arrangement that would not be possible in the case of an all-carbon linker. Aminopropoxy derivative 3 (IC50 4.2 μM, LE 0.24; Figure 1C) duly formed the desired salt bridge interaction, but this change resulted in a less ligand efficient molecule than 2. However, replacement of the substituted phenol group with 4-pyrazolyl 4 significantly improved potency and ligand efficiency (IC50 1.0 μM, LE 0.33). Pyrazole containing fragments have been reported as kinase hinge binders,32 so there was a perceived risk that the molecule would switch orientation and bind with the pyrazole adjacent to the hinge. However, the crystal structure (Figure 1D) clearly showed that such a reorientation did not occur; conversely, the binding mode of original hit 1 was recapitulated.
Further optimization was carried out in the solvent exposed region. Examination of the surface contacts around the THIQ moiety in the co-crystal structure of 1 showed that the saturated ring did not optimally fill the solvent exposed pocket. Docking studies suggested that expansion of the saturated ring to seven atoms would be advantageous, and accordingly, symmetrical benzazepine 5 was prepared. Gratifyingly this change was well tolerated (IC50 31 μM, LE 0.29), and the seven-membered ring showed excellent shape complementarity with the protein surface (Figure 2).
Figure 2.
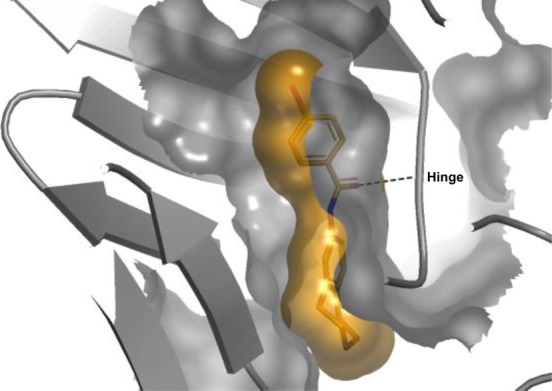
X-ray crystal structure of compound 5 bound to MELK viewed from the solvent exposed area, showing Connolly surfaces of ligand (orange) and protein (gray). The hydrogen bond between the amide carbonyl and the hinge is shown as a dotted line.
Replacement of the bromo substituent in 5 with 4-pyrazolyl then gave a 75-fold increase in potency, with 6 achieving a submicromolar IC50 value. A common feature observed in all crystal structures is the near-coplanar arrangement of the phenyl benzamide core. It is known from small molecule X-ray crystal structures that benzamides preferentially adopt a conformation where the amide is twisted by 30–40° out of the plane of the aryl ring.33 It was reasoned that introduction of a methoxy substituent ortho to the amide carbonyl in 6 would stabilize the desired planar conformation, resulting in synthesis of compound 7. This change resulted in a further 10-fold increase in affinity (IC50 37 nM) and a further improvement in LE. Figure 3 shows the binding mode of 7, wherein the pyrazole now forms a network of through-water H-bond interactions with Lys40 and Glu57.
Figure 3.
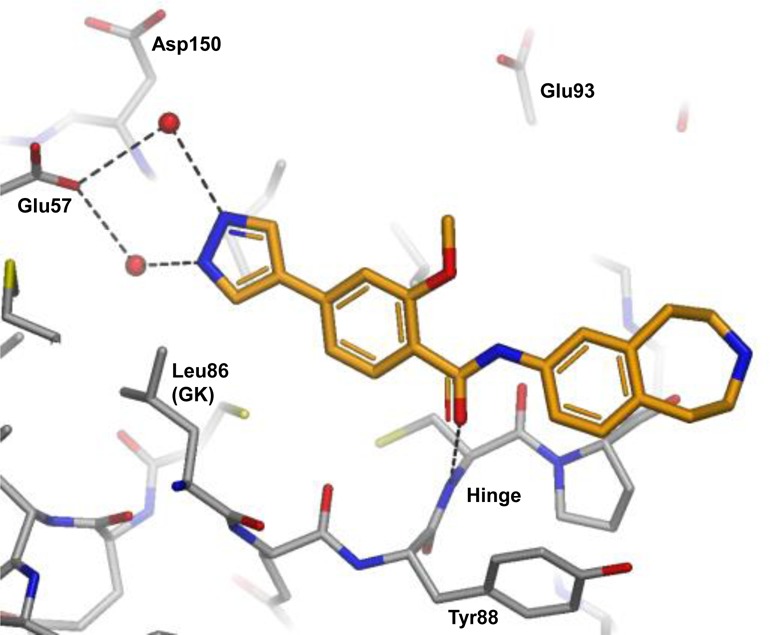
X-ray crystal structure of compound 7 bound to MELK. Hydrogen bond interactions are shown as dotted lines and water molecules as red spheres.
Compound 7 (MELK-T1) satisfied our potency criteria for a chemical probe, but it was necessary to establish whether the selectivity profile was appropriate and also provide evidence that the molecule was able to enter cells and thereby access the target. Across a panel of 235 kinases, compound 7 showed >50% inhibition at 1 μM against only six enzymes; of these, by far the most potent interactions were with Flt3 (IC50 18 nM) and MELK (IC50 23 nM) (Table 2). Importantly, no inhibitory activity was detected for the key antitarget AMPK, despite the close structural relationship with MELK.
Table 2. Kinase Selectivity of MELK-T1 (7).
| kinasea | % inh @ 1 μM (IC50 nM) |
|---|---|
| MELK | 97 (23) |
| Flt3 | 90 (18) |
| CAMKIIδ | 60 (810) |
| Mnk2 | 59 (760) |
| CAMKIIγ | 55 (1000) |
| MLCK | 54 (1000) |
| AMPKα1 | 5 |
| AMPKα2 | –12 |
Millipore kinase panel; for details, see Supporting Information
The potent inhibition of Flt3 provided a means of testing the cell permeability of 7 using engineered Flt3-driven Ba/F3 cell lines.34 In the Ba/F3-Flt3 assay, 7 showed inhibition of proliferation with IC50 1.5 μM in the absence of IL-3, while no inhibitory activity was observed in the presence of IL-3. In Ba/F3 cell lines transfected with either FGFR1, FGFR3, or KDR, compound 7 did not inhibit proliferation, either in the presence or absence of IL-3, thus confirming both intracellular target engagement and specificity for Flt3. Given such a demonstration of intracellular activity, compound 7 should therefore be a suitable chemical probe for elucidating cellular phenotypic effects arising from inhibition of MELK.
In conclusion, we have demonstrated that the choice of an atypical hinge binding motif as a starting point for FBDD can be a successful strategy for obtaining a kinase inhibitor with a good selectivity profile. In the case of MELK, fragment screening generated many high LE hits that fell into the category of “frequent hitter” hinge binding elements. Such frequent hitters are by definition predisposed to interact with multiple kinases, so here one may face the challenge of designing out off-target activity at each stage of the fragment growing process. The benefit of starting from a nonfrequent hitter is that selectivity is potentially built in at the outset, though there may be a trade-off in terms of lower on-target potency and/or LE, as in the MELK example here. A consequence of choosing a less ligand-efficient hinge binding fragment, such as 1, is the need to identify several high energy interactions at multiple sites in the binding pocket. This was successfully achieved for MELK, hence a notable feature of the optimization from 1 to 7 is the significant increase in LE with molecular size. The availability of multiple protein–ligand crystal structures enabled a highly efficient optimization process (∼35 compounds prepared in total), the end result being the discovery of dual-selective and cell penetrant chemical probe MELK-T1 7, suitable for the further elucidation of MELK biology and as a starting point for further optimization.
Acknowledgments
We would like to thank Prof. Jan Cools (Catholic University Leuven, Leuven, Belgium) for the gift of the Ba/F3 Flt3 cell line. We would also like to thank Luis Trabalón (VillaPharma, Parque tecnologico de Fuente Alamo, Ctra. El Estrecho-Lobosillo, Km. 2,5-Av. Azul, E-30320 Murcia, Spain) for scale-up synthesis of compound 7.
Glossary
Abbreviations Used
- DME
1,2-dimethoxyethane
- XPhos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
- Pd2dba3
tris(dibenzylidene-acetone)dipalladium(0)
Supporting Information Available
Assay details; structures of dorsomorphin and OTSSP167; fragment screen details; crystallographic details and PDB accession codes; experimental procedures for the preparation of novel compounds and intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ (P.B.) Institut de Chimie Organique et Analytique (ICOA), Université d’Orléans, UMR CNRS 7311, 45057 Orléans Cedex 2, France.
The authors declare no competing financial interest.
Supplementary Material
References
- Gil M.; Yang Y.; Lee Y.; Choi I.; Ha H. Cloning and expression of a cDNA encoding a novel protein serine/threonine kinase predominantly expressed in hematopoietic cells. Gene 1997, 195, 295–301. [DOI] [PubMed] [Google Scholar]
- Egan B.; Zierath J. R. Hunting for the SNARK in metabolic disease. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E969–E972. [DOI] [PubMed] [Google Scholar]
- Lizcano J. M.; Goransson O.; Toth R.; Deak M.; Morrice N. A.; Boudeau J.; Hawley S. A.; Udd L.; Makela T. P.; Hardie D. G.; Alessi D. R. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D.; Jubb A. M.; Hogue D.; Dowd P.; Kljavin N.; Yi S.; Bai W.; Frantz G.; Zhang Z.; Koeppen H.; de Sauvage F. J.; Davis D. P. Maternal embryonic leucine zipper kinase/murine protein serine-threonine kinase 38 is a promising therapeutic target for multiple cancers. Cancer Res. 2005, 65, 9751–9761. [DOI] [PubMed] [Google Scholar]
- Nakano I.; Masterman-Smith M.; Saigusa K.; Paucar A. A.; Horvath S.; Shoemaker L.; Watanabe M.; Negro A.; Bajpai R.; Howes A.; Lelievre V.; Waschek J. A.; Lazareff J. A.; Freije W. A.; Liau L. M.; Gilbertson R. J.; Cloughesy T. F.; Geschwind D. H.; Nelson S. F.; Mischel P. S.; Terskikh A. V.; Kornblum H. I. Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J. Neurosci. Res. 2008, 86, 48–60. [DOI] [PubMed] [Google Scholar]
- Pickard M. R.; Green A. R.; Ellis I. O.; Caldas C.; Hedge V. L.; Mourtada-Maarabouni M.; Williams G. T. Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res. 2009, 11, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.-L.; Park J.-H.; Toshihiko Nishidate T.; Nakamura Y.; Katagiri T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 2009, 11, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R.; Fälth M.; Pressinotti N. C.; Brase J. C.; Puig S. B.; Metzger J.; Gade S.; Schäfer G.; Bartsch G.; Steiner E.; Klocker H.; Sültmann H. The maternal embryonic leucine zipper kinase (MELK) is upregulated in high-grade prostate cancer. J. Mol. Med. 2013, 91, 237–248. [DOI] [PubMed] [Google Scholar]
- Ryu B.; Kim D. S.; DeLuca A. M.; Alani R. M. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One 2007, 2, e594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku J.-L.; Shin Y.-K.; Kim D.-W.; Kim K.-H.; Choi J.-S.; Hong S.-H.; Jeon Y.-K.; Kim S.-H.; Kim H.-S.; Park J.-H.; Kim I.-J.; Park J.-G. Establishment and characterization of 13 human colorectal carcinoma cell lines: mutations of genes and expressions of drug-sensitivity genes and cancer stem cell markers. Carcinogenesis 2010, 31, 1003–1009. [DOI] [PubMed] [Google Scholar]
- Joshi K.; Banasavadi-Siddegowa Y.; Mo X.; Kim S.-H.; Mao P.; Kig C.; Nardini D.; Sobol R. W.; Chow L. M. L.; Kornblum H. I.; Waclaw R.; Beullens M.; Nakano I. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 2013, 31, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C.; Banasavadi-Siddegowda Y. K.; Joshi K.; Nakamura Y.; Kurt H.; Gupta S.; Nakano I. Tumor-specific activation of the C-JUN/MELK pathway regulates glioma stem cell growth in a p53-dependent manner. Stem Cells 2013, 31, 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbard L. W.; Maurer J.; Miller A.; Lesperance J.; Hassell J.; Oshima R. G.; Terskikh A. V. Maternal embryonic leucine zipper kinase is upregulated and required in mammary tumor-initiating cells in vivo. Cancer Res. 2010, 70, 8863–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S.; Suzuki H.; Miyamoto T.; Takamatsu N.; Tatsuguchi A.; Ueda K.; Kijima K.; Nakamura Y.; Matsuo Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget 2012, 3, 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beullens M.; Vancauwenbergh S.; Morrice N.; Derua R.; Ceulemans H.; Waelkens E.; Bollen M. Substrate specificity and activity regulation of protein kinase MELK. J. Biol. Chem. 2005, 280, 40003–40011. [DOI] [PubMed] [Google Scholar]
- Kig C.; Beullens M.; Beke L.; Van Eynde A.; Linders J. T. M.; Brehmer D.; Bollen M. Maternal embryonic leucine-zipper kinase (MELK) reduces replication stress in glioblastoma cells. J. Biol. Chem. 2013, 288, 24223–24233. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bain J.; Plater L.; Elliott M.; Shapiro N.; Hastie C. J.; Mclaughlan H.; Klevernic I.; Arthur J. S. C.; Alessi D. R.; Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley M. E.; Rubino R. S.; Hoffman W. F.; Hambaugh S. R. Optimization of a pyrazolo[1,5-a]pyrimidine class of KDR kinase inhibitors: improvements in physical properties enhance cellular activity and pharmacokinetics. Bioorg. Med. Chem. Lett. 2002, 12, 3537–3541. [DOI] [PubMed] [Google Scholar]
- Zhou G.; Myers R.; Li Y.; Chen Y.; Shen X.; Fenyk-Melody J.; Wu M.; Ventre J.; Doebber T.; Fujii N.; Musi N.; Hirshman M. F.; Goodyear L. J.; Moller D. E. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N.; Takagi T.; Saijo S.; Kishishita S.; Takaya D.; Toyama M.; Terada T.; Shirouzu M.; Suzuki A.; Lee S.; Yamauchi T.; Okada-Iwabu M.; Iwabu M.; Kadowaki T.; Minokoshi Y.; Yokoyama S. Structural basis for compound C inhibition of the human AMP-activated protein kinase [alpha]2 subunit kinase domain. Acta Crystallogr. 2011, D67, 480–487. [DOI] [PubMed] [Google Scholar]
- Matsuo Y.; Hisada S.; Nakamura Y.; Ahamed F.; Huntley R.; Walker J. R.; Decornez H.. Quinoline derivatives and melk inhibitors containing the same. WO 2012016082.
- Matsuo Y.; Hisada S.; Nakamura Y.; Ahmed F.; Walker J. R.; Huntley R.. 1,5-Naphthyridine derivatives and melk inhibitors containing the same. WO 2013109388.
- Mahasenan K. V.; Li C. Novel inhibitor discovery through virtual screening against multiple protein conformations generated via ligand-directed modeling: a maternal embryonic leucine zipper kinase example. J. Chem. Inf. Model. 2012, 52, 1345–1355. [DOI] [PubMed] [Google Scholar]
- Kowalczyk P.; Wegryn P.; Edyta P.; Zarebski A.; Sabiniarz A.; Stefanska K.; Kwiek U.; Zawadzki P.; Krawczynska K. Zurawska M.; Trebacz A.; Prymula K.; Milik M.; Brzozka K.. Development of selective MELK kinase inhibitors for cancer treatments. AACR 2013, poster 2160. [Google Scholar]
- Canevari G.; Re Depaolini S.; Cucchi U.; Bertrand J. A.; Casale E.; Perrera C.; Forte B.; Carpinelli P.; Felder E. R. Structural insight into materal embryonic leucine zipper kinase (MELK) conformation and inhibition towards structure-based drug design. Biochemistry 2013, 52, 6380–6387. [DOI] [PubMed] [Google Scholar]
- Bollag G.; Tsai J.; Zhang J.; Zhang C.; Ibrahim P.; Nolop K.; Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 2012, 11, 873–886. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Cao L.-S.; Wang J.; Chen Y.; Deng H.; Wang Z.-X.; Wu J.-W. Structural basis for the regulation of maternal embryonic leucine zipper kinase. PLoS One 2013, 8, e70031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE = −ΔG/HAC ≈ −RT ln(IC50)/HAC.Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Kerkela R.; Woulfe K. C.; Durand J.-B.; Vagnozzi R.; David Kramer D.; Chu T. F.; Beahm C.; Chen M. H.; Force T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin. Transl. Sci. 2009, 2, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell R. M.; Bamborough P.; Cleasby A.; Cockerill S. G.; Jones K. L.; Mooney C. J.; Somers D. O.; Walker A. L. Biphenyl amide p38a kinase inhibitors 1: discovery and binding mode. Bioorg. Med. Chem. Lett. 2008, 18, 318–323. [DOI] [PubMed] [Google Scholar]
- Saxty G.; Woodhead S. J.; Berdini V.; Davies T. G.; Verdonk M. L.; Wyatt P. G.; Boyle R. G.; Barford D.; Downham R.; Garrett M. D.; Carr R. D. Identification of inhibitors of protein kinase B using fragment-based lead discovery. J. Med. Chem. 2007, 50, 2293–2296. [DOI] [PubMed] [Google Scholar]
- Schärfer C.; Schulz-Gasch T.; Ehrlich H.-C.; Guba W.; Rarey M.; Stahl M. Torsion angle preferences in druglike chemical space: a comprehensive guide. J. Med. Chem. 2013, 56, 2016–2028. [DOI] [PubMed] [Google Scholar]
- Warmuth M.; Kim S.; Gu X.-J.; Xia G.; Adrián F. Ba/F3 cells and their use in kinase drug discovery. Curr. Opin. Oncol. 2007, 19, 55–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.