

Abstract
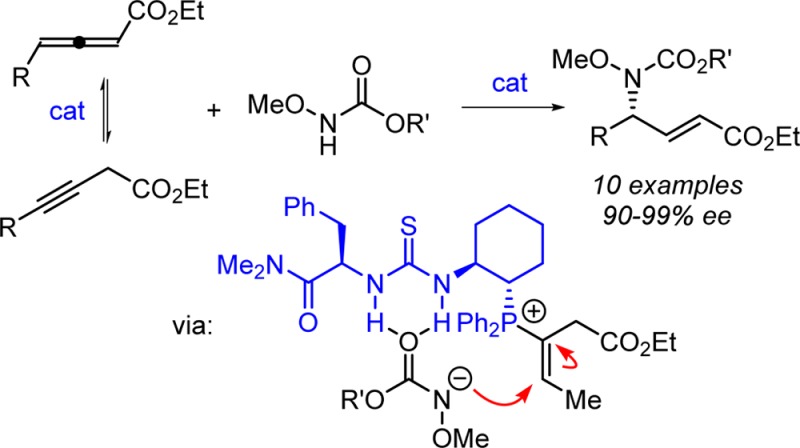
Bifunctional phosphinothiourea catalysts have been developed successfully for the highly regio- and enantioselective γ-hydroamination of allenyl and propargyl esters with N-methoxy carbamate nucleophiles to yield α,β-unsaturated γ-amino acid ester products. In the case of propargyl ester substrates, the reaction proceeds through reversible phosphinothiourea-catalyzed isomerization to the corresponding allenyl ester. The high enantioselectivity of the process is attributed to a cooperative conjugate addition of a thiourea-bound carbamate anion to a vinyl phosphonium ion resulting from covalent activation of the allenyl ester substrate.
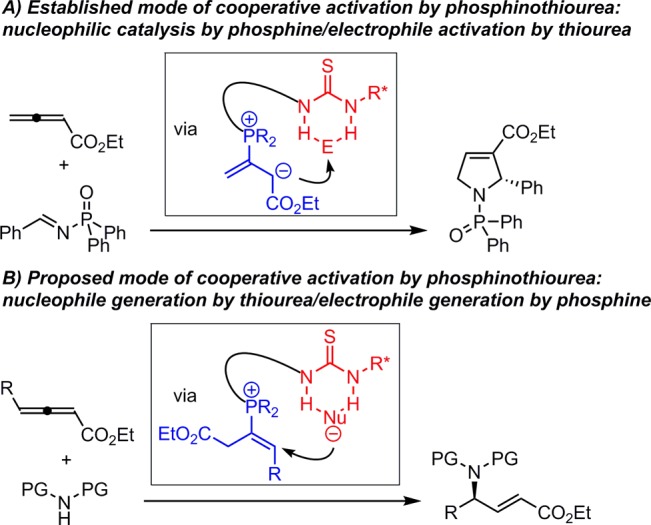
Small, polyfunctional molecules capable of cooperative activation and precise positioning of reacting partners hold tremendous potential in selective catalysis.1 Recently, we reported the development of a family of phosphinothiourea catalysts2 that promote imine-allene [3 + 2] cycloadditions via nucleophilic activation of the allene by the phosphine with simultaneous imine activation by hydrogen bonding to the thiourea (Scheme 1A).3,4 We were intrigued by the potential of a complementary reactivity mode with the same family of catalysts, wherein the H-bond donor would promote formation of a reactive nucleophile by anion binding,5,6 while the phosphine component could induce generation of an activated vinyl phosphonium electrophile (Scheme 1B).7,8 We report here the successful development of this strategy, with the application of this new type of cooperative activation to the highly regio- and enantioselective γ-hydroamination of allenyl and propargyl esters.9 This methodology provides practical access to synthetically valuable α,β-unsaturated γ-amino esters in highly enantioenriched form.10
Scheme 1. Cooperative Catalysis with Phosphinothioureas.
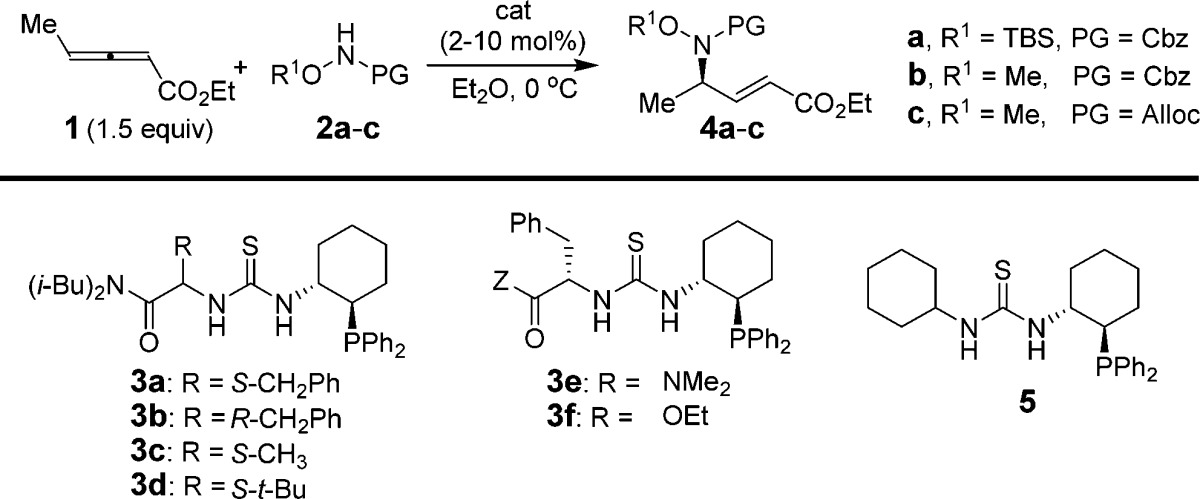
A survey of potential N-centered nucleophiles revealed that compounds with pKa values between 8 and 10 (in H2O) were suitable reacting partners in γ-additions to allenoate 1 catalyzed by phosphinothiourea 3a, whereas compounds with acidities lying outside that range were unreactive.11 Highest enantioselectivities were achieved with O-substituted hydroxylamine carbamate derivatives,12 and the reaction of 1 with 2a was selected as a model reaction for catalyst optimization studies (Table 1).
Table 1. Optimization Studies.
| entry | Nu | cat | loading (mol %) | time (h) | yield (%)a,c | ee (%) |
|---|---|---|---|---|---|---|
| 1 | 2a | 3a | 10 | 120 | 65 | 92 |
| 2 | 2a | 3b | 10 | 120 | 93 | 88 |
| 3 | 2a | 3c | 10 | 120 | 66 | 88 |
| 4 | 2a | 3d | 10 | 120 | 72 | 88 |
| 5 | 2b | 3a | 10 | 24 | >99b | 82 |
| 6 | 2c | 3a | 10 | 24 | >99b | 92 |
| 7 | 2c | 3a | 1 | 48 | 53 | 89 |
| 8 | 2c | 3e | 1 | 48 | 90 | 92 |
| 9 | 2c | 3f | 10 | 20 | 92d | 88 |
| 10 | 2c | 5 | 10 | 20 | 77 | 50 |
| 11 | 2c | CyPPh2 | 10 | 24 | 36e | |
| 12 | 2c | 3e | 2 | 24 | 96 | 93 |
| 13f | 2c | 3e | 2 | 24 | 99 | 94 |
| 14f | 2c | CyPPh2 | 10 | 24 | N.R. |
Isolated yield, unless noted otherwise.
Conversion.
In all cases except entries 9 and 11, the γ-adduct was the only detectable product (γ/α >100:1).
γ/α = 50:1.
γ/α = 20:1.
Propargyl ester 6 was used instead of allenyl ester 1.
Catalysts with the general structure depicted in 3 were generally effective in the model reaction, affording the γ-adduct 4a as the only detectable product. Variation of the identity or the stereochemistry of the α-amino acid component had little effect on enantioselectivity (entries 1–4), although the S-phenylalanine-R,R-amino-phosphinocyclohexane combination afforded 4a with the highest ee (entry 1).
Improved reactivity was observed in reactions of less sterically hindered carbamate nucleophiles, and the O-methyl alloc variant (2c) provided the best balance of reactivity and enantioselectivity (entry 6). In this manner, the loading of catalyst 3a could be reduced from 10 to 1 mol% with only a slight decrease in enantioselectivity (entries 6–7). Further improvement in reactivity was provided by reducing the steric demand of the amide component as in 3e (entry 8). Replacement of the amide with an ethyl ester (3f) had a negative effect on reaction rate, but very little effect on ee (entry 9). Overall, it appears that the amino acid-derived component of 3 plays an important, albeit subtle role in the mechanism of catalysis, particularly given the observation that simplified catalyst 5 promoted the γ-addition reaction with much lower enantioselectivity (entry 10). On the basis of this analysis, 3e was selected for further investigation.
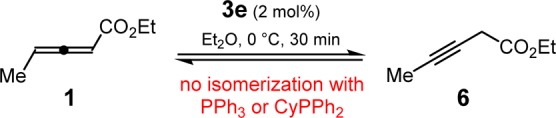
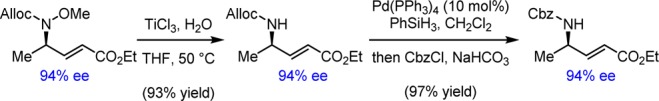
During the course of these optimization studies, unreacted allenyl ester 1 isolated after reactions catalyzed by the phosphinothiourea catalysts was always detected as a 1:1 mixture with propargyl ester 6. We subsequently found that phosphinothiourea 3e (2 mol%) catalyzed the rapid interconversion of 1 and 6 in the absence of carbamate, reaching equilibrium as a 1:1 mixture within 30 min (Scheme 2).13 It was thereby possible to engage propargyl ester 6 in the hydroamination reaction, with slightly improved yield and enantioselectivity compared to reactions employing allenyl ester 1 (Table 1, entries 12 vs 13). Simple phosphines such as triphenylphosphine and cyclohexyldiphenylphosphine were not observed to promote the isomerization between 1 and 6, even at higher catalyst loadings and elevated temperatures, nor did they promote hydroamination of 6 (entry 14). The use of propargyl esters such as 6 in the enantioselective reaction provides a practical advantage because they are readily prepared in a general manner and in one step by Fu’s copper-catalyzed reaction of terminal alkynes with diazoacetates.14
Scheme 2. Phosphinothiourea-Catalyzed Interconversion of Allenyl Ester 1 and Propargyl Ester 6.
The scope of the enantioselective γ-hydroamination reaction was examined under the optimized conditions identified above. Significant variation of the substitution on the propargyl ester was possible while maintaining excellent product yields and enantioselectivities (Table 2). In particular, primary alkyl chloride (entry 4), terminal alkynyl (entries 5 and 6), alkyl and silyl ether (entries 7 and 8), phthalimide (entry 10), and nitrile-containing (entry 11) propargyl esters all underwent clean reaction to afford the corresponding α,β-unsaturated γ-amino acid ester products in consistently high ee’s. However, the reaction proved sensitive to the local steric properties of the alkyne, with the isobutyl-substituted derivative requiring higher catalyst loadings (entry 9), and branched alkynes (or their corresponding allenyl esters) proving unreactive.11 The γ-addition products could be readily deprotected in high yields to afford the free amine with preservation of enantiomeric integrity (Scheme 3).11
Table 2. Enantioselective γ-Hydroaminations Catalyzed by 3e.
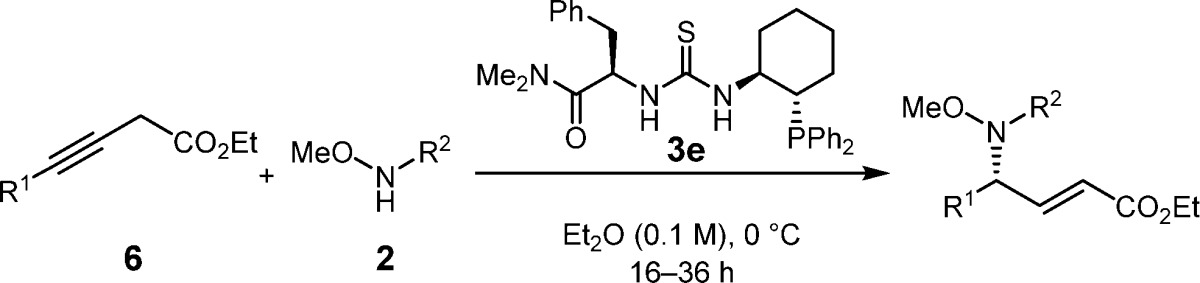
CH2Cl2 was used as solvent.
Scheme 3. Deprotection.
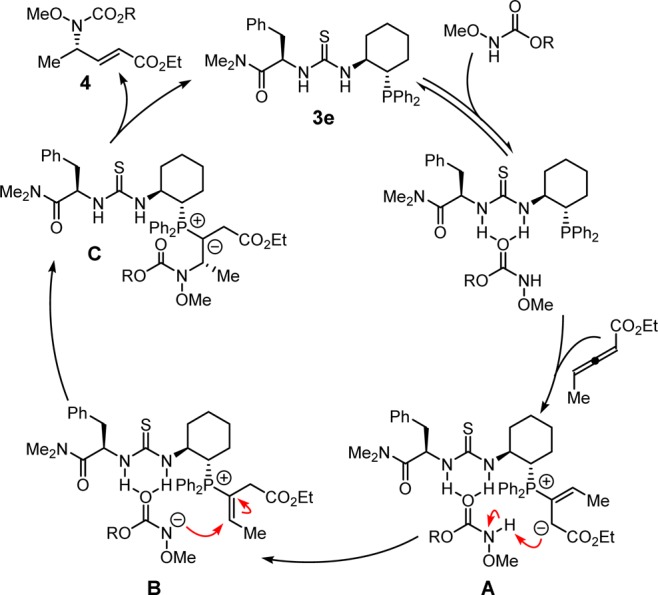
We propose a mechanism of catalysis for the γ-hydroamination reaction in Scheme 4. The loading of the carbamate and the allenyl ester onto the catalyst can occur in either order to generate intermediate A. The hydrogen-bond interaction with the carbamate is expected to lower the pKa of the N–H bond substantially,15 favoring proton transfer to the ion pair intermediate B. Addition of the catalyst-bound, deprotonated carbamate to the resulting vinyl phosphonium ion should be very facile, producing ylide intermediate C, which can liberate the product 4 and regenerate the active catalyst upon tautomerization and β-elimination.2
Scheme 4. Proposed Catalytic Cycle.
The utility of chiral phosphinothioureas has thus been extended into a new, anion-binding manifold with the highly enantioselective γ-hydroamination of allenyl esters. Further studies into the reactivity and selectivity of these versatile polyfunctional catalysts are ongoing.
Acknowledgments
This work was supported by the NIH (GM-43214). Y.-Q.F. gratefully acknowledges NSERC for postdoctoral fellowship support, and P.M.T. gratefully acknowledges the NIH for postdoctoral fellowship support.
Supporting Information Available
Nucleophile and catalyst screening information, product absolute stereochemistry assignment, complete experimental procedures, characterization data, and chromatographic analyses of the catalyst and the products. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For selected reviews, see:; a Allen A. E.; MacMillan D. W. C. Chem. Sci. 2012, 3, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shibasaki M.; Yoshikawa N. Chem. Rev. 2002, 102, 2187. [DOI] [PubMed] [Google Scholar]; c Paull D. H.; Abraham C. J.; Scerba M. T.; Alden-Danforth E.; Lectka T. Acc. Chem. Res. 2008, 41, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shibasaki M.; Kanai M.; Matsunaga S.; Kumagai N. Acc. Chem. Res. 2009, 42, 1117. [DOI] [PubMed] [Google Scholar]; e Park Y. J.; Park J.-W.; Jun C.-H. Acc. Chem. Res. 2008, 41, 222. [DOI] [PubMed] [Google Scholar]; f Stegbauer L.; Sladojevich F.; Dixon D. J. Chem. Sci. 2012, 3, 942. [Google Scholar]; g Ma J.-A.; Cahard D. Angew. Chem., Int. Ed. 2004, 43, 4566. [DOI] [PubMed] [Google Scholar]; h Knowles R. R.; Jacobsen E. N. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fang Y.-Q.; Jacobsen E. N. J. Am. Chem. Soc. 2008, 130, 5660. [DOI] [PubMed] [Google Scholar]; For a closely related system, see:; b Han X.; Zhong F.; Wang Y.; Lu Y. Angew. Chem., Int. Ed. 2012, 51, 767. [DOI] [PubMed] [Google Scholar]
- For recent reviews of polyfunctional phosphine organocatalysts, see:; a Wang S.-X.; Han X.; Zhong F.; Wang Y.; Lu Y. Synlett 2011, 2766. [Google Scholar]; b Marinetti A.; Voituriez A. Synlett 2010, 174. [Google Scholar]; c Zhao Q.-Y.; Lian Z.; Wei Y.; Shi M. Chem. Commun. 2012, 48, 1724. [DOI] [PubMed] [Google Scholar]; For selected, recent examples, see:; d Zhong F.; Dou X.; Han X.; Yao W.; Zhu Q.; Meng Y.; Lu Y. Angew. Chem., Int. Ed. 2013, 52, 943. [DOI] [PubMed] [Google Scholar]; e Yang Y.-L.; Pei C.-K.; Shi M. Org. Biomol. Chem. 2011, 9, 3349. [DOI] [PubMed] [Google Scholar]; f Takizawa S.; Kiriyama K.; Ieki K.; Sasai H. Chem. Commun. 2011, 47, 9227. [DOI] [PubMed] [Google Scholar]
- For reviews of asymmetric catalytic allenoate activation with polyfunctional catalysts, see:; a Fan Y. C.; Kwon O. Chem. Commun. 2013, 49, 11203. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cowen B. J.; Miller S. J. Chem. Soc. Rev. 2009, 38, 3102. [DOI] [PubMed] [Google Scholar]; For selected examples of phosphine-catalyzed asymmetric transformations of allenes, see:; c Cowen B. J.; Miller S. J. J. Am. Chem. Soc. 2007, 129, 10988. [DOI] [PubMed] [Google Scholar]; d Han X.; Wang Y.; Zhong F.; Lu Y. J. Am. Chem. Soc. 2011, 133, 1726. [DOI] [PubMed] [Google Scholar]; e Xiao H.; Chai Z.; Wang H.-F.; Wang X.-W.; Cao D.-D.; Liu W.; Lu Y.-P.; Yang Y.-Q.; Zhao G. Chem.—Eur. J. 2011, 17, 10562. [DOI] [PubMed] [Google Scholar]; f Zhong F.; Han X.; Wang Y.; Lu Y. Chem. Sci. 2012, 3, 1231. [Google Scholar]; g Xiao H.; Chai Z.; Zheng C.-W.; Yang Y.-Q.; Liu W.; Zhang J.-K.; Zhao G. Angew. Chem., Int. Ed. 2010, 49, 4467. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Mukherjee S. Chem. Commun. 2013, 49, 11203. [DOI] [PubMed] [Google Scholar]
- For reviews on anion-binding catalysis, see:; a Zhang Z.; Schreiner P. R. Chem. Soc. Rev. 2009, 38, 1187. [DOI] [PubMed] [Google Scholar]; b Phipps R. J.; Hamilton G. L.; Toste F. D. Nature Chem. 2012, 4, 603. [DOI] [PubMed] [Google Scholar]; c Brak K.; Jacobsen E. N. Angew. Chem., Int. Ed. 2013, 52, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Trost B. M.; Li C.-J. J. Am. Chem. Soc. 1994, 116, 3167. [Google Scholar]; b Trost B. M.; Li C.-J. J. Am. Chem. Soc. 1994, 116, 10819. [Google Scholar]; c Trost B. M.; Dake G. R. J. Am. Chem. Soc. 1997, 119, 7595. [Google Scholar]; d Trost B. M.; Dake G. R. J. Org. Chem. 1997, 62, 5670. [Google Scholar]
- Asymmetric variations using chiral phosphine catalysts:; a Lundgren R. J.; Wilsily A.; Marion N.; Ma C.; Chung Y. K.; Fu G. C. Angew. Chem., Int. Ed. 2013, 52, 2525. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhu G.; Chen Z.; Jiang Q.; Xiao D.; Cao P.; Zhang X. J. Am. Chem. Soc. 1997, 119, 3836. [Google Scholar]; c Chung Y. K.; Fu G. C. Angew. Chem., Int. Ed. 2009, 48, 2225. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Smith S. W.; Fu G. C. J. Am. Chem. Soc. 2009, 131, 14231. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sun J.; Fu G. C. J. Am. Chem. Soc. 2010, 132, 4568. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Sinisi R.; Sun J.; Fu G. C. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on hydroamination, see:; a Hannedouche J.; Schulz E. Chem.—Eur. J. 2013, 19, 4972. [DOI] [PubMed] [Google Scholar]; b Chemler S. R. Org. Biomol. Chem. 2009, 7, 3009. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Müller T. E.; Hultzsch K. C.; Yus M.; Foubelo F.; Tada M. Chem. Rev. 2008, 108, 3795. [DOI] [PubMed] [Google Scholar]
- For examples of the α,β-unsaturated γ-amino acid motif in natural products, see:Groll M.; Schellenberg B.; Bachmann A.; Archer C. R.; Huber R.; Powell T. K.; Lindow S.; Kaiser M.; Dudler R. Nature 2008, 452, 755. [DOI] [PubMed] [Google Scholar]
- See Supporting Information.
- Chen Y. K.; Yoshida M.; MacMillan D. W. C. J. Am. Chem. Soc. 2006, 128, 9328. [DOI] [PubMed] [Google Scholar]
- a Inokuma T.; Furukawa M.; Suzuki Y.; Kimachi T.; Kobayashi Y.; Takemoto Y. ChemCatChem 2012, 4, 983. [Google Scholar]; b Inokuma T.; Furukawa M.; Uno T.; Suzuki Y.; Yoshida K.; Yano Y.; Matsuzaki K.; Takemoto Y. Chem.—Eur. J. 2011, 17, 10470. [DOI] [PubMed] [Google Scholar]
- Suarez A.; Fu G. C. Angew. Chem., Int. Ed. 2004, 43, 3580. [DOI] [PubMed] [Google Scholar]
- Zuend S. J.; Jacobsen E. N. J. Am. Chem. Soc. 2009, 131, 15358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.