

Abstract
Flexibility of the glycine-rich flaps is known to be essential for catalytic activity of the HIV-1 protease, but their exact conformations at the different stages of the enzymatic pathway remain subject to much debate. While hundreds of crystal structures of protease-inhibitor complexes have been solved, only about a dozen inhibitor-free protease structures have been reported. These apo-structures reveal a large diversity of flap conformations, ranging from closed, to semi-open and wide-open. To evaluate the average structure in solution, we measured residual dipolar couplings (RDCs) and compared these to values calculated for crystal structures representative of the closed, semi-open and wide-open states. The RDC data clearly indicate that the inhibitor-free protease, on average, adopts a closed conformation in solution that is very similar to the inhibitor-bound state. By contrast, a highly drug-resistant protease mutant, PR20, adopts the wide-open flap conformation.
Keywords: active site access, flap dynamics, HIV protease, liquid crystal NMR, residual dipolar coupling
The human immunodeficiency virus type 1 (HIV-1) protease (PR) is an aspartic hydrolase that functions as an obligatory homodimer with 99 amino acids in each subunit. Each monomer contains a glycine rich flap segment (residues 44-57) that folds as an antiparallel β-sheet and covers the active site (Figure 1A). The flexibility of the flaps is believed to control the access of substrates and inhibitors to the active site and is therefore recognized as essential for the enzymatic activity.[1, 2] Despite intense research over the past 20 years, the detailed role of the flaps in the substrate binding mechanism remains subject to much debate. While most of the current mechanistic models propose that wide-opening of the flaps gives access to the active site,[3-5] a recent molecular dynamics (MD) simulation suggests a translational procession of the substrate into the active site while maintaining the flaps in closed or semi-open conformations.[6] The access pathway of substrate to the active site of HIV-1 PR is of fundamental interest for gaining improved understanding of how this class of enzymes channels its substrates, and is also particularly important for the design of high affinity future-generation inhibitors.
Figure 1.

X-ray structures of HIV-1 PR. A) Structure of PR with flaps in the closed conformation (PDB entry 3BVB), highlighting the flaps (yellow), the hinge regions (orange) and the active site (blue). B) Comparison of the different structural models used in this study, showing the flap conformations of PR in a closed (3BVB, yellow), semi-open (1HHP, green) and wide-open (1TW7, cyan) conformation, as well as the PR20 structures used as reference models for the closed (3UFC, dark cyan) and wide-open (3UF3, red) flap conformations. All models have been superimposed to yield a minimal Cα rmsd for the Core region (comprising residues 10-23, 62-73 and 87-93 of both monomers).
Several hundred crystal structures of protease-inhibitor complexes have been deposited in the protein database (PDB) and in all these complexes the flaps adopt closed conformations. On the other hand, far fewer structural data are available in the absence of inhibitor, in part due to the problems associated with autoproteolysis and decreased stability of PR in the absence of inhibitors. These inhibitor-free structures show a variety of flap conformations, ranging from closed to semi-open and wide-open, in which the flap hairpin moieties have moved apart by several angstroms compared to the closed conformation.[7] Interestingly, differences between the different subtypes of HIV have also been reported. For example, while the unbound PR isolated from subtype A HIV only has been crystallized in a closed conformation, for subtype B six inhibitor-free structures were found in a semi-open conformation, two in a wide-open and only a single one in the closed conformation.[8] These data cannot unambiguously distinguish whether this heterogeneity reflects the preferred flap conformations or results from crystal packing effects.
The present study addresses the question concerning the preferred flap orientations by using solution NMR spectroscopy to record residual dipolar couplings (RDCs) in a B-type active site mutant PR in the absence of inhibitor. These RDCs can be measured by imposing a very slight deviation from the random, isotropic distribution of macromolecules in an NMR sample and are very sensitive reporters on the time-averaged orientation of the corresponding internuclear vectors.[9, 10] RDCs for the backbone N-H amide vectors were measured here for an inhibitor-free PR and a symmetric inhibitor DMP323-bound complex[11], as well as for an unbound drug-resistant PR bearing 20 mutations (PR20) plus a D25N mutation at the active site.[12] Weak alignment of the NMR samples was obtained by the addition of squalamine, an antibiotic isolated from dogfish shark, which was recently shown to form a lyotropic liquid crystal at very small volume fractions in water.[13] The lack of any direct interaction between squalamine and PR was evidenced by the complete absence of any detectable change in chemical shifts in the 1H-15N chemical shift correlation spectrum, which is an exquisitely sensitive reporter of such interactions when present. Although amide RDCs alone are typically insufficient to build a protein structure de novo, they are exceptionally well suited to evaluate agreement between the actual state of the protein present in solution and the coordinates seen in different crystal structures, and that is the approach taken in our study.
Here, we choose three crystal structures as reference models for representing the three different conformations of the flaps: PDB entry 3BVB [14] for the closed conformation, 1HHP[15] as the most widely used model for the semi-open conformation, and 1TW7[16] for the wide-open conformation (Figure 1B). For evaluating movement of the flaps, we define the PR Core as the region of the protein dimer for which predicted amide N-H vector orientations, and thereby the predicted 1DNH RDCs, are most invariant between these three structural models. For this invariant Core region, excellent agreement between measured 1DNH RDCs and best-fitted values was obtained for all three X-ray structures and the experimental data collected for unbound and DMP-bound PR, as well as the inhibitor-free PR20 mutant, as reflected in Q-factors close to 20% (reported as Q-Core in Figure 2). This Core region comprises the first and last two β-sheets (residues 10-23 and 62-73) as well as the sole α-helix (residue 87-93). The five alignment tensor parameters determined for each structural model from a singular value decomposition (SVD) fit to the Core region were then fixed and used to predict 1DNH RDCs for the flap region (Table 1). Here, we define the flaps as extended regions that include both the T80 loop and G40 loop (Figure 1A), since these two hinge segments are also involved in the opening and closing of the flaps. They are relatively invariant between the different inhibitor-free and inhibitor-bound X-ray structures (Cα root mean square difference to average of <0.4 Å).
Figure 2.

Fit of the NH RDCs measured for the inhibitor-free and DMP-bound PR to the closed (A and B), semi-open (C and D) and wide-open (E and F) structural models. The fit of the Core region RDCs (residues 10-23, 62-73 and 87-93) are shown in black. RDCs corresponding to the flaps (residues 30-61 and 74-84) are displayed in red, with predicted values derived using the alignment tensor parameters of the Core region. The quality factor, Q, reflects the rmsd between observed RDCs and those predicted by the structure, normalized for the range of RDCs observed for each sample: Q = rms(Dcalc-Dobs)/rms(Dobs), where rms is the root-mean-square function, Dobs and Dcalc refer to the observed RDC value and the RDC calculated based on the atomic coordinates, when using a best-fitted alignment tensor[17]. To avoid an effect from the non-random distribution of the 15N-1H vectors in the protein, rms(Dobs) is actually calculated as {Da2[4+3R2]/5}1/2, where Da and R are the magnitude and rhombicity of the best-fitted alignment tensor[18]. The experimental uncertainty in Dobs is very small (±0.15 Hz), but due to the uncertainty in the exact positions of the hydrogens, used for deriving Dcalc, even for a perfect X-ray structure the lower limit on Q is ca 0.15.
Table 1.
SVD-derived alignment tensor parameters obtained by a fit of the 1DNH RDCs to the Core region of the corresponding structural models.a
| Da (Hz) | Rh | Euler(x) | Euler(y) | Euler(z) | |
|---|---|---|---|---|---|
| PR + DMP b
3BVB |
2.92 | 0.13 | 43.8 | −20.8 | 53.6 |
| PR + DMP b
1HHP |
2.91 | 0.12 | 40.2 | −17.5 | 56.3 |
| PR + DMP b
1TW7 |
2.84 | 0.15 | 43.3 | −21.1 | 54.9 |
| PR apo c
3BVB |
2.91 | 0.11 | 41.9 | −19.1 | 52.8 |
| PR apo c
1HHP |
2.98 | 0.11 | 39.0 | −14.2 | 53.8 |
| PR apo c
1TW7 |
2.79 | 0.12 | 41.2 | −20.0 | 51.5 |
| PR20 apo d
3UF3 |
3.12 | 0.17 | 40.5 | −18.8 | 51.6 |
| PR20 apo d
3UCB |
3.01 | 0.16 | 39.6 | −14.4 | 46.3 |
The Core region comprises the first and last two β-sheets (residues 10-23 and 62-73) as well as the C-terminal helix (residue 87-93). The magnitude of the alignment tensor obtained for the apo-PR and PR20 data sets was scaled up according to the 2H quadrupole splitting measured for the PR + DMP323 sample (25.6 Hz, compared to 22 Hz and 19 Hz for the apo PR and PR20 respectively).
RDC data were collected for PR bound to DMP323
RDC data collected for inhibitor-free PR
RDC data collected for inhibitor-free PR20
As expected for an inhibitor-bound complex, the flap RDCs measured for the PR in the presence of DMP323 agree much better with the coordinates of the closed X-ray structure, 3BVB, than those of the semi-open (1HHP) and wide-open (1TW7) reference structures, yielding Q-factors of 18.6 %, 32.3% and 75.8%, respectively (Figure 2). Interestingly, much better agreement is also obtained when comparing the RDCs of the inhibitor-free PR flaps to the closed structure, 3BVB (Q-flap=22.9%) than for the semi-open (Q-flap=42.9%) and wide-open (Q-flap=73.6%) references (Figure 2). For both the inhibitor-free and DMP-bound states, the hinge residues T80 and E35 display the largest deviations between the experimental and predicted values when the flap RDCs are compared to the wide-open structure 1TW7 (Figure 2E and 2F). In this respect, it is important to note that even for the very best X-ray structures of any protein, RDC Q factors rarely will fall below ca 15-20%, mostly limited by the fact that X-ray maps do not contain adequate electron density for defining the precise position of hydrogens, and these are typically added by model-building assuming idealized geometry[13, 18].
To verify that the backbone RDCs can capture a truly open flap conformation, we also measured RDCs for a protease (PR20) bearing 20 mutations, found clinically in a patient with high protease drug resistance[19]. PR20 exhibits a 3-fold higher dimer dissociation constant, a similar catalytic constant (kcat), ~13-fold higher Km for the hydrolysis of a synthetic substrate and >3 orders of magnitude decreased affinity for the clinical protease inhibitors darunavir and saquinavir relative to the wild-type PR.[12] The crystal structure of inhibitor-free PR20 reveals wide-open conformations of the flaps, similar to those seen in 1TW7, while in the inhibitor-bound PR20 the flaps were found in their usual, closed conformation.[20]
We also measured the 1DNH RDCs for unbound PR20 under the same conditions as for inhibitor-free and DMP-bound PR and used the two corresponding PR20 crystal structures 3UCB and 3UF3 as reference structures for the closed and wide-open conformations, respectively. As for PR, the alignment tensor parameters were determined by an SVD fit of the PR20 RDCs to the Core region, and these parameters were subsequently used to predict the RDCs for the extended flap regions (Table 1). The experimental RDCs show much closer agreement with values predicted for the wide-open PR20 conformation (Fig. 3A; Q-flap=21.9%) than those for the closed conformation (Fig. 3B; Q-flap=67.8%). Therefore, in contrast to inhibitor-free PR, the RDCs measured for PR20 clearly indicate that the flaps adopt a wide-open conformation in the unbound state. The large difference between the inhibitor-free conformation of PR and PR20 likely arises from the mutations in the flap hinge region (E35D, M36I, and S37N), which eliminate several native interactions that have been postulated to impact the orientation of the flaps.[20]
Figure 3.
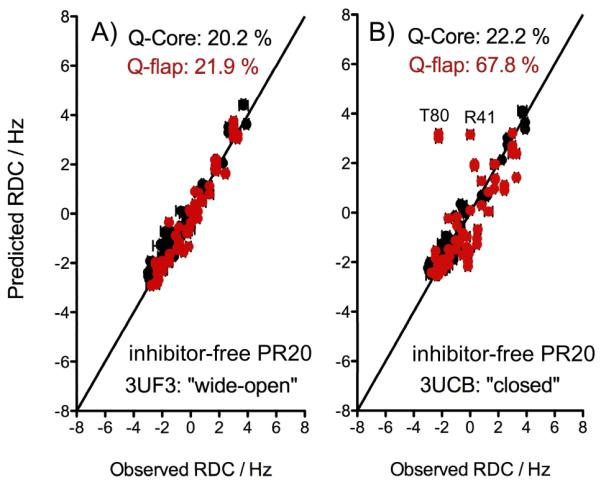
Correlation between 1DNH RDCs measured for the unbound PR20 and (A) wide-open or (B) closed structural models. RDCs corresponding to the Core region are shown in black and were used to determine the alignment tensor through SVD fitting, while flap RDCs are shown in red, with predicted values derived using alignment tensor parameters obtained for the Core residues.
NMR relaxation data measured for inhibitor-free PR revealed dynamic fluctuations of the flap tips on the μs-ms time scale,[21] which have been commonly interpreted as reflecting a dynamic equilibrium between semi-open and wide-open conformations.[3-5] Combining the crystallographic and NMR data with MD simulations, a popular model of the PR binding mechanism has emerged over the past years. In this model, the inhibitor-free protease adopts a semi-open conformation with rare and transient wide opening of the flaps that would allow smooth access of the natural poly-proteins into the active site. After the substrate binds to the active site, the flaps close and then adopt the conformation observed in the protease-inhibitor complexes.
However, it is important to realize that the NMR relaxation data cannot distinguish a semi-open to wide-open conformational transition from any other mode of structural fluctuation. The backbone RDCs measured in the present study clearly show that the average conformation of the inhibitor-free PR in solution is very similar to the closed conformation observed for PR-inhibitor complexes. The NMR relaxation data reported by Freedberg et al.[21] on the inhibitor-free PR therefore may be reinterpreted in view of the present data as reflecting transient switching to a small population of semi-open, or possibly wide-open conformations, rather than between semi-open and wide-open conformations. Indeed, a recent MD simulation study shows a transversal procession of the N-terminal cleavage site sequence (TFR/PR) through the active site, without wide opening of the flaps.[6] A complete opening of the flaps therefore may not be required for the catalytic mechanism of PR. In this regard, it is also interesting to note that the flap RDCs measured in the inhibitor-free state are not systematically reduced relative to those seen in the Core region, indicating that dynamic averaging of these RDCs, which is integrated over the time scale of milliseconds in such RDC measurements,[22, 23] is minimal. Our data therefore paint a picture in which the flaps, with the exception of their tips, have a structural rigidity that is comparable to the Core region of the dimer and predominantly adopt the closed conformation even in the absence of substrate or ligands. RDCs predicted for the semi-open state differ only modestly from those of the closed state, and our RDC data suggest an upper limit of at most ~25% for the semi-open state. If the alternate state is the wide-open configuration, for which the predicted RDCs differ substantially more from the closed state, an upper limit of ca 10% for the population of this state applies.
Our results demonstrate that measurement of RDCs provides a convenient method for identifying the loop conformations actually present in solution from a range of possibilities suggested by available crystal structures. This type of study is analogous to earlier work on other flexible protein systems, such as hemoglobin[24] and lysozyme.[25] For each of these, two different liquid crystalline media were used to prove that the state of the protein is not impacted by the alignment medium. For PR, it was difficult to find liquid crystalline media compatible with both the free and inhibitor-bound states of the protein, and we were only able to get high quality data in the newly discovered squalamine liquid crystal. However, the fact that we can observe the wide-open state for PR20, versus the closed state for inhibitor-free PR, indicates that the protein is not forced to adopt any particular state by the presence of the liquid crystal.
The use of paramagnetic NMR, pseudo-contact shifts in particular, provides an alternate method for probing the structure of proteins subject to dynamic rearrangement[26], such as the state of the HIV-1 protease flaps in solution. This method recently was used to investigate the structure of the flap covering the active site in the heterodimeric dengue virus protease NS2B-NS3, which surprisingly also was found to adopt the closed conformation in the inhibitor-free state[27]. This approach requires that paramagnetic lanthanide tags be linked at suitable positions to the protein, but obviates the need for finding a suitable orienting medium needed for measurement of RDCs. In favorable cases, introduction of the paramagnetic tag will also yield weak protein alignment without requiring an orienting medium, then enabling unambiguous evaluation of the dynamic properties of the protein[28]. Both RDC and pseudo-contact shift methods therefore are particularly useful for probing the state of dynamic regions in isotropic solution, under conditions that can closely mimic the relevant physiological environment.
Experimental Section
Uniformly 2H/13C/15N-enriched (>98%) protease samples were purified from inclusion bodies by size-exclusion chromatography under denaturing conditions followed by reverse-phase high-pressure liquid chromatography, as previously described.[29] Proteins were folded by dilution from a stock solution of 2 mg/ml in 12 mM HCL in 6.6 volumes of 5 mM acetate buffer pH 6.0, with or without DMP323 inhibitor and dialyzed extensively in 20 mM sodium phosphate, pH 5.7 and concentrated. The protease constructs used in this study contain an additional active-site D25N mutation to prevent autoproteolysis. The D25N mutation was introduced into PR20 by Quick-Change mutagenesis.
The 1DNH RDCs were derived from the difference in 1JNH + 1DNH splitting measured at 600 MHz using an ARTSY-HSQC experiment[30] on an isotropic sample and an aligned sample. All experiments were performed at 293 K. The alignment of the samples was obtained by the addition of 10 mg/ml squalamine and 5 mM hexanol, yielding a stable 2H quadrupole splitting of ~20 Hz. The average experimental error in the measured 1DNH RDCs is 0.15 Hz. Alignment tensor parameters are listed in Table 1.
Acknowledgements
We thank Annie Aniana, James L. Baber, and Jinfa Ying for technical assistance and Dennis A. Torchia for helpful discussions. We acknowledge support from the National Institute of Diabetes and Digestive and Kidney Diseases Advanced Mass Spectrometry Core. This work was funded by the NIH Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases and by the Intramural AIDS-Targeted Antiviral Program of the Office of the Director, NIH.
References
- [1].Louis JM, Ishima R, Torchia DA, Weber IT. Hiv-1: Molecular Biology and Pathogenesis: Viral Mechanisms. (2nd) 2007;55:261. [Google Scholar]
- [2].Torbeev VY, Raghuraman H, Hamelberg D, Tonelli M, Westler WM, Perozo E, Kent SBH. Proc. Natl. Acad. Sci. U. S. A. 2011;108:20982. doi: 10.1073/pnas.1111202108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hornak V, Okur A, Rizzo RC, Simmerling C. Proc. Natl. Acad. Sci. U. S. A. 2006;103:915. doi: 10.1073/pnas.0508452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Perryman AL, Lin JH, McCammon JA. Protein Sci. 2004;13:1434. doi: 10.1110/ps.03468904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hornak V, Simmerling C. Drug Discovery Today. 2007;12:132. doi: 10.1016/j.drudis.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kashif Sadiq S, Noe F, De Fabritiis G. Proc. Natl. Acad. Sci. U. S. A. 2012;109:20449. doi: 10.1073/pnas.1210983109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Heaslet H, Rosenfeld R, Giffin M, Lin Y-C, Tam K, Torbett BE, Elder JH, McRee DE, Stout CD. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2007;63:866. doi: 10.1107/S0907444907029125. [DOI] [PubMed] [Google Scholar]
- [8].Robbins AH, Coman RM, Bracho-Sanchez E, Fernandez MA, Gilliland CT, Li M, Agbandje-McKenna M, Wlodawer A, Dunn BM, McKenna R. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010;66:233. doi: 10.1107/S0907444909054298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Prestegard JH, Bougault CM, Kishore AI. Chem. Rev. 2004;104:3519. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- [10].Bax A, Grishaev A. Curr. Opin. Struct. Biol. 2005;15:563. doi: 10.1016/j.sbi.2005.08.006. [DOI] [PubMed] [Google Scholar]
- [11].Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang CH, Weber PC, Jackson DA, Sharpe TR, Ericksonviitanen S. Science. 1994;263:380. doi: 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- [12].Louis JM, Aniana A, Weber IT, Sayer JM. Proc. Natl. Acad. Sci. U. S. A. 2011;108:9072. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Maltsev AS, Grishaev A, Roche J, Zasloff M, Bax A. J. Am. Chem. Soc. 2014;136:3752. doi: 10.1021/ja4132642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sayer JM, Liu F, Ishima R, Weber IT, Louis JM. J. Biol. Chem. 2008;283:13459. doi: 10.1074/jbc.M708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Spinelli S, Liu QZ, Alzari PM, Hirel PH, Poljak RJ. Biochimie. 1991;73:1391. doi: 10.1016/0300-9084(91)90169-2. [DOI] [PubMed] [Google Scholar]
- [16].Martin P, Vickrey JF, Proteasa G, Jimenez YL, Wawrzak Z, Winters MA, Merigan TC, Kovari LC. Structure. 2005;13:1887. doi: 10.1016/j.str.2005.11.005. [DOI] [PubMed] [Google Scholar]
- [17].Cornilescu G, Marquardt JL, Ottiger M, Bax A. J. Am. Chem. Soc. 1998;120:6836. [Google Scholar]
- [18].Bax A. Protein Sci. 2003;12:1. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dierynck I, De Wit M, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K. J. Virol. 2007;81:13845. doi: 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Agniswamy J, Shen C-H, Aniana A, Sayer JM, Louis JM, Weber IT. Biochemistry. 2012;51:2819. doi: 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Freedberg DI, Ishima R, Jacob J, Wang YX, Kustanovich I, Louis JM, Torchia DA. Protein Sci. 2002;11:221. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tolman JR. J. Am. Chem. Soc. 2002;124:12020. doi: 10.1021/ja0261123. [DOI] [PubMed] [Google Scholar]
- [23].Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Science. 2008;320:1471. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- [24].Lukin JA, Kontaxis G, Simplaceanu V, Yuan Y, Bax A, Ho C. Proc. Natl. Acad. Sci. U. S. A. 2003;100:517. doi: 10.1073/pnas.232715799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Goto NK, Skrynnikov NR, Dahlquist FW, Kay LE. J. Mol. Biol. 2001;308:745. doi: 10.1006/jmbi.2001.4614. [DOI] [PubMed] [Google Scholar]
- [26].Cerofolini L, Fields GB, Fragai M, Geraldes CFGC, Luchinat C, Parigi G, Ravera E, Svergun DI, Teixeira JMC. J. Biol. Chem. 2013;288:30659. doi: 10.1074/jbc.M113.477240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].de la Cruz L, Thi HDN, Ozawa K, Shin J, Graham B, Huber T, Otting G. J. Am. Chem. Soc. 2011;133:19205. doi: 10.1021/ja208435s. [DOI] [PubMed] [Google Scholar]
- [28].Bertini I, Del Bianco C, Gelis I, Katsaros N, Luchinat C, Parigi G, Peana M, Provenzani A, Zoroddu MA. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6841. doi: 10.1073/pnas.0308641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ishima R, Torchia DA, Louis JM. J. Biol. Chem. 2007;282:17190. doi: 10.1074/jbc.M701304200. [DOI] [PubMed] [Google Scholar]
- [30].Fitzkee NC, Bax A. J. Biomol. NMR. 2010;48:65. doi: 10.1007/s10858-010-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]