

Abstract
Small/intermediate conductance KCa channels (KCa2/3) are Ca2+/calmodulin regulated K+ channels that produce membrane hyperpolarization and shape neurologic, epithelial, cardiovascular, and immunologic functions. Moreover, they emerged as therapeutic targets to treat cardiovascular disease, chronic inflammation, and some cancers. Here, we aimed to generate a new pharmacophore for negative-gating modulation of KCa2/3 channels. We synthesized a series of mono- and dibenzoates and identified three dibenzoates [1,3-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate) (RA-2), 1,2-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate), and 1,4-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate)] with inhibitory efficacy as determined by patch clamp. Among them, RA-2 was the most drug-like and inhibited human KCa3.1 with an IC50 of 17 nM and all three human KCa2 subtypes with similar potencies. RA-2 at 100 nM right-shifted the KCa3.1 concentration-response curve for Ca2+ activation. The positive-gating modulator naphtho[1,2-d]thiazol-2-ylamine (SKA-31) reversed channel inhibition at nanomolar RA-2 concentrations. RA-2 had no considerable blocking effects on distantly related large-conductance KCa1.1, Kv1.2/1.3, Kv7.4, hERG, or inwardly rectifying K+ channels. In isometric myography on porcine coronary arteries, RA-2 inhibited bradykinin-induced endothelium-derived hyperpolarization (EDH)–type relaxation in U46619-precontracted rings. Blood pressure telemetry in mice showed that intraperitoneal application of RA-2 (≤100 mg/kg) did not increase blood pressure or cause gross behavioral deficits. However, RA-2 decreased heart rate by ≈145 beats per minute, which was not seen in KCa3.1−/− mice. In conclusion, we identified the KCa2/3–negative-gating modulator, RA-2, as a new pharmacophore with nanomolar potency. RA-2 may be of use to generate structurally new types of negative-gating modulators that could help to define the physiologic and pathomechanistic roles of KCa2/3 in the vasculature, central nervous system, and during inflammation in vivo.
Introduction
Small-conductance Ca2+-activated K+ channels (KCa2) (Köhler et al., 1996; Adelman et al., 2012) and the intermediate-conductance Ca2+-activated K+ channel (KCa3.1) (Ishii et al., 1997) are Ca2+/calmodulin (CaM)-regulated and voltage-independent K+ channels (Wei et al., 2005). Their activation produces solid membrane hyperpolarization that in turn influences electrical excitability and shapes calcium entry through calcium-permeable channels. KCa2 channels (subtypes 2.1, 2.2, and 2.3) are expressed in excitable tissues such as neurons, skeletal muscle, adrenal gland, and heart, and some nonexcitable tissues such as the liver and vascular endothelium, with varying subtype-specific tissue expression profiles (Wei et al., 2005). In neurons, these channels underlie the apamin-sensitive medium afterhyperpolarization and regulate firing frequency as well as learning and memory (Adelman et al., 2012). In the cardiovascular system, KCa2 channels contribute to cardiac repolarization (Li et al., 2009; Diness et al., 2010), endothelium-derived hyperpolarization (EDH)–type arterial dilation in response to increased hemodynamics (Edwards et al., 2010; Milkau et al., 2010; Wulff and Köhler, 2013), and provide negative feedback on sympathetic tone (Taylor et al., 2003). With respect to pathophysiological relevant functions in humans, recent evidence suggests a role, particularly of KCa2.3, in lone atrial fibrillation (Diness et al., 2010; Ellinor et al., 2010), cancer cell migration and metastasis (Chantôme et al., 2013), and overactive bladder (Soder et al., 2013).
KCa3.1 channels are mostly expressed in nonexcitable tissues such as red and white blood cell lineages, secretory epithelia, and the vascular endothelium (Devor et al., 1996; Köhler et al., 2000; Wei et al., 2005; Wulff and Köhler, 2013). Here, hyperpolarization and K+ efflux through Ca2+-activated KCa3.1 channels regulate cell volume regulation (Vandorpe et al., 1998), fluid secretion (Devor et al., 1996), and—together with KCa2.3 channels—EDH-type arterial dilation, specifically to acetylcholine stimulation (Edwards et al., 2010; Wulff and Köhler, 2013). Initially, KCa3.1 was believed to be an exclusively nonneuronal channel (Ishii et al., 1997; Wei et al., 2005). However, recent evidence suggests possible expression in cerebellar Purkinje cells in rats (Engbers et al., 2012) and a role of the channel in behavior as suggested by the locomotor hyperactivity in KCa3.1−/− mice (Lambertsen et al., 2012). KCa3.1 channels have been patho-mechanistically implicated in human disease such as arterial endothelial dysfunction (Félétou et al., 2010); cancer growth; cancer cell migration, metastasis (D’Alessandro et al., 2013), and neo-angiogenesis (Grgic et al., 2005); organ fibrosis (Grgic et al., 2009); atherosclerosis (Toyama et al., 2008); neointima formation (Köhler et al., 2003; Tharp et al., 2008) and T cell responses (Wulff et al., 2007); and microglia activity (Kaushal et al., 2007). For extensive in-depth reviews, we refer the interested reader to Félétou et al. (2010), Wulff and Castle (2010), and Wulff and Köhler (2013). Considering the patho-mechanistic roles of the channels, blockers of KCa3.1 channels have emerged as potential drug candidates for the treatment of sickle-cell disease (Ataga and Stocker, 2009), immunosuppression (Wulff and Castle, 2010), asthma (Van Der Velden et al., 2013), fibrosis (Grgic et al., 2009), atherosclerosis (Toyama et al., 2008), and cancer (Ruggieri et al., 2012; D’Alessandro et al., 2013). Positive-gating modulators (activators) of KCa3.1, such as naphtho[1,2-d]thiazol-2-ylamine (SKA-31) (Sankaranarayanan et al., 2009) and 5-methylnaphtho[2,1-d]oxazol-2-amine (SKA-121) (Coleman et al., 2014), may improve endothelium-dependent vasodilation and lower blood pressure (Sankaranarayanan et al., 2009; Köhler et al., 2010; Damkjaer et al., 2012; Mishra et al., 2013; Radtke et al., 2013; Wulff and Köhler, 2013). Activators of KCa2 channels could be useful to treat epilepsy and ataxia (Shakkottai et al., 2011), while blockers may improve learning and memory. For a review, see Lam et al. (2013).
The existing small molecule blockers of KCa3.1 and KCa2 channels are mainly pore blockers—e.g., for KCa3.1: 5-[(2-chlorophenyl)(diphenyl)methyl]-1H-pyrazole (TRAM-34) (Wulff et al., 2001), 4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4-benzothiazin-3(4H)-one (NS6180) (Strøbæk et al., 2013), and ICA-17043 (Ataga and Stocker, 2009) that obstruct ion flow at the inner cavity of the channel or at the outer vestibule, i.e., for KCa2: 6,10-diaza-3(1,3)8,(1,4)-dibenzena-1,5(1,4)-diquinolinacyclodecaphane (UCL1684) (Rosa et al., 1998). However, a caveat is that they may enter the inner cavities of other channels and thereby exert unspecific effects at high dosage. At 10 μM, a concentration often used in cancer-related studies, NS6180 inhibits large-conductance KCa1.1 [maxi K], Kv1.3, and Kv11.1 (hERG) by more than 50%, while TRAM-34 blocks Kv1.3, Kv1.4, Kv7.2 + Kv7.3, and Nav1.4 (Strøbæk et al., 2013). Clotrimazole, a related KCa3.1 blocker and cytochrome P450 enzyme blocking antifungal inhibits TRPM8 channels (Meseguer et al., 2008) with submicromolar potencies, and has been shown to modulate TRPV1 and TRPA1 channels that act as receptors for noxius heat/pain and irritants, respectively, in nociceptive neurons (Meseguer et al., 2008), thus limiting the utility of these compounds for studying KCa3.1 functions at the organ or systemic levels. Therefore, negative-gating modulators of KCa2/3 channels, which interfere with channel gating, could be advantageous over the present pore blockers. To date, two negative-gating modulators, (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphthylamine (NS8593) (Jenkins et al., 2011) and (–)-N-{7-[1-(4-tert-butyl-phenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine [(−)-B-TPMF] (Hougaard et al., 2012) have been described for KCa2 channels. Recently, our screening of a series of phenols and polyphenols identified the synthetic fluoro-tri-benzoic ester, 13b (Lamoral-Theys et al., 2010) (Fig. 1A), as a negative-gating modulator of KCa2/3 channels (Oliván-Viguera et al., 2013). However, a disadvantage of 13b is that its high molecular weight (582) and its log P value of 6.0 violate the Lipinski et al. (2001) rule of five and make it unlikely that the compound will have oral bioavailability. Moreover, the structure-activity relationship accounting for channel inhibition has not been characterized yet. Therefore, we decided to explore the structure-activity relationship by synthesizing less lipophilic and smaller analogs. Three compounds [1,3-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate) (RA-2), 1,2-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate) (RA-3), and 1,4-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate) (RA-4)] (Fig. 1B) were identified as KCa2/3 pan inhibitors with negative-gating modulator properties and potencies in the nanomolar range. RA-2 inhibited EDH-type dilations in porcine coronary arteries (PCAs) and was found not to increase blood pressure in telemetry recordings despite reducing heart rate (HR).
Fig. 1.

Synthesis, structures, and selected properties of mono- and difluoro benzoates. (A) Structure of the parent compound, 13b. (B) Scheme of the synthesis and structures of RA-1 to RA-6, together with molecular weights and log P values.
Materials and Methods
Synthesis of Mono- and Dibenzoates.
As shown in Fig. 1B, monobenzoate benzyl 3-fluoro-4-hydroxybenzoate (RA-1) was obtained by reaction of benzylbromide with 3-fluoro-4-hydroxybenzoic acid. Dibenzoates RA-2, RA-3, and RA-4 were obtained by reaction of the corresponding bis(bromomethyl)benzene with 3-fluoro-4-hydroxybenzoic acid. The dibenzoate 1,3-phenylenebis(methylene) bis(4-acetamido-3-fluorobenzoate) (RA-5) was obtained by reaction of 1,3-bis(bromomethyl)benzene with 3-fluoro-4-acetamidobenzoic acid. The dibenzoate 5-(hydroxymethyl)-1,3-phenylene)bis(methylene) bis(3-fluoro-4-hydroxybenzoate) (RA-6) was obtained by reaction of 1,3-bis(bromomethyl)-5-hydroxymethylbenzene with 3-fluoro-4-hydroxybenzoic acid. 3-Fluoro-4-acetamidobenzoic acid was obtained by acetylation of 4-amino-3-fluorobenzoic acid. 1,3-bis(bromomethyl)-5-hydroxymethylbenzene was prepared following literature procedures (Díez-Barra et al., 2001). All reactions were performed in anhydrous dimethylformamide (DMF) at 105°C using sodium bicarbonate or potassium carbonate and yielded the corresponding mono- or dibenzoates in 33–60% yields. Log P values were calculated with the program ChemBioDraw-Ultra-13.0 (PerkinElmer Inc, Waltham, MA). Benzylbromide, 1,2-, 1,3-, and 1,4-bis(bromomethyl)benzene, 4-amino-3-fluorobenzoic acid, and 3-fluoro-4-hydroxybenzoic acid were purchased from Sigma-Aldrich (St. Louis, MO), Alfa-Aesar (Ward Hill, MA), or Fluorochem (Derbyshire, UK).
General Procedures and Physical Data.
Whenever possible, the reactions were monitored by thin layer chromatography, which was performed on precoated silica gel polyester plates. The products were visualized using UV light (254 nm) or ethanolic phosphomolybdic acid solution followed by heating. Column chromatography was performed using silica gel (Kieselgel 60, 230–400 mesh; Sigma-Aldrich). Melting points were determined in open capillaries using a Gallenkamp capillary melting point apparatus and were not corrected. Fourier transform infrared (IR) spectra were recorded as KBr pellets using a Thermo Nicolet Avatar 360 Fourier transform IR spectrometer (Thermo Fisher Scientific Inc., Waltham, MA); νmax values expressed in cm−1 are given for the main absorption bands. 1H NMR and 13C NMR spectra were acquired on a Bruker AV-400 (Wageningen UR, The Netherlands) spectrometer operating at 400 MHz for 1H NMR, 100 MHz for 13C NMR, and 376 MHz for 19F NMR at room temperature using a 5 mm probe. The chemical shifts (δ) are reported in parts per million and were referenced to the residual solvent peak. Coupling constants (J) are quoted in Hertz. The following abbreviations are used: s, singlet; d, doublet; m, multiplet; bs, broad singlet; and dd, doublet of doublets. High-resolution mass spectra (HRMS) were recorded using a Bruker Daltonics MicroToF-Q instrument from methanolic solutions unless otherwise indicated using the positive electrospray ionization (ESI+) mode.
Synthesis of 4-Acetamido-3-Fluorobenzoic Acid.
A mixture of 4-amino-3-fluorobenzoic acid (233 mg, 1.5 mmol) and acetic anhydride (459 mg, 425 µl, 4.5 mmol) in anhydrous pyridine (7 ml) was heated at 60°C overnight. Then, the mixture was concentrated in vacuo. Over the residue, water (6 ml) was added and then the aqueous solution was acidified with HCl 2N until pH = 1. The product precipitated and was collected by filtration and dried providing 281 mg (95%) of compound 4-acetamido-3-fluorobenzoic acid as a slightly brownish solid. 1H NMR (CD3OD, 400 MHz) δ 2.20 (s, 3H), 7.74 (dd, 1H, J = 11.5, J = 1.8), 7.80 (doublet of doublets of doublets, 1H, J = 8.5, J = 1.8, J = 0.9), 8.20 (dd, 1H, J = 8.1, J = 8.1); 13C NMR (CD3OD, 100 MHz) δ 24.2, 117.3 (d, 21.1), 123.6, 127.1 (d, 3.1), 128.2, 132.2 (d, 11.3), 153.8 (d, 243.9), 167.8 (d, 2.6), 171.5; 19F NMR (CD3OD, 376 MHz) δ -128.6.
Synthesis of RA-1.
A mixture of benzylbromide (171 mg, 1 mmol), 3-fluoro-4-hydroxybenzoic acid (156 mg, 1.0 mmol), and NaHCO3 sodium (101 mg, 1.2 mmol) in anhydrous DMF (15 ml) under an argon atmosphere was heated at 105°C under argon atmosphere overnight. The mixture was cooled and then saturated aqueous NaHCO3 (7 ml), saturated aqueous NaCl (10 ml), and AcOEt (50 ml) were added. The mixture was filtered through a pad of Celite and after decantation the aqueous layer was extracted with AcOEt (2 × 20 ml). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. Finally, the crude product was purified by silica gel column chromatography (eluent: AcOEt/hexane, 1:3) to afford 147 mg (60%) of compound RA-1 as a white solid. M.p. 93–94°C; IR (KBr): wave number (νmax) = 3330, 1684, 1621, 1592, 1522; 1H NMR (CDCl3, 400 MHz) δ 5.35 (s, 2H), 6.20 (bs, 1H), 7.00–7.07 (m, 1H), 7.32–7.48 (m, 5H), 7.78–7.83 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 66.9, 117.0 (d, J = 2.0), 117.2 (d, J = 19.6), 122.9 (d, J = 6.0), 127.2 (d, J = 3.2), 128.2, 128.3, 128.6, 135.8, 148.2 (d, J = 14.2), 150.4 (d, J = 237.6), 165.5 (d, J = 2.8); 19F NMR (CDCl3, 376 MHz) δ -139.7; HRMS (ESI+): calculated for C14H11FNaO3 [M + Na]+ 269.0584; found 269.0559.
General Procedure for the Synthesis of Bis(3-fluoro-4-hydroxybenzoates) from Dibromomethylbenzene Derivatives.
A mixture of the corresponding dibromide derivate (1.0 mmol), 3-fluoro-4-hydroxybenzoic acid (343 mg, 2.2 mmol), and NaHCO3 (210 mg, 2.5 mmol) in anhydrous DMF (15 ml) under an argon atmosphere was heated at 105°C overnight. The mixture was cooled and then saturated aqueous NaHCO3 (7 ml), saturated aqueous NaCl (10 ml), and AcOEt (50 ml) were added. The mixture was filtered through a pad of Celite and after decantation the aqueous layer was extracted with AcOEt (2 × 20 ml). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. Finally, the crude product was purified by silica gel column chromatography (eluent: AcOEt/hexane, 1:3).
RA-2.
Application of the general procedure to 1,3-bis(bromomethyl)benzene and 3-fluoro-4-hydroxybenzoic acid afforded 145 mg (35%) of compound RA-2 as a white solid. M.p. 165–166°C; IR (KBr): νmax = 3250, 1700, 1616, 1597, 1517; 1H NMR (CD3OD, 400 MHz) δ 5.27 (s, 4H), 6.85–6.95 (m, 2H), 7.34–7.35 (m, 3H), 7.46 (bs, 1H), 7.60–7.70 (m, 4H); 13C NMR (CD3OD, 100 MHz) δ 67.3, 118.3 (d, J = 20.0), 118.4 (d, J = 3.0), 122.6 (d, J = 6.0), 127.9 (d, J = 3.0), 128.6, 128.8, 129.9, 138.1, 151.3 (d, J = 12.9), 152.2 (d, J = 240.3), 166.8 (d, J = 2.7); 19F NMR (CD3OD, 376 MHz) δ -138.7; HRMS (ESI+): calculated for C22H16F2NaO6 [M + Na]+ 437.0807; found 437.0787.
RA-3.
Application of general procedure to 1,2-bis(bromomethyl)benzene and 3-fluoro-4-hydroxybenzoic acid afforded 156 mg (38%) of compound RA-3 as a white solid. M.p. 207–208°C; IR (KBr): νmax = 3281, 1703, 1619, 1523; 1H NMR (CD3OD, 400MHz) δ 5.45 (s, 4H), 6.80–6.90 (m, 2H), 7.35–7.45 (m, 2H), 7.48–7.53 (m, 2H), 7.54–7.60 (m, 4H); 13C NMR (CD3OD, 100 MHz) δ 65.5, 118.2 (d, J = 20.0), 118.4 (d, J = 3.0), 122.5 (d, J = 6.0), 127.9 (d, J = 3.0), 130.0, 131.4, 136.3, 151.5 (d, J = 12.9), 152.2 (d, J = 240.3), 166.8 (d, J = 2.7); 19F NMR (CD3OD, 376 MHz) δ -138.6; HRMS (ESI+): calculated for C22H16F2NaO6 [M+Na]+ 437.0807; found 437.0809.
RA-4.
Application of general procedure to 1,4-bis(bromomethyl)benzene and 3-fluoro-4-hydroxybenzoic acid afforded 138 mg (33%) of compound RA-4 as a white solid. M.p. 212–213°C; IR (KBr): νmax = 3269, 1684, 1617, 1600, 1530; 1H NMR (CD3OD, 400 MHz) δ 5.27 (s, 4H), 6.90–6.95 (m, 2H), 7.41 (s, 4H), 7.60–7.70 (m, 4H); 13C NMR (CD3OD, 100 MHz) δ 67.3, 118.3 (d, J = 19.9), 118.5 (d, J = 3.0), 122.6 (d, J = 6.0), 127.9 (d, J = 3.0), 129.4, 137.6, 151.4 (d, J = 12.8), 152.3 (d, J = 240.3), 166.9 (d, J = 2.6); 19F NMR (CD3OD, 376 MHz) δ -138.5; HRMS (ESI+): calculated for C22H16F2NaO6 [M+Na]+ 437.0807; found 437.0807.
RA-6.
Application of general procedure for the synthesis of esters from bromide derivatives to 1,3-bis(bromomethyl)-5-hydroxymethylbenzene and 3-fluoro-4-hydroxybenzoic acid afforded 125 mg (28%) of compound RA-6 as a white solid. 1H NMR (CD3OD, 400 MHz) δ 4.63 (s, 2H), 5.31 (s, 4H), 6.90–6.95 (m, 2H), 7.38–7.42 (m, 3H), 7.60–7.70 (m, 4H); 13C NMR (CD3OD, 100 MHz) δ 64.8, 67.4, 118.3 (d, J = 20.0), 118.5 (d, J = 3.0), 122.7 (d, J = 6.0), 127.4, 127.5, 128.0 (d, J = 3.0), 138.3, 143.8, 151.4 (d, J = 12.8), 152.3 (d, J = 240.3), 166.8 (d, J = 2.7); 19F NMR (CD3OD, 376 MHz) δ -138.5.
Synthesis of RA-5.
A mixture of 1,3-bis(bromomethyl)benzene (132 mg, 0.5 mmol), 4-acetamido-3-fluorobenzoic acid (217 mg, 1.1 mmol) and K2CO3 (173 mg, 1.25 mmol) in anhydrous DMF (7 ml) under an argon atmosphere was heated at 105°C overnight. The mixture was cooled and then saturated aqueous NaHCO3 (3 ml), saturated aqueous NaCl (5 ml), and AcOEt (25 ml) were added. The obtained mixture was filtered through a pad of Celite and after decantation the aqueous layer was extracted with AcOEt (2 × 10 ml). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. Recrystallization from methanol afforded 123 mg (50%) of the compound RA-5 as a brownish solid. M.p. 190–191°C; IR (KBr): νmax = 3305, 3262, 3192, 1719, 1676, 1620, 1607, 1541; 1H NMR (CD3OD, 400 MHz) δ 2.14 (s, 6H), 5.36 (s, 4H), 7.42–7.45 (m, 3H), 7.56 (bs, 1H), 7.70–7.80 (m, 4H), 8.23 (dd, 2H, J = 8.1, J = 8.1), 10.03 (bs, 2H); 13C NMR (CD3OD, 100 MHz) δ 23.7, 66.0, 115.9 (d, J = 21.2), 122.2, 124.9 (d, J = 6.9), 125.8 (d, J = 2.9), 127.2, 127.6, 128.7, 131.3 (d, J = 11.1), 136.3, 151.8 (d, J = 244.1), 164.2 (d, J = 2.5), 169.2; 19F NMR (CD3OD, 376 MHz) δ -124.7; HRMS (ESI+) (from DMF solution): calculated for C26H22F2N2NaO6 [M + Na]+ 519.1338; found 519.1307.
Cell Lines.
Human embryonic kidney cells (HEK293) stably expressing hKCa3.1 (a kind gift from Dr. Khaled M. Houamed, University of Chicago) (Cao and Houamed, 1999); hKv1.2-B82 cells (Werkman et al., 1992); mKv1.3-L929 cells (Grissmer et al., 1994); hKv7.4-HEK293 (a kind gift from Nicole Schmitt, University Copenhagen, Denmark); hERG-HEK293 (a kind gift from Craig January, University of Wisconsin, Madison, WI); hKCa2.1-HEK293 cells and rKCa2.2-HEK293 cells; hKCa2.3-COS7 cells (Sankaranarayanan et al., 2009); 3T3 fibroblasts (3T3-L1, ref# CL-173, American Type Culture Collection, Rockville, MD); U251 glioblastoma cells; and primary PCA endothelial cells (PCAECs) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and penicillin/streptomycin (all from Biochrom KG, Berlin, Germany). The PCAECs were isolated from hearts as described previously (Oliván-Viguera et al., 2013). Hearts were kindly provided by the local abattoir (Mercazaragoza, Zaragoza). Prior to patch-clamp experimentation, cells were trypsinized, seeded on cover slips in a NaCl bath solution (see below), and used for electrophysiological measurements within the same day.
Compounds and Chemicals.
Compounds for synthesis and experimentation were purchased from Sigma-Aldrich, Tocris (Bristol, United Kingdom), Fluorochem, or Alfa Aesar. TRAM-34 (Wulff et al., 2000) and SKA-31 (Sankaranarayanan et al., 2009) were synthesized in the Wulff laboratory (Pharmacology, University of California at Davis, Davis, CA). Stock solutions (at 1 or 10 mM) of all compounds were prepared with dimethylsulfoxide (DMSO). The final DMSO concentration did not exceed 0.5% in single experiments testing one or more compounds.
Patch-Clamp Electrophysiology.
Inside-out and whole-cell membrane currents were recorded using an EPC10-USB patch-clamp amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany), U-ramps (−100 to 100 mV, 1 second), and Patchmaster software (HEKA Electronics) as described in more detail previously (Oliván-Viguera et al., 2013). The amplitudes of K+-outward currents were measured at 0 mV. For measurements of hERG currents, we used a prepulse to −80 mV (1 second), a depolarizing pulse to +30 mV (1 second), and a pulse to −40 mV (1 second). Leak subtraction was omitted during data acquisition, although ohmic leak was subtracted if appropriate. In fast whole-cell experiments on KCa channels, the K-pipette solution was composed of (in mM): 140 KCl, 1 MgCl2, 2 EGTA, 1.71 CaCl2 (1 µM [Ca2+]free) and 5 HEPES (adjusted to pH 7.2 with KOH). For measuring voltage-gated K+ channels, the pipette solution contained 100 nM [Ca2+]free (2 mM EGTA, 0.7 mM CaCl2). The NaCl bath solution was composed of (mM): 140 NaCl, 5 KCl, 1 MgSO4, 1 CaCl2, 10 glucose, and 10 HEPES (adjusted to pH = 7.4 with NaOH). For calculation of the IC50 values, data points were fitted using the dose-response equation: or the Boltzmann equation: . The inside-out experiments on hKCa3.1 shown in Fig. 2D were performed in symmetrical K+. The extracellular solutions contained (in mM): 154 KCl, 10 HEPES (pH = 7.4), 2 CaCl2, 1 MgCl2. Solutions on the intracellular side contained (in mM): 154 KCl, 10 HEPES (pH = 7.2), 10 EGTA, 1.75 MgCl2, and CaCl2 to yield calculated free Ca2+ concentrations of 0.05, 0.1, 0.25, 0.3, 0.5, 1, 10, and 30 µM. Free Ca2+ concentrations were calculated with the MaxChelator program (http://maxchelator.stanford.edu/) assuming a temperature of 25°C, a pH of 7.2, and an ionic strength of 160 mM. Cells were clamped to a holding potential of 0 mV and KCa currents were measured using 200 millisecond voltage ramps from −80 to 80 mV applied every 5 seconds.
Fig. 2.
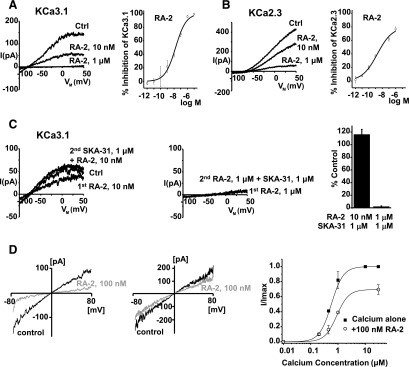
Negative-gating modulation of KCa2/3 channels by RA-2. (A) Left panel: inside-out patch-clamp experiments showing concentration-dependent inhibition of cloned hKCa3.1. (B) Left panel: inhibition of cloned hKCa2.3. Right panels (A and B): concentration-response curves. Data points (mean ± S.E.M.; KCa3.1: n = 4–10, experiments for each concentration; KCa2.3: n = 2–12, experiments each) were fitted with the Boltzmann equation (A) or the dose-response equation (B). (C) The positive-gating modulator SKA-31 (1 µM) reversed KCa3.1 channel inhibition at nanomolar (left panel) but not at micromolar concentrations of RA-2 (right panel). Lower panel: quantitative data (mean ± S.E.M.; n = 3, experiments for each concentration). (D) Left and middle panels: representative currents from inside-out patches in the presence of 500 nM (top) and 30 µM (bottom) intracellular Ca2+ before and after application of 100 nM of RA-2. Right panel: Ca2+-concentration response curve for KCa3.1 activation measured from inside-out patches in the absence or presence of 100 nM RA-2. Currents from individual patches were normalized to the effect of 10 µM Ca2+ in the absence of RA-2. Data are mean ± S.D. (n = 3, experiments per data point).
Myography on PCAs.
Isometric myography on PCA rings was done as described in detail previously (Alda et al., 2009; Valero et al., 2011). In brief, rings of arteries were mounted onto an isometric force transducer (Pioden UF1, Graham Bell House, Canterbury, UK). The bath containing Krebs buffer (37°C; equilibrated with 95% O2/5% CO2) consisted of (in mM): NaCl 120, NaHCO3 24.5, CaCl2 2.4, KCl 4.7, MgSO4 1.2, KH2PO4 1, and glucose 5.6, pH = 7.4. Rings were prestretched to an initial tension of 1 g (10 mN). Changes in force were registered using a Mac Laboratory System/8e program (AD Instruments, Inc., Milford, MA) at a sample rate of 0.5 seconds. To analyze EDH-type relaxation, the buffer contained the NO-synthase blocker, Nω-nitro-L-arginine (L-NNA, 300 µM), and the cyclooxygenase blocker, indomethacin (10 µM). Rings were precontracted with the vasospasmic thromboxane analog, (Z)-7-[(1S,4R,5R,6S)-5-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid (U46619) (0.2 µM) in the presence of RA-2 or its vehicle DMSO, followed by relaxation with BK (1 µM). Thereafter, rings were fully contracted with KCl (60 mM) buffer for 10 minutes, followed by addition of sodium nitroprusside (10 µM) to produce endothelium-independent relaxation.
Stock solutions of compounds were made in DMSO and appropriate amounts were added to the bath. Other compounds were dissolved in Milli-Q water. Data analysis: Relaxations were determined as percentage change of precontraction and relative to the totally relaxed state (absence of the contracting agent).
Blood Pressure Telemetry.
Telemetry was performed as described previously (Brähler et al., 2009; Radtke et al., 2013). Animal protocols were in accordance with Animal Research: Reporting of In Vivo Experiments guidelines and approved by the Institutional Animal Care and Use Committee of the University of Zaragoza and IACS (Comisión Ética Asesora [CEA]; permit no. PI01/13). In brief, TA11PA-C10 pressure transducers (Data Sciences International, St. Paul, MN) were implanted into the left carotid artery of four adult female wild-type (WT) and three adult female KCa3.1−/− (Brähler et al., 2009) under deep anesthesia as described previously (Radtke et al., 2013). Mice were allowed to recover for 10 days until reaching normal day night rhythm. Mice had free access to tap water and standard chow. Telemetry data were recorded over 1 minute every 10 minutes over 24 hours and averaged. Data were analyzed using the Data Sciences International software.
The compound or vehicle (peanut oil) was injected during the third hour of the dark phase (activity phase) and we collected telemetry data after 20–30 minutes after injection. To minimize stress and pain caused by intraperitoneal injections, mice were briefly anesthetized by isoflurane inhalation. After a first injection, animals were reused for injections of a higher dose of RA-2 or vehicle. Preparation and injection of RA-2: Appropriate amounts of RA-2 were dissolved in warmed peanut oil (Sigma-Aldrich) to give a dose of 3, 30, or 100 mg/kg. Injection volume was ≤600 µl.
Pharmacokinetics.
RA-2 was dissolved in peanut oil and 30 mg/kg were administered intraperitoneally to female C57Bl6J mice (20–30 g, 3–5 month old, n = 3 per time point) as described above. Mice were sacrificed at 1, 2, 4, 8, 24, or 48 hours after injection. Blood was taken by puncture of the right ventricle under CO2 anesthesia (permit no. PI01/13), transferred into EDTA-containing tubes, and stored on ice until further processing. EDTA-blood was centrifuged at 1000g for 10 minutes and 4°C and plasma was stored at −20°C. Tissue samples (liver, brain, and femoral skeletal muscle) were excised, frozen, and stored at −20°C until use.
Plasma (50 µl) was added to 950 µl acetonitrile, vortexed and centrifuged at 13,500g for 30 minutes at room temperature to precipitate the protein. The supernatant was transferred to a 4 ml vial, concentrated to dryness and reconstituted with acetonitrile to 150 µl. Tissue samples (200 mg) were homogenized in 1 ml of H2O with a Brinkman Kinematica PT 1600E homogenizer (Brinkman Kinematica, Eschbach, Germany) and protein precipitated with 1 ml of acetonitrile. The samples were then centrifuged at 13,500g for 30 minutes. The supernatants were concentrated to dryness and reconstituted in 200 µl of acetonitrile. Liquid chromatography and mass spectrometry analysis was performed with a Waters Acquity UPLC (Waters, New York) equipped with a Acquity UPLC BEH 1.7 µm RP-8, 2.1 × 150 mm column (Waters) interfaced to a TSQ Quantum Access Max mass spectrometer (ThermoFisher Scientific, Waltham, MA). Using an atmospheric pressure chemical ionization mass spectrometer and selective reaction monitoring (capillary temperature 327°C, vaporization temperature 350°C, collision energy 38 eV, negative ion mode), RA-2 was quantified by its base peak of 110.2 m/z and its concentration was calculated with a four-point calibration curve from 10 nM to 1 μM. The mobile phase consisted of acetonitrile and water, both containing 0.2% acetic acid with a flow rate of 0.300 ml/min. The gradient was ramped from 95/5 water/acetonitrile to 5/95 acetonitrile/water over 2 minutes and returned to 95/5 water/acetonitrile after 6 minutes. RA-2 was introduced onto the column with an injection volume of 6 µl. Using these conditions, RA-2 had a retention time of 3.78 minutes. Liver metabolites were analyzed under full scan mode without collision energy with a mass range of 150−1500 m/z. The mobile phase consisted of an isocratic gradient (90/10 acetonitrile/water). Under these conditions, RA-2 had a retention time of 1.76 minutes and liver metabolites eluted at 1.74, 2.71, and 3.11 minutes.
Statistics.
Data are given as mean ± S.E.M. if not stated otherwise. For comparison of data sets we used the unpaired Student’s t test or one-way analysis of variance followed by the Tukey post hoc test in the case of multiple comparisons. P values of <0.05 were considered significant.
Results
We previously identified 13b (Fig. 1A) as a negative-gating modulator of KCa2/3 channels (Oliván-Viguera et al., 2013). However, 13b’s drug likeness is poor because of its high molecular weight and high lipophilicity, limiting its in vivo bioavailability. Therefore, we synthesized a series of smaller and less lipophilic mono- and dibenzoates that met Lipinski’s rule of five for a drug-like compound (Lipinski et al., 2001) (Fig. 1). RA-1, RA-2, RA-3, RA-4, and RA-6 (Fig. 1B) were obtained from the corresponding mono- or dibromide-3-fluoro-4-hydroxybenzoic acid. RA-5 was obtained from 1,3-bis(bromomethyl)benzene and 4-acetamido-3-fluorobenzoic acid, which in turn was obtained by acetylation of 4-amino-3-fluorobenzoic acid.
Electrophysiology.
We performed inside-out and whole-cell patch-clamp experiments on cloned hKCa3.1 and hKCa2.3 as well as native KCa3.1/KCa2 in PCAECs and a series of distantly related K channels to test efficacy and selectivity.
Removal of lipophilic branches from the parent compound led to the following results: Compound RA-1 with only one benzoate ring had no considerable inhibitory activity on KCa3.1 (Table 1). The 1,3-dibenzoate RA-2 was a potent KCa3.1 and KCa2.3 inhibitor with IC50 values of 17 ± 3 nM and 2 ± 1 nM as determined in inside-out recordings on hKCa3.1 in HEK-293 cells and hKCa2.3 in COS7 cells (Fig. 2, A and B; for numeric data, see Supplemental Table 1 and Table 1). RA-2 similarly inhibited cloned hKCa2.1 and rKCa2.2 at nanomolar concentrations (Supplemental Table 1). We obtained similar IC50 values in whole-cell experiments on murine KCa3.1 expressed in 3T3-fibroblasts (Supplemental Fig. 1A). The 1,2- and 1,4-dibenzoate derivatives, RA-3 and RA-4, showed a similar potency with an IC50 value for RA-3 of 6 ± 2 nM (Supplemental Fig. 1B; Table 1). Compound RA-5, in which the hydroxyl group on the benzoic acid moiety had been substituted by an acetamido group (Fig. 1B), had no considerable inhibitory activity (Table 1). Introduction of a hydroxymethyl substituent in the 5-position of the central aromatic core (RA-6, Fig. 1B) led to a loss of activity (Table 1). It is noteworthy that 3-fluorobenzoic moieties (4-hydroxy-3-fluorobenzoic acid and 4-amino-3-fluorobenzoic acid) and 1,3-phenylenedimethanol as potential metabolites of RA-2/RA-5 hydrolysis, respectively, did not modify KCa3.1 currents.
TABLE 1.
Inhibitory efficacy of mono- and difluorobenzoates on hKCa3.1
Data are derived from inside-out patch-clamp experiments on hKCa3.1 stably expressed in HEK293 cells. Data are given as mean ± S.E.M., n ≥ 3.
| Compound |
IC50 |
Control at 1 μM |
Control at 5 μM |
Control at 10 μM |
|---|---|---|---|---|
| % | ||||
| 4-Amino-3-fluorobenzoic acid | NT | NT | 90 ± 3 | |
| 4-Hydroxy-3-fluorobenzoic acid | NT | NT | 80 ± 8 | |
| 1,3-Phenylenedimethanol | NT | NT | 106 ± 8 | |
| RA-1 | 94 ± 1 | 87 ± 2 | NT | |
| RA-2 | 17 ± 3 nM | 4 ± 1 | NT | NT |
| RA-3 | 6 ± 2 nM | 12 ± 2 | 2 ± 1 | NT |
| RA-4 | 15 ± 6 | NT | NT | |
| RA-5 | 97 ± 2 | 94 ± 1 | NT | |
| RA-6 | 83 ± 3 | 74 ± 11 | NT | |
NT, not tested.
Similar to our previous observations with the template 13b (Oliván-Viguera et al., 2013), we found that the positive-gating modulator, SKA-31, was capable of reversing the inhibition caused by nanomolar but not micromolar concentrations of RA-2 (Fig. 2C). This suggested (1) antagonism of the two compounds; (2) inhibitory actions of RA-2 similar to that of 13b; and (3) that RA-2 acted as a negative-gating modulator again similar to 13b (Oliván-Viguera et al., 2013).
To determine if the effects of RA-2 were Ca2+ dependent as was the case with previously described negative-gating modulators such as NS8593 and its derivatives (Jenkins et al., 2011), we performed inside-out experiments, in which we varied the intracellular [Ca2+]i concentration and investigated the ability of 100 nM of RA-2 to inhibit KCa3.1 currents. As shown in Fig. 2D, 100 nM of RA-2 nearly completely inhibited the current elicited by 500 nM [Ca2+]i, while the same concentration had a smaller effect in the presence of saturating 30 μM [Ca2+]i. Ca2+ concentration-response curves obtained in the presence and absence of 100 nM of RA-2 (Fig. 2D, right-hand side) revealed that RA-2 shifted the curve to the right but also reduced the maximal current at 30 μM, suggesting that the compound exerted negative-gating modulation of KCa3.1.
With respect to selectivity, we found that RA-2 had no considerable inhibitory or activating effects at a concentration of 1 μM on the distantly related human KCa1.1 channel in U251 glioblastoma cells; cloned Kv1.2, Kv1.3, and Kv7.4 channels; the important cardiac hERG channels (Kv11.1); or native inwardly rectifying K+ channels in U251 cells. The data are summarized in Supplemental Table 1 and representative recordings are shown in Supplemental Figure 2.
Patch Clamp on Porcine Coronary Endothelium and Isometric Myography.
Considering RA-2 as one of the drug-like compounds of this series, we continued evaluating functional activity in an ex vivo test system, i.e., PCAs (Oliván-Viguera et al., 2013), in which KCa2/3 channels have been suggested to initiate—at least in part (Ge et al., 2000)—the so-called EDH-type of endothelium-dependent vasorelaxation (Edwards et al., 2010). In the present study, we first measured endogenous KCa2 and KCa3.1 currents in freshly isolated PCAEC and we found that 1 μM RA-2 virtually abolished SKA-31–activated composite KCa2/KCa3.1 currents and also fully inhibited the TRAM-34–insensitive KCa2-mediated current in these cells (Fig. 3A).
Fig. 3.
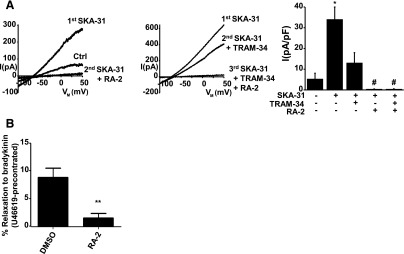
Negative-gating modulation of endogenous KCa3.1 and KCa2.3 channels in porcine endothelium and RA-2–evoked modulation of arterial contraction and relaxation of the PCA. (A) Left panel: potentiation of KCa2/KCa3.1 from preactivation levels (during infusion of 1 µM Ca2+) by 1 µM SKA-31 followed by current inhibition by 1 µM RA-2. Middle panel: partial inhibition of SKA-31–potentiated current by 1 µM TRAM-34 followed by complete inhibition of the TRAM-34–insensitive component by 1 µM RA-2. Right panel: summary data. Data points are mean ± S.E.M. (n = 3-8, arteries for each compound). *P < 0.01 SKA-31 versus Ctrl (without compounds); #P < 0.05; SKA-31 versus TRAM-34, RA-2, or TRAM-34 and RA-2; Student’s t test. (B) RA-2 inhibited BK (1 µM)-induced relaxation in rings strongly precontracted with U46619 (0.2 µM). Data points are mean ± S.E.M. (n = 5–6, arteries each); **P < 0.01.
Our isometric myography experiments in the presence of blockers of NO and prostacyclin synthesis (to specifically study EDH-type relaxation) showed that RA-2 at 1 μM did not modulate basal tone in these arteries (data not shown). However, RA-2 almost abolished the BK-induced relaxation in rings being strongly precontracted with a vasospasmic agent, the thromboxane analog, U46619 (Fig. 3B). RA-2 did not modulate sodium nitroprusside–induced and endothelium-independent relaxation of 60 mM KCl-contracted rings (data not shown). Taken together, RA-2 showed activity in PCAs by inhibiting EDH-type endothelium-dependent relaxation.
Systemic Cardiovascular Effects of RA-2.
In keeping with the expression of KCa3.1 and KCa2.3 channels in the vascular endothelium and their proposed roles in systemic cardiovascular regulation (Brähler et al., 2009), we next evaluated cardiovascular activity and selectivity of the pan-KCa2/3–negative-gating modulator, RA-2, by blood pressure telemetry in WT and KCa3.1−/− mice (Brähler et al., 2009). If compared with the vehicle, intraperitoneal injections of 3, 30, or 100 mg/kg RA-2 did not significantly change mean arterial blood pressure over 24 hours in the WT mice (Fig. 4A, for 30 mg/kg, and Supplemental Fig. 3A, for 3 and 100 mg/kg). However, RA-2 at 30 and 100 mg/kg significantly reduced HR. The reduction in HR started apparently 30–60 minutes after injection and reached lowest levels (≈Δ145 beats per minute) at the end of the activity phase and during the resting phase (Fig. 4B; Supplemental Fig. 3A, lower-right panel). RA-2 at the lower dose of 3 mg/kg produced a smaller reduction in HR (Supplemental Fig. 3A, lower-left panel). Besides the lower HR, wave forms were similar in the presence or absence of RA-2 and are shown in Supplemental Fig. 3B.
Fig. 4.

Systemic cardiovascular actions of RA-2 in mice. (A) Telemetric 24 hour measurements of mean arterial blood pressure (MAP) after intraperitoneal injections of 30 mg/kg (n = 4, experiments) RA-2 or vehicle (n = 6, experiments) into a total of 4 WT mice. (B) HR lowering effects of RA-2. (C) and (D) HR-lowering actions of 30 mg/kg RA-2 (n = 3, experiments on three mice) were absent in KCa3.1−/− mice. Black and white parts of the y-axis indicate dark and light phases. Arrow indicates time of injection (t = 0). Data points are mean ± S.E.M. Horizontal lines above or below data points indicate statistical significant difference from vehicle; *P < 0.05, Student’s t test.
We next evaluated to which extent the HR reducing effects of RA-2 in the WT mice depended on KCa3.1 channels and performed telemetry in KCa3.1−/− mice. Similar to WT mice, RA-2 at 30 mg/kg did not change the mean arterial blood pressure in the KCa3.1−/− mice (Fig. 4C, left-hand side). However, we did not find a reduction in HR in KCa3.1−/− mice (P < 0.05; Fig. 4D, right-hand side). Together, the telemetric monitoring revealed that RA-2 had no gross deleterious effects on blood pressure. Nonetheless, it is noteworthy that RA-2 reduced HR in a KCa3.1-dependent manner.
Pharmacokinetics of RA-2.
Analysis of tissue concentrations and distribution of RA-2 in plasma, brain, skeletal muscle, fat, and liver by a combination of ultra-high performance liquid chromatography and mass spectrometry (Fig. 5A) revealed that RA-2 plasma concentration was 136 nM at 1 hour after injection and 18 nM at 2 hours after injection, and that the compound was not detectable at later time points (Fig. 5B). RA-2 went rapidly into tissue (skeletal muscle, brain, and fat), where at 1 hour it reached concentrations of 1.7 µM (skeletal muscle), 91 nM (brain), and 6 µM (fat) (Fig. 5, C and D). As expected for an ester, RA-2 was rapidly cleared from the circulating blood within 1 hour (Fig. 5B), presumably via hydrolysis by plasma and hepatic esterases as indicated by the absence of RA-2 in liver and high amounts of possible metabolites in this tissue (Fig. 6). Compounds with the masses shown in Fig. 6 were identified in liver homogenate and were present in estimated concentrations of 1 to approximately 50 µM but could not be precisely quantified without reference compounds. The metabolites were likely eliminated via the bile since metabolites were not detectable in plasma at the same time point. In conclusion, intraperitoneal administration of RA-2 yielded tissue concentrations above the IC50 value for channel inhibition for 1–2 hours that fairly corresponded to the beginning of bradycardia (Fig. 4).
Fig. 5.

Pharmacokinetics of RA-2. (A) RA-2 plasma calibration curve with concentrations ranging from 10 nM to 1 µM (r2 = 0.9995). (B) RA-2 plasma concentrations following intraperitoneal application of 30 mg/kg (n = 3, mice). (C) Tissue concentrations 1 hour after intraperitoneal administration of RA-2 30 mg/kg (n = 3, mice). (D) Time course of tissue concentrations after intraperitoneal administration of RA-2 30 mg/kg (n = 3, mice). Data points are mean ± S.D.
Fig. 6.
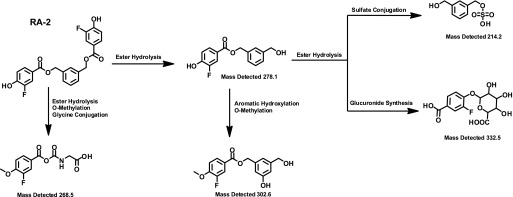
Possibly metabolites of RA-2 found in liver homogenate. Note that the parent compound, RA-2, was not found in the liver.
Discussion
The purpose of the present study was to synthesize potent and structurally novel pharmacophores for negative-gating modulation of KCa2/3 channels. We succeeded in identifying three fluoro-dibenzoates that inhibited KCa2/3 channels in the low nanomolar range and in a Ca2+-concentration-dependent manner and exhibited antagonism with a positive-gating modulator. The most drug-like compound RA-2 inhibited KCa2/3-initiated EDH-type relaxation and in vivo treatments did not show acute toxicity, although we found a KCa3.1-dependent reduction of HR.
Compared with 13b (Lamoral-Theys et al., 2010) that served as a template for structural modifications, compounds RA-2, RA-3, and RA-4, in which two of the three 3-fluoro-4-hydroxybenzoyloxymethyl substituents in 13b were conserved, showed inhibitory activity. RA-2 with both substituents in the 1,3 position was equally potent in inhibiting KCa3.1 (RA-2: 17 nM versus 13b: 19 nM) (Oliván-Viguera et al., 2013). In line with the notion that RA-2 is a pan-negative KCa2/3 modulator, all three subtypes of KCa2 channels were inhibited by RA-2 at nanomolar concentrations, although the IC50 value was higher for RA-2 (2 nM) than for 13b (360 pM) in the case of hKCa2.3. The structurally very similar compounds RA-3 and RA-4 with both substituents in the 1,2 and 1,4- positions, respectively, were also found to be potent KCa3.1 inhibitors. A major change in the structure such as the absence of an additional 3-fluoro-4-hydroxybenzoyloxymethyl substituent (as in RA-1), the substitution of the hydroxyl groups by acetamido groups on the benzoic acid moieties (as in RA-5), or the introduction of a hydroxymethyl substituent in the 5-position of the central aromatic core (as in RA-6) gave lower log P values but caused a loss of inhibitory efficacy. Thus, regarding the structure-activity relationship, this suggested that the intactness of the aromatic core as a lipophilic spacer and the presence of at least two 3-fluoro-4-hydroxybenzoyloxymethyl branches were critical for maintaining inhibitory efficacy. A change in the relative position of the two 3-fluoro-4-hydroxybenzoyloxymethyl branches had no major consequences with log P values (4.7) similar to RA-2.
Together, the three structurally similar compounds, RA-2, RA-3, and RA-4 were identified as potential pharmacophores for pan-negative modulation of KCa2/3 channels that had a better chemical profile with a lower molecular weight of 414 Da (RA-2, RA-3, and RA-4) and lower log P values of 4.7 than the starting compound 13b (mol. wt. = 582; log P = 6.0), suggesting in vivo utility.
Similar to 13b, RA-2 acted as a negative-gating modulator as concluded from the substantially more potent inhibition at nanomolar Ca2+ and the antagonism (relief of channel inhibition) by the positive-gating modulator, SKA-31. Structurally, the negative-gating modulation by RA-2 and its antagonism by the positive-gating modulator, SKA-31, could occur at the interface of CaM and the cytosolic C-terminal CaM-binding domain of the channels. Indeed, docking experiments using co-crystals of CaM and the C-terminal CaM-binding domain of KCa2.2 (Zhang et al., 2012) revealed that NS309 and 1-EBIO that are structurally related to SKA-31 bind in the interface between the CAM and the CAMBD to keep the channel in the open state. However, we do not wish to exclude that other allosteric effects may account for the functional antagonism observed here. Regarding selectivity, micromolar concentrations of RA-2 did not interfere with (block or activate) a series of members of distantly related K+ channel families, suggesting considerable selectivity for KCa2/3 over other K+ channels.
The utility of RA-2 as a new tool and/or drug candidate for in vivo use was demonstrated by the results of the present ex vivo and in vivo experiments on freshly isolated PCAEC and coronary arteries. The patch-clamp experiments showed that endogenous KCa2 and KCa3.1 currents in PCAEC were fully inhibited by RA-2, in this regard similar to 13b. The results from myography on PCAs showed that similar to 13b (Oliván-Viguera et al., 2013) RA-2 did not modulate basal tone, suggesting that KCa2/3 channels were not essentially involved in the control of basal arterial tone at least under these ex vivo conditions. Agonist-induced EDH-type relaxations independent of nitric oxide are known to require the activation of KCa2/3 channels and subsequent smooth muscle hyperpolarization in many vascular beds (Edwards et al., 2010). Here, we found that RA-2 inhibited BK-induced EDH-type relaxation in the presence of the vasospasmic agent, U46619. This inhibition was almost complete and the small RA-2–resistant relaxation could be explained by the contribution of other endothelium-derived relaxing factors such as eicosanoids, as shown previously (Fisslthaler et al., 1999). Nonetheless, these results showed that KCa2/3 activation by BK-induced endothelial stimulation was a critically step in EDH-type relaxation in strongly precontracted PCA, as also suggested previously by others (Ge et al., 2000). However, the specific contributions of KCa2/3 to this response were not elucidated thus far. Regarding the potency of inhibition by RA-2, it was noteworthy that RA-2 was more efficient than the more lipophilic template 13b that did not produce significant inhibition of BK-induced relaxation of PCAs in the presence of the vasospamic U46619 in our previous study (Oliván-Viguera et al., 2013).
Our telemetry in freely moving mice showed that the animals tolerated the RA-2 injections well at a dosage of up to 100 mg/kg per day (highest dose tested). There were no signs of acute toxicity. Alterations of arterial pressure were not evident, and therefore acute systemic KCa2/3 inhibition by RA-2 appeared to be safe. However, it should be noted that RA-2 was cleared from the circulation rapidly, presumably via hydrolysis by plasma and hepatic esterases. Nonetheless, RA-2 entered the brain and skeletal muscle where tissue concentrations reached values clearly above IC50 values within the first 2 hours after administration.
Albeit blood pressure changes were not evident, a single dose of RA-2 (30 or 100 mg/kg) produced an appreciable decrease in HR, which was apparent after injection and more pronounced during the end of the activity phase and the resting phase (≈Δ145 beats per minute). An obvious interpretation of these data is that this bradycardia might be an adaptation to an increase of total peripheral resistance—caused by endothelial KCa2/KCa3 inhibition in resistance size arteries—and baroreceptor activation to lower cardiac output and to maintain pressure constant.
In addition to expression in neurons and arteries, expression of KCa2 channels—in particular of the KCa2.2 subtype—have been reported in atrial myocytes (Tuteja et al., 2005; Li et al., 2009) and the atrioventricular node (Zhang et al., 2008). Indeed, blockers of KCa2 have been suggested to serve as antiarrhythmic drugs by prolonging repolarization times and thereby action potential duration, finally terminating atrial fibrillation (Diness et al., 2010). Thus, one alternative explanation for the lower HR might be that cardiac KCa2 inhibition by RA-2 caused prolongation of action potential duration in atrial tissues. An additional explanation could be that RA-2 altered transmission time at the level of the atrioventricular node.
Nonetheless, the precise mechanism by which RA-2 produced bradycardia remains to be identified in future studies. At present, we do not wish to exclude that by esterase-mediated hydrolysis RA-2 could be a source of diphenols with potential antioxidative properties. However, the observation that RA-2–induced bradycardia was not seen in KCa3.1-deficient mice suggested that an inhibition of KCa3.1 channels, but not of KCa2 channels, was mechanistically involved in the WT situation.
In conclusion, we identified RA-2 as a selective pan-negative-gating modulator of KCa2/3 channels with nanomolar potency and ex vivo and in vivo activity in coronary artery endothelium and in systemic cardiovascular regulation. We presumed RA-2 to be a novel pan-negative-gating modulator of KCa2/3 channels with a drug-like profile. Moreover, RA-2 can be considered a useful tool compound to study physiologic and pathophysiological roles of KCa2/3 channels in vitro or in vivo and may be of therapeutic utility to treat hypotension and undesired hyperemia as well as chronic inflammation and disorders characterized by abnormal cell proliferation.
Supplementary Material
Acknowledgments
The authors thank Dr. Eduardo Romanos-Alfonso and Susana Murillo-Pola from the Unit of Functional Evaluations of the IACS for excellent technical support (telemetry).
Abbreviations
- BK
bradykinin
- bs
broad singlet
- CaM
calmodulin
- d
doublet
- dd
doublet of doublets
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDH
endothelium-derived hyperpolarization
- ESI
electrospray ionization
- HEK
human embryonic kidney
- HR
heart rate
- HRMS
high-resolution mass spectra
- IR
infrared
- KCa
Ca2+-activated K+
- KCa2
small-conductance Ca2+-activated K+
- KCa3.1
intermediate-conductance Ca2+-activated K+
- m
multiplet
- PCA
porcine coronary artery
- PCAEC
porcine coronary artery endothelial cell
- s
singlet
- RA-1
benzyl 3-fluoro-4-hydroxybenzoate
- RA-2
1,3-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate)
- RA-3
1,2-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate)
- RA-4
1,4-phenylenebis(methylene) bis(3-fluoro-4-hydroxybenzoate)
- RA-5
1,3-phenylenebis(methylene) bis(4-acetamido-3-fluorobenzoate)
- RA-6
5-(hydroxymethyl)-1,3-phenylene)bis(methylene) bis(3-fluoro-4-hydroxybenzoate)
- SKA-31
naphtho[1,2-d]thiazol-2-ylamine
- TRAM-34
1-[(2-chlorophenyl) (diphenyl)methyl]-1H-pyrazole
- U46619
(Z)-7-[(1S,4R,5R,6S)-5-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid
- WT
wild-type
Authorship Contributions
Participated in research design: Oliván-Viguera, Wulff, Valero, Gálvez, Díaz-de-Villegas, Badorrey, Köhler.
Conducted experiments: Oliván-Viguera, Valero, Laría, Murillo, Coleman, Brown, Badorrey, Köhler.
Performed data analysis: Oliván-Viguera, Valero, Murillo, Badorrey, Köhler.
Wrote or contributed to the writing of the manuscript: Oliván-Viguera, Gálvez, Díaz-de-Villegas, Wulff, Badorrey, Köhler.
Footnotes
A.O.-V. and R.K. were supported by the Deutsche Forschungsgemeinschaft [KO1899/11-1]; European Community [FP7-PEOPLE-CIG-321721]; the Danish Hjerteforening, Department of Industry and Innovation, Government of Aragon [GIPASC-B105]; REFBIO Pyrenees Biomedical Network; and the Fondo de Investigación Sanitaria [Red HERACLES RD12/0042/0014]. J.A.G., M.D.D., and R.B. were supported from the Government of Aragón [GA E-102]. B.M.B. was supported by a NIGMS-funded Pharmacology Training Program [T32GM099608]. N.C. was supported by a NIHLB-funded Training Program in Basic and Translational Cardiovascular Science [T32HL086350]. H.W. was supported by the National Institute of Neurologic Disorders and Stroke (NINDS) [R21NS072585].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Adelman JP, Maylie J, Sah P. (2012) Small-conductance Ca2+-activated K+ channels: Form and function. Annu Rev Physiol 74:245–269. [DOI] [PubMed] [Google Scholar]
- Alda JO, Valero MS, Pereboom D, Gros P, Garay RP. (2009) Endothelium-independent vasorelaxation by the selective alpha estrogen receptor agonist propyl pyrazole triol in rat aortic smooth muscle. J Pharm Pharmacol 61:641–646. [DOI] [PubMed] [Google Scholar]
- Ataga KI, Stocker J. (2009) Senicapoc (ICA-17043): a potential therapy for the prevention and treatment of hemolysis-associated complications in sickle cell anemia. Expert Opin Investig Drugs 18:231–239. [DOI] [PubMed] [Google Scholar]
- Brähler S, Kaistha A, Schmidt VJ, Wölfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, et al. (2009) Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119:2323–2332. [DOI] [PubMed] [Google Scholar]
- Cao YJ, Houamed KM. (1999) Activation of recombinant human SK4 channels by metal cations. FEBS Lett 446:137–141. [DOI] [PubMed] [Google Scholar]
- Chantôme A, Potier-Cartereau M, Clarysse L, Fromont G, Marionneau-Lambot S, Guéguinou M, Pagès JC, Collin C, Oullier T, Girault A, et al. (2013) Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res 73:4852–4861. [DOI] [PubMed] [Google Scholar]
- Coleman N, Brown BM, Oliván-Viguera A, Singh V, Olmstead MM, Valero MS, Köhler R, Wulff H. (2014) New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1. Mol Pharmacol 86:342–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro G, Catalano M, Sciaccaluga M, Chece G, Cipriani R, Rosito M, Grimaldi A, Lauro C, Cantore G, Santoro A, et al. (2013) KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis 4:e773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damkjaer M, Nielsen G, Bodendiek S, Staehr M, Gramsbergen JB, de Wit C, Jensen BL, Simonsen U, Bie P, Wulff H, et al. (2012) Pharmacological activation of KCa3.1/KCa2.3 channels produces endothelial hyperpolarization and lowers blood pressure in conscious dogs. Br J Pharmacol 165:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Frizzell RA, Bridges RJ. (1996) Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca2+-dependent K+ channel. Am J Physiol 271:L775–L784. [DOI] [PubMed] [Google Scholar]
- Díez-Barra E, García-Martínez JC, Merino S, del Rey R, Rodríguez-López J, Sánchez-Verdú P, Tejeda J. (2001) Synthesis, characterization, and optical response of dipolar and non-dipolar poly(phenylenevinylene) dendrimers. J Org Chem 66:5664–5670. [DOI] [PubMed] [Google Scholar]
- Diness JG, Sørensen US, Nissen JD, Al-Shahib B, Jespersen T, Grunnet M, Hansen RS. (2010) Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol 3:380–390. [DOI] [PubMed] [Google Scholar]
- Edwards G, Félétou M, Weston AH. (2010) Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch 459:863–879. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, Sinner MF, de Bakker PI, Mueller M, Lubitz SA, et al. (2010) Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 42:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engbers JD, Anderson D, Asmara H, Rehak R, Mehaffey WH, Hameed S, McKay BE, Kruskic M, Zamponi GW, Turner RW. (2012) Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc Natl Acad Sci USA 109:2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M, Köhler R, Vanhoutte PM. (2010) Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep 12:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. (1999) Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401:493–497. [DOI] [PubMed] [Google Scholar]
- Ge ZD, Zhang XH, Fung PC, He GW. (2000) Endothelium-dependent hyperpolarization and relaxation resistance to N(G)-nitro-L-arginine and indomethacin in coronary circulation. Cardiovasc Res 46:547–556. [DOI] [PubMed] [Google Scholar]
- Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, Köhler R. (2005) Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol 25:704–709. [DOI] [PubMed] [Google Scholar]
- Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J, Müller A, Kaistha A, Schmidt C, Raman G, et al. (2009) Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc Natl Acad Sci USA 106:14518–14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. (1994) Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 45:1227–1234. [PubMed] [Google Scholar]
- Hougaard C, Hammami S, Eriksen BL, Sørensen US, Jensen ML, Strøbæk D, Christophersen P. (2012) Evidence for a common pharmacological interaction site on KCa2 channels providing both selective activation and selective inhibition of the human KCa2.1 subtype. Mol Pharmacol 81:210–219. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. (1997) A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci USA 94:11651–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DP, Strøbæk D, Hougaard C, Jensen ML, Hummel R, Sørensen US, Christophersen P, Wulff H. (2011) Negative gating modulation by (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphthylamine (NS8593) depends on residues in the inner pore vestibule: Pharmacological evidence of deep-pore gating of KCa2 channels. Mol Pharmacol 79:899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. (2007) The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci 27:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. (1996) Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273:1709–1714. [DOI] [PubMed] [Google Scholar]
- Köhler R, Degenhardt C, Kühn M, Runkel N, Paul M, Hoyer J. (2000) Expression and function of endothelial Ca2+-activated K+ channels in human mesenteric artery: A single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ Res 87:496–503. [DOI] [PubMed] [Google Scholar]
- Köhler R, Kaistha BP, Wulff H. (2010) Vascular KCa-channels as therapeutic targets in hypertension and restenosis disease. Expert Opin Ther Targets 14:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, Grgic I, Kämpfe D, Si H, Wibawa J, et al. (2003) Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation 108:1119–1125. [DOI] [PubMed] [Google Scholar]
- Lam J, Coleman N, Garing AL, Wulff H. (2013) The therapeutic potential of small-conductance KCa2 channels in neurodegenerative and psychiatric diseases. Expert Opin Ther Targets 17:1203–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Gramsbergen JB, Sivasaravanaparan M, Ditzel N, Sevelsted-Møller LM, Oliván-Viguera A, Rabjerg M, Wulff H, Köhler R. (2012) Genetic KCa3.1-deficiency produces locomotor hyperactivity and alterations in cerebral monoamine levels. PLoS ONE 7:e47744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoral-Theys D, Pottier L, Kerff F, Dufrasne F, Proutière F, Wauthoz N, Neven P, Ingrassia L, Antwerpen PV, Lefranc F, et al. (2010) Simple di- and trivanillates exhibit cytostatic properties toward cancer cells resistant to pro-apoptotic stimuli. Bioorg Med Chem 18:3823–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, Zhang Z, Singapuri A, Albert TR, Rajagopal AV, et al. (2009) Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol 587:1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. [DOI] [PubMed] [Google Scholar]
- Meseguer V, Karashima Y, Talavera K, D’Hoedt D, Donovan-Rodríguez T, Viana F, Nilius B, Voets T. (2008) Transient receptor potential channels in sensory neurons are targets of the antimycotic agent clotrimazole. J Neurosci 28:576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkau M, Köhler R, de Wit C. (2010) Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J 24:3572–3579. [DOI] [PubMed] [Google Scholar]
- Mishra RC, Belke D, Wulff H, Braun AP. (2013) SKA-31, a novel activator of SKCa and IKCa channels, increases coronary flow in male and female rat hearts. Cardiovasc Res 97:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliván-Viguera A, Valero MS, Murillo MD, Wulff H, García-Otín AL, Arbonés-Mainar JM, Köhler R. (2013) Novel phenolic inhibitors of small/intermediate-conductance Ca2+-activated K+ channels, KCa3.1 and KCa2.3. PLoS ONE 8:e58614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke J, Schmidt K, Wulff H, Köhler R, de Wit C. (2013) Activation of KCa3.1 by SKA-31 induces arteriolar dilatation and lowers blood pressure in normo- and hypertensive connexin40-deficient mice. Br J Pharmacol 170:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH. (1998) Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca2+-activated K+ channel. J Med Chem 41:2–5. [DOI] [PubMed] [Google Scholar]
- Ruggieri P, Mangino G, Fioretti B, Catacuzzeno L, Puca R, Ponti D, Miscusi M, Franciolini F, Ragona G, Calogero A. (2012) The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PLoS ONE 7:e47825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, Köhler R, Wulff H. (2009) Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol 75:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. (2011) Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 31:13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soder RP, Parajuli SP, Hristov KL, Rovner ES, Petkov GV. (2013) SK channel-selective opening by SKA-31 induces hyperpolarization and decreases contractility in human urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol 304:R155–R163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strøbæk D, Brown DT, Jenkins DP, Chen YJ, Coleman N, Ando Y, Chiu P, Jørgensen S, Demnitz J, Wulff H, et al. (2013) NS6180, a new KCa3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease. Br J Pharmacol 168:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. (2003) Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res 93:124–131. [DOI] [PubMed] [Google Scholar]
- Tharp DL, Wamhoff BR, Wulff H, Raman G, Cheong A, Bowles DK. (2008) Local delivery of the KCa3.1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler Thromb Vasc Biol 28:1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, Fujiwara Y, Mattson DL, Das S, Melvin JE, et al. (2008) The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest 118:3025–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vázquez AE, Young JN, et al. (2005) Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289:H2714–H2723. [DOI] [PubMed] [Google Scholar]
- Valero MS, Pereboom D, Barcelo-Batllory S, Brines L, Garay RP, Alda JO. (2011) Protein kinase A signalling is involved in the relaxant responses to the selective β-oestrogen receptor agonist diarylpropionitrile in rat aortic smooth muscle in vitro. J Pharm Pharmacol 63:222–229. [DOI] [PubMed] [Google Scholar]
- Van Der Velden J, Sum G, Barker D, Koumoundouros E, Barcham G, Wulff H, Castle N, Bradding P, Snibson K. (2013) KCa3.1 channel-blockade attenuates airway pathophysiology in a sheep model of chronic asthma. PLoS ONE 8:e66886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandorpe DH, Shmukler BE, Jiang L, Lim B, Maylie J, Adelman JP, de Franceschi L, Cappellini MD, Brugnara C, Alper SL. (1998) cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J Biol Chem 273:21542–21553. [DOI] [PubMed] [Google Scholar]
- Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. (2005) International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev 57:463–472. [DOI] [PubMed] [Google Scholar]
- Werkman TR, Kawamura T, Yokoyama S, Higashida H, Rogawski MA. (1992) Charybdotoxin, dendrotoxin and mast cell degranulating peptide block the voltage-activated K+ current of fibroblast cells stably transfected with NGK1 (Kv1.2) K+ channel complementary DNA. Neuroscience 50:935–946. [DOI] [PubMed] [Google Scholar]
- Wulff H, Castle NA. (2010) Therapeutic potential of KCa3.1 blockers: an overview of recent advances, and promising trends. Expert Rev Clin Pharmacol 3:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Gutman GA, Cahalan MD, Chandy KG. (2001) Delineation of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel, IKCa1. J Biol Chem 276:32040–32045. [DOI] [PubMed] [Google Scholar]
- Wulff H, Köhler R. (2013) Endothelial small- and intermediate-conductance KCa channels: an update on their pharmacology and usefulness as cardiovascular targets. J Cardiovasc Pharmacol 61:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. (2007) Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem 14:1437–1457. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. (2000) Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc Natl Acad Sci USA 97:8151–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Pascal JM, Schumann M, Armen RS, Zhang JF. (2012) Identification of the functional binding pocket for compounds targeting small-conductance Ca2+-activated potassium channels. Nat Commun 3:1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Timofeyev V, Lu L, Li N, Singapuri A, Long MK, Bond CT, Adelman JP, Chiamvimonvat N. (2008) Functional roles of a Ca2+-activated K+ (SK2) channel in atrioventricular nodes. Circ Res 102:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.