

Abstract
Background and Purpose
The Kvβ1.3 subunit modifies the gating and pharmacology of Kv1.5 channels in a PKC-dependent manner, decreasing channel sensitivity to bupivacaine- and quinidine-mediated blockade. Cardiac Kv1.5 channels associate with receptor for activated C kinase 1 (RACK1), the Kvβ1.3 subunit and different PKC isoforms, resulting in the formation of a functional channelosome. The aim of the present study was to investigate the effects of PKC inhibition on bupivacaine and quinidine block of Kv1.5 + Kvβ1.3 channels.
Experimental Approach
HEK293 cells were transfected with Kv1.5 + Kvβ1.3 channels, and currents were recorded using the whole-cell configuration of the patch-clamp technique. PKC inhibition was achieved by incubating the cells with either calphostin C or bisindolylmaleimide II and the effects of bupivacaine and quinidine were analysed.
Key Results
The voltage-dependent inactivation of Kv1.5 + Kvβ1.3 channels and their pharmacological behaviour after PKC inhibition with calphostin C were similar to those displayed by Kv1.5 channels alone. Indeed, the IC50 values for bupivacaine were similar in cells whose PKC was inhibited with calphostin C or bisindolylmaleimide II. Similar results were also observed in the presence of quinidine.
Conclusions and Implications
The finding that the voltage-dependence of inactivation and the pharmacology of Kv1.5 + Kvβ1.3 channels after PKC inhibition resembled that observed in Kv1.5 channels suggests that both processes are dependent on PKC-mediated phosphorylation. These results may have clinical relevance in diseases that are characterized by alterations in kinase activity.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| Kv1.5 channel | Bupivacaine |
| PKC | Quinidine |
| PKCβ | Calphostin C |
| α-adrenoceptor | |
| β-adrenoceptor | |
| PKA |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b,c).
Introduction
The activation of Kv1.5 channels generates the ultra-rapid outward potassium current (IKur), which plays an important role in human atrial repolarization and the maintenance of the resting membrane voltage in the pulmonary vascular system (Archer et al., 1998; Ravens and Wettwer, 2011). Indeed, mutations in and down-regulation of, Kv1.5 channels are involved in the development of atrial fibrillation and pulmonary arterial hypertension (PAH) (Yuan et al., 1998; Olson et al., 2006; Remillard et al., 2007; González et al., 2010). The slow and partial inactivation and the voltage-dependence of these channels underlie their important role in the regulation of the duration of atrial action potentials (Fedida et al., 1993; Snyders et al., 1993). This slow inactivation is modified by the assembly of Kv1.5 subunits with β subunits (Kvβ1.2, Kvβ1.3 and Kvβ2.1) that are present in the human myocardium (Uebele et al., 1996; 1998). The Kvβ1.3 subunit modifies the electrophysiological characteristics of Kv1.5 channels, inducing a fast and partial inactivation, a greater degree of slow inactivation, a shift of the activation curve towards more negative potentials, a decrease in the sensitivity of the channel to blockade induced by antiarrhythmic drugs and local anaesthetics, and a decrease in the degree of stereoselective blockage (González et al., 2002; Decher et al., 2005; Arias et al., 2007).
IKur is highly modulated by α- and β-adrenoceptor stimulation (Li et al., 1996) mediated by PKC and PKA, respectively (Murray et al., 1994; Kwak et al., 1999a,b). The expression levels of α- and β-adrenoceptors, as well as the release of catecholamines, can be modified in several cardiac pathologies (Schlaich et al., 2003; 2005). In fact, cardiac hypertrophy is associated with an up-regulation of different PKC isoforms (Takeishi et al., 1999; Braz et al., 2002; Kerkela et al., 2002; Hahn et al., 2003). During PAH-associated right ventricular hypertrophy, the activity of GPCR kinases modulates the β1-adrenoceptor-mediated response (Piao et al., 2012). Furthermore, one of the most effective treatments for atrial fibrillation is the administration of β-blockers (Kuhlkamp et al., 2000); pharmacological and electrical remodelling are induced in patients chronically treated with these drugs (Valenzuela, 2003). In addition, a decrease in myocyte death as a result of selective PKA inhibition has been proposed as one of the mechanisms responsible for the beneficial effects of β-blockers in heart failure patients (Sabbah, 2004; Zhang et al., 2013). PKC and PKA activities are also required for Kv1.5 channel modulation by the auxiliary subunits Kvβ1.2 and Kvβ1.3 (Kwak et al., 1999a,b; Williams et al., 2002). Indeed, calphostin C-mediated PKC inhibition removes many of the Kvβ1.3-dependent electrophysiological effects (Kwak et al., 1999b). The presence of a Kv1.5 channel complex (or channelosome) has recently been shown to be composed of several PKC isoforms (βI and βII), receptor for activated C kinase 1 (RACK1) and the Kvβ1.3 subunit in the rat ventricle, but not in the rat atria (David et al., 2012).
Kv1.5- or Kvβ1.3-phosphorylation is required for the Kvβ1.3-induced effects on Kv1.5 channels (Kwak et al., 1999b). Moreover, leucine at position 510 in the Kv1.5 channel is involved in the binding of both the Kvβ1.3 subunit and bupivacaine. In addition, bupivacaine and quinidine share a common receptor site at Kv1.5 channels, and this site is located at the S6 segment and involves both a polar and a hydrophobic interaction (through T507 and V514, respectively) (Yeola et al., 1996; Franqueza et al., 1997; González et al., 2002; Decher et al., 2005; Arias et al., 2007). Kv1.5 + Kvβ1.3 channels expressed in cells treated with calphostin C exhibit an inactivation curve that is shifted towards more positive potentials compared with Kv1.5 + Kvβ1.3 channels expressed in control conditions (David et al., 2012). Taken together, these results suggest that after PKC inhibition, both bupivacaine and quinidine should be more potent at inducing blockade.
In the present study, we analysed the electrophysiological and pharmacological consequences of PKC inhibition in Kv1.5 + Kvβ1.3 channels. The voltage-dependent activation of Kv1.5 + Kvβ1.3 channels after treatment with calphostin C or bisindolylmaleimide II was similar to that exhibited by Kv1.5 channels alone. Also, the voltage-dependent inactivation and pharmacological activity of Kv1.5 + Kvβ1.3 channels after calphostin C- or bisindolylmaleimide II-mediated PKC inhibition was similar to that exhibited by Kv1.5 channels alone. More importantly, the potency of bupivacaine and quinidine for blocking Kv1.5 + Kvβ1.3 channels expressed in cells treated with calphostin C or with bisindolylmaleimide II was more similar to that displayed by Kv1.5 channels than it was for that displayed by untreated Kv1.5 + Kvβ1.3 channels. In summary, PKC activity was found to regulate both the electrophysiological and pharmacological properties of Kv1.5 + Kvβ1.3 channels, indicating that PKC may be considered a new therapeutic target in some cardiac pathologies.
Methods
Expression plasmids, cell culture and transient transfection
Expression of human Kv1.5 channels and the Kvβ1.3 subunit (Alexander et al., 2013b) in pBK was performed as previously described (Uebele et al., 1998; David et al., 2012). Briefly, the human Kv1.5 channel (−22 to 1894 nt) and the Kvβ1.3 subunit (−53 to 1500 nt) were inserted into the same pBK vector, with the Kv1.5 channel placed 3′ to the Kvβ1.3 subunit and preceded by an internal ribosome entry sequence, thus generating a bicistronic messenger RNA, as previously described (Kwak et al., 1999b). Expression of human Kv1.5 channels in pBK was performed as previously described (Arias et al., 2007).
HEK293 cells were cultured in DMEM that had been supplemented with 10% FBS, 10 U·mL−1 of penicillin-streptomycin (Sigma-Aldrich, St. Louis, MO, USA), and 1% nonessential amino acids. For the electrophysiological experiments, cells plated in 35 mm dishes were transfected with Kv1.5 + Kvβ1.3 channels (0.8 μg) and a reporter plasmid expressing CD8 (1.8 μg) using Fugene 6 (at a 1:3 ratio of μg of DNA : μL of Fugene 6) (Promega, Madison, WI, USA). Prior to experimental use, the cells were incubated with polystyrene microbeads pre-coated with an anti-CD8 antibody (Dynabeads M450, Life Technologies, Grand Island, NY, USA), as previously described (González et al., 2001; 2002; Arias et al., 2007).
Electrophysiological recordings and data acquisition
The intracellular pipette filling solution contained the following (in mM): K-aspartate 80, KCl 42, phosphocreatine 3, KH2PO4 10, MgATP 3, HEPES-K 5 and EGTA-K 5 (adjusted to pH 7.25 with KOH). The bath solution contained the following (in mM): NaCl 140, KCl 4, CaCl2 1.8, MgCl2 1, HEPES-Na 10 and glucose 10 (adjusted to pH 7.40 with NaOH). Currents were recorded using the whole-cell configuration of the patch-clamp technique with a patch-clamp amplifier (Axopatch-200B Patch-clamp Amplifier; Molecular Devices Corp., Menlo Park, CA, USA) and were stored on a personal computer (TD Systems, Camarillo, CA, USA) with a Digidata 1440A analogue-to-digital converter (Molecular Devices Corp.). PClamp version 10 software was used for both data acquisition and analyses (Molecular Devices Corp.). Currents were recorded at room temperature (21–23°C) at a stimulation frequency of 0.1 Hz and were sampled at 4 kHz after antialias filtering at 2 kHz. The average pipette resistance ranged from 1 to 3 MΩ (n = 25). Gigaohm seal formation was achieved by suction (2–5 GΩ, n = 25). After seal formation, the cells were lifted from the bottom of the bath, and the membrane patch was ruptured with a brief additional suction. Microcal Origin 8.5 (OriginLab Co., Northampton, MA, USA) and the clampfit utility of pClamp 10 were used to perform least-squares fitting and data presentation. Experiments were excluded from analysis if the voltage error estimate exceeded 5 mV after series resistance compensation. Deactivation was fitted to a mono- or bi-exponential process using the following equations: y = Aexp(−t/τ) + C or y = A1exp(−t/τ1) + A2exp(−t/τ2) + C, respectively, where τx is the system time constant; Ax is the amplitude of the component of the exponential; and C is the baseline value. The voltage-dependence of the activation and inactivation curves was fitted using a Boltzmann equation: y = 1/{1 + exp [−(V − Vh)/s]}, where s represents the slope factor; V represents the membrane potential; and Vh represents the voltage at which 50% of the channels are open. In both cases, the maximum peak tail current or the maximum current generated during the application of the test pulse were plotted versus the previous membrane potential in order to generate the activation or inactivation curves respectively. Drug-induced block was measured at the end of 50 and 250 ms depolarizing pulses from −80 to +60 mV. The degree of inhibition obtained for each drug concentration was used to calculate the IC50 and nH values based on fitting these values to the Hill equation: 1/{1 + [IC50/(D)nH]}. Concentration–response curves with two components were fitted to a biphasic Hill equation.
Statistical analysis
The data are presented as the means ± SEM. One-way anova was used to compare more than two groups. Statistical significance was set at P < 0.05. The curve-fitting procedure used a nonlinear least-squares (Gauss–Newton) algorithm, and the results are displayed in linear and semilogarithmic formats with the difference plots. Goodness of fit was determined using the χ2 criterion and by inspecting systematic non-random trends in the difference plot.
Drugs
Racemic bupivacaine and quinidine (Sigma-Aldrich) were dissolved in distilled deionized water to yield stock solutions of 10 mM, from which further dilutions were made. Calphostin C, which was obtained from Calbiochem (Merck KGaA, Darmstadt, Germany), was dissolved in ethanol at a concentration of 100 μM for use as a stock solution. In all experiments, calphostin C was applied at a concentration of 3 μM for 2 h. Bisindolylmaleimide II (Sigma-Aldrich) was dissolved in DMSO at a concentration of 1 mM for use as a stock solution. In all experiments, bisindolylmaleimide II was applied at a concentration of 1 μM during 30 min. Hispidine from Calbiochem (Merck KGaA), was dissolved in DMSO at a concentration of 10 mM for use as a stock solution. In all experiments, hispidin was applied at a concentration of 5 μM for 30 min.
Results
Kvβ1.3-induced fast inactivation is abolished by PKC inhibition
Figure 1 shows the effects of the Kvβ1.3 subunit on the Kv1.5 channel, which induced a fast and partial inactivation, a greater degree of slow inactivation, a slower deactivation process and a shift of the activation curve towards more negative potentials (Figures 1A and B, and Table 1). PKC inhibition with calphostin C prevented Kvβ1.3-induced fast inactivation of Kv1.5 channels (Murray et al., 1994; Kwak et al., 1999a,b) as a consequence of a shift in the inactivation midpoint towards more positive potentials (David et al., 2012). Under these experimental conditions, Kv1.5 + Kvβ1.3-calphostin C channels exhibited an activation curve intermediate between that observed for the Kv1.5 and Kv1.5 + Kvβ1.3 channels (Table 1), and similar deactivation kinetics to Kv1.5 + Kvβ1.3 channels (Figures 1B and C). Also, PKC inhibition induced by bisindolylmaleimide II prevented Kvβ1.3-induced fast inactivation (Figure 1D), and like calphostin C, shifted the activation curve towards more positive potentials relative to Kv1.5 + Kvβ1.3 channels. A series of experiments were performed with hispidin (a selective PKCβI and PKCβII inhibitor). Hispidin only eliminated the Kvβ1.3-induced fast inactivation in 30% of the cells analysed, consistent with previous studies (David et al., 2012).
Figure 1.

Original recordings obtained upon depolarization from a holding potential of −80 to +60 mV in 10 mV steps and upon repolarization to −40 mV. (A) Current recordings obtained from the activation of Kv1.5 channels. (B) Current recordings of Kv1.5 channels in the presence of the Kvβ1.3 subunit. (C) Current recordings of Kv1.5 channels in the presence of a Kvβ1.3 subunit in cells that had been previously treated with calphostin C. (D) Current recordings of Kv1.5 channels in the presence of a Kvβ1.3 subunit in cells that had been previously treated with bisindolylmaleimide II. In all cases, the current (top), the deactivation process recorded at −40 mV after a pulse to +60 mV (middle) and the I–V relationship and activation curves (dashed line) obtained (bottom) are shown. When PKC is inhibited, the linearity of the I–V is recovered, and the activation process is shifted towards a more negative potential when the Kvβ1.3 subunit is present.
Table 1.
Voltage-dependent activation and inactivation parameters of Kv1.5, Kv1.5 + Kvβ1.3 and Kv1.5 + Kvβ1.3-calphostin C channels obtained after applying 250 ms prepulses
| Activation | Inactivation | ||||
|---|---|---|---|---|---|
| Vh (mV) | s (mV) | Vh (mV) | s (mV) | Degree of inactivation (%) | |
| Kv1.5 | −6.0 ± 0.7 | 5.2 ± 0.4 | −1.3 ± 3.4 | 9.8 ± 1.6 | 18.0 ± 3.4 |
| Kv1.5 + Kvβ1.3 control | −23.6 ± 2.4a | 3.9 ± 0.2 | −26.5 | 4.2 | 48.0 ± 3.0 |
| Kv1.5 + Kvβ1.3-calphostin C | −15.3 ± 1.2b,c | 6.0 ± 0.6b,c | −6.5 ± 1.81998 | 10.1 ± 1.11998 | 33.8 ± 2.2b,c |
s, slope of the Boltzmann equation; Vh, half-inactivation voltage. Data represent the mean ± SEM of n > 7 experiments.
Statistically significant difference between Kv1.5 + Kvβ1.3 and Kv1.5.
Statistically significant difference between Kv1.5 + Kvβ1-calphostin C and Kv1.5 + Kvβ1.3-control channels.
Statistically significant difference between Kv1.5 + Kvβ1.3-calphostin C and Kv1.5 channels.
To analyse the electrophysiological characteristics of the inactivation process of Kv1.5 + Kvβ1.3 channels expressed in cells treated with calphostin C (Kv1.5 + Kvβ1.3-calphostin C) we applied the double-pulse protocol shown at the top of Figure 2. Figure 2A shows the inactivation curves of Kv1.5, Kv1.5 + Kvβ1.3, Kv1.5 + Kvβ1.3-calphostin C. PKC inhibition induced by calphostin C shifted the inactivation curve towards more positive potentials, which was similar to the Vh observed in Kv1.5 channels (Table 1). Although the degree of inactivation of Kv1.5 + Kvβ1.3-calphostin C was decreased in comparison with Kv1.5 + Kvβ1.3 channels, it had not fully reverted back to that observed in Kv1.5 channels (Figures 1 and 2, and Table 1). The degree of inactivation of Kv1.5 + Kvβ1.3 channels continued to increase as the membrane potential became more positive, indicating that Kvβ1.3-mediated fast inactivation was voltage-dependent, as previously reported (Uebele et al., 1998). However, the inactivation of Kv1.5 + Kvβ1.3-calphostin C channels did not increase, similar to that observed with Kv1.5 channels (Figure 2A).
Figure 2.

Voltage-dependence of Kv1.5 + Kvβ1.3 channel inactivation before and after treatment with calphostin C. (A) Voltage-dependence of the inactivation following a 250 ms in duration prepulse. (B) Voltage-dependence of the inactivation following a 10 ms in duration prepulse.
To isolate the fast component of the inactivation induced by the Kvβ1.3 subunit, a double-pulse protocol using a 10 ms prepulse from −80 to +150 mV in 10 mV steps was applied (Uebele et al., 1998). Figure 2B shows the inactivation curve obtained after applying this double-pulse protocol. After applying this short prepulse, Kv1.5 channels were not inactivated, because they exhibited a slow inactivation with a time constant of ∼200 ms. In contrast, Kv1.5 + Kvβ1.3 channels that exhibited a fast inactivation with a time constant of ∼3 ms displayed an inactivation curve with a Vh = −5.2 ± 2.5 mV and s = 5.3 ± 0.9 mV (n = 4). This inactivation curve only represents the voltage-dependence of Kvβ1.3-induced fast inactivation; when cells were treated with calphostin C, this process was abolished. Figure 2B also shows that when this pulse protocol was applied, the fractional inactivation of Kv1.5 + Kvβ1.3 channels did not increase with membrane potential, exhibiting similar behaviour to that observed when a 250 ms in duration prepulse was applied in calphostin C-treated cells (Figure 2B and Table 1). However, when PKC was inhibited, the fast inactivation process was completely abolished, exhibiting the same phenotype as Kv1.5 channels alone.
Pharmacological properties of Kv1.5 + Kvβ1.3 channels after calphostin C treatment mimic those in the absence of the Kvβ1.3 subunit
In previous studies, we demonstrated that mutations in Kv1.5 channel or their assembly with the Kvβ1.3 subunits modify the pharmacological characteristics of the channel (Franqueza et al., 1997; González et al., 2002; Arias et al., 2007). Therefore, we studied the bupivacaine- and quinidine-induced blockade of Kv1.5 + Kvβ1.3 channels after PKC inhibition. Current recordings through Kv1.5 + Kvβ1.3-calphostin C channels were obtained in the absence and presence of bupivacaine (50 μM) or quinidine (30 μM) (Figure 3A, B). The current–voltage (I–V) relationships were obtained after plotting the current amplitude measured at the end of 250 ms depolarizing pulses at different membrane potentials between −80 and +60 mV in the absence and presence of either drug (Figure 3). Both compounds decreased the magnitude of the current at all membrane potentials tested.
Figure 3.

Effects of bupivacaine (50 μM) and quinidine (30 μM) on Kv1.5 + Kvβ1.3 channels in cells that have been treated with calphostin C or bisindolylmaleimide II. (A) Current recordings and I–V relationships obtained in the absence and presence of bupivacaine in cells pretreated with calphostin C. (B) Current recordings and I–V relationships obtained in the absence and presence of quinidine in cells pretreated with calphostin C. (C) Current recordings and I–V relationships obtained in the absence and presence of bupivacaine in cells pretreated with bisindolylmaleimide II. (D) Current recordings and I–V relationships obtained in the absence and presence of quinidine in cells pretreated with bisindolylmaleimide II. The blockade induced by both drugs was measured at the end of the pulse at +60 mV. Each point represents the mean ± SEM of six and five experiments respectively.
We also analysed the effects of bupivacaine and quinidine in Kv1.5 + Kvβ1.3 channels expressed in cells treated with bisindolylmaleimide II (Kv1.5 + Kvβ1.3-bisindolylmaleimide II) and in cells in which hispidin suppressed the Kvβ1.3-induced fast inactivation. Current recordings through Kv1.5 + Kvβ1.3-bisindolylmaleimide II channels were obtained in the absence and presence of bupivacaine (30 μM) or quinidine (30 μM) (Figure 3C, D). The I–V relationships were obtained after plotting the current amplitude measured at the end of 250 ms depolarizing pulses at different membrane potentials between −80 and +60 mV in the absence and presence of either drug (Figure 3). Both compounds decreased the magnitude of the current at all membrane potentials tested.
The concentration–response curves obtained for the inhibitory effects of bupivacaine and quinidine on Kv1.5, Kv1.5 + Kvβ1.3, Kv1.5 + Kvβ1.3-calphostin C, Kv1.5 + Kvβ1.3-bisindolylmaleimide II and Kv1.5 + Kvβ1.3-hispidin channels are shown in Figure 4 and Supporting Information Fig. S1. The suppression of the current at the end of 50 or 250 ms depolarizing pulses to +60 mV was used as an index of block (Figure 4, Supporting Information Figs S1–S4). In contrast to the blockade previously reported for Kv1.5 + Kvβ1.3 channels (González et al., 2002), bupivacaine-induced blockade of Kv1.5 + Kvβ1.3-calphostin C channels showed a biphasic curve, with an IC50(1) of 0.2 ± 0.1 nM and an IC50(2) of 21.4 ± 4.1 μM (n = 41). Note that the IC50(2) was similar to that of Kv1.5 channels (González et al., 2002). PKC inhibition did not modify the nH value (0.74 ± 0.06 vs. 0.71 ± 0.08 for bupivacaine-produced blockade of Kv1.5 + Kvβ1.3 and Kv1.5 + Kvβ1.3-calphostin C channels, respectively) (González et al., 2002) (Figure 4A). Similarly, quinidine-induced blockade of Kv1.5 + Kvβ1.3 channels was modified by calphostin C treatment (Figure 4B). Indeed, the concentration–response curve was also a biphasic concentration–response curve, with IC50(1) and IC50(2) values of 3.4 ± 5.4 μM and 70.4 ± 15.4 μM (n = 50) respectively. Interestingly, the IC50(1) value was similar to that reported for the quinidine-induced blockade of Kv1.5 channels (Yeola et al., 1996), whereas the IC50(2) value was similar to the value reported for blockade of Kv1.5 + Kvβ1.3 channels (González et al., 2002). The nH(1) and nH(2) values for this concentration–response curve were 0.34 ± 0.07 and 1.36 ± 0.46 respectively. The nH(2) value was similar to that observed in the concentration–response curve of quinidine-induced blockade of Kv1.5 + Kvβ1.3 channels (González et al., 2002). In contrast to that observed in Kv1.5 + Kvβ1.3-calphostin C, the concentration–response curves for bupivacaine and quinidine of Kv1.5 + Kvβ1.3-bisindolylmaleimide II channels were monophasic with IC50 values of 12.4 ± 1.8 μM and 7.2 ± 1.2 μM respectively. These values resemble those previously reported for bupivacaine and quinidine to block Kv1.5 channels (13.5 ± 1.4 μM and 4 μM, respectively). Intermediate results were obtained after inhibition of PKC with hispidin (Supporting Information Fig. S1). IC50 values were similar when block was measured after 50 ms or 250 ms of depolarizing pulses under all experimental conditions (Supporting Information Figs S1–S3).
Figure 4.
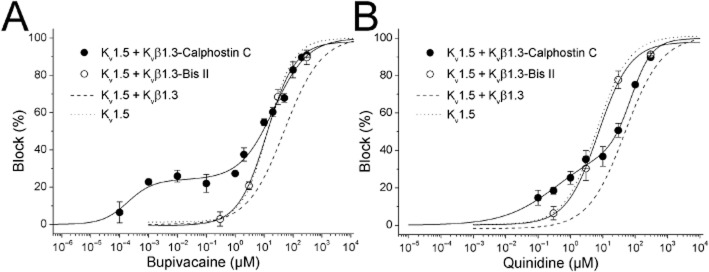
Concentration-dependence of bupivacaine-induced (A) and quinine-induced (B) blockade of calphostin C- or bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. The dotted and dashed lines represent the dose–response curves obtained for the bupivacaine- and quinidine-produced blockade of Kv1.5 channels alone and Kv1.5 + Kvβ1.3 channels, respectively (taken from González et al., 2002). The reduction in the current (relative to the control) at the end of depolarizing steps from −80 to +60 mV was used as an index of blockade. Concentration–response curves for bupivacaine and quinidine in calphostin C-treated Kv1.5 + Kvβ1.3 channels and in bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels are shown. Each point represents the mean ± SEM of three to eight experiments. The continuous line represents the fit of the experimental data to a biphasic or monophasic Hill equation.
Bupivacaine- and quinidine-produced blockade of Kv1.5 + Kvβ1.3-calphostin C channels was time-dependent, as indicated by the normalized current in the insets of the left panels of Figure 5A and B. To analyse the kinetics of blockade induced by both drugs, we plotted the ratio of the drug-sensitive current and the current under control conditions [(Icontrol − Idrug)/Icontrol] during the first 40 ms in the presence of 20, 50 and 100 μM bupivacaine and 10, 30 and 100 μM quinidine (right panel of Figure 5A and B, inset). The blockade exponentially increased during depolarization, and the time constant of this process was faster at higher drug concentrations. Thus, the time constant of this process was considered to be a good index of the development of blockade (τblock). Based on the τblock values obtained at different bupivacaine concentrations, the association (k) and dissociation (l) rate constants were derived, averaging 5.9 ± 0.5 μM−1·s−1 and 252.3 ± 33.0 s−1 versus 3.8 ± 0.3 μM−1·s−1 and 108.3 ± 14.3 s−1 (n = 14) in Kv1.5 + Kvβ1.3 and Kv1.5 + Kvβ1.3-calphostin C channels, respectively (Figure 5A) (González et al., 2002). In the case of quinidine, the k and l values for Kv1.5 + Kvβ1.3 channels were 5.1 ± 0.51 μM−1·s−1 and 454.7 ± 38.05 s−1, while the k and l values for Kv1.5 + Kvβ1.3-calphostin C channels were 5.0 ± 0.02 μM−1·s−1 and 196.4 ± 1.1 s−1 (n = 11) (Figure 5B). Similar changes in the association and dissociation rate constants were observed when the bupivacaine and quinidine kinetics of block were studied in Kv1.5 + Kvβ1.3-bisindolylmaleimide II channels (Figure 5C, D). Although the blockade induced by both drugs was time-dependent, as observed during depolarization pulses from −80 to +60 mV, this time dependence was not evident in the tail currents recorded at −40 mV under any experimental condition, probably because of the changes in the association and dissociation kinetics and/or to changes in the gating of the channel (Figure 6).
Figure 5.
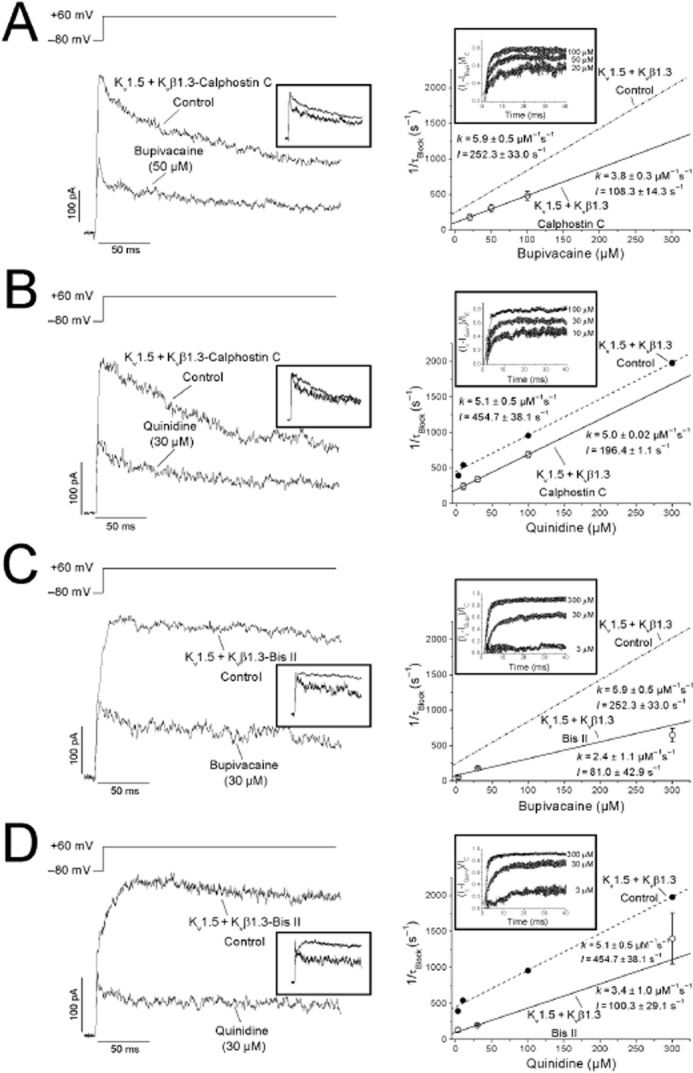
Time-dependence of bupivacaine- and quinidine-induced blockade of calphostin C- and bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. Left: Superimposed traces for the steps from −80 to +60 mV and those normalized to the control values (inset). Right: Relationship between 1/τB values at different concentrations of bupivacaine (A, C) and quinidine (B, D). Each point represents the mean ± SEM of three to six experiments. For a first-order blocking scheme, a relationship is expected: 1/τB = k × [Drug] + l. The lines represent the fit from which the apparent binding (k) and unbinding (l) rate constants were obtained under control (dashed) and PKC-inhibited (solid) conditions. The relationship between the sensitive current and depolarizing time [(Icontrol − Idrug)/Icontrol] at three different drug concentrations is plotted in the inset of each panel.
Figure 6.
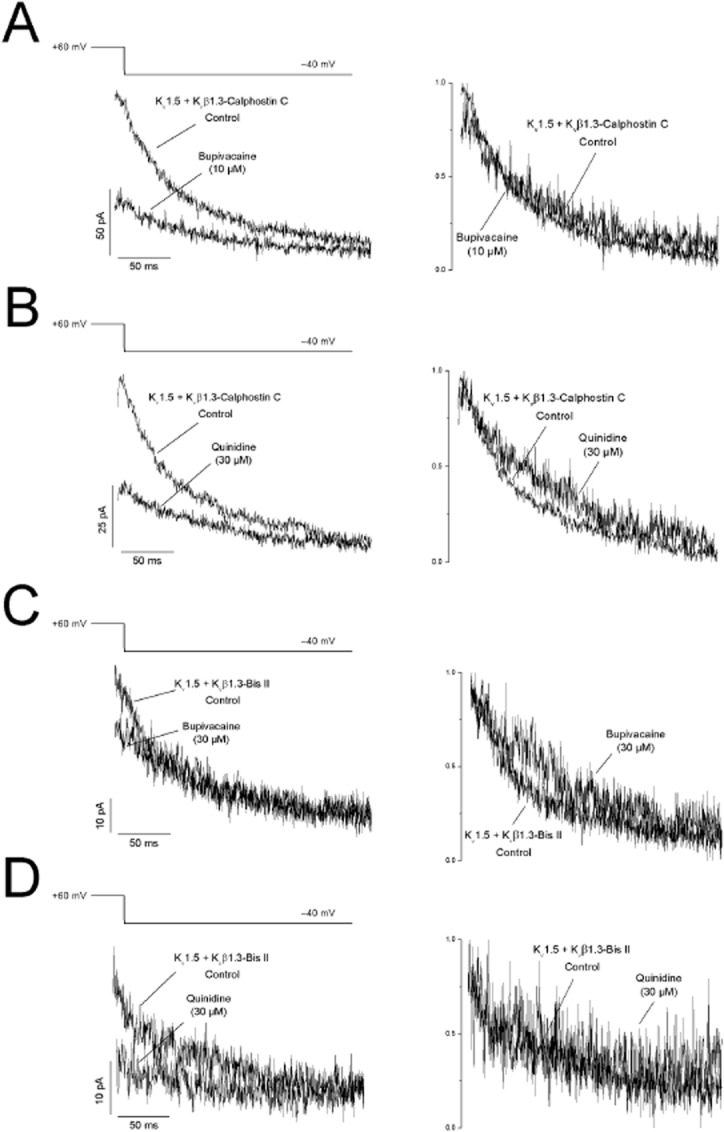
(A,C) Bupivacaine- and (B, D) quinidine-induced effects on the deactivation kinetics of (A, B) calphostin C- and (C, D) bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. The currents obtained after applying the protocol outlined at the top of the figure are shown. The tails were superimposed (top) and normalized (bottom) to show the similar deactivation kinetics in the absence and presence of the drugs.
Discussion and conclusions
In the present study, we analysed the electrophysiological and pharmacological consequences of PKC inhibition using calphostin C and bisindolylmaleimide II in Kv1.5 + Kvβ1.3 channels. We demonstrated that the voltage-dependence of Kv1.5 + Kvβ1.3 channel inactivation after PKC inhibition resembles that observed in Kv1.5 channels alone. Inhibition of PKC using calphostin C or bisindolylmaleimide II partially reversed the changes in the pharmacological properties induced by the assembly of the Kvβ1.3 subunit with Kv1.5 channels (González et al., 2002; Decher et al., 2005) in such a way that the pharmacology of these channels in cells pretreated with calphostin C or bisindolylmaleimide II resembled that exhibited by Kv1.5 channels in the absence of the Kvβ1.3 subunit (Snyders et al., 1992; Valenzuela et al., 1995).
Calphostin C-mediated PKC inhibition results in the abolishment of Kvβ1.3-induced fast inactivation because of a shift in the inactivation midpoint of the Kv1.5 + Kvβ1.3 channels (David et al., 2012). Calphostin C produces a marked down-regulation that can be due to an abnormal recycling of the Kv1.5 channels to the membrane. Surprisingly, the voltage-dependence of inactivation of Kv1.5 + Kvβ1.3-calphostin C and that of Kv1.5 channels were similar. These results suggest not only that the voltage-dependent inactivation induced by this β subunit is abolished, but also that the complex behaves as if the Kvβ1.3 subunit was not assembled with the Kv1.5 channels. The voltage-dependence of Kvβ1.3-mediated fast inactivation differs from that of N-type inactivation in Shaker channels and with the fast inactivation conferred by Kvβ1.2 on Kv1.5 channels, both of which are insensitive to membrane potential (Zagotta et al., 1989; De Biasi et al., 1997; Uebele et al., 1998). The voltage dependence of Kvβ1.3-mediated inactivation may be due to the charge within the blocking particle sensing the transmembrane potential as well as to some voltage-dependent changes in the Kv1.5 channel that are involved in the interaction with the Kvβ1.3 subunit. To discriminate between these possibilities, experiments using a pulse protocol with 10 ms depolarizing pre-pulses were performed. These experiments allowed us to isolate the fast inactivation induced by the Kvβ1.3 subunit from the slow inactivation that is characteristic of Kv1.5 channels. Indeed, inactivation curves obtained from Kv1.5 + Kvβ1.3 and Kv1.5 + Kvβ1.3-calphostin C channels were similar after applying 10 and 250 ms pre-pulse protocols respectively. Taken together, these data suggest that the voltage-dependence of Kvβ1.3-mediated inactivation is independent of the properties of the Kv1.5 channel. However, we cannot rule out the possibility that a conformational change in Kvβ1.3, due to dephosphorylation, occurs and could be transmitted to the S6 in Kv1.5 subunit, changing the interaction between the receptor site at the Kv1.5 and the Kvβ1.3 inactivation ‘ball’.
Bupivacaine- and quinidine-induced blockade of Kv1.5 channels result from their binding to external and internal receptor sites (Yeola et al., 1996; Franqueza et al., 1997; Longobardo et al., 2000; 2001). Although the molecular determinants of the external binding site remain unknown, both drugs share a common internal receptor site, which is located at the S6 segment and involves a polar interaction with T507 and two hydrophobic interactions with L510 and V514 (Yeola et al., 1996; Franqueza et al., 1997; Arias et al., 2007). Moreover, the bupivacaine binding site on Kv1.5 channels overlaps with the binding site of the Kvβ1.3 subunit in such a way that bupivacaine blocks the Kv1.5 + Kvβ1.3 channel to a lesser extent than Kv1.5 channels alone (Arias et al., 2007). The pharmacology of the Kv1.5 + Kvβ1.3 channels was modified after PKC inhibition, decreasing the IC50 values for both bupivacaine and quinidine. These results can be explained by the decrease in the dissociation rate values obtained in Kv1.5 + Kvβ1.3-calphostin C and Kv1.5 + Kvβ1.3-bisindolylmaleimide II channels with bupivacaine and quinidine, which indicates that the drug-channel complex is more stable than in Kv1.5 + Kvβ1.3 channels, but less stable than that observed in Kv1.5 channels. However, the association rate constants remained nearly unchanged in comparison with those reported for Kv1.5 and Kv1.5 + Kvβ1.3 channels (González et al., 2002; Arias et al., 2007). This result also corroborates the finding that the phenotype of Kv1.5 + Kvβ1.3-calphostin C or Kv1.5 + Kvβ1.3-bisindolylmaleimide II channels is closer to that of Kv1.5 channels alone (Arias et al., 2007). This gain of potency is a very important result if we keep in mind that the expression levels of α- and β-adrenoceptors, as well as the release of catecholamines, can be modified in several cardiac pathologies (Schlaich et al., 2003; 2005), and may have important consequences in cardiac pharmacology (Longobardo et al., 1998; González et al., 2002; 2010; Ravens and Wettwer, 2011). Interestingly, the concentration–response curves for bupivacaine- and quinidine-induced block in cells in which PKC activity was inhibited with calphostin C was biphasic, while these curves were monophasic when bisindolylmalinmide II was used to inhibit PKC. If drug binding at the site with highest affinity saturates at around 25%, then we have to assume that drug binding only partially blocks ionic flow, somewhat similar to the ‘turret’ block of Kv11.1 channels by some toxins, like CnErg1 (Hill et al., 2007). Therefore, block induced by very low drug concentrations can be attributed to rapid fluctuations between the drug-blocked channel and a full conductance drug-channel encounter complex. In fact, similar to the block produced by CnErg1 that induces an incomplete block at very high concentrations, bupivacaine and quinidine were not able to completely abolish the current even at very high concentrations (Hill et al., 2007).
In summary, we have characterized the functional contribution of PKC activity to the modulation of the Kv1.5 channelosome from an electrophysiological and pharmacological perspective. Importantly, PKC inhibition changes the electrophysiological and pharmacological characteristics of Kv1.5 + Kvβ1.3 channels, causing them to be more similar to those observed in the absence of the Kvβ1.3 subunit. This functional characterization of the Kv1.5 channelosome opens up a variety of possible mechanisms that may elucidate the variations underlying the development of cardiac hypertrophy or any other cardiovascular disease that involves modifications in PKC activity.
Acknowledgments
This study was supported by MINECO (SAF2010-14916 and SAF2013-45800-R), FIS-RECAVA (RD06/0014/0006) and FIS-RIC (RD12/0042/0019) grants to C. V. A. M. and A. P. hold JAE-Predoc and FPI fellowships respectively. A. dlC. and T. G. hold RIC and R&C contracts respectively.
Glossary
- PAH
pulmonary arterial hypertension
Author contributions
C. V. designed research. A. M., A. P., A. dlC. and D. A. P. performed and analysed experiments. A. M., T. G., M. M. T. and C. V. analysed the data. C. V. and A. M. wrote the paper.
Conflict of interest
The authors declare that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the ublisher's web-site: http://dx.doi.org/10.1111/bph.12822
Figure S1 Concentration dependence of bupivacaine-induced blockade of hispidin-treated Kv1.5 + Kvβ1.3 channels. (A) The dashed and dotted lines represent the dose–response curves obtained for the bupivacaine-induced blockade of Kv1.5 and Kv1.5 + Kvβ1.3 channels, respectively (taken from González et al., 2002). Reduction in the current (relative to the control) at the end of depolarizing steps from −80 to +60 mV was used as an index of blockade. (●): Concentration–response curves for bupivacaine in bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels and in hispidin-treated Kv1.5 + Kvβ1.3 channels (○). (B) Reduction in the current (relative to the control) at 50 ms (○) and at 250 ms (●) depolarizing steps from −80 to +60 mV. Each point represents the mean ± SEM of three to four experiments. The lines represent the fit of the experimental data to a monophasic Hill equation.
Figure S2 Concentration dependence of bupivacaine- (A) and quinidine-induced (B) blockade of calphostine C-treated Kv1.5 + Kvβ1.3 channels. The dashed and continuous lines represent the dose–response curves obtained for the bupivacaine- or quinidine-induced blockade at 50 ms (○) and at 250 ms (●) depolarizing steps from −80 to +60 mV respectively. Each point represents the mean ± SEM of three to eight experiments. The continuous line represents the fit of the experimental data to a biphasic Hill equation.
Figure S3 Concentration dependence of bupivacaine- (A) and quinidine-induced (B) blockade of bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. The dashed and continuous lines represent the dose–response curves obtained for the bupivacaine- or quinidine-induced blockade at 50 ms (○) and at 250 ms (●) depolarizing steps from −80 to +60 mV respectively. Each point represents the mean ± SEM of three to eight experiments. The continuous line represents the fit of the experimental data to a monophsic Hill equation.
Figure S4 Absolute values for bupivacaine- and quinidine- blockade of calphostin C- and bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. Magnitude of calphostin CKv1.5 + Kvβ1.3 currents in the absence and in the presence of different bupivacaine (A) and quinidine (B) concentrations. Magnitude of bisindolylmaleimide II-Kv1.5 + Kvβ1.3 currents in the absence and in the presence of different bupivacaine (C) and quinidine (D) concentrations. Each point represents the mean ± SEM of three to eight experiments.
Table S1 IC50 and nH from the of the concentration–response curves to bupivacaine in Kv1.5 + Kvβ1.3 channels expressed in cells treated with hispidin and bisindolylmaleimide II in comparison with those obtained of bupivacaine on Kv1.5 and Kv1.5 + Kvβ1.3 channels.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14:Ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Souil E, Dinh Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, et al. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–2330. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C, Guizy M, David M, Marzian S, González T, Decher N, et al. KVβ1.3 reduces the degree of stereoselective bupivacaine block of KV1.5 channels. Anesthesiology. 2007;107:641–651. doi: 10.1097/01.anes.0000282100.32923.5c. [DOI] [PubMed] [Google Scholar]
- Braz JC, Bueno OF, De Windt LJ, Molkentin JD. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2) J Cell Biol. 2002;156:905–919. doi: 10.1083/jcb.200108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Macias A, Moreno C, Prieto A, Martinez-Marmol R, Vicente R, et al. PKC activity regulates functional effects of KVβ1.3 on KV1.5 channels. Identification of a cardiac KV1.5 channelosome. J Biol Chem. 2012;287:21416–21428. doi: 10.1074/jbc.M111.328278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi M, Wang Z, Accili E, Wible B, Fedida D. Open channel block of human heart hKV1.5 by the beta-subunit hKVbeta1.2. Am J Physiol. 1997;272:H2932–H2941. doi: 10.1152/ajpheart.1997.272.6.H2932. [DOI] [PubMed] [Google Scholar]
- Decher N, Kumar P, Gonzalez T, Renigunta V, Sanguinetti MC. Structural basis for competition between drug binding and KVbeta1.3 accessory subunit-induced N-type inactivation of KV1.5 channels. Mol Pharmacol. 2005;68:995–1005. doi: 10.1124/mol.105.011668. [DOI] [PubMed] [Google Scholar]
- Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S, et al. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ Res. 1993;73:210–216. doi: 10.1161/01.res.73.1.210. [DOI] [PubMed] [Google Scholar]
- Franqueza L, Longobardo M, Vicente J, Delpon E, Tamkun MM, Tamargo J, et al. Molecular determinants of stereoselective bupivacaine block of hKv1.5 channels. Circ Res. 1997;81:1053–1064. doi: 10.1161/01.res.81.6.1053. [DOI] [PubMed] [Google Scholar]
- González T, Longobardo M, Caballero R, Delpon E, Tamargo J, Valenzuela C. Effects of bupivacaine and a novel local anesthetic, IQB-9302, on human cardiac K+ channels. J Pharmacol Exp Ther. 2001;296:573–583. [PubMed] [Google Scholar]
- González T, Navarro-Polanco R, Arias C, Caballero R, Moreno I, Delpon E, et al. Assembly with the Kvβ1.3 subunit modulates drug block of hKv1.5 channels. Mol Pharmacol. 2002;62:1456–1463. doi: 10.1124/mol.62.6.1456. [DOI] [PubMed] [Google Scholar]
- González T, David M, Moreno C, Macias A, Valenzuela C. Kv1.5-Kv beta interactions: molecular determinants and pharmacological consequences. Mini Rev Med Chem. 2010;10:635–642. doi: 10.2174/138955710791384018. [DOI] [PubMed] [Google Scholar]
- Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, et al. Protein kinase Calpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res. 2003;93:1111–1119. doi: 10.1161/01.RES.0000105087.79373.17. [DOI] [PubMed] [Google Scholar]
- Hill AP, Sunde M, Campbell TJ, Vandenberg JI. Mechanism of block of the hERG K+ channel by the scorpion toxin CnErg1. Biophys J. 2007;92:3915–3929. doi: 10.1529/biophysj.106.101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkela R, Ilves M, Pikkarainen S, Tokola H, Ronkainen J, Vuolteenaho O, et al. Identification of PKCalpha isoform-specific effects in cardiac myocytes using antisense phosphorothioate oligonucleotides. Mol Pharmacol. 2002;62:1482–1491. doi: 10.1124/mol.62.6.1482. [DOI] [PubMed] [Google Scholar]
- Kuhlkamp V, Schirdewan A, Stangl K, Homberg M, Ploch M, Beck OA. Use of metoprolol CR/XL to maintain sinus rhythm after conversion from persistent atrial fibrillation: a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2000;36:139–146. doi: 10.1016/s0735-1097(00)00693-8. [DOI] [PubMed] [Google Scholar]
- Kwak YG, Hu N, Wei J, George AL, Jr, Grobaski TD, Tamkun MM, et al. Protein kinase A phosphorylation alters Kvβ1.3 subunit- mediated inactivation of the Kv1.5 potassium channel. J Biol Chem. 1999a;274:13928–13932. doi: 10.1074/jbc.274.20.13928. [DOI] [PubMed] [Google Scholar]
- Kwak YG, Navarro-Polanco R, Grobaski T, Gallagher DJ, Tamkun MM. Phosphorylation is required for alteration of Kv1.5 K+ channel function by the Kvβ1.3 subunit. J Biol Chem. 1999b;274:25355–25361. doi: 10.1074/jbc.274.36.25355. [DOI] [PubMed] [Google Scholar]
- Li GR, Feng J, Wang Z, Fermini B, Nattel S. Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circ Res. 1996;78:903–915. doi: 10.1161/01.res.78.5.903. [DOI] [PubMed] [Google Scholar]
- Longobardo M, Delpon E, Caballero R, Tamargo J, Valenzuela C. Structural determinants of potency and stereoselective block of hKv1.5 channels induced by local anesthetics. Mol Pharmacol. 1998;54:162–169. doi: 10.1124/mol.54.1.162. [DOI] [PubMed] [Google Scholar]
- Longobardo M, González T, Navarro-Polanco R, Caballero R, Delpon E, Tamargo J, et al. Effects of a quaternary bupivacaine derivative on delayed rectifier K+ currents. Br J Pharmacol. 2000;130:391–401. doi: 10.1038/sj.bjp.0703334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longobardo M, González T, Caballero R, Delpon E, Tamargo J, Valenzuela C. Bupivacaine effects on hKv1.5 channels are dependent on extracellular pH. Br J Pharmacol. 2001;134:359–369. doi: 10.1038/sj.bjp.0704251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KT, Fahrig SA, Deal KK, Po SS, Hu NN, Snyders DJ, et al. Modulation of an inactivating human cardiac K+ channel by protein kinase C. Circ Res. 1994;75:999–1005. doi: 10.1161/01.res.75.6.999. [DOI] [PubMed] [Google Scholar]
- Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao L, Fang YH, Parikh KS, Ryan JJ, D'Souza KM, Theccanat T, et al. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012;126:2859–2869. doi: 10.1161/CIRCULATIONAHA.112.109868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res. 2011;89:776–785. doi: 10.1093/cvr/cvq398. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C1837–C1853. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- Sabbah HN. Biologic rationale for the use of beta-blockers in the treatment of heart failure. Heart Fail Rev. 2004;9:91–97. doi: 10.1023/B:HREV.0000046363.59374.23. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Kaye DM, Lambert E, Sommerville M, Socratous F, Esler MD. Relation between cardiac sympathetic activity and hypertensive left ventricular hypertrophy. Circulation. 2003;108:560–565. doi: 10.1161/01.CIR.0000081775.72651.B6. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Kaye DM, Lambert E, Hastings J, Campbell DJ, Lambert G, et al. Angiotensin II and norepinephrine release: interaction and effects on the heart. J Hypertens. 2005;23:1077–1082. doi: 10.1097/01.hjh.0000166850.80344.cf. [DOI] [PubMed] [Google Scholar]
- Snyders DJ, Knoth KM, Roberds SL, Tamkun MM. Time-, voltage-, and state-dependent block by quinidine of a cloned human cardiac potassium channel. Mol Pharmacol. 1992;41:322–330. [PubMed] [Google Scholar]
- Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J Gen Physiol. 1993;101:513–543. doi: 10.1085/jgp.101.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeishi Y, Jalili T, Ball NA, Walsh RA. Responses of cardiac protein kinase C isoforms to distinct pathological stimuli are differentially regulated. Circ Res. 1999;85:264–271. doi: 10.1161/01.res.85.3.264. [DOI] [PubMed] [Google Scholar]
- Uebele VN, England SK, Chaudhary A, Tamkun MM, Snyders DJ. Functional differences in Kv1.5 currents expressed in mammalian cell lines are due to the presence of endogenous Kvβ2.1 subunits. J Biol Chem. 1996;271:2406–2412. doi: 10.1074/jbc.271.5.2406. [DOI] [PubMed] [Google Scholar]
- Uebele VN, England SK, Gallagher DJ, Snyders DJ, Bennett PB, Tamkun MM. Distinct domains of the voltage-gated K+ channel Kvβ1.3 β-subunit affect voltage-dependent gating. Am J Physiol. 1998;274:C1485–C1495. doi: 10.1152/ajpcell.1998.274.6.C1485. [DOI] [PubMed] [Google Scholar]
- Valenzuela C. Pharmacological electrical remodelling in human atria induced by chronic beta-blockade. Cardiovasc Res. 2003;58:498–500. doi: 10.1016/s0008-6363(03)00370-5. [DOI] [PubMed] [Google Scholar]
- Valenzuela C, Delpon E, Tamkun MM, Tamargo J, Snyders DJ. Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys J. 1995;69:418–427. doi: 10.1016/S0006-3495(95)79914-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CP, Hu N, Shen W, Mashburn AB, Murray KT. Modulation of the human Kv1.5 channel by protein kinase C activation: role of the Kvbeta1.2 subunit. J Pharmacol Exp Ther. 2002;302:545–550. doi: 10.1124/jpet.102.033357. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, et al. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc Natl Acad Sci U S A. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, et al. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res. 2013;112:498–509. doi: 10.1161/CIRCRESAHA.112.273896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Concentration dependence of bupivacaine-induced blockade of hispidin-treated Kv1.5 + Kvβ1.3 channels. (A) The dashed and dotted lines represent the dose–response curves obtained for the bupivacaine-induced blockade of Kv1.5 and Kv1.5 + Kvβ1.3 channels, respectively (taken from González et al., 2002). Reduction in the current (relative to the control) at the end of depolarizing steps from −80 to +60 mV was used as an index of blockade. (●): Concentration–response curves for bupivacaine in bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels and in hispidin-treated Kv1.5 + Kvβ1.3 channels (○). (B) Reduction in the current (relative to the control) at 50 ms (○) and at 250 ms (●) depolarizing steps from −80 to +60 mV. Each point represents the mean ± SEM of three to four experiments. The lines represent the fit of the experimental data to a monophasic Hill equation.
Figure S2 Concentration dependence of bupivacaine- (A) and quinidine-induced (B) blockade of calphostine C-treated Kv1.5 + Kvβ1.3 channels. The dashed and continuous lines represent the dose–response curves obtained for the bupivacaine- or quinidine-induced blockade at 50 ms (○) and at 250 ms (●) depolarizing steps from −80 to +60 mV respectively. Each point represents the mean ± SEM of three to eight experiments. The continuous line represents the fit of the experimental data to a biphasic Hill equation.
Figure S3 Concentration dependence of bupivacaine- (A) and quinidine-induced (B) blockade of bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. The dashed and continuous lines represent the dose–response curves obtained for the bupivacaine- or quinidine-induced blockade at 50 ms (○) and at 250 ms (●) depolarizing steps from −80 to +60 mV respectively. Each point represents the mean ± SEM of three to eight experiments. The continuous line represents the fit of the experimental data to a monophsic Hill equation.
Figure S4 Absolute values for bupivacaine- and quinidine- blockade of calphostin C- and bisindolylmaleimide II-treated Kv1.5 + Kvβ1.3 channels. Magnitude of calphostin CKv1.5 + Kvβ1.3 currents in the absence and in the presence of different bupivacaine (A) and quinidine (B) concentrations. Magnitude of bisindolylmaleimide II-Kv1.5 + Kvβ1.3 currents in the absence and in the presence of different bupivacaine (C) and quinidine (D) concentrations. Each point represents the mean ± SEM of three to eight experiments.
Table S1 IC50 and nH from the of the concentration–response curves to bupivacaine in Kv1.5 + Kvβ1.3 channels expressed in cells treated with hispidin and bisindolylmaleimide II in comparison with those obtained of bupivacaine on Kv1.5 and Kv1.5 + Kvβ1.3 channels.