

Abstract
Background and Purpose
The OX40–OX40L protein–protein interaction (PPI) is an important cell-surface signalling co-stimulatory regulator within the TNFR superfamily (TNFRSF) and a promising therapeutic target for immunomodulation. PPIs are difficult to modulate using small-molecules. Here, we describe the identification of a small-molecule OX40 modulator and confirm its partial agonist character.
Experimental Approach
Cell-free screening assays were developed and used to identify OX40–OX40L inhibitors. Modified versions of this assay were used to elucidate the binding partner and the binding nature of active compounds. OX40-transfected sensor cells with NF-κB reporters were constructed and used to confirm and characterize activity and specificity. Immunomodulatory activity and partial agonist nature were further confirmed by ex vivo T-cell polarization assays.
Key Results
Several compounds that concentration-dependently affected OX40-OX40L were identified. Cell assays indicated that they were partial agonists with low micromolar potency and adequate selectivity. Under polarizing conditions based on TGF-β, the most promising compound mimicked the effect of an agonistic anti-OX40 antibody in suppressing regulatory T-cell generation and diverting CD4+CD62L+Foxp3− cells to TH9 phenotype in vitro.
Conclusions and Implications
We identified, to our knowledge, the first small-molecule compounds able to interfere with OX40–OX40L binding and, more importantly, to act as partial agonists of OX40. This is particularly interesting, as small-molecule agonism or activation of PPIs is considered unusually challenging and there are only few known examples. These results provide proof-of-principle evidence for the feasibility of small-molecule modulation of the OX40–OX40L interaction and for the existence of partial agonists for TNFRSF-PPIs.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| OX40 (TNFRSF4) | OX40L |
| CD40 (TNFRSF5) | CD40L |
| RANK (TNFRSF11A) | RANKL |
| 4-1BB (TNFRSF9) | 4-1BBL |
| TNFR1 (TNFRSF1A) | TNF-α |
| TGF-β1 | |
| IL-2 | |
| IL-4 |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
OX40 (ACT35, CD134, TNFRSF4), a surface type 1 transmembrane glycoprotein expressed on a variety of cell types, is one of the most important and widely studied members of the TNF receptor superfamily (TNFRSF). The biological and pathological responses initiated upon OX40 ligand (OX40L, CD252, CD134L, gp34, TNFSF4) binding have been thoroughly studied, and modulation of the OX40–OX40L co-stimulatory interaction is a particularly promising therapeutic target for immunomodulation (Sugamura et al., 2004; Croft et al., 2009; Croft, 2010; Ishii et al., 2010). Either inhibition or stimulation of OX40 signalling could be beneficial depending on the nature of the physiological and pathological conditions targeted, and biological drugs targeting this pathway are in clinical trials for asthma and prostate cancer respectively (Tansey and Szymkowski, 2009; Sheridan, 2013; Yao et al., 2013). Of particular interest, inhibition of the OX40–OX40L co-stimulatory interaction at week 12 has been shown to significantly reduce the incidence of autoimmune diabetes in NOD mice, a common animal model of type 1 diabetes (T1D) (Pakala et al., 2004). OX40 has also been shown to be involved in controlling allograft tolerance (Kinnear et al., 2013), including islet allograft tolerance (Chen et al., 2008). In contrast, the positive modulation of OX40 expressed on both CD4+ and CD8+ T-cells could lead to promising immunostimulatory candidates for tumour therapy (Kjaergaard et al., 2000; Weinberg et al., 2000; Piconese et al., 2008) or as antigen-specific adjuvants in T1D (Bresson et al., 2011). Augmentation of anti-tumour T-cell effector activity or suppression of regulatory T-cell function has been observed on injection of agonistic OX40 antibodies (OX86), OX40L-Ig fusion proteins and RNA aptamers that bind OX40 in many animal models (Weinberg et al., 2000; Dollins et al., 2008; Piconese et al., 2008). Recently, OX86 has also been shown to synergize with the antigen-specific prevention of T1D in vivo in NOD mice (Bresson et al., 2011). Thus, small-molecule agonists or antagonists of OX40 could become not only valuable tools for studying the immunomodulatory role of this interaction but also potential therapeutics of interest for tumour suppression or immune diseases, including T1D and other autoimmune disorders.
The OX40–OX40L interaction is a typical protein–protein interaction (PPI) that, along with all other PPIs, was considered undruggable by small molecules for a long time because the relatively large protein interfaces involved in these interactions generally lack well-defined binding pockets as compared to most of the traditional targets such as enzymes or GPCRs (Hopkins and Groom, 2002; Smith et al., 2006; Cheng et al., 2007). Nevertheless, within the last few years, several sufficiently effective and even clinically promising small-molecule PPI inhibitors (PPIIs) have been identified for a number of different targets (Cochran, 2000; Arkin and Wells, 2004; Whitty and Kumaravel, 2006; Wells and McClendon, 2007; Buchwald, 2010). By focusing on the chemical space of organic dyes, we have recently identified the first small-molecule inhibitors of the CD40–CD40L co-stimulatory PPI (Margolles-Clark et al., 2009a,b) as well as what seem to be the first promiscuous small-molecule PPIIs (Ganesan et al., 2011). By all accounts, the identification of small-molecule PPI stimulators is even more challenging than that of PPI inhibitors since they, in addition to binding, also need to trigger the downstream activation cascade. Only a very limited number of small-molecule PPI ‘agonists’ (i.e., enhancers or stabilizers) have been identified to date; published examples include stabilizers of some of the PPIs in which protein 14-3-3 is involved (Rose et al., 2010; Milroy et al., 2013; Ottmann, 2013) and a possible small-molecule activator of the TNF-related apoptosis-inducing ligand receptor DR5 (also known as TNFRSF10B; Wang et al., 2013).
Here, we report results of our ongoing search for small-molecule co-stimulatory modulators focusing on the OX40–OX40L interaction. We have identified several compounds with relatively well-defined structural scaffolds capable of interfering with the OX40–OX40L PPI with low micromolar potency in cell-free screening assays. Notably, subsequent cell-based assays revealed that several of them are capable of stimulatory activity at OX40 and mechanistic studies indicated that they act as partial agonists.
Methods
PPI binding assays
Microtitre plates (Nunc FMaxisorp; 96-well) were coated overnight at 4°C with 100 µL per well of Fc-conjugated receptors diluted in PBS 7.2. This was followed by blocking with 200 µL per well of blocking solution (PBS 7.2, 0.05% Tween-20, 1% BSA) for 1 h at room temperature (RT). Then, plates were washed twice using washing solution (PBS 7.4, 0.05% Tween-20) and tapped dry before the addition of the appropriate FLAG-tagged/biotinylated ligands along with different concentrations of tested compounds diluted in binding buffer (100 mM HEPES, 0.005% BSA, pH 7.2) to give a total volume of 100 µL per well. After 1 h of incubation, three washes were conducted, and anti-FLAG HRP conjugate was used to detect the bound FLAG-tagged ligand. Plates were washed three times before the addition of 120 µL per well of HRP substrate TMB (3,3′,5,5′-tetramethylbenzidine) and kept in the dark for 15–30 min. The reaction was stopped using 30 µL of 1 M H2SO4, and the absorbance value was read at 450 nm. The concentrations of receptors used were 0.3 µg mL−1 for CD40, TNFR1 (also known as TNFRSF1A) and RANK (TNFRSF11A); 0.6 µg mL−1 for OX40 and 4-1BB (TNFRSF9). The concentrations of the ligands were fixed at 0.02 µg mL−1 for CD40L, TNF-α and RANKL; 0.06 µg mL−1 for OX40L and 0.1 µg mL−1 for 4-1BBL. These values were selected following preliminary testing to optimize response (i.e., to produce a high-enough signal at conditions close to half-maximal response, EC50).
Identification of the binding partner
To determine whether these compounds bind to the receptor OX40 or OX40L, 96-well plates coated with OX40:Fc or OX40L:Fc were first incubated with different concentrations of compounds for 1 h at RT. Then, after three washes with washing solution, the plates were incubated for 1 h with the corresponding tagged ligands (OX40L or OX40 respectively). Plates were then washed three times with washing solution, and the amount of bound partner was detected with secondary antibody anti-tag-peroxidase HRP conjugate followed by reaction with HRP substrate TMB, as described earlier.
Schild analysis
Schild analysis was performed as described previously (Ganesan et al., 2011) using the same set-up described for the PPI binding assay. The plot was constructed by fixing the concentration of OX40 at 0.6 µg mL−1. The concentration of OX40L was varied from 0.001 to 10 µg mL−1 by serial dilution to obtain concentration–response curves in the absence or the presence of increasing concentrations (0.4–50.0 µM) of test compound. Binding data were fitted with a unified model as described below in the data fitting subsection.
BrdU incorporation and DAPI exclusion assays
For the BrdU [bromodeoxyuridine (5-bromo-2′-deoxyuridine)] assay, THP-1 human myeloid cells obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) with 10% FBS (v v−1; Invitrogen) and 1% penicillin-streptomycin (v v−1; Invitrogen). THP-1 human myeloid cells were centrifuged and re-suspended in the same medium without FBS and added to a 96-well microtitre plate at a density of 50 000 cells per well in the absence or presence of various concentrations of compounds diluted in the same media. The plate was incubated at 37°C for 24 h. BrdU (5-bromo-2′-deoxyuridine) incorporation level was determined using the BrdU cell proliferation kit from Roche (Mannheim, Germany) according to the manufacturer's protocol. Briefly, BrdU was added to the culture after treatments, and cells were incubated at 37°C for another 6 h. Then, fixing/denaturing buffer was added followed by the addition of HRP-conjugated detection anti-BrdU antibody. BrdU levels were measured using a plate reader at 450 nm after colour development with TMB solution and addition of a stopping solution of H2SO4. For the 4′,6-diamidino-2-phenylindol (DAPI) exclusion assay, THP-1 cells were seeded to a density of 500 000 cells mL−1 in the absence or presence of test compounds for 24 h. Viability upon treatment was determined using a BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA, USA) and the software FlowJo version 7.2.2 (Ashland, OR, USA). The number of live cells was quantified after gating out DAPI and cell debris assessed on the basis of forward and side-scatter properties of the untreated samples as reference.
Cell transfection
HEK293 cells containing a reporter gene under the control of a minimal promoter fused to NF-κB binding sites (HEK-Blue TNFα/IL-1β cells) were acquired from InvivoGen (San Diego, CA, USA) and further modified to express native or hybrid TNFRSF of interest such as CD40 or OX40. The coding regions of the CD40 and OX40 genes were amplified by PCR using as template cDNA of activated CD19+ B cells and activated CD4+ T-cells respectively. Primers were designed from published nucleic acid sequences of CD40 (BC012419) and OX40 (BC105072). Hybrids of receptors OX40 and CD40 were generated via overlapping PCR by fusing the corresponding extracellular regions including signal sequences of OX40 (amino acids 1–214) to a region of the CD40 containing the transmembrane and intracellular domains (amino acids 193–277). After cloning the amplified sequences into the vector pcDNA 3.3-TOPO TA (Invitrogen), the resulting plasmids were transfected into HEK-Blue TNF-α/IL-1β cells. Stable lines resistant to 0.6 mg mL−1 of Geneticin (G418; Invitrogen) were analysed by flow cytometry for expression of TNF receptors.
Sensor cell assay
TNFR1, OX40 and CD40 expressing sensor cells were maintained in DMEM at 80% confluence for each experiment. Cells were trypsinized and re-suspended in the same medium with 1% FBS and seeded on 96-well microtiter plates at a density of 100 000 cells per well in the absence and presence of various concentrations of compounds diluted in the same media. For ligand-mediated stimulation, final concentrations of recombinant human TNF-α (20 ng mL−1), CD40L (20 ng mL−1) or OX40L (40 ng mL−1), which have been selected following preliminary testing to optimize response, were maintained in the wells for this purpose. After 18 h of incubation at 37°C, 20 µL of supernatant of each well was taken and added to another 96-well microtitre plate containing 180 µL per well of QUANTI-Blue (InvivoGen). The level of secreted embryonic alkaline phosphatase (SEAP) was determined after 30 min of incubation at 37°C by reading at 625 nm using a spectrophotometer.
Mice
Foxp3GFP mice were obtained from Dr. A. Y. Rudensky (Memorial Sloan-Kettering Cancer Center, NY, USA) and maintained at the University of Miami. All animal studies were carried out under protocols approved by the University of Miami Institutional Animal Care and Use Committee.
Polarization of naïve CD4+ T-cells in vitro
Naïve CD4+ T-cells (CD4+CD62L+Foxp3−) obtained from GFP-Foxp3 reporter mice were sorted by flow cytometry, then 100 000 cells per well were activated with anti-CD3 and anti-CD28 conjugated Dynabeads (3 µL per well; Invitrogen). The induction of iTreg cells and TH9 cells was carried out in the presence of 3 ng mL−1 human TGF-β1 (R&D Systems) and 10 ng mL−1 IL-2 (PeproTech, Rocky Hill, NJ, USA) for induction of iTreg cells. For TH9 polarization, 3 ng mL−1 human TGF-β1 and 10 ng mL−1 IL-4 (PeproTech) were used. In addition, various concentrations of test compound 4 (CVN) diluted in the same media were added along with the Dynabeads and cytokines. CD4+ T-cells cultured for 3 days under these polarizing conditions were collected, and cell polarization was assessed by intracellular staining and flow cytometry.
For intracellular cytokine staining, CD4+ T-cells activated under TH9 polarizing conditions were re-stimulated for 4 h with phorbol 12-myristate 13-acetate (50 ng mL−1) and ionomycin (500 ng mL−1; Sigma-Aldrich) in the presence of GolgiStop (BioLegend, San Diego, CA, USA). After being stained with Live/Dead yellow (Sigma-Aldrich), cells were fixed and made permeable with Cytofix/Cytoperm solution (BD Biosciences) and were stained with fluorochrome-conjugated anti-IL-9 (RM9A4; BioLegend). In addition, for the iTreg cell conversion experiment, DAPI staining (Sigma-Aldrich) was used to distinguish live/dead cells, and iTreg cells were identified by the expression of GFP. All samples were acquired with BD LSR II flow cytometer (BD Biosciences), and data were analysed with FACSDiva software (BD Biosciences).
Data fitting and statistics
All binding analyses were carried out in duplicate per plate and repeated at least three times; the averaged data were normalized and used for data fitting and analysis. Binding data were fitted using the standard log inhibitor versus response model in GraphPad Prism (GraphPad, La Jolla, CA, USA):
| Table 1 |
For the Schild analysis, a common Gaddum/Schild EC50-shift model was used to fit all binding data obtained at five different inhibitor concentrations:
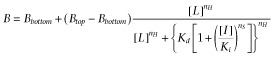 |
Table 2 |
All fittings were carried out with GraphPad Prism. Cytotoxicity and reporter assay data were analysed by one-way repeated-measures anova followed by Dunnett's multiple comparison post hoc test for individual differences using GraphPad Prism and a significance level of P < 0.05 for all comparisons.
The NF-κB activation data obtained in the sensor cell assays were fitted with a general quantitative modelling of activation for competitive partial agonists obtained using the minimal ‘two-state theory’ (del Castillo–Katz) model for receptor activation (Del Castillo and Katz, 1957; Jenkinson, 2003; Bodor and Buchwald, 2012) [mathematically equivalent to the Black and Leff operational model (Black and Leff, 1983; Kenakin, 2006)] when two ligands (L1, L2) of different affinities (Kd1, Kd2) and efficacies (ε1, ε2) are present simultaneously. Details of the model are discussed in Supporting Information Appendix S1; the final equation used for fitting was:
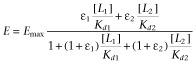 |
Table 3 |
Materials
The monoclonal anti-human OX40L (clone 159403), anti-human CD40L (clone 40804) and anti-human TNF-α antibodies (clone 1825) were purchased from R&D Systems (Minneapolis, MN, USA). Purified recombinant Fc-conjugated receptors (CD40, TNFR1, RANK, OX40, muOX40 and 4-1BB) and their corresponding FLAG-tagged ligands (CD40L, TNF-α, RANKL, OX40L, muOX40L and 4-1BBL) were obtained from Enzo Life Science (San Diego, CA, USA). OX40L:Fc and OX40-His were from Imgenex (San Diego, CA, USA) and Life Technologies (Carlsbad, CA, USA) respectively. Small-molecule compounds of interest are denoted as follows – 1: DB36 (direct black 36); 2: DB71 (direct blue 71, CAS 4399-55-7); 3: DB15 (direct blue 15, CAS 2429-74-5); and 4: CVN (chlorazol violet N, direct violet 1, CAS 2586-60-9). CVN was from TCI America (Portland, OR, USA); all other chemicals and reagents used were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Results
Effects on the OX40–OX40L binding
Following our recent success in identifying the first small-molecule inhibitors of the CD40-CD40L co-stimulatory interaction within an organic dye-focused library (Margolles-Clark et al., 2009b; 2010; Ganesan et al., 2011), we performed pre-screening assays with this library of several hundred compounds representative of this chemical space for a number of TNFRSF–ligand interactions and identified several compounds (Figure 1) with promising inhibitory activity on the OX40-OX40L binding. Activities were quantified using cell-free in vitro assays measuring the amount of soluble human OX40L bound to plate-coated OX40 in the presence of increasing concentrations of test compounds as described previously (Margolles-Clark et al., 2010; Ganesan et al., 2011). Compounds 1–4 were selected as some of the most promising ones that concentration-dependently inhibited the OX40-OX40L binding with low micromolar potency (Figure 1). As before, tartrazine was included as a negative control in all assays since it is a polysulfonated dye with a chemical structure somewhat related to those of 1–4 that, however, consistently showed no activity in any of these assays. Median inhibitory concentrations (IC50s) obtained by fitting the data with a standard binding model were 1.2, 2.7, 1.5 and 1.9 µM for 1, 2, 3 and 4, respectively, and are summarized in Table 2013. All binding assays were performed at room temperature (25°C), but for verification purposes, we have also performed them at body temperature (37°C) for compounds 1–4 and observed no significant differences (i.e., no IC50 changed by more than 35%, within the experimental error; data not shown). As before for the CD40-CD40L system (Margolles-Clark et al., 2009b), in addition to the human proteins, the binding assay was also performed with mouse OX40 and OX40L. Whereas the human antibody inhibited only the human system, the small molecules showed similar inhibitory activity in both systems (data not shown).
Figure 1.

Concentration-dependent inhibition of the OX40–OX40L and other TNFSF ligand–receptor bindings by the small-molecule compounds (1–4) of the present study with structures as shown (see Table 2013 for the corresponding IC50s). Data are average ± SD (normalized to % binding) for n = 3 independent experiments with duplicate for each condition.
Median inhibitory concentrations (IC50) and their 95% confidence intervals (CI) for compounds 1–4 for different TNFRSF-ligand interactions as indicated
| Compound | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| OX40–OX40L | RANK–RANKL | 4-1BB–4-1BBL | CD40–CD40L | TNFR1–TNF-α | OX40 binding*1983 | |
| Human | Human | Human | Human | Human | Human | |
| 1 (DB36) | 1.2 | 10.4 | 52.9 | 27.4 | 43.6 | 0.4 |
| [1.0–2.6] | [8.7–12.6] | [39.3–71.1] | [21.8–34.4] | [33.0–57.5] | [0.2–0.7] | |
| 2 (DB71) | 2.7 | 5.3 | 47.1 | 3.4 | 97.3 | 32.0 |
| [2.2–3.2] | [4.7–6.0] | [34.4–64.6] | [1.9–6.0] | [71.6–132.4] | [23.7–43.1] | |
| 3 (DB15) | 1.5 | 2.0 | 30.2 | 11.1 | 38.4 | 2.0 |
| [1.2–2.0] | [1.6–2.4] | [25.5–35.7] | [7.4–16.8] | [30.0–49.3] | [1.2–3.2] | |
| 4 (CVN) | 1.9 | 9.2 | 41.4 | 23.6 | 48.4 | 6.6 |
| [1.5–2.6] | [7.6–11.1] | [33.2–51.6] | [19.1–29.3] | [42.4–55.3] | [4.2–10.5] | |
| TZ | >2000 | >2000 | >2000 | >2000 | >2000 | >2000 |
| ErB† | 1.7 | 1.7 | 10.8 | 3.1 | 4.6 | 1.7 |
| [1.2–2.6] | [1.4–2.2] | [8.7–13.4] | [2.5–3.8] | [3.6–5.7] | [1.3–2.1] | |
Notation for small-molecule compounds – 1: DB36 (direct black 36); 2: DB71 (direct blue 71); 3: DB15 (direct blue 15); 4: CVN (chlorazol violet N, direct violet 1.
IC50 values obtained in the binding partner assay. CI shown in italic font and in square parentheses.
Values obtained in Ganesan et al. (2011), except for the last column, which was measured here.
TZ, tartrazine; ErB, erythrosine B.
Specificity (within TNFSF) and cytotoxicity
Besides activity, selectivity towards the desired target also has to be defined. Accordingly, for compounds of interest, in addition to OX40-OX40L, we quantified inhibitory activities towards several other ligand–receptor pairs within the TNF superfamily, such as TNFR1-TNFα, CD40-CD40L, RANK-RANKL and 4-1BB-4-1BBL. While 1 and 4 showed acceptable (>5–20-fold) selectivity towards OX40–OX40L interaction when compared to all other TNFSF interactions tested, 2 and 3 showed similar activity toward CD40-CD40L and RANK-RANKL, respectively, as they inhibited these interactions with IC50s comparable to that for OX40-OX40L (Figure 1; Table 2013). For comparison, data for erythrosine B, a promiscuous PPI inhibitor we have identified earlier that shows no specificity and inhibits a variety of interaction with IC50s consistently in the 2–20 µM range (Ganesan et al., 2011), were also included in Table 2013.
For the most promising compounds, we also performed brief cytotoxicity evaluations in THP-1 (human monocytic leukaemia) cells. Toxic effects were assessed by a standard cell proliferation assay (BrdU incorporation) as well as a viability assay (DAPI exclusion). In the BrdU assay, none of the compounds showed cytotoxicity with IC50s less than 100 µM (well above the range of OX40-OX40L inhibitory activities); however, 2 inhibited cell proliferation at the highest concentration of 500 µM (Supporting Information Figure S1). Interestingly, some compounds such as 1 and 3 increased BrdU incorporation indicating a possibly increased proliferation rate. Toxic effects were also assessed by an additional viability assay (DAPI exclusion analysed by flow cytometry), and results confirmed well the observations of the BrdU assay with only 2 showing some cytotoxicity at the higher concentrations (Supporting Information Figure S2).
Binding partner and nature of OX40-OX40L binding inhibition
To elucidate the mechanism of the observed binding inhibition, we first performed experiments to assess whether these compounds bind to the receptor (OX40) or its ligand partner (OX40L). Similar experiments with our previously identified CD40-CD40L PPIIs showed that the most active compounds in that assay bind to the surface of CD40L, but not CD40 (Margolles-Clark et al., 2009b). Plate-coated proteins (OX40:Fc or OX40L:Fc, respectively) were incubated for 1 h with increasing concentrations of the test compounds, and after three washes to eliminate the unbound small molecules, the abilities to still bind the corresponding protein partner (OX40L or OX40, respectively) were assessed. Since our compounds bound with low micromolar affinities, we conducted preliminary assessments of the binding kinetics, which indicated that the binding was sufficiently long-lasting to make this assay feasible (data not shown). Results (Figure 2) indicate OX40 as the binding site for all of these compounds as they showed inhibition with IC50s in general agreement with those obtained for their previous interaction inhibitory activity (Figure 1; Table 2013). None of the compounds tested bound OX40L, except for the promiscuous PPI inhibitor erythrosine B (Ganesan et al., 2011).
Figure 2.
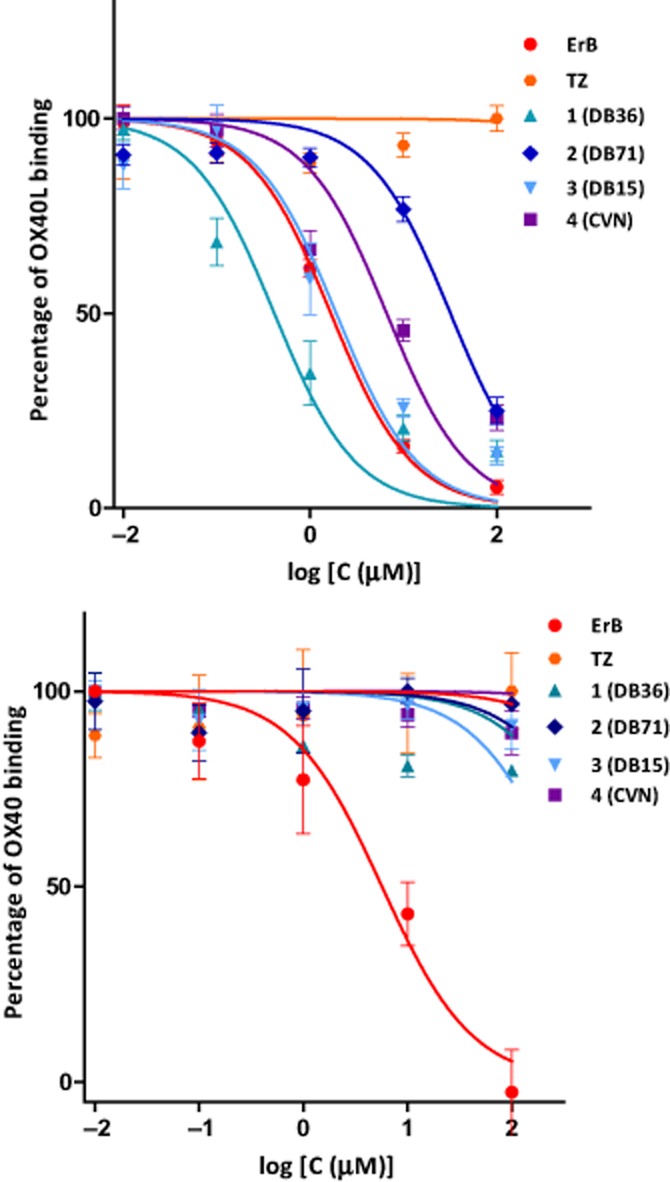
Identification of the binding partner (OX40 or OX40L) by assessing the amount of OX40L (top) or OX40 receptors (bottom) bound after incubations of the candidate compounds with OX40 receptors or OX40L, respectively, and addition of the protein binding partner (OX40L or OX40) only after a wash-out. All tested compounds bind to OX40 receptors, while only the promiscuous PPI inhibitor eythrosine B (ErB) binds to OX40L. Data are average ± SD (normalized to percent binding) for n = 3 independent experiments with triplicate for each condition.
As a further mechanistic evaluation, we performed a Schild analysis (Kenakin, 2006; Ganesan et al., 2011) with 4 as our most promising representative compound to assess the competitive/reversible nature of this binding. Concentration-dependency curves were generated by using a fixed concentration of OX40 and varying concentrations of OX40L in the presence of increasing concentrations of 4. Data from this complex binding assay (Supporting Information Figure S3) could be fitted well with the unified Gaddum–Schild model (Eq. 2004), indicating that 4 interferes with the OX40-OX40L binding in a manner that is consistent with a competitive binding interaction. The fit indicated a Kd of approximately 5 nM for the binding of the protein receptor–ligand pair (OX40-OX40L) and a pKB value of 5.9 (corresponding to KB of 1.2 µM; 95% CI: 0.6–2.4 µM) for the binding of 4, in good agreement with data obtained in the plain binding experiment (1.9 µM, Table 2013).
Effect in sensor cells with NF-κB reporter
To confirm the activity of the identified most promising compounds in a cell-based model, we used sensor cells containing a SEAP reporter gene under the control of a promoter fused to NF-κB binding site and transfected them with several receptors within the TNFRSF, including OX40, as described in the Methods section. Thus, in these cells the secretion of SEAP is specifically induced by the binding of a particular TNFSF ligand to its corresponding cell-surface receptor that all have a shared downstream activation of the NF-κB pathway. We confirmed that the OX40-transfected cells are indeed activated by OX40L in a concentration-dependent manner (Supporting Information Figure S4). Intriguingly enough, for compounds 1–4, which all inhibited OX40-OX40L binding in the cell-free binding assay, we could not see inhibition. Rather, they all showed a concentration-dependent enhancement of the OX40-linked activation of NF-κB in these sensor cells in the presence of a fixed concentration (100 ng mL−1 ≈ 3.3 nM) of recombinant OX40L with 1 and 4 showing the most potency (Supporting Information Figure S3). This, in combination with the previous results, suggested the possibility that these tested compounds cannot only compete for binding to OX40 with its innate OX40 ligand, but might also have the potential to activate the OX40 signalling downstream. To test whether the presence of OX40 ligand is required for the stimulation of OX40 signalling by the small-molecule compounds, we incubated the sensor cells with increasing concentrations of 1 and 4 and quantified NF-κB activation via SEAP. Interestingly, both compounds clearly increased SEAP secretion in concentration-dependent manner even without the presence of recombinant OX40L; however, they could not produce the same maximum activation. Furthermore, the activation by 1 and 4 was target specific as they did not activate the downstream NF-κB pathway when incubated with either TNFR1 or CD40 expressing sensor cells (Figure 3).
Figure 3.
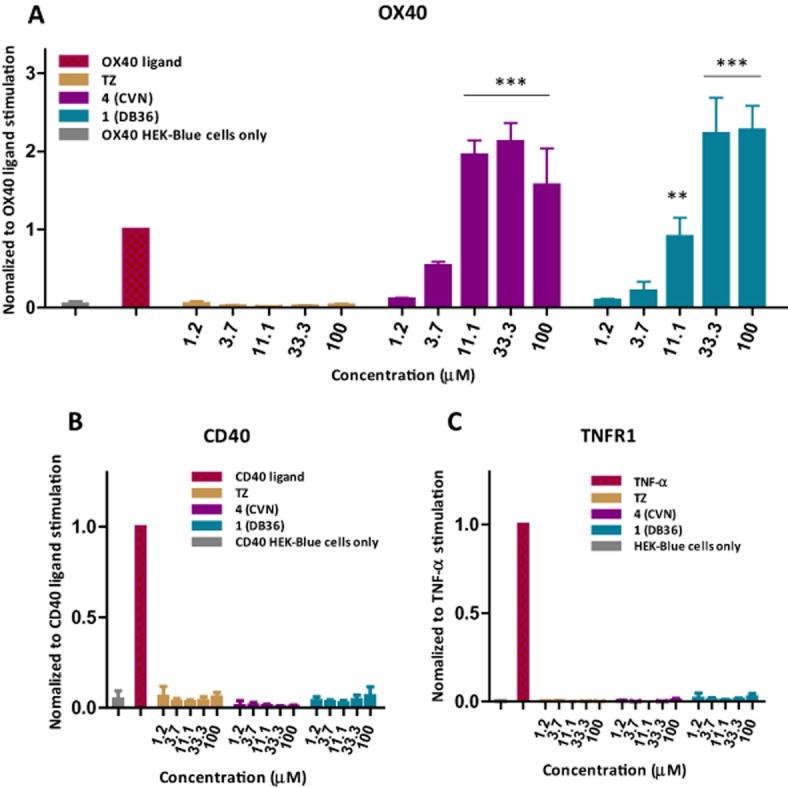
Selective stimulatory activity of 1 (DB36) and 4 (CVN) in OX40-transfected NF-κB sensor cells (A) as compared to total lack of such effect in similar CD40 (B) and TNFR1 (C) sensor cells. Data (average ± SD for n = 3 independent experiments with triplicate for each condition) were analysed by anova with Dunnett's multiple comparison post hoc test. **P < 0.01 and ***P < 0.001 as compared with control (cells incubated without any stimulation).
As a next step, to clarify the agonistic nature of these compounds, we quantified in detail the concentration dependence of the NF-κB activation caused in these sensor cells by combinations of 4 and the natural ligand OX40L at various concentrations. In the absence of OX40L (or in the presence of low concentrations of OX40L), 4 was able to produce concentration-dependent increase in NF-κB activation, but with a maximum that even at saturation was only a fraction of that produced by the natural ligand OX40L (Figure 4). In the presence of sufficiently high OX40L (1000 ng mL−1 ≈ 33 nM), 4 actually produced a slight decrease of activation in a concentration-dependent manner (Figure 4), a behaviour typical for partial agonists (Jenkinson, 2003; Kenakin, 2006). To verify this, we performed a quantitative modelling of the activation using a generalization of the minimal ‘two-state theory’ (del Castillo–Katz) model for receptor activation (Del Castillo and Katz, 1957; Jenkinson, 2003; Bodor and Buchwald, 2012) [mathematically equivalent with the Black and Leff operational model (Black and Leff, 1983; Kenakin, 2006)] for the case of two ligands (L1, L2) of different affinities (Kd1, Kd2) and efficacies (ε1, ε2) simultaneously present (eq. 1983, see Supporting Information Appendix S1 for full details). Fitting of the model to the data (Figure 5) shows that assumptions of a partial agonist with an efficacy of about 70% of that of OX40L can indeed account well for the behaviour seen, including the concentration-dependent decrease of activation produced in the presence of the highest concentration of the natural ligand OX40L. The affinity (Kd) values derived from this cell-based assay, as fitted with a unified model, are somewhat lower but still in acceptable agreement with those obtained in previous cell-free assays (approximately 11 nM for OX40L and 17 µM for 4 obtained by fitting an equation equivalent to eq. 1983, but with efficacies incorporated into different Emaxs).
Figure 4.
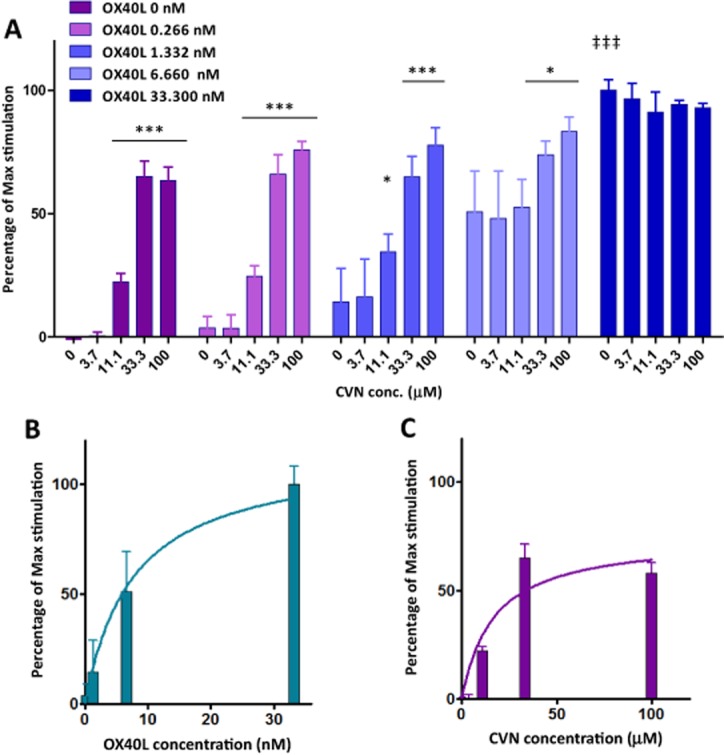
Activation of OX40 NF-κB reporter cells by OX40L and 4 combined at various concentrations. (A) OX40 sensor cells with NF-κB-induced SEAP were incubated overnight with a combination of both serial diluted OX40L and 4. Results are represented as the absorbance value of collected cell supernatant at the wavelength of 650 nm. Asterisks denote statistically significant differences compared to CVN 0 within each OX40L group (*P < 0.05, ***P < 0.001); double daggers denote ‡‡‡P < 0.001 between the maxima of OX40L alone (OX40L 33.3 nM, CVN 0) versus CVN alone (OX40L 0, CVN 100 µM) (anova with Dunnett's multiple comparison post hoc test). As compared to OX40L (B), 4 (CVN) alone (C) only achieved a maximum activation of the OX40-triggered response that was about 70%, indicating a partial agonist type response in this system.
Figure 5.

Three-dimensional surface representation of the activation effect on OX40 by a combination of full agonist OX40L and partial agonist 4 (CVN) in sensor cells with an NF-κB reporter (A). Purple dots represent experimental data and the surface calculated values by the corresponding fitted model (Eq. 1983). Concentrations are on the horizontal axes and are on log scales. The bottom figure (B) shows a corresponding two-dimensional representation (essentially equivalent with a cross-sectional view of the top figure from left, front) with a linear concentration scale.
Effect on T-cells (Treg and TH9)
Regulatory T-cells (Tregs) are known to play important immunomodulatory roles, and OX40 agonist signalling has been shown to suppress the generation of induced regulatory T-cells (iTregs) (So and Croft, 2007; Xiao et al., 2012). To validate the stimulatory activity on OX40 signalling of our small-molecule partial agonists, we performed iTreg conversion experiments using flow cytometry sorted naïve CD4+ Tconv cells (CD62L+ Foxp3−) from Foxp3-GFP reporter mice with our most promising representative compound (4, CVN). They were activated in vitro under iTreg polarizing conditions (TGF-β plus IL-2) and then assessed for Foxp3 induction. After 3 days of incubation, approximately 30% of CD4+ Tconv cells were converted into Foxp3+ iTreg cells in the presence of anti-CD3 and anti-CD28 magnetic beads (Dynabeads) plus TGF-β and IL-2. The agonistic anti-OX40 monoclonal antibody (mAb) (OX86) used as a positive control indeed inhibited iTreg conversion, and small-molecule 4, a partial agonist, also showed a less potent, but concentration-dependent inhibition with significant inhibition at 50 µM (Figure 6).
Figure 6.

Compound 4 inhibits iTreg generation. (A) Naïve wild-type CD4+ T-cells were activated for three days (Dynabeads plus iTreg polarizing cytokines, IL-2 + TGF-β) by increasing concentrations of 4 (CVN). Agonist OX40 antibody (OX86) and tartrazine (TZ) were added as positive and negative controls respectively. Inhibitory effect on the induction of iTreg cells were accessed by gating on the live CD4+ T-cell population. (B) Summary of all experiments showing average ± SD of three independent replicates. Data were analysed by anova with Dunnett's post hoc test with *P < 0.05 and **P < 0.01 as compared with Treg polarizing conditions (IL-2 + TGF-β).
Stimulation on OX40 has also been shown to enhance TH9 cell generation in vitro (Xiao et al., 2012). Therefore, as a further test of the OX40 partial agonist nature, we incubated 4 with naive CD4+ Tconv cells in the presence of anti-CD3 and anti-CD28 conjugated beads plus TH9 cell polarization conditions (TGF-β plus IL-4) for 3 days, re-stimulated with PMA/ionomycin for 4 h in the presence of Golgi stop, and assessed IL-9 levels by intracellular staining. The OX40 agonistic antibody (OX86) significantly enhanced TH9 cell percentage (by approximately 35%; Figure 7). Compound 4 also caused less prominent, but statistically significant enhancement of the TH9 cell population: approximately 15% at 50 µM versus approximately 5% of control (TGF-β plus IL-4 only) (Figure 7).
Figure 7.

Compound 4 enhances the induction of TH9 cells. (A) Naïve wild-type CD4+ T-cells were activated for three days (Dynabeads plus iTreg polarizing cytokines, IL-4 + TGF-β) by increasing concentrations of 4 (CVN). Agonist OX40 antibody (OX86) and tartrazine (TZ) were added as positive and negative controls, respectively. Stimulatory effects on the induction of TH9 cells were accessed by gating on the live CD4+ T-cell population. (B) Summary of all experiments showing average ± SD. Data were analysed by anova with Dunnett's post hoc test with *P < 0.05 and ***P < 0.001 as compared with TH9 polarizing conditions (IL-4 + TGF-β).
Discussion
As part of our ongoing search for potential therapeutically useful small-molecule modulators of co-stimulatory signalling, we have now identified the first small-molecule compounds capable of interfering with the OX40-OX40L PPI that also seem to act as partial agonists at the OX40 receptor. This is remarkable not only because PPIs have been traditionally considered untreatable with small-molecule inhibitors [with some success achieved only within the last decade or so (Arkin and Wells, 2004; Whitty and Kumaravel, 2006; Wells and McClendon, 2007; Buchwald, 2010)], but also because small-molecule PPI agonists are considered even less likely to exist since, in addition to binding, they also need to be able to trigger the downstream activation. Not surprisingly, only a very limited number of small-molecule PPI ‘agonists’ (i.e., enhancers or stabilizers) have been published to date (Rose et al., 2010; Milroy et al., 2013; Ottmann, 2013; Wang et al., 2013). The TNFRSF family has many high-value therapeutic targets, and biological compounds targeting almost all of the more than twenty known ligand–receptor pairs in this superfamily are now in clinical development for autoimmune diseases and cancer respectively (Tansey and Szymkowski, 2009; Croft et al., 2013; Yao et al., 2013). In the meantime, only very few small-molecule PPI inhibitors have been identified for TNFSF; they include one for TNFR1-TNF-α (SP307) (He et al., 2005) as well as some polyaromatic inhibitors for the CD40-CD40L co-stimulatory interaction including those by our group (Margolles-Clark et al., 2009b; 2010) and another, quite different scaffold (BIO8898) from Biogen (Silvian et al., 2011). SP307 was found to bind the TNF-α trimer and facilitate its dissociation (He et al., 2005), whereas BIO8898 was found to bind between two subunits of the CD40L trimer and distort it sufficiently to disrupt its binding with CD40 (Silvian et al., 2011).
By starting from the chemical space of organic dyes that provided our first CD40-CD40L PPIIs, we have now identified what seem to be the first small-molecule disruptors of the OX40-OX40L PPI that also act as partial agonists of the OX40 receptor with low micromolar potency. Detailed quantitative data support its partial agonist nature indicating that it causes agonism when no other agonists are present, but antagonism of more powerful (full) agonists (here, OX40L) if they are present. The activity of 4 (CVN, Figure Figure 1) has been confirmed in sensor cell assays (Figure 4) as well as in T-cells, including Treg and TH9 cells (Figure 6, Figure 7). Compared with the binding assay, activities in the cell-based assays are lower – probably, partly due to nonspecific protein binding in the presence of protein-rich media, as observed previously with similar compounds by us (Margolles-Clark et al., 2009b; 2010), and partly due to the interactions with other cell-surface TNFRSF and other receptors confounding the effect especially as the OX40 receptor is only transiently expressed in T-cells. OX40 (CD134), a member of the TNFRSF superfamily, is mainly expressed on activated T-cells. Conflicting data regarding the role of OX40 agonists on the persistence and function of regulatory T-cells (Tregs) have revealed that OX40 signalling plays a complex role in the physiology of Tregs (Takeda et al., 2004; Valzasina et al., 2005; Ito et al., 2006; Kroemer et al., 2007; So and Croft, 2007; Vu et al., 2007; Duan et al., 2008; Piconese et al., 2008; Redmond et al., 2009; Ruby et al., 2009; Griseri et al., 2010; Kinnear et al., 2013). Nevertheless, there is compelling evidence supporting a suppressive effect of OX40 agonism on the development of Tregs in vitro. In addition, under an appropriate cytokine microenvironment (in the presence of TGF-β and IL-4), OX40 stimulation has been demonstrated to induce TH9 cell generation (Xiao et al., 2012). Consistent with these findings, our small-molecule OX40 partial agonist 4 showed less prominent, but similar activity to that of the OX40 agonistic mAb (OX86) when tested in these models relying upon the induction of Treg and TH9 cells from naive CD4+ T-cells.
Preclinical data from tumour models and clinical data from breast cancer patients have suggested the beneficial effect of OX40 agonists (either OX40L:Ig fusion protein or OX40 agonist antibody) on tumour progression (Weinberg et al., 2011). Both CD4+ and CD8+ T-cells seem to be involved in the therapeutic efficacy of OX40 agonists. Due to the inducible and time-limited expression of OX40 on these cells, therapies targeting OX40 might produce less toxicity and reduced systemic side effects than existing immunomodulatory therapies in clinical use. Consequently, several biological candidates targeting this pathway are in various stages of clinical development (Tansey and Szymkowski, 2009; Croft et al., 2013; Sheridan, 2013; Yao et al., 2013). Furthermore, and of particular interest for our autoimmune disease-focused work, recently it has been shown that OX40 stimulation can promote Treg expansion in vivo in both mouse models of diabetes and colitis, which, in turn, prevented diabetes onset and suppressed colitis (Griseri et al., 2010; Bresson et al., 2011). The dual function of OX40 agonists to increase T-cell effector function as well as regulate Treg function selectively in different pathological conditions renders OX40 agonism a promising therapeutic possibility for the prevention of these diseases. Our proof-of-principle results identifying the first small molecules that can act as disruptors of the OX40-OX40L PPI as well as partial OX40 agonists represent a first step towards identifying new pharmacological tools to investigate this interaction that could also lead to possible therapeutic alternatives complementing the current biological drugs under development.
Acknowledgments
This work was supported by a grant from the National Institutes of Health National Institute of Allergy and Infectious Diseases (1R01AI101041-02, PI: P. B.).
Glossary
- BrdU
bromodeoxyuridine (5-bromo-2′-deoxyuridine)
- CVN
chlorazol violet N
- OX40L
OX40 ligand (also known as TNFSF4)
- PPI
protein–protein interaction
- SEAP
secreted embryonic alkaline phosphatase
- T1D
type 1 diabetes
- TNFRSF
tumour necrosis factor receptor superfamily
Author contributions
Y. S. performed the experiments, collected data, analysed results and wrote the manuscript. E. M.-C. prepared the transfected lines and performed experiment. A. B. and Y. S. designed the mouse immunology experiments and analysed the results. P. B. conceived the study, designed the experiments, analysed data and results, and wrote the manuscript. All authors reviewed the final manuscript.
Conflict of interest
The authors declare that they have no competing financial interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12819
Figure S1 Cell toxicities of the present compounds (1–4) assessed by a standard proliferation assay (BrdU) in THP-1 cells.
Figure S2 Cell toxicities of the present compounds (1–4 plus tartrazine, TZ, as a negative control and erythrosine B, ErB, as a positive control) assessed by standard apoptosis assay (DAPI exclusion analysed by flow cytometry) in THP-1 cells.
Figure S3 Schild analysis of the interference with the OX40-OX40L binding activity for compound 4 (CVN).
Figure S4 Concentration dependence of the ligand-induced activation in CD40- and OX40-expressing NF-κB reporter sensor cells used in the present work.
Figure S5 Confirmation of activity for the present compounds of interest in OX40-expressing NF-κB reporter sensor cells.
Appendix S1 Quantification of receptor activation for competitive partial agonists in the presence of a full agonist using the minimal two-state receptor model.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013;170:1676–1703. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff J. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Bodor N, Buchwald P. Retrometabolic Drug Design and Targeting. 1st edn. Hoboken, NJ: Wiley; 2012. [Google Scholar]
- Bresson D, Fousteri G, Manenkova Y, Croft M, von Herrath M. Antigen-specific prevention of type 1 diabetes in NOD mice is ameliorated by OX40 agonist treatment. J Autoimmun. 2011;37:342–351. doi: 10.1016/j.jaut.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life. 2010;62:724–731. doi: 10.1002/iub.383. [DOI] [PubMed] [Google Scholar]
- Chen M, Xiao X, Demirci G, Li XC. OX40 controls islet allograft tolerance in CD154 deficient mice by regulating FOXP3+ Tregs. Transplantation. 2008;85:1659–1662. doi: 10.1097/TP.0b013e3181726987. [DOI] [PubMed] [Google Scholar]
- Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 2007;25:71–75. doi: 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- Cochran AG. Antagonists of protein-protein interactions. Chem Biol. 2000;7:R85–R94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annu Rev Immunol. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. 2009;229:173–191. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov. 2013;12:147–168. doi: 10.1038/nrd3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Interaction at end-plate receptors between different choline derivatives. Proc R Soc Lond B Biol Sci. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- Dollins CM, Nair S, Boczkowski D, Lee J, Layzer JM, Gilboa E, et al. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem Biol. 2008;15:675–682. doi: 10.1016/j.chembiol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, So T, Croft M. Antagonism of airway tolerance by endotoxin/lipopolysaccharide through promoting OX40L and suppressing antigen-specific Foxp3+ T regulatory cells. J Immunol. 2008;181:8650–8659. doi: 10.4049/jimmunol.181.12.8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan L, Margolles-Clark E, Song Y, Buchwald P. The food colorant erythrosine is a promiscuous protein-protein interaction inhibitor. Biochem Pharmacol. 2011;81:810–818. doi: 10.1016/j.bcp.2010.12.020. [DOI] [PubMed] [Google Scholar]
- Griseri T, Asquith M, Thompson C, Powrie F. OX40 is required for regulatory T cell-mediated control of colitis. J Exp Med. 2010;207:699–709. doi: 10.1084/jem.20091618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, et al. Small-molecule inhibition of TNF-α. Science. 2005;310:1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Ishii N, Takahashi T, Soroosh P, Sugamura K. OX40-OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv Immunol. 2010;105:63–98. doi: 10.1016/S0065-2776(10)05003-0. [DOI] [PubMed] [Google Scholar]
- Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci USA. 2006;103:13138–13143. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson DH. Classical approaches to the study of drug-receptor interactions. In: Foreman JC, Johansen T, editors. Textbook of Receptor Pharmacology. 2nd edn. Boca Raton, FL: CRC Press; 2003. pp. 3–80. [Google Scholar]
- Kenakin TP. A Pharmacology Primer: Theory, Applications, and Methods. 2nd edn. San Diego, CA: Academic Press; 2006. [Google Scholar]
- Kinnear G, Wood KJ, Fallah-Arani F, Jones ND. A diametric role for OX40 in the response of effector/memory CD4+ T cells and regulatory T cells to alloantigen. J Immunol. 2013;191:1465–1475. doi: 10.4049/jimmunol.1300553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000;60:5514–5521. [PubMed] [Google Scholar]
- Kroemer A, Xiao X, Vu MD, Gao W, Minamimura K, Chen M, et al. OX40 controls functionally different T cell subsets and their resistance to depletion therapy. J Immunol. 2007;179:5584–5591. doi: 10.4049/jimmunol.179.8.5584. [DOI] [PubMed] [Google Scholar]
- Margolles-Clark E, Jacques-Silva MC, Ganesan L, Umland O, Kenyon NS, Ricordi C, et al. Suramin inhibits the CD40-CD154 costimulatory interaction: a possible mechanism for immunosuppressive effects. Biochem Pharmacol. 2009a;77:1236–1245. doi: 10.1016/j.bcp.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Margolles-Clark E, Umland O, Kenyon NS, Ricordi C, Buchwald P. Small molecule costimulatory blockade: organic dye inhibitors of the CD40-CD154 interaction. J Mol Med. 2009b;87:1133–1143. doi: 10.1007/s00109-009-0519-3. [DOI] [PubMed] [Google Scholar]
- Margolles-Clark E, Kenyon NS, Ricordi C, Buchwald P. Effective and specific inhibition of the CD40-CD154 costimulatory interaction by a naphthalenesulphonic acid derivative. Chem Biol Drug Des. 2010;76:305–313. doi: 10.1111/j.1747-0285.2010.01014.x. [DOI] [PubMed] [Google Scholar]
- Milroy LG, Brunsveld L, Ottmann C. Stabilization and inhibition of protein-protein interactions: the 14-3-3 case study. ACS Chem Biol. 2013;8:27–35. doi: 10.1021/cb300599t. [DOI] [PubMed] [Google Scholar]
- Ottmann C. Small-molecule modulators of 14-3-3 protein-protein interactions. Bioorg Med Chem. 2013;21:4058–4062. doi: 10.1016/j.bmc.2012.11.028. [DOI] [PubMed] [Google Scholar]
- Pakala SV, Bansal-Pakala P, Halteman BS, Croft M. Prevention of diabetes in NOD mice at a late stage by targeting OX40/OX40 ligand interactions. Eur J Immunol. 2004;34:3039–3046. doi: 10.1002/eji.200425141. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29:187–201. doi: 10.1615/critrevimmunol.v29.i3.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R, Erdmann S, Bovens S, Wolf A, Rose M, Hennig S, et al. Identification and structure of small-molecule stabilizers of 14-3-3 protein-protein interactions. Angew Chem Int Ed Engl. 2010;49:4129–4132. doi: 10.1002/anie.200907203. [DOI] [PubMed] [Google Scholar]
- Ruby CE, Yates MA, Hirschhorn-Cymerman D, Chlebeck P, Wolchok JD, Houghton AN, et al. Cutting edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J Immunol. 2009;183:4853–4857. doi: 10.4049/jimmunol.0901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C. Industry pursues co-stimulatory receptor immunomodulators to treat cancer. Nat Biotechnol. 2013;31:181–183. doi: 10.1038/nbt0313-181. [DOI] [PubMed] [Google Scholar]
- Silvian LF, Friedman JE, Strauch K, Cachero TG, Day ES, Qian F, et al. Small molecule inhibition of the TNF family cytokine CD40 ligand through a subunit fracture mechanism. ACS Chem Biol. 2011;6:636–647. doi: 10.1021/cb2000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Hu L, Falkner JA, Benson ML, Nerothin JP, Carlson HA. Exploring protein-ligand recognition with binding MOAD. J Mol Graph Model. 2006;24:414–425. doi: 10.1016/j.jmgm.2005.08.002. [DOI] [PubMed] [Google Scholar]
- So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+ Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172:3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Szymkowski DE. The TNF superfamily in 2009: new pathways, new indications, and new drugs. Drug Discov Today. 2009;14:1082–1088. doi: 10.1016/j.drudis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang X, Yu H, Wei S, Williams N, Holmes DL, et al. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat Chem Biol. 2013;9:84–89. doi: 10.1038/nchembio.1153. [DOI] [PubMed] [Google Scholar]
- Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164:2160–2169. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- Weinberg AD, Morris NP, Kovacsovics-Bankowski M, Urba WJ, Curti BD. Science gone translational: the OX40 agonist story. Immunol Rev. 2011;244:218–231. doi: 10.1111/j.1600-065X.2011.01069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Whitty A, Kumaravel G. Between a rock and a hard place? Nat Chem Biol. 2006;2:112–118. doi: 10.1038/nchembio0306-112. [DOI] [PubMed] [Google Scholar]
- Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol. 2012;13:981–990. doi: 10.1038/ni.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov. 2013;12:130–146. doi: 10.1038/nrd3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cell toxicities of the present compounds (1–4) assessed by a standard proliferation assay (BrdU) in THP-1 cells.
Figure S2 Cell toxicities of the present compounds (1–4 plus tartrazine, TZ, as a negative control and erythrosine B, ErB, as a positive control) assessed by standard apoptosis assay (DAPI exclusion analysed by flow cytometry) in THP-1 cells.
Figure S3 Schild analysis of the interference with the OX40-OX40L binding activity for compound 4 (CVN).
Figure S4 Concentration dependence of the ligand-induced activation in CD40- and OX40-expressing NF-κB reporter sensor cells used in the present work.
Figure S5 Confirmation of activity for the present compounds of interest in OX40-expressing NF-κB reporter sensor cells.
Appendix S1 Quantification of receptor activation for competitive partial agonists in the presence of a full agonist using the minimal two-state receptor model.