

Background: Receptor trafficking and cross-talk regulate 7TM/GPCR signaling capacity.
Results: Inhibition of GLP-1R internalization in the presence of GIPR reduces GLP-1R signaling.
Conclusion: GLP-1R internalization is essential for full receptor functionality and is abrogated upon cross-talk with GIPR.
Significance: GLP-1R cross-talk with GIPR has functional consequences for GLP-1R signaling.
Keywords: Fluorescence Resonance Energy Transfer (FRET), G-protein-coupled Receptor (GPCR), Intracellular Trafficking, Receptor Internalization, Signal Transduction, Type 2 Diabetes, Glucagon-like Peptide-1, Glucose-dependent Insulinotropic Polypeptide, Receptor Cross-talk, Seven Transmembrane-spanning Receptor
Abstract
The signaling capacity of seven-transmembrane/G-protein-coupled receptors (7TM/GPCRs) can be regulated through ligand-mediated receptor trafficking. Classically, the recycling of internalized receptors is associated with resensitization, whereas receptor degradation terminates signaling. We have shown previously that the incretin glucagon-like peptide-1 receptor (GLP-1R) internalizes fast and is primarily resensitized through recycling back to the cell surface. GLP-1R is expressed in pancreatic islets together with the closely related glucose-dependent insulinotropic polypeptide (GIPR) and glucagon (GCGR) receptors. The interaction and cross-talk between coexpressed receptors is a wide phenomenon of the 7TM/GPCR superfamily. Numerous reports show functional consequences for signaling and trafficking of the involved receptors. On the basis of the high structural similarity and tissue coexpression, we here investigated the potential cross-talk between GLP-1R and GIPR or GCGR in both trafficking and signaling pathways. Using a real-time time-resolved FRET-based internalization assay, we show that GLP-1R, GIPR, and GCGR internalize with differential properties. Remarkably, upon coexpression of the internalizing GLP-1R and the non-internalizing GIPR, GLP-1-mediated GLP-1R internalization was impaired in a GIPR concentration-dependent manner. As a functional consequence of such impaired internalization capability, GLP-1-mediated GLP-1R signaling was abrogated. A similar compromised signaling was found when GLP-1R internalization was abrogated by a dominant-negative version of dynamin (dynamin-1 K44E), which provides a mechanistic link between GLP-1R trafficking and signaling. This study highlights the importance of receptor internalization for full functionality of GLP-1R. Moreover, cross-talk between the two incretin receptors GLP-1R and GIPR is shown to alter receptor trafficking with functional consequences for GLP-1R signaling.
Introduction
The importance of the incretin hormone glucagon-like peptide-1 (GLP-1)3 in regulating post-prandial blood glucose levels has been known for decades (1). Likewise, the impaired incretin function in type 2 diabetes is commonly accepted and has led to the development of stable GLP-1 analogs for the treatment of type 2 diabetes (2). On the pancreatic β-cell, GLP-1 exhibits its insulinotropic function via the GLP-1 receptor (GLP-1R), which is a member of the family B seven-transmembrane/G-protein-coupled receptors (7TM/GPCRs), together with the incretin glucose-dependent insulinotropic polypeptide (GIPR) and glucagon (GCGR) receptors (3). Insulin secretion is known to result from GLP-1-mediated activation of GLP-1R, whereby numerous intracellular second messenger pathways are initiated, i.e. the Gαs-mediated intracellular cAMP pathway, calcium mobilization, and MAPK pathway (4–6).
The signaling capacity of a 7TM/GPCR can be regulated through ligand-mediated receptor internalization and subsequent intracellular receptor sorting, i.e. either recycling back to the cell surface or degradation (7). Classically, 7TM/GPCR internalization is initiated by ligand-mediated phosphorylation of the activated receptor, followed by recruitment of β-arrestins, which uncouple the receptor from the G-protein (desensitization). The receptor then internalizes through either clathrin-mediated (8) or non-clathrin-mediated (9) pathways, thereby terminating receptor signaling. However, this classical view has recently been challenged because persistent signaling from internalized receptors of both families A (10) and B (11, 12) has been reported. This suggests that the trafficking properties of a 7TM/GPCR dictate the signaling proficiencies of a given receptor in a much more complicated manner than previously anticipated. We (13) and others (14) have previously reported that GLP-1R is a fast internalizing and recycling receptor, yet the functional consequences of GLP-1R trafficking are still largely unknown.
The signaling capacity of an individual 7TM/GPCR can moreover be regulated through cross-talk with other receptors. For instance, the physical interaction of 7TM/GPCRs in homo- and heteromeric receptor complexes has been shown to be a broad and general phenomenon within family B (15). Heteromerization between GLP-1R and the closely related GIPR has been reported to result in cross-talk with functional consequences for GLP-1R signaling (16, 17). In addition, heteromerization between GLP-1R and GCGR has been described (18). Because GLP-1R, GCGR, and GIPR are all (i) members of the family B 7TM/GPCRs based on their structural and sequential similarity, (ii) involved in blood glucose homeostasis regulation, and (iii) expressed in pancreatic islets (3), a functional cross-talk between these receptors could be anticipated. In this study, we investigated the functional consequences of GLP-1R cross-talk with the closely related GCGR and GIPR.
EXPERIMENTAL PROCEDURES
Materials
Cell culture medium (DMEM) with GlutaMAX-I and 4.5 g/liter d-glucose, DMEM/nutrient mixture F-12 with l-glutamine and 2.438 g/liter sodium bicarbonate, Dulbecco's PBS without CaCl2 and MgCl2, penicillin/streptomycin, Versene, Opti-MEM I with GlutaMAX-I, Lipofectamine 2000, Hanks' balanced salt solution with CaCl2 and MgCl2 (HBSS), FBS, and 10% Pluronic F-68 were purchased from Invitrogen. N-terminally SNAP-tagged human GLP-1R, SNAP-tagged human GIPR, SNAP-tagged human GCGR, Tag-lite SNAP-Lumi4-Tb, and the cAMP dynamic 2 and Cellul'erk (phospho-ERK1/2) kits were purchased from Cisbio Bioassays (Codolet, France). White opaque OptiPlate-96 and OptiPlate-384 and sterile and tissue culture-treated white opaque 96-well microplates were purchased from PerkinElmer Life Sciences. HEPES, fluorescein, and probenecid were purchased from Sigma-Aldrich. The FLIPR calcium 5 assay kit and FLIPR Tetra 384-well clear non-sterile pipette tips were purchased from Molecular Devices. Corning BioCoat poly-d-lysine black clear-bottom 384-well plates were purchased from Fisher Scientific. Polypropylene V-shaped 384-well microplates were purchased from Greiner Bio One. Human embryonic kidney cells (HEK293) were purchased from American Type Culture Collection. Coelenterazine 400A (DeepBlueC) was purchased from Biotium. COS-7 cells were kindly provided by Professor Ulrik Gether (University of Copenhagen, Copenhagen, Denmark). All ligands were prepared in-house using standard methods. The dynamin-1 K44E-HA plasmid was kindly provided by Professor Jennifer Whistler (University of California, San Francisco). Plasmids encoding GCGR, GIPR, HA-β2-adrenergic receptor (β2AR), GLP-1R-RLuc8, β-arrestin-2-GFP, and pcDNA3.1 were available in-house.
Cell Culture and Transfection
HEK293 and COS-7 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and in DMEM/nutrient mixture F-12 (1:1) supplemented with 10% heat-inactivated FBS plus 1% penicillin/streptomycin, respectively, in a humidified 5% CO2 air incubator at 37 °C. For all experiments, cells were transiently transfected 2 days prior to the assay using Lipofectamine 2000.
Real-time Receptor Internalization
Real-time internalization experiments were carried out using a time-resolved FRET-based assay as described previously (13). For studies of single-receptor internalization, HEK293 cells were transfected in 25-cm2 culture flasks with equal amounts (1.5 μg) of SNAP-GLP-1R, SNAP-GIPR, or SNAP-GCGR plasmid. COS-7 cells were transfected in 96-well plates (0.05 μg/well). The SNAP-GLP-1R construct has previously been reported to retain full binding to GLP-1 (13). Likewise, the SNAP tags on both the GCGRs and GIPRs did not compromise ligand binding (data not shown). HEK293 cells used for studying cross-talk or internalization inhibition were cotransfected in 25-cm2 culture flasks with (i) SNAP-GLP-1R and pcDNA3.1, GCGR, GIPR, or HA-β2AR plasmid in a 1:1 (1.5:1.5 μg) DNA ratio; (ii) SNAP-GLP-1R and GIPR plasmids in varying DNA ratios ranging from 1:0 (0.5:0 μg) to 1:1 (0.5:0.5 μg); or (iii) SNAP-GLP-1R and dynamin-1 K44E-HA plasmids in a 1:2 (1.5:3.0 μg) DNA ratio. In short, transiently transfected cells were seeded in sterile collagen-coated 96-well microplates and cultured overnight. Surface-expressed N-terminally SNAP-tagged receptors were labeled with 100 nm Tag-lite SNAP-Lumi4-Tb (donor) in Opti-MEM I for 60 min at 37 °C. Subsequently, cells were washed with HBSS supplemented with 10 mm HEPES and 0.1% Pluronic F-68 (HBSS buffer) and supplemented with 100 μm preheated fluorescein (acceptor). The cells were stimulated with preheated ligands diluted in HBSS buffer, and receptor internalization was measured every 6 min at 37 °C in an EnVision 2104 multilabel reader (PerkinElmer Life Sciences). Real-time traces of receptor internalization were assessed by plotting the donor/acceptor ratio versus time. For concentration-response curves, the area under the curve (AUC) of the real-time traces was calculated and plotted against ligand concentration. For coexpression studies, the data were baseline-corrected to compensate for the varying response levels resulting from repeated transient transfections. In addition, data were normalized to the maximum response generated from the SNAP-GLP-1R + pcDNA3.1 cotransfection for each experimental repeat.
β-Arrestin-2 Recruitment Assay
Bioluminescence resonance energy transfer saturation experiments for measuring β-arrestin-2 recruitment were carried out as described previously (19). In short, HEK293 cells were transiently cotransfected in 25-cm2 culture flasks with β-arrestin-2-GFP, GLP-1R-RLuc8, and pcDNA3.1, GCGR, GIPR, or HA-β2AR plasmid in a 6:1:1 (8.5:1.5:1.5 μg) DNA ratio. The cells were cultured for 2 days prior to harvesting and thorough resuspension in HBSS buffer. Ligands were diluted in HBSS, and the cells were stimulated for 15 min at room temperature in the dark. The bioluminescence and fluorescence signals were measured using a Mithras LB 940 plate reader (Berthold Technologies, Bad Wildbad, Germany) following automatic addition of the luciferase substrate DeepBlueC diluted in 30% EtOH. The data were plotted as acceptor/donor ratio (GFP/DeepBlueC) versus concentration and normalized to the maximum response generated from the SNAP-GLP-1R + pcDNA3.1 cotransfection.
cAMP and Phospho-ERK Signaling Assays
cAMP and phospho-ERK (pERK) accumulation was measured using the cAMP dynamic 2 and Cellul'erk (phospho-ERK1/2) kits, respectively. HEK293 cells were transiently cotransfected in 25-cm2 culture flasks with (i) SNAP-GLP-1R and pcDNA3.1, GCGR, GIPR, or HA-β2AR plasmid in a 1:1 (1.5:1.5 μg) DNA ratio; (ii) SNAP-GLP-1R and GIPR plasmids in varying DNA ratios ranging from 1:0 (0.5:0 μg) to 1:1 (0.5:0.5 μg; for cAMP measurements only); or (iii) SNAP-GLP-1R and dynamin-1 K44E-HA plasmids in a 1:2 (1.5:3.0 μg) DNA ratio. Cells were seeded in sterile polylysine-coated 96-well microplates and cultured overnight. The next day, cells were washed twice with HBSS buffer prior to incubation and reconstitution in HBSS buffer at 37 °C for 30 min or 2 h for cAMP or pERK experiments, respectively. For cAMP experiments, the buffer was supplemented with 500 μm isobutylmethylxanthine. The reconstituted cells were stimulated with preheated ligands diluted in HBSS buffer at 37 °C for 30 min or 5 min for cAMP or pERK experiments, respectively. The ligands were then aspirated, and the cells were lysed immediately in cold lysis buffer for 30 min. The lysates were transferred to a 384-well plate and incubated with the fluorescently labeled antibodies provided in the kit for 2 h in the dark at room temperature prior to reading with the EnVision 2104 multilabel reader. For cAMP experiments, the concentration of cAMP produced was interpolated from a standard curve. The data were plotted as concentration-response curves and normalized to the maximum response generated from the SNAP-GLP-1R + pcDNA3.1 cotransfection.
Calcium Signaling Assay
Calcium signaling was measured in real-time using the FLIPR calcium 5 assay kit. HEK293 were transiently cotransfected in 25-cm2 culture flasks with (i) SNAP-GLP-1R and pcDNA3.1, GCGR, or GIPR plasmid in a 1:1 (1.5:1.5 μg) DNA ratio or (ii) SNAP-GLP-1R and dynamin-1 K44E-HA plasmids in a 1:2 (1.5:3.0 μg) DNA ratio. Cells were seeded in sterile 384-well microplates and cultured overnight. The next day, the cells were incubated for 1 h at 37 °C with component A dissolved in HBSS buffer supplemented with 250 mm probenecid. Cells were then stimulated with the ligands diluted in HBSS buffer, and the signal was read every 2 s using the FLIPR Tetra system. From the real-time calcium signaling traces, the maximum response for each concentration was calculated and baseline-subtracted, and the data were normalized to the maximum response generated from the SNAP-GLP-1R + pcDNA3.1 cotransfection. Data were then plotted as a concentration-response curve.
Receptor Expression
Receptor expression was measured either as emission from terbium-labeled SNAP-tagged surface-expressed receptors or using a whole cell radioligand binding assay. For receptor expression measured by fluorescence, HEK293 and COS-7 cells were transiently transfected with equal amounts of SNAP-GLP-1R/SNAP-GIPR/SNAP-GCGR (1.5 μg in 25-cm2 culture flasks) or SNAP-GLP-1R/SNAP-GIPR (0.05 μg in 96-well plates) plasmids, respectively. As in the internalization assay, surface-expressed N-terminally SNAP-tagged receptors were labeled with 100 nm Tag-lite SNAP-Lumi4-Tb in Opti-MEM I for 60 min at 37 °C. Subsequently, cells were washed with HBSS buffer, and donor emission was measured in the absence of ligand using the EnVision 2104 multilabel reader during the internalization assay. Receptor expression is presented as the integrated AUC from these curves. For receptor expression measured by whole cell radioligand binding, HEK293 cells were transiently cotransfected in 25-cm2 culture flasks with (i) SNAP-GLP-1R and pcDNA3.1, GCGR, GIPR, or HA-β2AR plasmid in a 1:1 (1.5:1.5 μg) ratio; (ii) SNAP-GLP-1R and GIPR plasmids in varying DNA ratios ranging from 1:0 (0.5:0 μg) to 1:1 (0.5:0.5 μg); or (iii) SNAP-GLP-1R and GIPR plasmids in a 1:1 (1.5:1.5 μg) ratio. Cells were cultured in 96-well plates and incubated overnight at 4 °C with 240 pm 125I-GLP-1 or 125I-GIP and increasing concentrations of unlabeled GLP-1 or GIP diluted in ice-cold HBSS buffer. The following day, unbound ligand was washed off, and the cells were lysed in 0.1 m NaOH prior to addition of MicroScint-40. The radioactivity was measured as cpm in a TopCount NXT gamma counter (PerkinElmer Life Sciences) and plotted as (i + ii) Bmax in cpm or (iii) receptor number (in millions)/cell (calculated as described previously (20)).
Data and Statistical Analysis
Concentration-response curves were fitted to a simple log (agonist) versus response regression in GraphPad Prism (version 6.00, GraphPad Software, La Jolla, CA). Statistical analyses were performed in GraphPad Prism using a two-sided Student's t test for comparison of two samples or one-way analysis of variance, followed by Dunnett's post-test for comparisons of two or more sample groups with a control group. p < 0.05 was considered statistically significant.
RESULTS
Real-time Internalization of GCGR and GIPR in Comparison with GLP-1R
Utilizing a novel time-resolved FRET-based real-time internalization assay, we previously showed that GLP-1R is a potent and fast internalizing receptor (13). GLP-1R shares >40% sequence similarity with GCGR and GIPR. Due to the impact of trafficking on 7TM/GPCR signaling capacity, we first investigated the internalization properties of GCGR and GIPR (Fig. 1). SNAP-GLP-1R (Fig. 1A), SNAP-GCGR (Fig. 1B), or SNAP-GIPR (Fig. 1C) was transiently expressed in HEK293 cells, and real-time receptor internalization was measured in response to increasing concentrations of GLP-1, glucagon, or GIP, respectively. In contrast to the potent GLP-1-induced GLP-1R internalization (Fig. 1A), GCGR internalization was induced only by high glucagon concentrations (≥1 μm) (Fig. 1B). Notably, GIP concentrations even in the μm range were not sufficient to induce significant GIPR internalization (Fig. 1C). These different internalization properties were not caused by different expression levels of the three receptors (Fig. 1C, inset). To rule out any cell line-dependent differences in receptor internalization properties, we repeated the internalization experiment in COS-7 cells. As observed in HEK293 cells, GLP-1 potently induced GLP-1R internalization (Fig. 1D), whereas GIP failed to induce GIPR internalization (Fig. 1E). Similar to HEK293 cells, these differences were not caused by different receptor expression levels (Fig. 1E, inset). Thus, these data reveal diverse trafficking properties of the three otherwise highly related family B receptors.
FIGURE 1.

Diverse internalization profiles of GLP-1R, GCGR, and GIPR. Real-time receptor internalization in response to increasing concentrations of ligand (log10 (m)) was measured in HEK293 cells (A–C) and COS-7 cells (D and E) transfected with equal amounts of N-terminally SNAP-tagged receptor plasmids. A and D, GLP-1-stimulated SNAP-GLP-1R internalization. B, glucagon-stimulated SNAP-GCGR internalization. C and E, GIP-stimulated SNAP-GIPR internalization. C and E insets, no differences in receptor expression of GLP-1R (white bars), GCGR (striped bar), and GIPR (black bars) were observed, measured as total donor signal from surface-expressed receptors in the absence of ligand and plotted as AUC of the donor signal over time. Data represent means ± S.D. from one representative (A–E) or means ± S.E.M. (C and E insets) out of three individual experiments carried out in triplicates.
Coexpression of GLP-1R with GCGR or GIPR Alters GLP-1R Internalization
The interaction of structurally related 7TM/GPCRs in homo- and heteromers can alter the function of the involved receptors through receptor cross-talk. For example, heteromerization of the fast internalizing β2AR with the non-internalizing κ-opioid receptor inhibits β2AR internalization and furthermore decreases β2AR-mediated MAPK signaling (21). Oligomerization of the family B 7TM/GPCRs is a general phenomenon (15), and heteromerization of GLP-1R with both GIPR and GCGR has been reported (16–18). On the basis of the differences in GLP-1R, GCGR, and GIPR internalization abilities (Fig. 1), we hypothesized that cross-talk between the potent internalizing GLP-1R with the weakly internalizing GCGR or the non-internalizing GIPR would alter GLP-1R internalization. Real-time GLP-1R internalization was examined in HEK293 cells upon coexpression of pcDNA3.1 (Fig. 2A, transfection control), GCGR (Fig. 2B), GIPR (Fig. 2C), or the unrelated β2AR (Fig. 2D, negative control). In the presence of GCGR, GLP-1R internalization efficacy was significantly reduced (Emax = 43.1 ± 5.7%, p < 0.0001) (Fig. 2, B and E) in comparison with when GLP-1R was expressed alone (Emax = 100 ± 0.0%) (Fig. 2A). Coexpression of GLP-1R with GIPR inhibited GLP-1R internalization (Emax = 11.1 ± 2.1%, p < 0.0001) (Fig. 2, C and E). In contrast, the presence of the unrelated β2AR did not alter GLP-1R internalization (Emax = 90.0 ± 8.1%) (Fig. 2, D and E), indicating that altered GLP-1R trafficking is not a result of cotransfection in itself. Likewise, GLP-1R expression levels remained unchanged upon the respective cotransfections (Fig. 2F). Taken together, these data indicate that the presence of both GCGR and GIPR impairs GLP-1R internalization.
FIGURE 2.

GLP-1R internalization is impaired in the presence of GCGR or GIPR. GLP-1R internalization in response to increasing concentrations of GLP-1 (log10 (m)) was measured in real-time in HEK293 cells cotransfected with a 1:1 ratio of SNAP-GLP-1R and pcDNA3.1 (A), GCGR (B), GIPR (C), or β2AR (D). E, concentration-response curves of AUC from the real-time internalization traces. Cotransfection of SNAP-GLP-1R with either GCGR (■) or GIPR (▴) significantly reduced receptor internalization compared with cotransfection with pcDNA3.1 (○) or β2AR (♦) (p < 0.0001). F, no differences in GLP-1R expression were observed upon coexpression with pcDNA3.1 (white bar), GCGR (striped bar), GIPR (black bar), or β2AR (dotted bar), measured as radioligand binding to whole cells and plotted as Bmax in cpm. Data represent means ± S.D. from one representative (A–D) or means ± S.E.M. (E and F) from a minimum of three independent experiments carried out in triplicates.
β-Arrestin-2 Recruitment to GLP-1R Is Altered upon Receptor Cross-talk
The classical mechanism of 7TM/GPCR internalization involves C-terminal receptor phosphorylation, followed by recruitment and binding of β-arrestins (8). In line with this, GLP-1R activation has been reported to mediate β-arrestin recruitment (19, 22).
Because cross-talk between GLP-1R and GCGR or GIPR alters GLP-1R internalization (Fig. 2), we next determined whether coexpression of GLP-1R with GCGR or GIPR also affects β-arrestin-2 recruitment to GLP-1R. HEK293 cells were transfected with GLP-1R-RLuc8, β-arrestin-2-GFP, and pcDNA3.1, GCGR, GIPR, or the unrelated β2AR. GLP-1-induced recruitment of β-arrestin-2 to GLP-1R was measured in bioluminescence resonance energy transfer saturation experiments (Fig. 3). Cotransfection of GLP-1R with GCGR (Emax = 87.0 ± 3.0%) or β2AR (Emax = 86.1 ± 3.7%) did not significantly alter recruitment of β-arrestin-2 to GLP-1R compared with cotransfection with pcDNA3.1 (Emax = 100 ± 0.0%). In contrast, the presence of GIPR (Emax = 63.0 ± 6.3%) significantly reduced the efficacy of GLP-1-mediated recruitment of β-arrestin-2 to GLP-1R (p < 0.001) (Fig. 3). These data suggest reduced recruitment of β-arrestin-2 to GLP-1R mediated by the presence of GIPR. This correlates well with the observation that GLP-1R internalization was impaired upon coexpression with GIPR.
FIGURE 3.
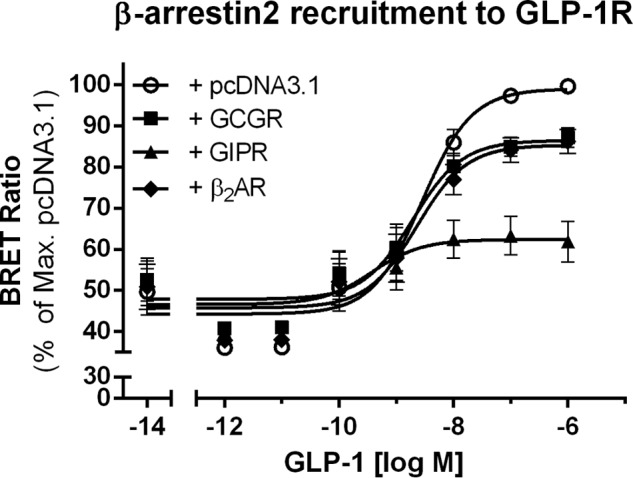
β-Arrestin-2 recruitment to GLP-1R is impaired in the presence of GIPR. GLP-1-stimulated recruitment of β-arrestin-2 to GLP-1R was measured using a bioluminescence resonance energy transfer (BRET) saturation assay. HEK293 cells were cotransfected with a 6:1:1 ratio of β-arrestin-2-GFP, GLP-1R-RLuc8, and pcDNA3.1 (○), GCGR (■), GIPR (▴), or β2AR (♦) and stimulated with increasing concentrations of GLP-1 for 15 min prior to measurement. Cotransfection of GLP-1R with GIPR significantly reduced recruitment of β-arrestin-2 to GLP-1R (p < 0.001). Data represent means ± S.E.M. from five independent experiments carried out in duplicates.
Cross-talk between GLP-1R and GCGR or GIPR Has Functional Consequences on GLP-1R Signaling
GLP-1R is coupled mainly to Gαs, and GLP-1-mediated cAMP production is an important prerequisite for insulin secretion from pancreatic β-cells (1). Because (i) 7TM/GPCR trafficking is thought to be an important regulator of receptor signaling and (ii) GLP-1R internalization is altered in the presence of GCGR or GIPR (Fig. 2), we next examined whether the presence of either GCGR or GIPR could alter the downstream signaling from GLP-1R.
cAMP accumulation was measured in HEK293 cells transfected with GLP-1R and either GCGR or GIPR following stimulation with increasing concentrations of GLP-1 for 30 min (Fig. 4A). Coexpression of GLP-1R with GIPR significantly reduced GLP-1-mediated cAMP accumulation (p < 0.05) (Fig. 4A). In comparison, the presence of GCGR did not affect GLP-1R cAMP accumulation (Fig. 4A and Table 1).
FIGURE 4.
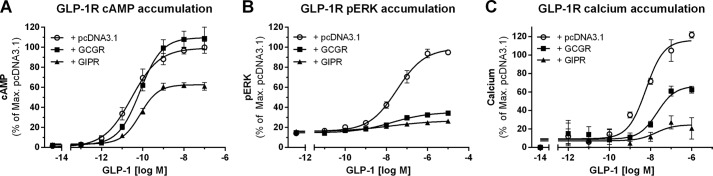
Impaired GLP-1R signaling in the presence of GCGR or GIPR. HEK293 cells were cotransfected with a 1:1 ratio of SNAP-GLP-1R and pcDNA3.1 (○), GCGR (■), or GIPR (▴) and stimulated for 30 min prior to measurement of cAMP production (A) or 5 min prior to measurement of ERK accumulation (B). C, calcium accumulation was measured in real-time and is plotted as concentration-response curves of the maximum response from the real-time signaling traces. cAMP production was significantly impaired in the presence of GIPR (p < 0.05). ERK accumulation was significantly impaired in the presence of GIPR and GCGR (p < 0.0001). Calcium accumulation was significantly impaired in the presence of GCGR (p < 0.01) and even further impaired in the presence of GIPR (p < 0.001). Data represent means ± S.E.M. from a minimum of three independent experiments carried out in triplicates.
TABLE 1.
GLP-1R signaling is impaired in the presence of GCGR and GIPR
The efficacy of GLP-1R signaling upon coexpression with pcDNA3.1, GCGR, or GIPR is presented as a percentage of the maximum signal obtained from cotransfection with pcDNA3.1. Values are means ± S.E.M. from a minimum of three individual experiments carried out in triplicates.
|
Emax |
|||
|---|---|---|---|
| cAMP | pERK | Calcium | |
| % | |||
| pcDNA3.1 | 100 ± 0.0 | 100 ± 0.0 | 116 ± 6.6 |
| GCGR | 111 ± 14.6 | 35.8 ± 2.5 | 66.4 ± 4.2 |
| GIPR | 63.0 ± 4.7 | 26.9 ± 2.2 | 25.0 ± 8.2 |
In addition to cAMP, several other second messengers have been reported to be activated in response to GLP-1 stimulation in β-cells, including proteins of the MAPK pathway and calcium mobilization (4–6). Hence, we next measured the effects of GIPR and GCGR coexpression on GLP-1R-mediated pERK and calcium signaling. GLP-1R-mediated ERK phosphorylation was measured in HEK293 cells upon cotransfection with either GCGR or GIPR in response to 5 min of stimulation with increasing concentrations of GLP-1 (Fig. 4B). Coexpression of GLP-1R with either GCGR or GIPR significantly impaired GLP-1R pERK signaling (p < 0.0001) (Fig. 4B and Table 1). GLP-1R-mediated calcium accumulation was measured in real-time in HEK293 cells upon cotransfection with either GCGR or GIPR and is presented as the maximum response as a function of GLP-1 concentration (Fig. 4C). Upon cotransfection with GCGR, a significant decrease in GLP-1R efficacy was observed (p < 0.01) (Fig. 4C). Notably, GLP-1R calcium signaling was even further impaired in the presence of GIPR (p < 0.001) (Fig. 4C and Table 1). Taken together, these data suggest substantial functional consequences of cross-talk between GLP-1R and GIPR, thereby resulting in decreased cAMP, pERK, and calcium accumulation. In comparison, GCGR did not affect cAMP accumulation emanating from GLP-1R, but reduced GLP-1R-mediated pERK and calcium signaling.
Cross-talk between GLP-1R and GIPR at Various Receptor Ratios
Our data suggest that the presence of GIPR has a negative impact on GLP-1R internalization (Fig. 2), β-arrestin recruitment (Fig. 3), and signaling (Fig. 4). Because GLP-1R and GIPR are physiologically coexpressed in β-cells (23) and have been reported to heteromerize (16, 17), we further investigated the cross-talk between GLP-1R and GIPR. To closer mimic the natural expression of these receptors in β-cells, we repeated the internalization and cAMP accumulation measurements with a decreased concentration of plasmid used for transfection and a varied ratio between GLP-1R and GIPR ranging from 1:0 to 1:1. Upon titration with increasing concentrations of GIPR, a concentration-dependent decrease in GLP-1R internalization was observed (Fig. 5, A–D), which was significant at a 1:0.25 GLP-1R/GIPR ratio (p < 0.01) (Fig. 5E). Likewise, a tendency for a concentration-dependent decrease in GLP-1R-mediated cAMP accumulation was observed in the presence of increasing GIPR concentrations (Fig. 5F). In addition, we observed that GLP-1R expression decreased upon coexpression with increasing GIPR concentrations, which was significant at a 1:0.5 GLP-1R/GIPR ratio (p < 0.05) (Fig. 5G). However, at comparable GLP-1R expression levels (pcDNA3.1 Bmax = 570 ± 74 cpm (Fig. 2A) versus 1:1 Bmax = 570 ± 32 cpm (Fig. 5D)), GLP-1R exhibited full internalization when expressed alone (Fig. 2A), whereas GLP-1R internalization was significantly inhibited in the presence of GIPR (Fig. 5D). Taken together, these data support a GIPR-mediated decrease in GLP-1R internalization and cAMP accumulation in a GIPR concentration-dependent manner. In addition, a GIPR concentration-dependent decrease in GLP-1R expression was observed. This suggests that GIPR has a dual role in regulating the function of GLP-1R by altering both its expression and internalization properties.
FIGURE 5.

Concentration-dependent decrease in GLP-1R function in the presence of GIPR. GLP-1R internalization in response to increasing concentrations of GLP-1 (log10 (m)) was measured in real-time in HEK293 cells cotransfected with varying SNAP-GLP-1R/GIPR ratios: 1:0 (A), 1:0.25 (B), 1:0.5 (C), or 1:1 (D). E, concentration-response curves of AUC from the real-time internalization traces. Internalization of SNAP-GLP-1R (1:0, ○) was significantly reduced upon cotransfection with GIPR in ratios of 1:0.25 (■; p < 0.01), 1:0.5 (♦; p < 0.01), and 1:1 (▴; p < 0.01). Data represent means ± S.D. from one representative (A–D) or means ± S.E.M. (E) out of five independent experiments carried out in triplicates. F, GLP-1R-mediated cAMP accumulation was measured in response to increasing concentrations of GLP-1 for 30 min and is plotted as the maximum response upon coexpression with GIPR in ratios of 1:0 (white bar), 1:0.25 (striped bar), 1:0.5 (dotted bar), and 1:1 (black bar) (p < 0.05). Data represent means ± S.E.M. from three independent experiments carried out in triplicates. G, expression of SNAP-GLP-1R (white bar) was unaffected by cotransfection with GIPR in a 1:0.25 ratio (striped bar), but was significantly decreased upon cotransfection with GIPR in a 1:0.5 ratio (dotted bar; *, p < 0.05) and a 1:1 ratio (black bar; *, p < 0.05), measured as radioligand binding to whole cells and plotted as Bmax in cpm. Data represent means ± S.E.M. from two independent experiments carried out in triplicates.
Consequences of GLP-1R and GIPR Cross-talk
Next, we examined any differences in signaling of the structurally similar GLP-1R and GIPR, as well as any cross-activation of the receptors. We first examined the main signaling pathway for both receptors (i.e. cAMP production) upon individual expression or coexpression of the two receptors.
cAMP production mediated by GLP-1R or GIPR upon individual expression in HEK293 cells was measured in response to GLP-1 or GIP stimulation, respectively (Fig. 6A). In comparison with GLP-1-stimulated GLP-1R cAMP production, GIP-stimulated GIPR cAMP production exhibited a significantly reduced efficacy (p < 0.0001) and potency (p < 0.01) (Fig. 6A and Table 2). These differences were not caused by different expression levels of the two receptors (Fig. 6D). As a control, no cross-activation of GLP-1R by GIP or of GIPR by GLP-1 was measured (Fig. 6A and Table 2), in correlation with previous studies (24). Upon coexpression of GLP-1R with GIPR, GIP-stimulated GIPR cAMP production was unaffected (Fig. 6B). In contrast, GLP-1-stimulated GLP-1R cAMP production was significantly impaired with regard to both efficacy (p < 0.0001) and potency (p < 0.0001) (Fig. 6B and Table 2), thereby mimicking the signaling profile of GIPR.
FIGURE 6.

Reduced cAMP production and internalization properties of GLP-1R in the presence of GIPR are not influenced by ligand co-stimulation. cAMP production mediated by GLP-1R and GIPR following 30 min of ligand stimulation was measured upon individual expression (A) or coexpression (B) in HEK293 cells transfected with equal amounts of SNAP-GLP-1R and GIPR plasmids. A, upon individual receptor expression, GLP-1 stimulation of GLP-1R (○) resulted in significantly higher cAMP production compared with GIP-stimulated GIPR (▵; p < 0.0001). No cross-activation of GLP-1R by GIP (⟐) or of GIPR by GLP-1 (♢) was observed. B, upon coexpression of GLP-1R and GIPR, GIP-stimulated GIPR signaling (▴) was unaffected, whereas GLP-1-stimulated GLP-1R signaling (●) was significantly reduced (p < 0.0001). Co-stimulation with GLP-1 and 100 nm GIP (♦) did not rescue the impaired GLP-1R signaling. Data represent means ± S.E.M. from a minimum of two independent experiments carried out in duplicates. C, ligand-stimulated GLP-1R internalization was measured in real-time in HEK293 cells transfected with SNAP-GLP-1R or cotransfected with SNAP-GLP-1R and GIPR in a 1:1 ratio. In comparison with GLP-1-stimulated GLP-1R internalization upon individual expression (○), GLP-1R internalization was significantly reduced upon coexpression with GIPR (●; p < 0.0001). Co-stimulation with GLP-1 and 1 μm GIP (♦) did not rescue the impaired GLP-1R internalization. Data represent means ± S.E.M. from three independent experiments carried out in triplicates. D, no differences in GLP-1R expression (white bar) and GIPR expression (black bar) were measured using radioligand binding to whole cells. Data are plotted as million receptor number/cell (Million no. pr. cell). Data represent means ± S.E.M. from three independent experiments carried out in duplicates.
TABLE 2.
GLP-1R and GIPR cAMP accumulation upon individual expression or coexpression
The efficacy and potency of GLP-1R and GIPR cAMP accumulation upon individual receptor expression or coexpression were normalized to the maximum signal obtained from GLP-1R upon individual expression. Efficacies are presented as a percentage of maximum GLP-1R signaling, and potencies are presented as EC50 values. Values are means ± S.E.M. from a minimum of two individual experiments carried out in duplicates. NA, data not available.
| GLP-1 |
GIP |
GLP-1 + GIP |
||||
|---|---|---|---|---|---|---|
| Emax | EC50 | Emax | EC50 | Emax | EC50 | |
| % | pm | % | pm | % | pm | |
| GLP-1R | 100 ± 0.01 | 23.0 ± 0.7 | 1.9 ± 0.1 | NA | NA | NA |
| GIPR | 3.5 ± 0.4 | NA | 49.1 ± 3.2 | 38.4 ± 1.7 | NA | NA |
| GLP-1R + GIPR | 44.2 ± 4.9 | 58.9 ± 2.6 | 44.4 ± 2.8 | 38.7 ± 4.9 | 44.2 ± 3.5 | NA |
In a recent study, GIP stimulation was reported to disrupt the heteromerization between GLP-1R and GIPR (16). Thus, we next tested whether co-stimulation of GLP-1 and GIP alters the GIPR-mediated impairment of GLP-1R cAMP production (Figs. 4A and 6B) and internalization (Fig. 2). Upon coexpression of GLP-1R with GIPR, co-stimulation with increasing concentrations of GLP-1 and a constant saturating concentration of GIP did not influence the impaired GLP-1R cAMP production (Fig. 6B and Table 2). Likewise, the reduced capacity of GLP-1R to internalize upon coexpression of GIPR (Emax = 10.9 ± 1.80%, p < 0.0001) was not affected when the receptors were co-stimulated with increasing concentrations of GLP-1 in the presence of a saturating concentration of GIP (Emax = 12.1 ± 2.92%, p < 0.0001) (Fig. 6C).
These data suggest that GLP-1R-mediated cAMP production in general is more efficient and potent compared with GIPR cAMP production. Upon coexpression of the two receptors, GLP-1-induced GLP-1R cAMP production was reduced and mimicked GIPR signaling with a lower efficacy and potency. Co-stimulation with both GLP-1 and GIP did not influence the impaired GLP-1R cAMP production and internalization mediated by the presence of GIPR.
GLP-1R Internalization Is Essential for Full Receptor Functionality
7TM/GPCR internalization is classically thought to be linked to receptor desensitization (8). In line with this, inhibition of receptor internalization is suggested to result in increased signaling. However, in this study, we have shown that GIPR-mediated loss of GLP-1R internalization (Fig. 2) was correlated with decreased cAMP, calcium, and pERK accumulation (Fig. 4), which is in conflict with the classical view of internalization-mediated desensitization. However, recent studies suggest a potential new paradigm with signaling from internalized functional 7TM/GPCRs (25, 26). Thus, we wanted to investigate whether altering GLP-1R internalization would affect receptor signaling. Like other 7TM/GPCRs, GLP-1R is expected to internalize either through clathrin-dependent or clathrin-independent (27) pathways, both of which rely on dynamin activity (28). We thus tested whether a dominant-negative mutant of dynamin, dynamin-1 K44E (29), could inhibit GLP-1R internalization. HEK293 cells were cotransfected with SNAP-GLP-1R and either pcDNA3.1 (Fig. 7A, transfection control) or dynamin-1 K44E (Fig. 7B), and receptor internalization was measured in real-time in response to increasing GLP-1 concentrations. GLP-1-mediated GLP-1R internalization was fully inhibited upon cotransfection with dynamin-1 K44E (p < 0.0001) (Fig. 7, B and C). Thus, GLP-1R internalization can be blocked by inhibiting dynamin-mediated endocytic pathways.
FIGURE 7.

Inhibition of GLP-1R internalization results in impaired signaling. GLP-1R internalization in response to increasing concentrations of GLP-1 (log10 (m)) was measured in real-time in HEK293 cells cotransfected with SNAP-GLP-1R and either pcDNA3.1 in a 1:1 ratio (A) or dynamin-1 K44E in a 1:2 ratio (B). C, concentration-response curves of AUC from real-time internalization traces. GLP-1R internalization was significantly inhibited upon cotransfection with dynamin-1 K44E (♦; p < 0.0001) compared with cotransfection with pcDNA3.1 (○). Data represent means ± S.D. from one representative (A and B) or means ± S.E.M. (C) out of four independent experiments carried out in triplicates. D–F, GLP-1-stimulated GLP-1R signaling was measured in HEK293 cells cotransfected with SNAP-GLP-1R and either pcDNA3.1 in a 1:1 ratio (○) or dynamin-1 K44E in a 1:2 ratio (♦). GLP-1R-mediated cAMP accumulation (D; p < 0.05), pERK accumulation (E; p < 0.0001), and calcium accumulation (F; p < 0.01) were significantly impaired in the presence of dynamin-1 K44E compared with pcDNA3.1 Data represent means ± S.E.M. from a minimum of three independent experiments carried out in triplicates.
Next, we tested whether inhibition of GLP-1R internalization has a functional consequence on receptor signaling. GLP-1-mediated cAMP production was measured in HEK293 cells in the presence or absence of dynamin-1 K44E (Fig. 7D). In its presence, cAMP production efficacy was significantly decreased (Emax = 59.5 ± 3.4%) compared with GLP-1R signaling upon coexpression with pcDNA3.1 (Emax = 100 ± 0.0%, p < 0.05). Likewise, both GLP-1R-induced ERK phosphorylation (Emax = 100 ± 0.0%) (Fig. 7E) and calcium signaling (Emax = 116 ± 6.6%) (Fig. 7F) were significantly reduced in the presence of dynamin-1 K44E (Emax = 48.8 ± 3.6%, p < 0.0001; and Emax = 65.1 ± 8.5%, p < 0.01, respectively). These data show that inhibition of GLP-1R internalization results in reduced receptor signaling, thereby indicating the importance of GLP-1R internalization for full receptor functionality.
DISCUSSION
In this study, we investigated the implications of cross-talk between GLP-1R and GIPR or GCGR, with a focus on the functional consequences of receptor trafficking and signaling. The potent internalizing GLP-1R shares high structural as well as functional similarity with GCGR and GIPR. Here, we found that despite these similarities, the three receptors exhibit differential internalization properties (Fig. 1). Previous studies have reported GCGR internalization but with varying potencies (30–32). In addition, two studies reported GIPR internalization at varying degrees, ranging from 10 to 60% (16, 33).
Interestingly, our data show that coexpression of GLP-1R with GCGR or GIPR impaired GLP-1R internalization to a degree corresponding to the internalization properties of the individual receptors (Fig. 2). In line with this, β-arrestin-2 recruitment to GLP-1R was reduced in the presence of GIPR (Fig. 3). Previous studies have reported altered trafficking of 7TM/GPCRs upon heteromerization (34). For example, β2AR heteromerization with either the internalizing δ-opioid receptor or the non-internalizing κ-opioid receptor alters β2AR internalization without affecting ligand binding properties. Thus, β2AR and the δ-opioid receptor co-internalize upon activation of either of the receptors, whereas β2AR heteromerization with the κ-opioid receptor completely blocks the internalizing properties of β2AR (21). From our data, it can be speculated that heteromerization of the internalizing GLP-1R with the non-internalizing GIPR traps the heteromer on the cell surface, hereby inhibiting co-internalization of the two receptors in a similar manner to the β2AR/κ-opioid receptor heteromer.
On the basis of the general consensus regarding a functional importance of 7TM/GPCR internalization, we hypothesized that the inhibiting effect of GCGR and GIPR on GLP-1R internalization would also affect GLP-1R signaling. Indeed, upon coexpression of GLP-1R with GIPR, we observed inhibition of cAMP, pERK, and calcium accumulation. In contrast, coexpression of GLP-1R with GCGR significantly inhibited only pERK and calcium signaling but not cAMP production (Fig. 4). The higher impact of GCGR on GLP-1R-mediated ERK phosphorylation compared with cAMP accumulation may be due to the per se lower capacity of GLP-1 to stimulate ERK phosphorylation. Overall, GIPR had a more prominent impact on GLP-1R signaling compared with GCGR. However, GIPR also had a stronger effect on GLP-1R internalization and β-arrestin-2 recruitment, again suggesting a direct link between receptor internalization and signaling. For receptor cross-talk to have physiological relevance, receptor coexpression in endogenous cells is essential. GLP-1R and GIPR coexpress on pancreatic β-cells (23). To our knowledge, there are no concluding reports suggesting that GLP-1R and GCGR are coexpressed endogenously in any cell type, although there is an ongoing debate regarding the presence of both GLP-1Rs and GCGRs on β-cells (23, 35–38). Thus, theoretically, GIPR resembles a more physiological relevant interaction partner for GLP-1R. When varying the ratio between GLP-1R and GIPR, we observed a GIPR concentration-dependent decrease in internalization and a similar tendency for GLP-1R-mediated cAMP accumulation. This supports a functional impact of cross-talk between GLP-1R and GIPR.
Coexpression of GLP-1R and GIPR has been reported to rescue the expression of a dysfunctional GIPR mutant. Hence, the physical assembly of GLP-1R and GIPR in a heteromeric complex was suggested to take place during the receptor maturation process in a ligand-independent manner (17). In comparison, Schelshorn et al. (16) reported a ligand-dependent GLP-1R/GIPR heteromer that was induced by GLP-1 and disrupted by GIP stimulation. Along with these findings, impaired β-arrestin recruitment to GLP-1R mediated by the presence of GIPR was rescued upon co-stimulation with GLP-1 and GIP. In our studies, co-stimulation with GLP-1 and GIP did not rescue either the impaired GLP-1R cAMP accumulation or the inhibited receptor internalization seen upon coexpression of the two receptors (Fig. 6). Hence, our data suggest a ligand (i.e. GIP)-independent cross-talk between GLP-1R and GIPR. Additionally, this study emphasizes a correlation between GIPR-mediated inhibition of GLP-1R internalization and altered signaling. We showed that inhibition of GLP-1R internalization using dynamin-1 K44E resulted in significantly reduced GLP-1-stimulated cAMP accumulation via this receptor (Fig. 7). A similar reduction in cAMP accumulation (and also insulin secretion) upon inhibition of GLP-1R internalization was recently reported by Kuna et al. (39). Hence, we suggest that GLP-1R internalization is an important mechanism for full receptor functionality.
In conclusion, this study highlights a previously unappreciated cross-talk between the two main incretin receptors, GLP-1R and GIPR, which are responsible for regulating glucose-dependent insulin secretion. Furthermore, our data emphasize the importance of GLP-1R internalization for full receptor functionality. Due to the coexpression of the two incretin receptors in pancreatic β-cells, the inhibitory effect of GIPR on GLP-1R function could be highly physiologically relevant. In fact, GIPR knock-out mice show an increased sensitivity to GLP-1R function in islets, which is independent of a compensatory up-regulation of either GLP-1 secretion or GLP-1R expression (40). Based on our data, it could be speculated that in the absence of GIPRs in GIPR knock-out mice, GLP-1R is a fully internalizing and thus highly functional receptor. In line with this, changes in the GLP-1R versus GIPR expression ratio (e.g. during the development of diabetes) can be anticipated to modify the cross-talk between these two receptors. Further studies are needed to determine the interplay between GLP-1R and GIPR in primary cells and in vivo.
Acknowledgments
We thank Professors Jennifer Whistler and Ulrik Gether for kindly providing the dynamin-1 K44E-HA plasmid and COS-7 cells, respectively; Nikolaj Kulahin for fruitful discussion; and Karen Arevad for excellent technical support.
Sarah Noerklit Roed and Sanne Moeller Knudsen own Novo Nordisk A/S stocks.
- GLP-1
- glucagon-like peptide-1
- GLP-1R
- GLP-1 receptor
- 7TM/GPCR
- seven-transmembrane/G-protein-coupled receptor
- GIPR
- glucose-dependent insulinotropic polypeptide receptor
- GCGR
- glucagon receptor
- HBSS
- Hanks' balanced salt solution
- SNAP
- soluble N-ethylmaleimide-sensitive factor attachment protein
- β2AR
- β2-adrenergic receptor
- AUC
- area under the curve
- pERK
- phospho-ERK.
REFERENCES
- 1. Holst J. J. (2007) The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439 [DOI] [PubMed] [Google Scholar]
- 2. Holst J. J., Deacon C. F. (2007) New horizons in diabetes therapy. Immunol. Endocr. Metab. Agents Med. Chem. 7, 49–55 [Google Scholar]
- 3. Mayo K. E., Miller L. J., Bataille D., Dalle S., Göke B., Thorens B., Drucker D. J. (2003) International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 55, 167–194 [DOI] [PubMed] [Google Scholar]
- 4. Doyle M. E., Egan J. M. (2007) Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 113, 546–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Montrose-Rafizadeh C., Avdonin P., Garant M. J., Rodgers B. D., Kole S., Yang H., Levine M. A., Schwindinger W., Bernier M. (1999) Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology 140, 1132–1140 [DOI] [PubMed] [Google Scholar]
- 6. Quoyer J., Longuet C., Broca C., Linck N., Costes S., Varin E., Bockaert J., Bertrand G., Dalle S. (2010) GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a β-arrestin 1-mediated ERK1/2 activation in pancreatic β-cells. J. Biol. Chem. 285, 1989–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanyaloglu A. C., von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 8. Ferguson S. S. (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24 [PubMed] [Google Scholar]
- 9. Chini B., Parenti M. (2004) G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J. Mol. Endocrinol. 32, 325–338 [DOI] [PubMed] [Google Scholar]
- 10. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Calebiro D., Nikolaev V. O., Gagliani M. C., de Filippis T., Dees C., Tacchetti C., Persani L., Lohse M. J. (2009). Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 7, e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferrandon S., Feinstein T. N., Castro M., Wang B., Bouley R., Potts J. T., Gardella T. J., Vilardaga J. P. (2009) Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 5, 734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roed S. N., Wismann P., Underwood C. R., Kulahin N., Iversen H., Cappelen K. A., Schäffer L., Lehtonen J., Hecksher-Soerensen J., Secher A., Mathiesen J. M., Bräuner-Osborne H., Whistler J. L., Knudsen S. M., Waldhoer M. (2014) Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol. Cell. Endocrinol. 382, 938–949 [DOI] [PubMed] [Google Scholar]
- 14. Widmann C., Dolci W., Thorens B. (1995) Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem. J. 310, 203–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harikumar K. G., Morfis M. M., Sexton P. M., Miller L. J. (2008) Pattern of intra-family hetero-oligomerization involving the G-protein-coupled secretin receptor. J. Mol. Neurosci. 36, 279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schelshorn D., Joly F., Mutel S., Hampe C., Breton B., Mutel V., Lütjens R. (2012) Lateral allosterism in the glucagon receptor family: GLP-1 induces GPCR heteromer formation. Mol. Pharmacol. 81, 309–318 [DOI] [PubMed] [Google Scholar]
- 17. Whitaker G. M., Lynn F. C., McIntosh C. H., Accili E. A. (2012) Regulation of GIP and GLP1 receptor cell surface expression by N-glycosylation and receptor heteromerization. PLoS ONE 7, e32675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roed S. N., Orgaard A., Jorgensen R., De Meyts P. (2012) Receptor oligomerization in family B1 of G-protein-coupled receptors: focus on BRET investigations and the link between GPCR oligomerization and binding cooperativity. Front. Endocrinol. 3, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jorgensen R., Norklit Roed S., Heding A., Elling C. E. (2011) Beta-arrestin-2 as a competitor for GRK2 interaction with the GLP-1 receptor upon receptor activation. Pharmacology 88, 174–181 [DOI] [PubMed] [Google Scholar]
- 20. DeBlasi A., O'Reilly K., Motulsky H. J. (1989) Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol. Sci. 10, 227–229 [DOI] [PubMed] [Google Scholar]
- 21. Jordan B. A., Trapaidze N., Gomes I., Nivarthi R., Devi L. A. (2001) Oligomerization of opioid receptors with β2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. U.S.A. 98, 343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jorgensen R., Kubale V., Vrecl M., Schwartz T. W., Elling C. E. (2007) Oxyntomodulin differentially affects glucagon-like peptide-1 receptor β-arrestin recruitment and signaling through Gα. J. Pharmacol. Exp. Ther. 322, 148–154 [DOI] [PubMed] [Google Scholar]
- 23. Moens K., Heimberg H., Flamez D., Huypens P., Quartier E., Ling Z., Pipeleers D., Gremlich S., Thorens B., Schuit F. (1996) Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 45, 257–261 [DOI] [PubMed] [Google Scholar]
- 24. Gallwitz B., Witt M., Fölsch U. R., Creutzfeldt W., Schmidt W. E. (1993) Binding specificity and signal transduction of receptors for glucagon-like peptide-1(7–36)amide and gastric inhibitory polypeptide on RINm5F insulinoma cells. J. Mol. Endocrinol. 10, 259–268 [DOI] [PubMed] [Google Scholar]
- 25. Irannejad R., von Zastrow M. (2014) GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 27, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calebiro D., Nikolaev V. O., Persani L., Lohse M. J. (2010) Signaling by internalized G-protein-coupled receptors. Trends Pharmacol. Sci. 31, 221–228 [DOI] [PubMed] [Google Scholar]
- 27. Syme C. A., Zhang L., Bisello A. (2006) Caveolin-1 regulates cellular trafficking and function of the glucagon-like peptide 1 receptor. Mol. Endocrinol. 20, 3400–3411 [DOI] [PubMed] [Google Scholar]
- 28. Harper C. B., Popoff M. R., McCluskey A., Robinson P. J., Meunier F. A. (2013) Targeting membrane trafficking in infection prophylaxis: dynamin inhibitors. Trends Cell Biol. 23, 90–101 [DOI] [PubMed] [Google Scholar]
- 29. Herskovits J. S., Burgess C. C., Obar R. A., Vallee R. B. (1993) Effects of mutant rat dynamin on endocytosis. J. Cell Biol. 122, 565–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buggy J. J., Heurich R. O., MacDougall M., Kelley K. A., Livingston J. N., Yoo-Warren H., Rossomando A. J. (1997) Role of the glucagon receptor COOH-terminal domain in glucagon-mediated signaling and receptor internalization. Diabetes 46, 1400–1405 [DOI] [PubMed] [Google Scholar]
- 31. Krilov L., Nguyen A., Miyazaki T., Unson C. G., Williams R., Lee N. H., Ceryak S., Bouscarel B. (2011) Dual mode of glucagon receptor internalization: role of PKCα, GRKs and β-arrestins. Exp. Cell Res. 317, 2981–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Merlen C., Fabrega S., Desbuquois B., Unson C. G., Authier F. (2006) Glucagon-mediated internalization of serine-phosphorylated glucagon receptor and Gsα in rat liver. FEBS Lett. 580, 5697–5704 [DOI] [PubMed] [Google Scholar]
- 33. Wheeler M. B., Gelling R. W., Hinke S. A., Tu B., Pederson R. A., Lynn F., Ehses J., McIntosh C. H. (1999) Characterization of the carboxyl-terminal domain of the rat glucose-dependent insulinotropic polypeptide (GIP) receptor. A role for serines 426 and 427 in regulating the rate of internalization. J. Biol. Chem. 274, 24593–24601 [DOI] [PubMed] [Google Scholar]
- 34. van Rijn R. M., Whistler J. L., Waldhoer M. (2010) Opioid-receptor-heteromer-specific trafficking and pharmacology. Curr. Opin. Pharmacol. 10, 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kedees M. H., Grigoryan M., Guz Y., Teitelman G. (2009) Differential expression of glucagon and glucagon-like peptide 1 receptors in mouse pancreatic alpha and beta cells in two models of alpha cell hyperplasia. Mol. Cell. Endocrinol. 311, 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moens K., Flamez D., Van Schravendijk C., Ling Z., Pipeleers D., Schuit F. (1998) Dual glucagon recognition by pancreatic β-cells via glucagon and glucagon-like peptide 1 receptors. Diabetes 47, 66–72 [DOI] [PubMed] [Google Scholar]
- 37. Kieffer T. J., Heller R. S., Unson C. G., Weir G. C., Habener J. F. (1996) Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology 137, 5119–5125 [DOI] [PubMed] [Google Scholar]
- 38. Gromada J., Ding W. G., Barg S., Renström E., Rorsman P. (1997) Multisite regulation of insulin secretion by cAMP-increasing agonists: evidence that glucagon-like peptide 1 and glucagon act via distinct receptors. Pflugers Arch. 434, 515–524 [DOI] [PubMed] [Google Scholar]
- 39. Kuna R. S., Girada S. B., Asalla S., Vallentyne J., Maddika S., Patterson J. T., Smiley D. L., DiMarchi R. D., Mitra P. (2013) Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 305, E161–E170 [DOI] [PubMed] [Google Scholar]
- 40. Pamir N., Lynn F. C., Buchan A. M., Ehses J., Hinke S. A., Pospisilik J. A., Miyawaki K., Yamada Y., Seino Y., McIntosh C. H., Pederson R. A. (2003) Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am. J. Physiol. Endocrinol. Metab. 284, E931–E939 [DOI] [PubMed] [Google Scholar]