

This study shows the tight interplay of regulation among signaling pathways within the cilium. While anterograde and retrograde transport distinctly regulate PDGFRα protein levels, disruption of either leads to PDGF-AA–insensitive Akt phosphorylation. It is shown that this is due to ciliary transport decreasing PP2A activity and increasing mammalian target of rapamycin signaling.
Abstract
Primary cilia are built and maintained by intraflagellar transport (IFT), whereby the two IFT complexes, IFTA and IFTB, carry cargo via kinesin and dynein motors for anterograde and retrograde transport, respectively. Many signaling pathways, including platelet- derived growth factor (PDGF)-AA/αα, are linked to primary cilia. Active PDGF-AA/αα signaling results in phosphorylation of Akt at two residues: P-AktT308 and P-AktS473, and previous work showed decreased P-AktS473 in response to PDGF-AA upon anterograde transport disruption. In this study, we investigated PDGF-AA/αα signaling via P-AktT308 and P-AktS473 in distinct ciliary transport mutants. We found increased Akt phosphorylation in the absence of PDGF-AA stimulation, which we show is due to impaired dephosphorylation resulting from diminished PP2A activity toward P-AktT308. Anterograde transport mutants display low platelet-derived growth factor receptor (PDGFR)α levels, whereas retrograde mutants exhibit normal PDGFRα levels. Despite this, neither shows an increase in P-AktS473 or P-AktT308 upon PDGF-AA stimulation. Because mammalian target of rapamycin complex 1 (mTORC1) signaling is increased in ciliary transport mutant cells and mTOR signaling inhibits PDGFRα levels, we demonstrate that inhibition of mTORC1 rescues PDGFRα levels as well as PDGF-AA–dependent phosphorylation of AktS473 and AktT308 in ciliary transport mutant MEFs. Taken together, our data indicate that the regulation of mTORC1 signaling and PP2A activity by ciliary transport plays key roles in PDGF-AA/αα signaling.
INTRODUCTION
Primary cilia, the microtubule-based projections found on the eukaryotic cell surface, are linked to a number of signaling pathways (Goetz and Anderson, 2010). Primary cilia are built and maintained by intraflagellar transport (IFT), whereby the two IFT complexes, IFTA and IFTB, carry cargo via kinesin and dynein motors for anterograde and retrograde transport, respectively. Many signaling pathways, including Sonic hedgehog (Shh), Wnt, PDGF, and mammalian target of rapamycin complex 1 (mTORC1), are linked to primary cilia, because mutations in IFTA, IFTB, kinesins, or dyneins alter the signaling response (Huangfu et al., 2003; Schneider et al., 2005; Corbit et al., 2008; Boehlke et al., 2010). The relationship between IFT and signaling can be complex; for example, Shh response is reduced in IFTB, dynein, and kinesin mutants, but elevated in IFTA mutants (Huangfu et al., 2003; May et al., 2005; Tran et al., 2008).
Altering IFT leads to distinct changes in ciliary structure, depending on which IFT or motor protein function is disrupted. For example, primary cilia do not form when anterograde transport is abolished via loss of IFTB or kinesin components. In contrast, cilia become swollen when either IFTA or dynein components are lost, preventing retrograde IFT (Goetz and Anderson, 2010). Several other classes of proteins play roles in trafficking and ciliary structure, including the small GTPase, ADP-ribosylation factor-like 13B (ARL13B; Larkins et al., 2011). Loss of ARL13B disrupts the microtubule structure of primary cilia and the localization and distribution of proteins within cilia (Caspary et al., 2007; Larkins et al., 2011).
Distinct signaling pathways require cilia for their activity. The best characterized of these is Shh signal transduction, which does not occur in the absence of cilia; dynamic ciliary movement of the Shh receptor and other pathway components is necessary to elicit an Shh response (Corbit et al., 2005; Haycraft et al., 2005; May et al., 2005; Rohatgi et al., 2007). The relationship between PDGF signaling and cilia is less understood. PDGF-AA acts through PDGFRαα homodimers to activate downstream targets, including Akt kinase. PDGFRα is up-regulated and enriched in primary cilia in growth-arrested fibroblasts (Schneider et al., 2005), and PDGF-AA/αα signaling is critical to induce G0 cells into the cell cycle (Stiles et al., 1979; Pledger et al., 1981; Greenberg and Ziff, 1983). Moreover, PDGF-AA cannot signal in fibroblasts lacking cilia, suggesting ciliary localization of the receptor may be critical to its function. However, cells lacking cilia display decreased PDGFRα protein levels, which could also explain the lowered signaling (Schneider et al., 2005, 2010).
Akt is fully activated via phosphorylation of T308 (P-AktT308) and S473 phosphorylation (P-AktS473). Dephosphorylation of P-AktT308 is mediated by the phosphatase PP2A. PP2A regulates the activity of a variety of additional substrates and is negatively regulated by the mammalian target of rapamycin complex 1 (mTORC1) (Andjelkovic´ et al., 1996; Zhao et al., 2003; Vereshchagina et al., 2008; Boehlke et al., 2010; McBride et al., 2010; Jackson and Pallas, 2012). Interestingly, the activities of both Akt and mTORC1 are linked to primary cilia (Schneider et al., 2005, 2010; Boehlke et al., 2010; Clement et al., 2013). Here, using mouse embryonic fibroblasts (MEFs) deficient for distinct aspects of ciliary transport, we investigate the role of ciliary transport in PDGF-AA/αα signaling along with the mechanistic connections to PP2A activity and mTORC1 signaling. Our data highlight the intricate connections among signaling pathways as they relate to cilia, emphasizing the role of the primary cilium as a signaling hub.
RESULTS
Abnormal response to PDGF-AA signaling in ciliary transport mutant MEFs
PDGF-AA/αα stimulation induces activation of Akt through phosphorylation on two residues, T308 and S473 (Bellacosa et al., 1998). Previous work has shown that PDGF-AA stimulation fails to increase P-AktS473 in Ift88orpk MEFs carrying a hypomorphic mutation in the IFTB component (Schneider et al., 2005, 2010). To investigate the role of additional ciliary transport proteins in PDGF-AA/αα signaling, including phosphorylation of both T308 and S473 residues, we derived control and mutant MEFs from mouse embryos carrying the following alleles of genes encoding ciliary proteins important for transport: Ift172wim and Ift172avc, an IFTB component involved in anterograde trafficking; Ift122sopb, an IFTA component involved in retrograde trafficking; and Arl13bhnn, a small, ciliary GTPase (Huangfu et al., 2003; Caspary et al., 2007; Friedland-Little et al., 2011; Qin et al., 2011). Ift172wim and Ift172avc represent null and hypomorphic alleles, respectively, in an IFTB complex protein, so Ift172wim MEFs lack cilia, while Ift172avc MEFs have truncated cilia; Ift122sopb MEFs have disrupted retrograde transport and display swollen or bulgy cilia; and Arl13bhnn MEFs have short cilia with an abnormal ultrastructure and mislocalized ciliary proteins. With the exception of Ift172wim MEFs, which cannot form cilia, the other MEFs can be induced to form cilia through serum starvation. We grew control, Ift172wim, Ift172avc, Ift122sopb, and Arl13bhnn MEFs in serum-supplemented media until confluent, serum starved them for 48 h, stimulated them with PDGF-AA ligand, and then harvested the cells.
We first examined PDGFRα levels in serum-starved control, Ift172wim, Ift122sopb, and Arl13bhnn MEFs. Similar to the published results in Ift88orpk MEFs, we found low PDGFRα levels in Ift172wim MEFs, suggesting the phenotype is shared when IFTB components are disrupted (Figure 1). However, PDGFRα levels in Ift122sopb and Arl13bhnn MEFs are similar to control MEFs (Figure 1 and Supplemental Figure S1). We assessed phosphorylation of PDGFRα on Y742 (P-PDGFRαY742), which occurs upon PDGF-AA binding to PDGFRα and initiates signaling (Yu et al., 1991, 1994). In the absence of PDGF-AA stimulation, we found a slight elevation in basal levels of P-PDGFRαY742 in Arl13bhnn and Ift122sopb MEFs compared with control MEFs, indicating inappropriate activation of PDGFRα in the absence of PDGF-AA ligand (Figure 1 and Supplemental Figure S1). In response to PDGF-AA stimulation, we found P-PDGFRαY742 levels increased in control, Arl13bhnn, and Ift122sopb MEFs, while there was no P-PDGFRαY742 in Ift172wim MEFs (Figure 1 and Supplemental Figure S1). Thus PDGFRα levels and response to PDGF-AA stimulation are disrupted in Ift172wim anterograde mutants. In Arl13bhnn and Ift122sopb mutants, despite the slightly elevated basal P-PDGFRαY742, the increase in P-PDGFRαY742 upon PDGF-AA stimulation indicates PDGFRα, when present, could be activated.
FIGURE 1:
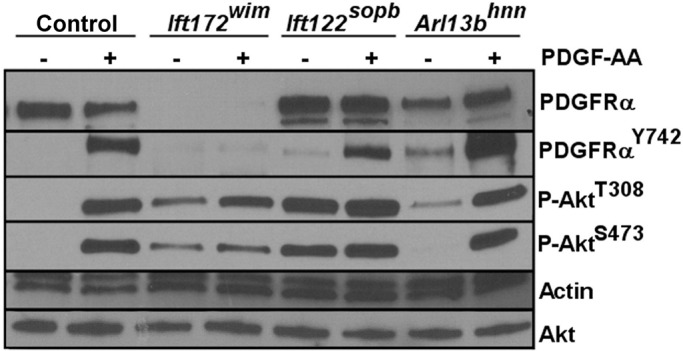
Response to PDGF-AA stimulation is misregulated in ciliary transport mutant MEFs. Comparison of PDGFRα, P-PDGFRαY742, P-AktT308, and P-AktS473 in control and ciliary transport mutant MEFs in the presence or absence of PDGF-AA ligand stimulation (n = 3).
To measure downstream pathway activation, we analyzed both P-AktT308 and P-AktS473 levels in serum-starved control, Ift172wim, Ift122sopb, and Arl13bhnn MEFs. We saw increased P-AktT308 in all ciliary transport mutant MEFs compared with control MEFs, while P-AktS473 was detectable only in Ift172wim and Ift122sopb MEFs (Figure 1 and Supplemental Figure S1). On PDGF-AA stimulation, we saw increased levels of P-AktT308 and P-AktS473 in control and Arl13bhnn MEFs; however, there was no further increase in P-AktT308 or P-AktS473 levels upon PDGF-AA stimulation in Ift172wim or Ift122sopb MEFs (Figure 1 and Supplemental Figure S1). These data indicate that control and Arl13bhnn MEFs respond to PDGF-AA by increasing phosphorylation of Akt, whereas Ift172wim and Ift122sopb MEFs do not. The insensitivity of P-AktT308 or P-AktS473 levels to PDGF-AA stimulation in Ift172wim and Ift122sopb MEFs is especially striking, because the mutations affected PDGFRα distinctly, pointing to PDGFRα-independent influences on either the phosphorylation or dephosphorylation of Akt.
P-AktT308 is increased in ciliary transport mutant MEFs
Because Akt phosphorylation is stimulated through phosphoinositide 3-kinase (PI3K) signaling, we wanted to determine whether the increased P-AktT308 levels were PI3K-dependent by treating the ciliary transport mutant MEFs with the PI3K inhibitor LY294002 (Andjelkovic´ et al., 1996). We could not detect P-AktT308 in LY294002-treated control, Ift172avc, Ift122sopb, or Arl13bhnn MEFs, confirming the T308 phosphorylation we saw was PI3K dependent (Figure 2, A and E). Notably, P-AktT308 remained elevated in Ift172wim MEFs after LY294002 treatment (Figure 2, A and E). This could be due either to a PI3K-independent pathway phosphorylating P-AktT308 or to a lack of P-AktT308 dephosphorylation.
FIGURE 2:

P-AktT308 is increased in ciliary transport mutant MEFs and the P-AktT308 phosphatase component PP2A localizes to the basal body. (A) Serum-starved control and ciliary transport mutant MEFs were lysed and processed for Western blot analysis of P-AktT308 under the indicated experimental conditions (no serum, +LY294002, +OA, and +OA +LY294002). Note the presence of P-AktT308 in Ift172wim MEFs during LY294002 treatment. Lanes shown are rearranged from a single gel. (B) Serum-starved control and ciliary transport mutant MEFs were immunolabeled for the catalytic subunit of PP2A PP2Ac (green) and for the basal body using γ-tubulin (red). (C) Serum-starved control MEFs were immunolabeled for the catalytic subunit of PP2A PP2Ac (green), for the basal body using γ-tubulin (red), and for cilia using Arl13b (red and blue). (D) Serum-starved control and Ift172wim MEFs were treated with a PP2A agonist, FTY720, and then lysed and processed for Western blot analysis of P-AktT308. FTY720 agonizes PP2A activity by inhibiting the PP2A inhibitor I2PP2A/SET1. Note the absence of P-AktT308 in Ift172wim MEFs. (E) Average densitometry values of P-AktT308 in control and ciliary transport mutant MEFs under the different experimental conditions. Error bars show SD (n = 3). Ciliary transport mutants were compared with their respective control littermates, under the same treatments, for statistical analyses (n = 3; *, p < 0.05; **, p < 0.01).
PP2Ac localizes to the basal body of MEFs
PP2A, a serine-threonine phosphatase, regulates the activity of Akt by dephosphorylating Akt on T308 and localizes to the flagella of Chlamydomonas reinhardtii (Yang et al., 2000). PP2A also localizes to the centrosome of mammalian cells (Horn et al., 2007; Flegg et al., 2010). The localization of PP2A relative to vertebrate cilia is unknown, so we first sought to determine the localization of PP2A relative to the cilium in serum-starved, control MEFs. To do so, we costained control MEFs with γ-tubulin and Arl13b and with an antibody for the catalytic subunit of PP2A (PP2Ac), and found PP2Ac localized to the basal body (Figure 2, B and C). Because P-AktS473 and AktT308 also localize to the basal body of ciliated cells (Zhu et al., 2009; Schneider et al., 2010), we examined P-AktT308 localization relative to cilia and found P-AktT308 localized to a single centriole of the basal body of control MEFs (Supplemental Figure S2). Notably, we saw no change in PP2Ac or P-AktT308 localization at the basal body when we examined serum-starved ciliary transport mutant MEFs (Figure 2B and Supplemental Figure S1). Thus PP2Ac and its substrate, P-AktT308, localize to the basal body of ciliated control and ciliary transport mutant MEFs.
PP2A activity toward P-AktT308 is disrupted in Ift172wim mutant MEFs
To determine whether the loss of P-AktT308 seen in control, Ift172avc, Ift122sopb, and Arl13bhnn MEFs after LY294002 treatment is PP2A dependent, we pharmacologically inhibited PP2A. Okadaic acid (OA) is a PP2A inhibitor, and treatment with OA increases P-AktT308 in a variety of cell lines in vitro (Cohen et al., 1989; Edelstein and Rockwell, 2012; Li et al., 2013). If PP2A is responsible for the loss of P-AktT308, then treatment with LY294002 and OA should lead to increased P-AktT308. We performed our analysis in the presence of OA alone or in the presence of both OA and LY294002. Under either treatment, we detected P-AktT308 in control, Ift172wim, Ift172avc, Ift122sopb, and Arl13bhnn MEFs (Figure 2, A and E), indicating that disruption of PP2A activity can lead to increased P-AktT308 levels, even in the absence of PI3K signaling.
The observations that P-AktT308 is increased in the absence of serum but lost during PI3K inhibition indicate that dephosphorylation of P-AktT308 is disrupted but still functional in Ift172avc, Ift122sopb, and Arl13bhnn MEFs. The observation that P-AktT308 is increased in Ift172wim MEFs even during PI3K inhibition suggests that dephosphorylation of P-AktT308 is more severely impaired compared with the other ciliary transport mutants. Additionally, there may be aberrant signaling through other PI3K-independent pathways in the ciliary transport mutant MEFs that could contribute to the increased P-AktT308. However, as PP2A is localized to the basal body of ciliated MEFs, we propose that disruption of ciliary transport affects PP2A activity toward P-AktT308.
PP2A activity is regulated by endogenous inhibitors, such as I2PP2A/SET, which localizes to primary cilia in human retinal pigment epithelial cells (Li et al., 1996; Wang and Brautigan, 2008; Saddoughi et al., 2013). To directly test whether loss of PP2A activity is responsible for the persistence of P-AktT308 in Ift172wim MEFs, we treated the cells with FTY720, the inhibitor of I2PP2A/SET, which thereby functioned as a PP2A agonist. We found P-AktT308 levels in Ift172wim MEFs with FTY720 treatment were equivalent to those in control MEFs, indicating that the defect in Ift172wim MEFs is due to lack of PP2A activity (Figure 2D). Taken together, our data argue that lack of anterograde transport, as in Ift172wim MEFs, results in an absence of PP2A activity, whereas diminished anterograde transport, as in the Ift172avc MEFs, or abnormal transport, as in the Ift122sopb and Arl13bhnn MEFs, leads to decreased PP2A activity (Figure 2E).
Additionally, we examined Dync2h1lln MEFs, which carry a null allele of the retrograde dynein motor. Dync2h1lln MEFs do not survive in serum-free conditions, requiring us to grow the cells in 0.5% serum, which introduces confounding signaling (Ocbina and Anderson, 2008; Ocbina et al., 2011; Supplemental Figure S3A). Nevertheless, P-AktT308 was absent following PI3K inhibition (LY294002) and present following PP2A inhibition (OA) (Supplemental Figure S3A). We investigated the Dync2h1lln MEFs in all subsequent assays and found the results were consistent with results from Ift122sopb, the other mutant that primarily affects retrograde transport (Supplemental Figure S2). Because Ift122sopb cells grow in the absence of sera, we focused on them.
Total PP2A activity is similar in control and ciliary transport mutant MEFs
PP2A has dozens of substrates that belong to a variety of pathways (Andjelkovic´ et al., 1996; Janssens and Goris, 2001; Vereshchagina et al., 2008; Basu, 2011). To determine whether overall PP2A activity is altered in the ciliary transport mutant MEFs, we used two distinct assays: 1) a commercially available PP2A activity assay; and 2) Western blot analysis of the inactive, unmethylated PP2A pool. For the phosphatase activity assay, PP2A was immunoprecipitated from the cell lysates and its activity monitored on a specific phosphopeptide substrate (K-R-pT-I-R-R; Saddoughi et al., 2013). While our control showed reduced PP2A activity by inhibition with OA in control and ciliary transport mutant lysates (Figure 3A), we found no difference in PP2A activity toward the phosphopeptide in any of the ciliary transport mutant MEFs compared with control MEFs. For the second assay, we monitored the methylation state of PP2Ac. Active PP2Ac is methylated, so unmethylated PP2Ac represents inactive PP2A (Turowski et al., 1995; Jackson and Pallas, 2012). We measured both total and unmethylated PP2Ac expression by Western blotting and found no differences in the level of total or unmethylated PP2Ac expression in control, Ift172wim, Ift122sopb, or Arl13bhnn MEFs (Figure 3B). These data indicate that ciliary transport does not alter overall PP2A activity and suggest that a subset of PP2A activity is affected.
FIGURE 3:

PP2A phosphatase activity and levels of total and unmethylated PP2Ac are unchanged in ciliary transport mutant MEFs. (A) PP2A activity was measured in control and ciliary transport mutant MEFs. Bars show the average of three independent experiments. Error bars show SD (n = 3; *, p < 0.05; **, p < 0.01). (B) Serum-starved control and ciliary transport mutant MEFs were lysed and processed for Western blot analysis of total and unmethylated PP2Ac levels. Lanes shown are rearranged from a single gel. Bar graphs show average densitometry values of unmethylated and total PP2Ac of control and ciliary transport mutant MEFs (n = 3).
mTORC1 pathway activity is increased in ciliary transport mutant MEFs
mTORC1 signaling and PP2A negatively regulate one another, and loss of cilia can result in increased mTORC1 signaling (Ballou et al., 1988; Peterson et al., 1999; Petritsch et al., 2000; Boehlke et al., 2010; Li et al., 2013). Thus we wanted to determine whether mTORC1 signaling is increased in the ciliary transport mutant MEFs. We assayed mTORC1 pathway activity by monitoring phosphorylation of the mTORC1 subunit, mTOR (P-mTORS2448), and mTORC1's substrate, p70 S6K (P-p70 S6KST389), through Western analysis. mTOR is the core component of mTORC1 and is phosphorylated on S2448 (P-mTORS2448) via Akt signaling (Inoki et al., 2002). Active mTORC1 phosphorylates p70 S6K (P-p70 S6KST389; Ballou, 1988; Jeno, 1988; Ferrari et al., 1991; Burnett et al., 1998; Isotani et al., 1999). Notably, P-p70 S6KT389 is a PP2A substrate (Ballou et al., 1988). We found elevated expression of P-mTORS2448 and of P-p70 S6KST389 in Ift172wim, Ift172avc, Ift122sopb, and Arl13bhnn MEFs compared with control MEFs, indicating mTORC1 signaling is up-regulated when ciliary transport is disrupted (Figure 4A).
FIGURE 4:

mTORC1 signaling is increased in ciliary transport mutant MEFs, and response to PDGF-AA stimulation is restored by rapamycin treatment. (A) Phosphorylation of mTOR (P-mTORS2448) (n = 2; control vs. Ift172wim: p < 0.01; control vs. Ift172sopb and control vs. Arl13bhnn: p < 0.05) and of p70 S6K (P-p70 S6KT389) (n = 2; control vs. Ift172wim and control vs. Ift172sopb: p < 0.001; control vs. Arl13bhnn: p = 0.0587) are increased in ciliary transport mutant MEFs. Lanes shown are rearranged from a single gel. (B) Average densitometry values of P-mTORS2448 and P-p70 S6KT389 in control and ciliary transport mutant MEFs. Error bars show SD (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (C) Rapamycin treatment of control and ciliary transport mutant MEFs in the absence of serum, with or without PDGF-AA ligand stimulation. (D) P-AktT308 levels of control and Ift172wim MEFs treated with rapamycin, PDGF-AA and an increasing concentration of PDGFRα blocking antibody (n = 3). (E) Average densitometry values of P-AktT308 of control and Ift172wim MEFs treated with rapamycin, PDGF-AA, and an increasing concentration of PDGFRα blocking antibody (n = 3; *, p < 0.05; **, p < 0.01).
Rapamycin treatment restores PDGFRα levels and response to PDGF-AA stimulation
mTORC1 signaling can also inhibit PDGFRα levels, thereby decreasing PDGF-AA/αα signaling (Zhang et al., 2003, 2007). To test whether the aberrant PDGF-AA response in Ift172wim MEFs was the result of increased mTORC1 inhibiting PDGFRα levels, we treated the cells with the mTORC1 inhibitor rapamycin and examined PDGFRα expression and PDGF-AA response. We first showed that rapamycin is able to inhibit mTORC1 signaling, with or without PDGF-AA, in wild-type and ciliary transport mutant MEFs (Supplemental Figure S4D). Despite an absence of cilia (Supplemental Figure S4C), we found total PDGFRα expression levels, by Western blotting, were restored by rapamycin in Ift172wim MEFs (Figure 4C; compare with Figure 1 and Supplemental Figure S1). While PDGFRα is localized to the cilia of serum-starved wild-type MEFs, we did not see distinct regions of PDGFRα staining in Ift172wim MEFs, with or without rapamycin (Supplemental Figure S4). We also saw induction of P-PDGFRαY742, P-AktT308, and P-AktS473 expression upon PDGF-AA stimulation, indicating that limited PDGFRα levels could explain the low PDGF-AA/αα response of IFT mutants (Figure 4C; compare with Figure 1).
The increased basal P-AktT308 in rapamycin-treated control MEFs highlights the feedback among mTORC1 signaling, PP2A activity, and PDGF-AA/αα signaling. P-AktT308 levels reflect the relative activities of PDGF-AA/αα-induced phosphorylation of Akt and of PP2A-mediated dephosphorylation of Akt. Previous reports showed that rapamycin can induce Akt phosphorylation (because loss of mTORC1 signaling increases PDGFRα protein expression and downstream signaling) and can increase PP2A activity (via loss of mTORC1's inhibition of PP2A) (Rodrik-Outmezguine et al., 2011; Ho et al., 2012; Li et al., 2013). Indeed, when we treated control MEFs with rapamycin and increasing concentrations of PDGFRα blocking antibody and then stimulated with PDGF-AA, the P-AktT308 levels dropped as PDGFRα blocking antibody concentration increased (Figure 4, D and E). We next tested whether the feedback equilibrium depends on IFT172 by repeating this treatment regimen in Ift172wim MEFs. We found that P-AktT308 levels were unchanged in Ift172wim MEFs in contrast to control MEFs (Figure 4, D and E). Together these data argue that the role of mTORC1 signaling in regulating the phosphorylation of Akt via PDGF-AA/αα signaling and PP2A activity depends on Ift172.
DISCUSSION
In this paper, we extend our understanding of the links between cilia and PDGF-AA/αα signaling by investigating two phosphorylation sites known to activate Akt in the context of distinct ciliary transport mutations. Consistent with previous data, we found low levels of P-AktS473 when anterograde transport is disrupted (Schneider et al., 2005, 2010). Moreover, we found that when either anterograde or retrograde ciliary transport is disrupted, both P-AktT308 and P-AktS473 levels are insensitive to PDGF-AA stimulation. Through pharmacological manipulation, we found that the aberrant P-AktT308 results from problems with its dephosphorylation due to decreased PP2A activity, indicating that ciliary transport regulates a subset of PP2A activity. Defects in ciliary transport are linked to increased mTORC1 signaling, which is also known to inhibit both PP2A activity and PDGFRα protein levels. By inhibiting mTORC1 signaling, we restored PDGFRα protein levels in cells lacking cilia and rescued the PDGF-AA ligand response in cells deficient in ciliary transport. Thus our work reveals that ciliary transport regulates PDGF-AA/αα signaling through elevated mTOR signaling and diminished PP2A activity.
PDGFRα is up-regulated and enriched in primary cilia in growth-arrested fibroblasts (Lih et al., 1996; Schneider et al., 2005), and PDGF-AA/αα signaling is critical to induce G0 cells into the cell cycle (Stiles et al., 1979; Pledger et al., 1981; Greenberg and Ziff, 1983). Because PDGF-AA/αα response is restored in rapamycin-treated MEFs lacking cilia (Ift172wim), this implies that PDGFRα, although normally localized to cilia in vitro, need not be there to function. Such a model posits that cilia provide a site for efficient PDGF-AA/αα signaling and that functional ciliary transport helps fine-tune PDGF-AA/αα signaling. Consistent with this, the other ciliary transport mutants also showed restoration of PDGF-AA/αα response (measured by increased P-Akt levels) upon rapamycin treatment, suggesting the lack of response in the ciliary transport mutants is due to elevated mTOR signaling.
Our finding of abnormally elevated P-AktT308 levels in MEFs with aberrant ciliary transport indicates disrupted PP2A activity toward P-AktT308, with the most severe defects in MEFs deficient for anterograde transport (Ift172wim). Indeed, in the presence of the PI3K inhibitor LY294002, the anterograde mutants show P-AktT308 levels consistent with complete inhibition of PP2A activity; however, these cells do lack cilia, making it impossible to distinguish whether the lack of cilia or the absence of anterograde traffic underlies the diminished PP2A activity. In the remaining ciliary transport mutant MEFs, PP2A is active, albeit less efficient than in control. In fact, Ift172avc, Ift122sopb, Arl13bhnn, and Dync2h1lln MEFs required PI3K inhibition for T308 to be completely dephosphorylated as in control MEFs.
Our data show that total PP2A activity is not changed, suggesting that the effects on P-AktT308 levels are due to misregulation of a specific pool of PP2A. We cannot pinpoint an exact mechanism for this—perhaps PP2A activity is affected only in specific subcellular compartments or perhaps only specific holoenzyme combinations of PP2A are affected. Several ciliary transport proteins, including Arl13b, can function outside the cilium, so perhaps they regulate PP2A activation in such a specific location (Robert et al., 2007; Sedmak and Wolfrum, 2010; Delaval et al., 2011; Barral et al., 2012; Casalou et al., 2014). Nonetheless, if only a subset of PP2A activity is decreased, it would explain why P-AktT308 is increased but total in vitro PP2A activity is not altered in the ciliary transport mutant MEFs. Given that PP2Ac and P-AktT308 are both localized at the base of cilia, we examined their signal intensity in the ciliary transport mutants but were unable to see any changes. This could be due to the changes being below the level of detection, which seems unlikely, because we could observe the differences by Western blotting. Alternatively, PP2A and Akt are both constantly trafficked around the cell, so their activity state need not be linked to their physical presence at the ciliary base. Our data suggest ciliary transport regulates PP2A activity and the Akt substrate's presence at the ciliary base facilitates Akt regulation.
There are several possibilities to explain how ciliary transport regulates PP2A activity, but our present work does not address whether this is via a direct or an indirect mechanism. One direct model would be that PP2A subunits or the assembled holoenzyme may require ciliary transport for activation, either by being physically brought into the cilium for activation or by an activator being transported into the cilium. Our observation that PP2Ac remained at the basal body in all ciliary transport mutant MEFs suggests the entire holoenzyme is not transported into the cilium. Alternatively, it is possible that an inhibitor of PP2A activity is regulated by ciliary transport. The rescue of P-AktT308 dephosphorylation in Ift172wim MEFs upon treatment with FTY720 is consistent with this possibility, because FTY720 inhibits an endogenous inhibitor of PP2A, I2PP2A/SET (Li et al., 1996; Saddoughi et al., 2013). I2PP2A/SET localizes to cilia in at least one cell type, suggesting that ciliary transport controls the kinetics of I2PP2A/SET inhibition of PP2A, thereby regulating a subset of PP2A activity, which can lead to indirect misregulation of the pathways PP2A regulates. The LY294002 sensitivity of Ift172avc MEFs compared with the insensitivity of Ift172wim MEFs would be consistent with such a model, because it suggests that the kinetics of ciliary transport are critical in regulating PP2A activity.
The feed-forward and feedback loops between mTORC1, PDGF-AA/αα, and PP2A highlight the intricate connections between these pathways and provide the scaffold upon which more indirect models can be built. It is worth noting that the pharmacological approaches we used shift the finely tuned balance among mTOR, PP2A, and PDGF-AA/αα, but do not cleanly reset all the pathways in the context of the others. Thus our data raise but cannot answer the question of whether the primary defect in the ciliary transport mutants is the increase in mTORC1 signaling or the diminished PP2A activity. Indeed, we observed increased basal P-PDGFRαY742 in both Arl13bhnn and Ift122sopb MEFs, which disappeared upon rapamycin treatment. The most parsimonious model favors the notion that disruption of ciliary transport leads to deregulation of a subset of PP2A activity, which in turn indirectly increases mTORC1 signaling. Normally, mTORC1 signaling results in phosphorylation of p70 S6KT389, and P-p70 S6KT389 is a PP2A substrate (Ballou et al., 1988; Hahn et al., 2010). Thus, diminished PP2A activity can explain the increased mTORC1 signaling and the aberrant P-AktT308 levels we see in ciliary transport mutant MEFs.
An alternative model, that the increased mTORC1 signaling causes the decreased PP2A activity, predicts that total PP2A activity would be lowered. Because total PP2A activity is not decreased in Ift172wim MEFs, where P-AktT308 levels are elevated under multiple conditions, this model is less likely. Furthermore, experiments that manipulate ciliary transport or various PP2A subunits in vivo also indicate that other PP2A functions are spared in ciliary transport mutants. PP2A Cα−/− embryos die around embryonic day 6.5 (e6.5) and fail to form mesoderm, underscoring the essential role of PP2A early in development (Götz et al., 1998). In contrast, Ift172wim, Ift172avc, Ift122sopb, Arl13bhnn, and Dync2h1lln embryos form mesoderm and survive through midgestation (Huangfu et al., 2003; Caspary et al., 2007; Ocbina and Anderson, 2008; Friedland-Little et al., 2011; Qin et al., 2011).
The past decade has witnessed a flurry of research into the primary cilium as a fundamental signaling organelle. Indeed, mutations in many cilia-related genes lead to the human ciliopathies, which are linked to diverse signaling pathways. Our data show that ciliary transport plays a role in the relationship among these pathways. Rescue of the PDGF-AA response in cells lacking cilia raise the possibility that some pathways localized in cilia may be there for efficiency; perhaps the cilium provides highly regulated real estate that facilitates pathway transduction. The next steps will distinguish between direct and indirect mechanisms for the relationships among the signal transduction pathways linked to cilia and will address when the receptors can and do act.
MATERIALS AND METHODS
Cell culture
Primary cultures of control, Arl13bhnn, Ift172wim, Ift172avc, and Ift122sopb mouse embryonic fibroblasts were isolated from e11.5–e12.5 embryos and maintained in DMEM/F12 supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (Corning, Manassas, VA: 10-092). Control MEFs were wild-type or heterozygous for Arl13bhnn, Ift172wim, Ift172avc, or Ift122sopb, and were paired with their respective ciliary transport mutant littermates for experiments. Primary Dync2h1lln and Ift172avc MEFs were provided by Kathryn Anderson (Memorial Sloan Kettering Cancer Center) and Ift122sopb MEFs by Jonathan Eggenschwiler (University of Georgia). MEFs were grown until confluent on plates precoated with 0.1% gelatin and then serum starved for 48 h before lysis and processing for Western blot analysis. Ciliary transport mutant cells, except Ift172wim, which lack cilia, were ciliated to roughly similar percentages, just below 20%. Depending on the experiment, cells were treated at the following doses/times: 50 ng/ml PDGF-AA for 10 min (R&D Systems, Minneapolis, MN: 221-AA); 50 μM LY294002 for 1 h (Cell Signaling, Danvers, MA: 9901); 10 nM rapamycin for 24 h (MP Biomedicals, Santa Ana, California: 159346); 200 nM OA for 3 h (Cell Signaling: 5934); and FTY720 for 5 h (Sigma-Aldrich, St. Louis, MO: SML0700-5MG).
PP2A immunoprecipitation phosphatase assay
The kit was purchased from Millipore (17-313). Experiments were performed following the manufacturer's protocol with 500 μg protein lysate, with or without 1 μM OA (Cell Signaling: 5934) during the phosphopeptide incubation.
Immunofluorescence (IF)
MEFs were grown and prepared for IF as previously described (Larkins et al., 2011). Fluorescence was visualized on either an Olympus FluoView 100 confocal IX81 inverted microscope, a Nikon Structured Illumination Microscope, or a Leica DM600B upright microscope. Images were processed using Fiji software (Schindelin et al., 2012).
SDS–PAGE and Western blot analysis
MEFs were rinsed with cold phosphate-buffered saline, scraped off the plate with a cell scraper, and lysed in 150 μl of RIPA buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich: P5726; Roche: 1697498001 and 04906845001). For experiments examining demethylated PP2Ac, lysates were prepared as described below, along with an additional treatment to demethylate PP2Ac (Jackson and Pallas, 2012). MEF lysate was passed through a 255/8 gauge needle 10 times and spun down at 16,000 relative centrifugal force for 10 min at 4°C, after which the supernatant was transferred to a clean tube. Protein concentrations were determined with a BCA protein kit (Pierce, Rockford, IL; 23221) or by Coomassie blue staining. MEF lysates were mixed with loading buffer, separated by SDS–PAGE on 4–20% Mini-PROTEAN TGX Precast Gels (456-1096EDU) with SDS running buffer, and transferred to Trans-Blot Turbo nitrocellulose membranes (Bio-Rad: 170-4059) using the Trans-Blot Turbo system (Bio-Rad: 170-4155). Membranes were blocked for 10 min at room temperature and incubated with primary antibodies in blocking buffer (Pierce: 37538) overnight at 4°C. Primary antibodies were detected using horseradish peroxidase–conjugated secondary antibodies (GE Healthcare Life Sciences, Pittsburgh, PA; NA934, and NA931) in 5% wt/vol nonfat milk in 0.1% Tris buffered saline with 0.1% Tween (TBST) at room temperature for 1 h and visualized with Amersham ECL Prime (GE Healthcare Life Sciences: RPN2232). Within each experiment, samples are from the same blot, originally performed in duplicate or in triplicate, and edited to show one representative band for each experimental condition, with white dotted lines drawn between lanes for ease of viewing. Band intensities from two to three biological replicates per condition were measured with Fiji software and normalized to actin loading control before averaging (McLean and Bennett, 2013). Bar graphs show the average of two or three experimental bands (as indicated) normalized to actin, with error bars showing SD. Data were analyzed using unpaired two-tailed t tests (Supplemental Table S1).
Antibodies
We used the following antibodies at the indicated dilutions for Western blotting (WB) or IF: from Cell Signaling, Akt (WB 1:1000, IF 1:200, #9272), P-AktS473 (WB 1:1000, IF 1:500, #587F11), P-AktT308 (WB 1:1000, IF 1:500, #2965), mTOR (WB 1:2000, #2983), P-mTORS2448 (WB 1:2000, #5536), P70 S6K (WB 1:2000, #2708), P-P70 S6KS371 (WB 1:2000, #9208), S6 (WB 1:2000, #2217), and P-S6 (WB 1:2000, #2211). From BD Pharmingen (San Diego, CA), PDGFRα (IF 1:200, #558774). From Santa Cruz Biotech (Dallas, TX), PDGFRα (WB 1:1000). From Sigma-Aldrich, acetylated tubulin (IF 1:2500, #T6793), actin (WB 1:1000, #A5060), α-rabbit γ-tubulin (IF 1:1000, #T5192), α-mouse γ-tubulin (IF 1:1000, #T6557), and P-PDGFRαY742 (WB 1:1000, #P8246). PP2Ac antibody was kindly provided by David Pallas (Emory University; WB 1:10,000, IF 1:200; BD Transduction Laboratories: 610555).
Supplementary Material
Acknowledgments
We thank Kathryn Anderson, Jonathan Eggenschwiler, and David Pallas for providing reagents; members of the Caspary lab for comments; and Cheryl Timms Strauss for editing. This research project was supported with research funding from the Muscular Dystrophy Association (131071), National Institute of Neurological Disorders and Stroke (NINDS; R01 NS056380), and Emory Neuroscience NINDS Core Facilities (P30NS055077—Emory University Integrated Cellular Imaging Microscopy Core). N.L.U. was supported by an Emory PRISM Graduate Teaching Fellows in K–12 Education Fellowship (DGE0536941), an Emory Genetics and Molecular Biology Graduate Program Training Grant (T32GM008490), and an Emory Human Disease Genetics Training Fellowship (T32MH087977).
Abbreviations used:
- ARL13B
ADP-ribosylation factor-like 13B
- IF
immunofluorescence
- IFT
intraflagellar transport
- MEF
mouse embryonic fibroblast
- mTORC1
mammalian target of rapamycin complex 1
- OA
okadaic acid
- PI3K
phosphatidylinositol 3 kinase
- Shh
Sonic hedgehog
- WB
Western blotting.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-0952) on November 12, 2014.
The authors have no conflicts to report.
REFERENCES
- Andjelkovic´ M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci USA. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou LM. S6 kinase in quiescent Swiss mouse 3T3 cells is activated by phosphorylation in response to serum treatment. Proc Natl Acad Sci USA. 1988;85:7154–7158. doi: 10.1073/pnas.85.19.7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou LM, Jenö P, Thomas G. Protein phosphatase 2A inactivates the mitogen-stimulated S6 kinase from Swiss mouse 3T3 cells. J Biol Chem. 1988;263:1188–1194. [PubMed] [Google Scholar]
- Barral DC, Garg S, Casalou C, Watts GFM, Sandoval JL, Ramalho JS, Hsu VW, Brenner MB. Arl13b regulates endocytic recycling traffic. Proc Natl Acad Sci USA. 2012;109:21354–21359. doi: 10.1073/pnas.1218272110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S. PP2A in the regulation of cell motility and invasion. Curr Protein Pept Sci. 2011;12:3–11. doi: 10.2174/138920311795659443. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Gödel M, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalou C, Seixas C, Portelinha A, Pintado P, Barros M, Ramalho JS, Lopes SS, Barral DC. Arl13b and the non-muscle myosin heavy chain IIA are required for circular dorsal ruffle formation and cell migration. J Cell Sci. 2014;127:2709–2722. doi: 10.1242/jcs.143446. [DOI] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Clement DL, Mally S, Stock C, Lethan M, Satir P, Schwab A, Pedersen SF, Christensen ST. PDGFRα signaling in the primary cilium regulates NHE1-dependent fibroblast migration via coordinated differential activity of MEK1/2-ERK1/2-p90RSK and AKT signaling pathways. J Cell Sci. 2013;126:953–965. doi: 10.1242/jcs.116426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen M-H, Chuang P-T, Reiter JF. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- Delaval B, Bright A, Lawson ND, Doxsey S. The cilia protein IFT88 is required for spindle orientation in mitosis. Nat Cell Biol. 2011;13:461–468. doi: 10.1038/ncb2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein J, Rockwell P. Okadaic acid induces Akt hyperphosphorylation and an oxidative stress-mediated cell death in serum starved SK-N-SH human neuroblastoma cells that are augmented by rapamycin. Neurosci Lett. 2012;531:74–79. doi: 10.1016/j.neulet.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J Biol Chem. 1991;266:22770–22775. [PubMed] [Google Scholar]
- Flegg CP, Sharma M, Medina-Palazon C, Jamieson C, Galea M, Brocardo MG, Mills K, Henderson BR. Nuclear export and centrosome targeting of the protein phosphatase 2A subunit B56α: role of B56α in nuclear export of the catalytic subunit. J Biol Chem. 2010;285:18144–18154. doi: 10.1074/jbc.M109.093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland-Little JM, Hoffmann AD, Ocbina PJR, Peterson MA, Bosman JD, Chen Y, Cheng SY, Anderson K V, Moskowitz IP. A novel murine allele of intraflagellar transport protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum Mol Genet. 2011;20:3725–3737. doi: 10.1093/hmg/ddr241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson K V. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Cα. Proc Natl Acad Sci USA. 1998;95:12370–12375. doi: 10.1073/pnas.95.21.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1983;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- Hahn K, Miranda M, Francis VA, Vendrell J, Zorzano A, Teleman AA. PP2A regulatory subunit PP2A-B′ counteracts S6K phosphorylation. Cell Metab. 2010;11:438–444. doi: 10.1016/j.cmet.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn V, Thelu J, Garcia A, Albiges-Rizo C, Block MR, Viallet J. Functional interaction of Aurora-A and PP2A during mitosis. Mol Biol Cell. 2007;18:1233–1241. doi: 10.1091/mbc.E06-12-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AL, Vasudeva SD, Laé M, Saito T, Barbashina V, Antonescu CR, Ladanyi M, Schwartz GK. PDGF receptor alpha is an alternative mediator of rapamycin-induced Akt activation: implications for combination targeted therapy of synovial sarcoma. Cancer Res. 2012;72:4515–4525. doi: 10.1158/0008-5472.CAN-12-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan K-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Isotani S, Hara K, Tokunaga C, Inoue H, Avruch J, Yonezawa K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J Biol Chem. 1999;274:34493–34498. doi: 10.1074/jbc.274.48.34493. [DOI] [PubMed] [Google Scholar]
- Jackson JB, Pallas DC. Circumventing cellular control of PP2A by methylation promotes transformation in an Akt-dependent manner. Neoplasia. 2012;14:585–599. doi: 10.1593/neo.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeno P. Identification and characterization of a mitogen-activated s6 kinase. Proc Natl Acad Sci USA. 1988;85:406–410. doi: 10.1073/pnas.85.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkins CE, Aviles GDG, East MP, Kahn RA, Caspary T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell. 2011;22:4694–4703. doi: 10.1091/mbc.E10-12-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang X, Yue P, Tao H, Ramalingam SS, Owonikoko TK, Deng X, Wang Y, Fu H, Khuri FR, et al. Protein phosphatase 2A and DNA-dependent protein kinase are involved in mediating rapamycin-induced Akt phosphorylation. J Biol Chem. 2013;288:13215–13224. doi: 10.1074/jbc.M113.463679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lih CJ, Cohen SN, Wang C, Lin-Chao S. The platelet-derived growth factor α-receptor is encoded by a growth-arrest-specific (gas) gene. Proc Natl Acad Sci USA. 1996;93:4617–4622. doi: 10.1073/pnas.93.10.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- McBride SM, Perez DA, Polley MY, Vandenberg SR, Smith JS, Zheng S, Lamborn KR, Wiencke JK, Chang SM, Prados MD, et al. Activation of PI3K/mTOR pathway occurs in most adult low-grade gliomas and predicts patient survival. J Neurooncol. 2010;97:33–40. doi: 10.1007/s11060-009-0004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean AC, Bennett SAL. A protocol for densitometric quantification of Western blots using Image J. 2013 [weblog entry]. Neural Regeneration Laboratory, Resources, University of Ottawa. Available at http://137.122.232.177/resources.html. [Google Scholar]
- Ocbina PJR, Anderson K V. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn. 2008;237:2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina PJR, Eggenschwiler JT, Moskowitz I, Anderson K V. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci USA. 1999;96:4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petritsch C, Beug H, Balmain A, Oft M. TGF-β inhibits p70 S6 kinase via protein phosphatase 2A to induce G1 arrest. Genes Dev. 2000;14:3093–3101. doi: 10.1101/gad.854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pledger WJ, Hart CA, Locatell KL, Scher CD. Platelet-derived growth factor-modulated proteins: constitutive synthesis by a transformed cell line. Proc Natl Acad Sci USA. 1981;78:4358–4362. doi: 10.1073/pnas.78.7.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci USA. 2011;108:1456–1461. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Margall-Ducos G, Guidotti J-E, Brégerie O, Celati C, Bréchot C, Desdouets C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J Cell Sci. 2007;120:628–637. doi: 10.1242/jcs.03366. [DOI] [PubMed] [Google Scholar]
- Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J, Guichard S, Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248–259. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 2013;5:105–121. doi: 10.1002/emmm.201201283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR, Stock C, Hoffmann EK, Yoder BK, Schwab A, et al. Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell Physiol Biochem. 2010;25:279–292. doi: 10.1159/000276562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Sedmak T, Wolfrum U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J Cell Biol. 2010;189:171–186. doi: 10.1083/jcb.200911095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles CD, Capone GT, Scher CD, Antoniades HN, Van Wyk JJ, Pledger WJ. Dual control of cell growth by somatomedins and platelet-derived growth factor. Proc Natl Acad Sci USA. 1979;76:1279–1283. doi: 10.1073/pnas.76.3.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski P, Fernandez a, Favre B, Lamb NJ, Hemmings Ba. Differential methylation and altered conformation of cytoplasmic and nuclear forms of protein phosphatase 2A during cell cycle progression. J Cell Biol. 1995;129:397–410. doi: 10.1083/jcb.129.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereshchagina N, Ramel M-C, Bitoun E, Wilson C. The protein phosphatase PP2A-B′ subunit Widerborst is a negative regulator of cytoplasmic activated Akt and lipid metabolism in Drosophila. J Cell Sci. 2008;121:3383–3392. doi: 10.1242/jcs.035220. [DOI] [PubMed] [Google Scholar]
- Wang W, Brautigan DL. Phosphatase inhibitor 2 promotes acetylation of tubulin in the primary cilium of human retinal epithelial cells. BMC Cell Biol. 2008;9:62. doi: 10.1186/1471-2121-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Fox L, Colbran RJ, Sale WS. Protein phosphatases PP1 and PP2A are located in distinct positions in the Chlamydomonas flagellar axoneme. J Cell Sci. 2000;113:91–102. doi: 10.1242/jcs.113.1.91. [DOI] [PubMed] [Google Scholar]
- Yu JC, Gutkind JS, Mahadevan D, Li W, Meyers KA, Pierce JH, Heidaran MA. Biological function of PDGF-induced PI-3 kinase activity: its role in alpha PDGF receptor-mediated mitogenic signaling. J Cell Biol. 1994;127:479–487. doi: 10.1083/jcb.127.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JC, Heidaran MA, Pierce JH, Gutkind JS, Lombardi D, Ruggiero M, Aaronson SA. Tyrosine mutations within the alpha platelet-derived growth factor receptor kinase insert domain abrogate receptor-associated phosphatidylinositol-3 kinase activity without affecting mitogenic or chemotactic signal transduction. Mol Cell Biol. 1991;11:3780–3785. doi: 10.1128/mcb.11.7.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Zhu D, Shi S, Wang H, Liao K. Growth arrest induces primary-cilium formation and sensitizes IGF-1-receptor signaling during differentiation induction of 3T3-L1 preadipocytes. J Cell Sci. 2009;122:2760–2768. doi: 10.1242/jcs.046276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.