

Abstract
Chronic graft versus host disease (cGVHD) continues to be a common complication of allogeneic hematopoietic stem cell transplantation (HSCT). Unlike acute GVHD, which is mediated almost entirely by donor T cells, the immune pathology of cGVHD is more complex and donor B cells have also been found to play an important role. Recent studies from several laboratories have enhanced our understanding of how donor B cells contribute to this clinical syndrome and this has led to new therapeutic opportunities. Here, Dr. Sarantopoulos reviews some of the important mechanisms responsible for persistent B cell activation and loss of B cell tolerance in patients with cGVHD. Dr. Blazar describes recent studies in preclinical models that have identified novel B cell directed agents that may be effective for prevention or treatment of cGVHD. Some B cell directed therapies have already been tested in patients with cGVHD and Dr. Cutler reviews the results of these studies documenting the potential efficacy of this approach. Supported by studies mechanistic studies in patients and preclinical models, new B cell directed therapies for cGVHD will now be evaluated in clinical trials.
Introduction
Chronic graft versus host disease (cGVHD) after allogeneic hematopoietic stem cell transplantation (HSCT) continues to be a common, debilitating and deadly complication of therapy. Despite improved tools for diagnosis and clinical assessment of disease activity, cGVHD pathophysiology remains ill-defined and this has hampered the development of effective new therapies [1, 2]. In this regard, analysis of patient blood and tissue samples and new murine models of cGVHD have expanded our knowledge of disease pathogenesis and the complexity of mechanisms that lead to tissue damage [3]. Although donor T cells clearly play a critical role in the initiation and maintenance of allo-immunity, many laboratory and clinical studies have shown that donor B cells also play an important role in the pathophysiology of cGVHD [4–6]. Importantly, therapeutic strategies targeting B cells can provide clinical benefit in many patients with active cGVHD [7].
This review will focus on recent advances in our understanding of the role of B cells in cGVHD. A series of new studies in HSCT patients and murine models have begun to elucidate the role of B cells in the pathogenesis and persistence of cGVHD and this has led to the evaluation of new therapeutic approaches specifically targeting aspects of B cell reconstitution and function after HSCT. As these new therapeutic approaches are evaluated and integrated with other established therapies, we anticipate that new therapeutic agents will lead to significant improvement in the long-term outcome of patients with cGVHD.
B Cell Activation Pathways in Chronic GVHD
In healthy individuals, B cell development is a dynamic, daily process with a high propensity for the formation of self-reactive B cells. Despite central B cell tolerance mechanisms, a remarkably large pool of polyreactive and potentially autoreactive B cells arise at a constant rate from bone marrow precursor cells [8]. Receptor editing, deletion, and anergy induction in the bone marrow [9–11] do not eliminate all potentially auto-reactive B cell clones, and it has been estimated that 55–75% of transitional B cells emerging from bone marrow in healthy adults are self-reactive [8, 12]. The maintenance of normal B cell immunity therefore requires deletion of auto-reactive clones coupled with positive selection following encounter with microbes (or other foreign antigens) [13]. In conjunction with BCR signaling, B cell activating factor (BAFF) plays an important role in determining B cell fate/survival. In acquired autoimmune diseases, abnormally high levels of BAFF subvert the development of B cell tolerance by attenuating B cell receptor (BCR)-triggered apoptosis of polyreactive B cells. In self-reactive BCR transgenic (Tg) murine models, limiting amounts of BAFF are required to promote B cell turnover and avoidance of autoreactivity [14, 15]. Early after HSCT, the peripheral B cell compartment is likely comprised of recent bone marrow emigrants consisting of short-lived transitional cells. While these cells are capable of primary immune reactions and generate short-lived plasma cells, they do not take part in the germinal center (GC) reaction. This likely explains why B cell populations post-HSCT have a relatively low diversity of antigen binding sites (i.e., BCRs) with a high frequency of low-affinity, potentially allo- or auto-reactive antibodies. Since BAFF levels are high after HSCT, B cells that are not deleted through negative selection are likely positively selected during B cell recovery. While specific antigen targets remain largely unknown, high-throughput BCR sequencing of B cell subsets suggests that the IgG CDR3s comprise poly and auto-reactive characteristics [16]. These data, along with frequent production of auto-antibodies [17–19] suggest a critical breakdown in peripheral B cell tolerance in patients with cGVHD. Taken together, these findings suggest a model for aberrant persistence of allo and auto-reactive B cells after transplant shown in Figure 1. In this model, we hypothesize that as B cells develop after HSCT, there is a failure of normal B cell tolerance checkpoints at least in part due to high levels of BAFF. As a result, there is persistence of donor B cells reactive to a variety of recipient antigens and secretion of pathologic allo- and auto-antibodies.
Figure 1. BAFF and antigen driven B cell activation.
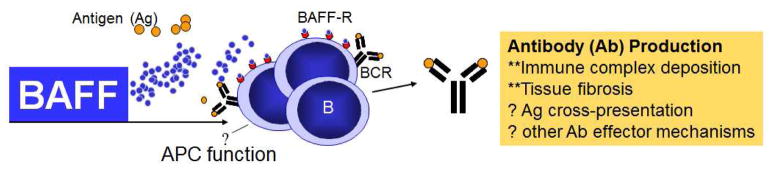
In cGVHD, B cells are promoted by antigen and excessive BAFF levels to survive and produce antibodies. Constitutive antibody production by certain B cell subsets in cGVHD may directly mediate pathology. B cells may also serve as antigen presenting cells (APC).
In the presence of a large, diverse mature B cell pool, normal B cells consume and sequester BAFF and auto-reactive B cell clones that require high levels of BAFF are unable to survive [20]. After allo-HSCT, persistently elevated BAFF levels are associated with B lymphopenia and cGVHD development [5, 21]. By contrast supranormal B cell numbers are found in patients without cGVHD [21, 22]. Since B cells in patients without cGVHD have high levels of BAFF receptors, these cells are able to sequester BAFF and prevent high levels of BAFF from promoting autoreactive B cell clones [23]. This hypothesis is supported by the finding that patients unable to robustly recover their B cell compartment may not respond to rituximab, and bone marrow production of B cells early after HCT may be critical for prevention of cGVHD [21, 22, 24, 25]. In addition to mature B cells that compete for BAFF, IL-10 producing regulatory B cells may play an important role in the prevention of cGVHD [26]. IL-10 producing cells are present in higher proportions in patients without cGVHD [27]. Our data indicate a role for altered B cell homeostasis in cGVHD pathogenesis and reveal a limitation of anti-CD20, rituximab, therapy after HCT [28]. How the B cell compartment is skewed toward robust mature B cell recovery and perhaps IL-10 production requires further study.
To determine if B cells are activated in cGVHD, we examined unstimulated B cell lysates for signaling pathways known to be downstream of BAFF. B cells in cGVHD showed evidence of increased metabolic activity and survival, with enhanced signaling via the AKT and ERK pathways [29]. Bim knockout mice have an autoimmune phenotype that is independent of BAFF, and BIM counteracts BAFF signaling [30]. Accordingly, excess BAFF reduces BIM levels in transgenic BCR autoreactive murine B cells [31]. While increased AKT and ERK signaling may be BCR-dependent, pro-apoptotic BIM is also increased after BCR engagement unless counteracted by BAFF. Consistent with BAFF-associated AKT and ERK signaling, we found BIM isoforms were decreased in cGVHD B cells [32]. Thus, similar to what is found in autoimmune mice, high BAFF levels may rescue intermediate affinity, self-reactive B cells during recovery after HCT by attenuating apoptosis of re-circulating B cells after GC reactions [14, 15]. Chronic exposure to BAFF results in hypermetabolic, enlarged B cells, capable of constitutive autoantibody production [33]. It is therefore tempting to directly link excessive BAFF levels to B cell activation in cGVHD pathophysiology, but this link remains to be determined.
In addition to BAFF, B cell activation and survival depends on the affinity and specificity for antigen and on co-receptor molecule binding for conveyance of positive or negative BCR signals [34]. We have begun to investigate this further in cGVHD to identify potential therapeutic molecular targets. In healthy mice, BAFF sets the threshold for BCR-mediated selection among naïve, mature B cells [35]. In the absence of BAFF, tonic or chronic stimulation with antigen results in constant signaling through the BCR leading to anergy or death [34]. In the setting of constant exposure to foreign (or allo) antigens and excessive BAFF, B cell hyper-responsiveness to antigen may contribute to cGVHD pathogenesis. Using anti-IgM as a surrogate for soluble antigen, we found that cGVHD B cells not only responded more robustly after exposure, but that this response was associated with increased protein levels of the proximal BCR machinery molecules SYK and BLNK [36]. Proliferation after antigen-engagement has been mechanistically linked to both BAFF-R and BLNK [37, 38]. Signaling after BCR engagement by antigen entails phosphorylation and activation of the tyrosine kinase SYK, which subsequently phosphorylates and activates the tyrosine-rich adapter protein BLNK that is then recruited to the BCR signalosome. BLNK is central to further downstream signaling as it also acts as a docking protein for Bruton’s tyrosine kinase (BTK), PLCγ2 and the other crucial BCR complex signaling molecules. We found that this heightened activation via BCR was abrogated by the SYK inhibitor fostamatinib [36], suggesting potential clinical utility of this agent. Further experiments in murine models of cGVHD in collaboration with Dr. Blazar’s lab have also shown the importance of BCR signaling and the ability of SYK inhibition to reverse tissue damage associated with cGVHD. Together our data suggest that BCR signaling after antigen engagement during B cell reconstitution following HSCT is likely critical for persistence of auto-reactive B cells and selective inhibition of BCR signaling may provide a new therapeutic target in patients with cGVHD.
While genetic disparity between donor and recipient must exist for cGVHD to develop, transferable T cell auto-reactivity occurs following allo-reactivity in murine models [39–41]. In patients, antibodies to both allo-antigens and to non-polymorphic (‘auto’) antigens are also found. For example, a coordinated T-B response to disparate epitopes on the mHA DBY has been described [42, 43]. T cells remain crucial effector cells in this disease [44], and while B-T coordinated responses have been delineated [42, 43], whether B cells drive T cell responses remains unclear. Antibodies to polymorphic and non-polymorphic recipient antigens have been associated with cGVHD [4, 17, 18]. In addition to other B cell functions [40, 45, 46], secreted antibodies likely play a role in cGVHD tissue destruction, since Ig deposits have been observed in lesional skin tissue [6, 47]. Likewise, antibody production was recently shown in a murine model to be required for cGVHD development [6]. Studies in other diseases demonstrate the role of antibodies in cross-presentation to T effector (Teff) cells [48, 49]. Thus, allo- and auto-antibodies may facilitate the cross-presentation of antigens to Teff, thereby amplifying T cell responses to minor histocompatibility antigens (mHA) or autoantigens [49, 50]. Unrestrained T follicular helper (TFH) cells have been associated with loss of B cell tolerance, potentially in abnormal GC reactions [51, 52]. In mice, TFH cells are vital for increased GC responses and for cGVHD genesis [53]. In patients, GC composition is altered, but whether TFH cells promote aberrant B cells is an area of active investigation. Cell surface CD27 increases after B cells encounter antigen. We found that circulating CD27+ B cells from cGVHD patients constitutively produce IgG without the requirement of further BCR or second signal stimulation [5]. These cells also have a distinct gene expression profile compared to healthy CD27+ B cell counterparts [32]. In patients, both pre-GC and post-GC peripheral blood B cell subsets occur in increased proportions, and these increases were associated with augmented levels of plasma BAFF [5]. Thus, unlike the anti-microbial memory response of healthy individuals, cGVHD CD27+ B cells potentially mediate anti-normal recipient tissue responses, although further study is required to affirm and quantify the pathologic potential of these cells [54, 55].
Together, our data have allowed us to develop a working model for potentially disease-mediating B cell development after HCT (Figure 2). Since aberrantly activated cells represent potential therapeutic targets, further studies of B cell signaling in patients with cGVHD are clearly needed. Pursuit of hypothesis-driven studies of human B cell function will advance our understanding of cGVHD pathophysiology and will lead to development of clinical trials aimed at prevention and treatment.
Figure 2. Development and persistence of potentially pathological cGVHD B cells.
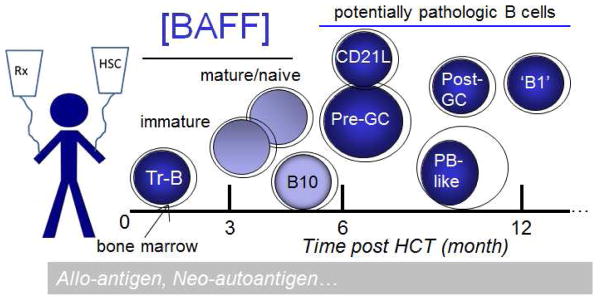
Altered B cell homeostasis with high BAFF to naive B cell ratios after HCT leads to persistence of aberrantly activated B cells. Decreased bone marrow output and production of immature (and potentially of B10) B cells leads to excessive levels of BAFF, sufficient to support potentially pathological B cell subsets.
Preclinical models evaluating novel B cell directed therapies for chronic GVHD
Preclinical development of new therapeutic modalities for cGVHD has been hindered by models with pathologic findings that may not simulate the development of human cGVHD. We developed a multi-organ system model of cGVHD induced by allogeneic HSCT after a conditioning regimen of cyclophosphamide and total-body radiation. In this model, cGVHD manifestations are observed in a wide spectrum of target organs (lung, liver, spleen, thymus, colon, tongue) [6, 56]. Fibrosis was demonstrated in the lung and liver and was associated with CD4+ T cells and B220+ B cell infiltration and IgG2c class-switched antibody deposition [6]. Lung fibrosis resulted in pulmonary dysfunction and airway obliteration, which leads to bronchiolitis obliterans syndrome (BOS), pathognomonic for cGVHD of the lung. Antibody secretion from bone marrow (BM)-derived B cells was essential for cGVHD as evidenced by the lack of disease when using BM incapable of producing B cells or secreted polyclonal IgG. Germinal centers (GCs), critical for efficient class switching and antibody secretion, were significantly increased in size and number in cGVHD mice [6]. cGVHD maintenance was dependent upon GC formation as infusion of a lymphotoxin-receptor-immunoglobulin fusion protein into mice with established cGVHD reduced GCs and suppressed BO measured by pulmonary function tests.
GCs are formed by cooperation between a subset of T and B cells known as T follicular helper (TFH) cells and GC B cells, respectively. Recently, we reported that increased frequency of TFH cells correlated with increased GC B cells, cGVHD, and BOS. In patients, the anti-CD20 mAb, rituximab, can prevent steroid requiring chronic GVHD as assessed in a phase 2 trial [57] and results in amelioration of cGVHD in patients especially with skin and mucosal involvement [7]. However, not all patients respond and not all responses are permanent. We have found that administering a highly depleting anti-CD20 monoclonal antibody (mAb) to mice with established cGVHD resulted in peripheral B cell depletion, but B cells remained in the lung, BOS was not reversed [53] and cGVHD manifestations remained [58]. Instead, BOS could be treated by eliminating production of interleukin-21 (IL-21) by donor T cells or IL-21 receptor (IL-21R) signaling of donor B cells. Blocking mAbs for TFH:GC B cell interactions including IL-21/IL-21R, inducible T cell costimulator (ICOS)/ICOS ligand, and CD40L/CD40 hindered GC formation and cGVHD, indicating a role for TFH and GC B cells and suggesting a new line of therapy to reverse cGVHD.
To study transcriptional pathways and develop targeted therapeutics for cGVHD, we studied the effects of emerging therapeutic agents targeting GCs. BCL6 is a master regulatory transcription factor which facilitates GC development in a cooperative manner with emerging chromatin-associated factors, namely the EZH2 lysine methyltransferase and the BRD4 epigenetic reader protein [59, 60]. To determine whether strategies that disrupt GC integrity could be used to treat cGVHD we targeted BCL6 using a direct-acting ligand (79-6) [61, 62], EZH2 using three structurally distinct inhibitors (JQ-E, UNC1999 [63] and DZNep [64]), and a first-in-class BRD4 inhibitor (JQ1) [60, 65]. In T cells, Bcl6 is selectively expressed in TFH and GC Tregs. We observed a 10-fold increase in % of splenic BCL6+ T cells in cGVHD mice. The small-molecule BCL6 inhibitor 79-6 was synthesized and used to treat mice with active cGVHD. Treatment reversed BOS and resulted in a decrease in GC B cells and lung collagen. EZH2 catalyzes the methylation of lysine 27 on histone 3 (H3K4me3) silencing genes to a transcriptionally repressive state. EZH2 is markedly upregulated during the GC reaction, and prevents GC B cell terminal differentiation, allowing affinity maturation to occur [62, 66, 67]. Selective EZH2 deletion in either BM-derived B cells or splenic T cells completely prevented cGVHD with a decrease in GC frequency. Whereas naïve T cells express low EZH2 levels, EZH2 is rapidly upregulated upon allo-stimulation. We compared 3 drugs that reduce H3K4me3, two pyridinone inhibitors (JQ-E and UNC1999) and DZNep, which destabilizes EZH2 complexes. At established doses, DZNep and UNC1999 were ineffective or toxic, respectively. In contrast, JQ-E fully reversed cGVHD lung dysfunction and fibrosis around the bronchioles was significantly decreased. JQ1 is a first in class epigenetic reader that recognizes histone modifications and has been shown to reduce B cell lymphomas via inhibiting super-enhancer-associated transcripts and to treat cardiac failure-induced fibrosis. JQ1 administration to cGVHD mice significantly inhibited BO and collagen deposition. Taken together, these data demonstrate for the first time the critical role of BCL6 and targeting of histones that affect the transcriptional repressive states via EZH2 and a BET bromodomain epigenetic reader. These data also provide a strong foundation for clinical trials of inhibitors that directly or epigenetic modifiers and readers that indirectly target BCL6.
Given our data on TFH produced IL-21 in cGVHD, we tested a Rho-associated kinase 2 (ROCK2) inhibitor, KD025, that blocks IL-21 production [68, 69] and has been shown safe in normal healthy volunteers. KD025 inhibits STAT3 phosphorylation, which supports Th17 generation and IL-21 production, each of which we have linked to cGVHD generation in our model. Concurrently, KD025 increases STAT5 phosphorylation and regulatory T cell (Treg) suppressor function in a dose-responsive fashion. KD025 treatment shifts Th17/Treg balance. KD025 treated cGVHD mice had a dose dependent decrease in the development of pathogenic pulmonary function, which correlated with a marked reduction of antibody deposition in the lungs comparable to non-cGVHD controls along with a decrease in collagen deposition in the lungs and GCs. We demonstrated that mice transplanted with inducible STAT3 deficient T cells or BM cells had pulmonary function comparable to the healthy controls, suggesting that STAT3 is a potential therapeutic target in both T and B cells and is necessary for the development of cGVHD.
Many of the cellular activation and effector functions of Th17 and TFH cells can be attributed to Bruton’s tyrosine kinase (BTK) and IL-2 inducible kinase (ITK) [70, 71] and antibody production by GC B cells depends upon BTK [71]. Ibrutinib is a first-in-class irreversible inhibitor of BTK and ITK that blocks downstream immune receptor activation [72–74]. Since ibrutinib can block the activation of B cells via BTK inhibition as well as specific T-helper subsets that drive the development of cGVHD via ITK inhibition, we hypothesized that it may be ideally suited to the treatment of cGVHD. We show that ibrutinib treatment reversed TFH:GC dependent cGVHD and BOS, associated with blockade of GC formation, reduction of antibody deposition and fibrosis. Additionally, ibrutinib ameliorated the progression of cGVHD in a minor antigen, T cell dependent murine model of sclerodermatous cGVHD, reducing skin lesions, hair loss, and lymphohistiocytic infiltration [75]. These data strongly support the clinical investigation of ibrutinib as a novel therapeutic strategy for the treatment of cGVHD.
Spleen tyrosine kinase (SYK) is activated by BCR engagement. After antigen-BCR engagement, SYK is phosphorylated at Y348, allowing for B cell survival and proliferation. Increased proximal BCR signaling was recently described in cGVHD patient B cells [36]. We now demonstrate that SYK was hyperactive in B cells during cGVHD and that SYK expression was necessary in B but not T cells for murine cGVHD progression. We found that total B cell frequency in mice receiving SYK KO BM was decreased 8-fold, while T cell frequency was unaffected. These data are consistent with the dependency of activated B cells on SYK for proliferation and survival, and a requirement for activated donor-derived BM B cells in cGVHD pathogenesis [6, 53]. Fostamatinib is a potent small molecule inhibitor of SYK. Studies in rheumatoid arthritis have demonstrated efficacy with fostamatinib in randomized phase II clinical trials [76, 77]. Mice receiving fostamatinib for cGVHD treatment had restoration of pulmonary function, similar to the healthy transplanted controls along with a reduced number of GC reactions in the spleen associated with a decrease in frequency of splenic GC B cells. To determine if human cGVHD B cells are more susceptible to SYK blockade, B cells were purified from human peripheral blood were treated in vitro with R406, the active form of R788. B cells from patients with cGVHD had increased apoptosis compared to patients without cGVHD after R406, consistent with work by Allen et al [36] reveal that R406 preferentially kills cGVHD B cells via apoptosis. Together, these data now demonstrate that constitutively activated B cells can be selectively targeted in cGVHD.
In summary, we have developed an array of antibody and drug targeting approaches based upon preventing effective TFH:GC interactions (Table 1). As many of these reagents have been tested in patients, these data lay the foundation for repurposing such reagents to treat steroid-refractory cGVHD in the clinic.
Table 1.
Monoclonal antibodies, drugs and their targets used to treat established cGVHD in mice.
| Agent | Target |
|---|---|
| Anti-IL21 | IL21/IL21R |
| Anti-ICOS | ICOS/ICOSL |
| Anti-CD40L | CD40/CD40L |
| 79-6 | BCL6 peptidomimetic |
| JQ-E | EZH2 |
| JQ-1 | BET bromodomain |
| KD025 | ROCK2 |
| Ibrutinib | BTK, ITK |
| Fostamatinib | SYK |
B cell directed therapy for treatment and prevention of chronic GVHD
Standard immune suppressive therapies for established cGVHD have primarily targeted the T cell compartment. With this approach, responses tend to be incomplete, and the toxicity of long-term administration of corticosteroids is prohibitive. Second-line anti-T cell therapy results in even poorer outcomes. The first anecdotal evidence that targeting B cells could be effective in the treatment of established GVHD was provided by Ratanatharathorn et al, [78] and Miklos et al [4, 79] were the first to associate the formation of specific anti-HY antibodies in a sex-mismatched model with the occurrence of cGVHD (and protection from relapse). Since then, a number of B cell-dependent processes have been implicated in the pathogenesis of cGVHD, and numerous clinical studies have provided evidence that targeting B cells can be effective in the management of and prevention of cGVHD.
Treatment of Corticosteroid-Refractory cGVHD
Several groups have reported the consistent effectiveness of B cell depletion as therapy for established advanced cGVHD [80]. In a review of the published literature on the effectiveness of rituximab for the treatment of corticosteroid-refractory cGVHD, response rates ranging from 43% to 83% were reported with a pooled overall response rate of 66% (95%CI 57%–74%) [81]. Common to all reported studies, rituximab administration in subjects with established cGVHD is relatively safe with a low incidence of adverse events. However, long-term replacement immunoglobulin therapy is often required. There appears to be a predilection for higher response rates in individuals with sclerodermatous or lichenoid cutaneous disease [82], and so a randomized trial comparing rituximab with imatinib, another agent with activity against cutaneous sclerosis, has recently been completed by the Chronic GVHD Consortium.
Initial therapy of cGVHD
Sahaf et al have performed a pilot study of rituximab in combination with corticosteroids as initial therapy of cGVHD [83]. 35 subjects with new-onset cGVHD that required corticosteroid treatment received four weekly infusions of rituximab (375 mg/m2/week) in combination with prednisone (1 mg/kg/day). A second course was allowed for incomplete responses. Response, defined as a complete or partial response with a prednisone dose of <0.25 mg/kg/day, was noted in 40% of subjects at 6 and 12 months, suggesting promising but incomplete activity. While there were 6 deaths from cGVHD within 12 months of rituximab therapy, only 2 subjects suffered a relapse of their malignancy [83]. In this trial, cGVHD responses were predicted by a naïve (CD19+CD38−IgD+CD27−) B cell phenotype (p=0.03). 15 cGVHD subjects included had undergone F→M HSCT, and 6 (38%) were H-Y IgM positive while 9 (56%) were IgG positive at cGVHD diagnosis. Among the 15 F→M HCT recipients with cGVHD treated with rituximab, 11 responded, and both H-Y IgM and IgG became undetectable for at least 6 months following rituximab. In contrast, the 4 F→M subjects who did not respond remained H-Y IgG positive. Moreover, 2 subjects that redeveloped H-Y IgG later had recurrent cGVHD. This suggests that either the allo-reactive B cell or the antibodies elucidated by plasma-cell derivatives from these cells may actually be pathogenic.
Prevention of cGVHD
Given the promising role for B cell depletion as therapy for established cGVHD, we explored the utility of rituximab therapy for primary prevention of cGVHD [57]. In a phase II trial, rituximab (375 mg/m2) was administered at 100 days, 6, 9 and 12 months after HSCT to 65 subjects without evidence of cGVHD at study onset. The cumulative incidence of cGVHD and systemic corticosteroid-requiring cGVHD at 2 years from HCT were 48% and 31% respectively, lower than rates in a concurrent control cohort (60%, p=0.1 and 48.5%, p=0.015, respectively). In related donors, the incidence of cGVHD and corticosteroid-requiring cGVHD at 2 years was 35% and 23%, whereas the corresponding incidence in unrelated donors was 59% and 38%. Rituximab was safe, with no severe infusion-related events and a 2 year cumulative incidence of grade 3–5 infections of 15%. There was no difference in relapse incidence (34% vs. 28%, p=0.79), but treatment-related mortality at 4 years from HSCT was significantly lower in rituximab-treated subjects when compared with controls (5.1% vs. 19.0%, p=0.02), and overall survival was superior at 4 years (71% vs. 56%, p=0.05). In a multivariable regression model, only rituximab use (HR 0.56, 95% CI 0.31–1.00, p=0.048) and high/very high disease risk index (HR 1.90, 95% CI 1.02–3.53, p=0.04) were significantly associated with overall survival. B cell leukopenia was profound and persisted through 18 months from HSCT, when evidence of B cell reconstitution was evident in some subjects. The BAFF/B cell ratio was significantly higher in cGVHD subjects in comparison with cGVHD-free subjects at 2 years (p=0.039). This trial included 13 F→M recipients, none of whom developed anti-DBY antibodies between 6 months and 1 year from HCT, whereas 4 of 10 F→M control recipients developed anti-DBY antibodies in the same time period (p=0.02).
In a similar experience, Arai et al also tested the role of rituximab as primary prevention of cGVHD after HSCT using a TLI/ATG preparative regimen [84]. In small trial of 35 subjects, rituximab (375 mg/m2) was infused weekly on days 56, 63, 70, and 77 after HSCT. The cumulative incidence of cGVHD was 20%, which is slightly lower than the published rates of cGVHD following TLI/ATG based transplantation [85]. In 10 F→M recipients enrolled in this trial, there was complete prevention of allo-reactive H-Y antibody development, and none of these subjects developed cGVHD. In comparison, among 25 F→M HCT patients who used the same TLI/ATG conditioning regimen but without prophylactic rituximab during this time period, 14 of the 25 (56%) developed H-Y antibodies, with 13 of the 25 (52%) developing cGVHD (p=0.01).
Most recently, Glass et al reported the results of a randomized trial in which 42 subjects with NHL were randomized to receive rituximab (375 mg/m2) on 8 separate days between weeks 3 and 28 after transplantation [86]. 36 subjects actually received one or more doses, but in an intention to treat analysis, there was no difference in the incidence of cGVHD (33% in rituximab-treated patients vs. 41% in control patients, p = 0.28). However, in this trial, ATG was used, reducing the global rates of cGVHD, and the relapse rate was high early after transplantation (10 subjects in each group relapsed at a median of 88 days and 90 days in the rituximab and control groups respectfully), making a competing risk cumulative risk analysis less statistically powerful to detect differences.
Earlier attempts to prevent GVHD in single arm studies (generally to prevent acute GVHD, reviewed in Kharfan-Dabaja and Cutler [7]) as well as a recent experience with B cell depletion to prevent EBV reactivation [87] did not demonstrate a marked effect on the incidence of cGVHD, however in the majority of these experiences, B cell depletion was administered early in the peri-transplant period, prior to active B cell reconstitution.
Conclusions and Other Considerations
Anti-B cell therapy has been firmly established as being partially effective in the treatment of established cGVHD and recent studies suggest that anti-B cell therapy may also play a role in the primary prevention of cGVHD. However, control and prevention with anti-CD20 monoclonal antibody, rituximab, is incomplete, and novel strategies are required to improve outcomes and prevent the recurrent cGVHD that can occur with B cell reconstitution. Novel second generation anti-CD20 monoclonal antibodies may provide more effective B cell depletion and elimination of pathogenic allo- and auto-reactive antibodies in vivo. However, as demonstrated in preclinical models, small molecule inhibitors that target the B cell receptor signaling complex may be more effective than rituximab. Clinical studies evaluating this approach will require testing in prospective clinical trials and have recently begun. Additionally, anti-B cell therapy in combination with T cell directed therapies may ultimately be the most effective strategy.
Highlights.
Donor B cells contribute to immune pathology in chronic GVHD
High levels of BAFF after allogeneic HSCT promote the survival of allo- and auto-reactive B cells
Persistent activation of B cell signaling pathways is evident in chronic GVHD
Inhibition of B cell signaling can prevent or reverse tissue injury in murine models of chronic GVHD exhibit
B cell directed therapies can provide new clinical approaches for prevention or treatment of chronic GVHD
Acknowledgments
The authors thank Jing Du, Katelyn Goodman, and Drs Ryan Flynn, Jay Bradner, Jun Qi, Ari Melnick, Raphael Clynes, Alexandra Zanin-Zhorov, Jason Dubovsky, John Byrd and Lawrence Elias for performing experiments, providing reagents and/or helpful discussions. We thank Rigel Pharmaceuticals for providing fostamatinib, Kadmon Pharmaceuticals for KD025 and Pharmacyclics for providing ibrutinib for laboratory studies. Studies described in this review were supported by NIH grants CA142106, K08HL107756 and AI056299, DOD grant CA100254, Kadmon Pharmaceuticals Inc and a translational grant from the Leukemia and Lymphoma Society of America
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pidala J, Sarwal M, Roedder S, Lee SJ. Biologic markers of chronic GVHD. Bone Marrow Transplant. 2014;49:324–31. doi: 10.1038/bmt.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duarte RF, Greinix H, Rabin B, Mitchell SA, Basak G, Wolff D, et al. Uptake and use of recommendations for the diagnosis, severity scoring and management of chronic GVHD: an international survey of the EBMT-NCI Chronic GVHD Task Force. Bone Marrow Transplant. 2014;49:49–54. doi: 10.1038/bmt.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Socie G, Ritz J. Current issues in chronic graft-versus-host disease. Blood. 2014;124:374–84. doi: 10.1182/blood-2014-01-514752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miklos DB, Kim HT, Miller KH, Guo L, Zorn E, Lee SJ, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. 2005;105:2973–8. doi: 10.1182/blood-2004-09-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarantopoulos S, Stevenson KE, Kim HT, Cutler CS, Bhuiya NS, Schowalter M, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. 2009;113:3865–74. doi: 10.1182/blood-2008-09-177840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srinivasan M, Flynn R, Price A, Ranger A, Browning JL, Taylor PA, et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood. 2012;119:1570–80. doi: 10.1182/blood-2011-07-364414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kharfan-Dabaja MA, Cutler CS. Rituximab for prevention and treatment of graft-versus-host disease. Int J Hematol. 2011;93:578–85. doi: 10.1007/s12185-011-0855-2. [DOI] [PubMed] [Google Scholar]
- 8.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 9.Nemazee D. Does immunological tolerance explain the waste in the B-lymphocyte immune system? Experiment and theory. Ann N Y Acad Sci. 1995;764:397–401. doi: 10.1111/j.1749-6632.1995.tb55854.x. [DOI] [PubMed] [Google Scholar]
- 10.Goodnow CC. Balancing immunity and tolerance: deleting and tuning lymphocyte repertoires. Proc Natl Acad Sci U S A. 1996;93:2264–71. doi: 10.1073/pnas.93.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meffre E, Davis E, Schiff C, Cunningham-Rundles C, Ivashkiv LB, Staudt LM, et al. Circulating human B cells that express surrogate light chains and edited receptors. Nat Immunol. 2000;1:207–13. doi: 10.1038/79739. [DOI] [PubMed] [Google Scholar]
- 12.Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201:703–11. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancro MP. Peripheral B-cell maturation: the intersection of selection and homeostasis. Immunol Rev. 2004;197:89–101. doi: 10.1111/j.0105-2896.2004.0099.x. [DOI] [PubMed] [Google Scholar]
- 14.Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–98. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–53. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 16.Tata PV, BGV, P-FK, CJ, JS, SS High-throughput immunoglobulin heavy chain variable region repertoire analysis of CD27+ B cell subsets from patients with chronic GVHD. ASH Annual Meeting Abstracts. 2013;122:2055. [Google Scholar]
- 17.Patriarca F, Skert C, Sperotto A, Zaja F, Falleti E, Mestroni R, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389–96. doi: 10.1016/j.exphem.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood. 2007;110:237–41. doi: 10.1182/blood-2007-01-071043. [DOI] [PubMed] [Google Scholar]
- 19.Kier P, Penner E, Bakos S, Kalhs P, Lechner K, Volc-Platzer B, et al. Autoantibodies in chronic GVHD: high prevalence of antinucleolar antibodies. Bone Marrow Transplant. 1990;6:93–6. [PubMed] [Google Scholar]
- 20.Cyster JG, Hartley SB, Goodnow CC. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 1994;371:389–95. doi: 10.1038/371389a0. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson CA, Sun L, Kim HT, McDonough SM, Reynolds CG, Schowalter M, et al. Post-transplantation B cell activating factor and B cell recovery before onset of chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2014;20:668–75. doi: 10.1016/j.bbmt.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fedoriw Y, Samulski TD, Deal AM, Dunphy CH, Sharf A, Shea TC, et al. Bone marrow B cell precursor number after allogeneic stem cell transplantation and GVHD development. Biol Blood Marrow Transplant. 2012;18:968–73. doi: 10.1016/j.bbmt.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarantopoulos S, Stevenson KE, Kim HT, Bhuiya NS, Cutler CS, Soiffer RJ, et al. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin Cancer Res. 2007;13:6107–14. doi: 10.1158/1078-0432.CCR-07-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storek J, Witherspoon RP, Webb D, Storb R. Lack of B cells precursors in marrow transplant recipients with chronic graft-versus-host disease. Am J Hematol. 1996;52:82–9. doi: 10.1002/(SICI)1096-8652(199606)52:2<82::AID-AJH3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Glauzy S, Soret J, Fournier I, Douay C, Moins-Teisserenc H, Peffault de Latour R, et al. Impact of acute and chronic graft-versus-host disease on human B-cell generation and replication. Blood. 2014;124:2459–62. doi: 10.1182/blood-2014-05-573303. [DOI] [PubMed] [Google Scholar]
- 26.Le Huu D, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, et al. Donor-derived regulatory B cells are important for suppression of murine sclerodermatous chronic graft-versus-host disease. Blood. 2013;121:3274–83. doi: 10.1182/blood-2012-11-465658. [DOI] [PubMed] [Google Scholar]
- 27.Khoder A, Sarvaria A, Alsuliman A, Chew C, Sekine T, Cooper N, et al. Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood. 2014;124:2034–45. doi: 10.1182/blood-2014-04-571125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarantopoulos S, Stevenson KE, Kim HT, Washel WS, Bhuiya NS, Cutler CS, et al. Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood. 2011;117:2275–83. doi: 10.1182/blood-2010-10-307819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen JL, Wooten J, Fore M, Bhuiya NS, Armistead P, Coghill JM, et al. B cells from patients with chronic GVHD signal via the AKT-driven survival and metabolic fitness pathway. BMT Tandem Meeting Abstracts. 2012;18:S208. [Google Scholar]
- 30.Oliver PM, Vass T, Kappler J, Marrack P. Loss of the proapoptotic protein, Bim, breaks B cell anergy. J Exp Med. 2006;203:731–41. doi: 10.1084/jem.20051407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J Exp Med. 2003;198:1119–26. doi: 10.1084/jem.20030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen JL, Fore MS, Wooten J, Roehrs PA, Bhuiya NS, Hoffert T, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood. 2012;120:2529–36. doi: 10.1182/blood-2012-06-438911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192:3626–36. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancro MP. Signalling crosstalk in B cells: managing worth and need. Nat Rev Immunol. 2009;9:657–61. doi: 10.1038/nri2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patke A, Mecklenbrauker I, Erdjument-Bromage H, Tempst P, Tarakhovsky A. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J Exp Med. 2006;203:2551–62. doi: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allen JL, Tata PV, Fore MS, Wooten J, Rudra S, Deal AM, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood. 2014;123:2108–15. doi: 10.1182/blood-2013-10-533562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan JE, Wong SC, Gan SK, Xu S, Lam KP. The adaptor protein BLNK is required for b cell antigen receptor-induced activation of nuclear factor-kappa B and cell cycle entry and survival of B lymphocytes. J Biol Chem. 2001;276:20055–63. doi: 10.1074/jbc.M010800200. [DOI] [PubMed] [Google Scholar]
- 38.Fu L, Lin-Lee YC, Pham LV, Tamayo AT, Yoshimura LC, Ford RJ. BAFF-R promotes cell proliferation and survival through interaction with IKKbeta and NF-kappaB/c-Rel in the nucleus of normal and neoplastic B-lymphoid cells. Blood. 2009;113:4627–36. doi: 10.1182/blood-2008-10-183467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tivol E, Komorowski R, Drobyski WR. Emergent autoimmunity in graft-versus-host disease. Blood. 2005;105:4885–91. doi: 10.1182/blood-2004-12-4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young JS, Wu T, Chen Y, Zhao D, Liu H, Yi T, et al. Donor B cells in transplants augment clonal expansion and survival of pathogenic CD4+ T cells that mediate autoimmune-like chronic graft-versus-host disease. J Immunol. 2012;189:222–33. doi: 10.4049/jimmunol.1200677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao D, Young JS, Chen YH, Shen E, Yi T, Todorov I, et al. Alloimmune response results in expansion of autoreactive donor CD4+ T cells in transplants that can mediate chronic graft-versus-host disease. J Immunol. 2011;186:856–68. doi: 10.4049/jimmunol.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zorn E, Miklos DB, Floyd BH, Mattes-Ritz A, Guo L, Soiffer RJ, et al. Minor histocompatibility antigen DBY elicits a coordinated B and T cell response after allogeneic stem cell transplantation. J Exp Med. 2004;199:1133–42. doi: 10.1084/jem.20031560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porcheray F, Miklos DB, Floyd BH, Sarantopoulos S, Bellucci R, Soiffer RJ, et al. Combined CD4 T-cell and antibody response to human minor histocompatibility antigen DBY after allogeneic stem-cell transplantation. Transplantation. 2011;92:359–65. doi: 10.1097/TP.0b013e3182244cc3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biedermann BC, Sahner S, Gregor M, Tsakiris DA, Jeanneret C, Pober JS, et al. Endothelial injury mediated by cytotoxic T lymphocytes and loss of microvessels in chronic graft versus host disease. Lancet. 2002;359:2078–83. doi: 10.1016/S0140-6736(02)08907-9. [DOI] [PubMed] [Google Scholar]
- 45.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14:573–82. doi: 10.1016/s1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 46.Hon H, Oran A, Brocker T, Jacob J. B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. J Immunol. 2005;174:5233–42. doi: 10.4049/jimmunol.174.9.5233. [DOI] [PubMed] [Google Scholar]
- 47.Tsoi MS, Storb R, Jones E, Weiden PL, Shulman H, Witherspoon R, et al. Deposition of IgM and complement at the dermoepidermal junction in acute and chronic cutaneous graft-vs-host disease in man. J Immunol. 1978;120:1485–92. [PubMed] [Google Scholar]
- 48.Groh V, Li YQ, Cioca D, Hunder NN, Wang W, Riddell SR, et al. Efficient cross-priming of tumor antigen-specific T cells by dendritic cells sensitized with diverse anti-MICA opsonized tumor cells. Proc Natl Acad Sci U S A. 2005;102:6461–6. doi: 10.1073/pnas.0501953102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kita H, Lian ZX, Van de Water J, He XS, Matsumura S, Kaplan M, et al. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–23. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rafiq K, Bergtold A, Clynes R. Immune complex-mediated antigen presentation induces tumor immunity. J Clin Invest. 2002;110:71–9. doi: 10.1172/JCI15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62:234–44. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 52.Weinstein JS, Hernandez SG, Craft J. T cells that promote B-Cell maturation in systemic autoimmunity. Immunol Rev. 2012;247:160–71. doi: 10.1111/j.1600-065X.2012.01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood. 2014;123:3988–98. doi: 10.1182/blood-2014-03-562231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Treml LS, Crowley JE, Cancro MP. BLyS receptor signatures resolve homeostatically independent compartments among naive and antigen-experienced B cells. Semin Immunol. 2006;18:297–304. doi: 10.1016/j.smim.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 55.Brink R. Regulation of B cell self-tolerance by BAFF. Semin Immunol. 2006;18:276–83. doi: 10.1016/j.smim.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 56.Panoskaltsis-Mortari A, Tram KV, Price AP, Wendt CH, Blazar BR. A new murine model for bronchiolitis obliterans post-bone marrow transplant. Am J Respir Crit Care Med. 2007;176:713–23. doi: 10.1164/rccm.200702-335OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cutler C, Kim HT, Bindra B, Sarantopoulos S, Ho VT, Chen YB, et al. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation: results of a phase 2 trial. Blood. 2013;122:1510–7. doi: 10.1182/blood-2013-04-495895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnston HF, Xu Y, Racine JJ, Cassady K, Ni X, Wu T, et al. Administration of anti-CD20 mAb is highly effective in preventing but ineffective in treating chronic graft-versus-host disease while preserving strong graft-versus-leukemia effects. Biol Blood Marrow Transplant. 2014;20:1089–103. doi: 10.1016/j.bbmt.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bunting KL, Melnick AM. New effector functions and regulatory mechanisms of BCL6 in normal and malignant lymphocytes. Curr Opin Immunol. 2013;25:339–46. doi: 10.1016/j.coi.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498:246–50. doi: 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerchietti LC, Yang SN, Shaknovich R, Hatzi K, Polo JM, Chadburn A, et al. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood. 2009;113:3397–405. doi: 10.1182/blood-2008-07-168773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duy C, Yu JJ, Nahar R, Swaminathan S, Kweon SM, Polo JM, et al. BCL6 is critical for the development of a diverse primary B cell repertoire. J Exp Med. 2010;207:1209–21. doi: 10.1084/jem.20091299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–34. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He S, Wang J, Kato K, Xie F, Varambally S, Mineishi S, et al. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood. 2012;119:1274–82. doi: 10.1182/blood-2011-06-364422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. 2012;247:172–83. doi: 10.1111/j.1600-065X.2012.01112.x. [DOI] [PubMed] [Google Scholar]
- 67.Ci W, Polo JM, Cerchietti L, Shaknovich R, Wang L, Yang SN, et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood. 2009;113:5536–48. doi: 10.1182/blood-2008-12-193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin. Blood Coagul Fibrinolysis. 2008;19:709–18. doi: 10.1097/MBC.0b013e32830b2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zanin-Zhorov A, Mo R, Scher J, Nyudzefe M, Depoli D, Koslow M, et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells. 2013 doi: 10.1073/pnas.1414189111. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol. 2005;23:549–600. doi: 10.1146/annurev.immunol.22.012703.104743. [DOI] [PubMed] [Google Scholar]
- 71.Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–7. [PubMed] [Google Scholar]
- 72.Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, et al. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011;13:R115. doi: 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1 selective pressure in T-lymphocytes. Blood. 2013;122:2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hamilton BL, Parkman R. Acute and chronic graft-versus-host disease induced by minor histocompatibility antigens in mice. Transplantation. 1983;36:150–5. doi: 10.1097/00007890-198308000-00008. [DOI] [PubMed] [Google Scholar]
- 76.Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, et al. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three-month randomized, placebo-controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum. 2011;63:337–45. doi: 10.1002/art.30114. [DOI] [PubMed] [Google Scholar]
- 77.Weinblatt ME, Kavanaugh A, Burgos-Vargas R, Dikranian AH, Medrano-Ramirez G, Morales-Torres JL, et al. Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis Rheum. 2008;58:3309–18. doi: 10.1002/art.23992. [DOI] [PubMed] [Google Scholar]
- 78.Ratanatharathorn V, Carson E, Reynolds C, Ayash LJ, Levine J, Yanik G, et al. Anti-CD20 chimeric monoclonal antibody treatment of refractory immune-mediated thrombocytopenia in a patient with chronic graft-versus-host disease. Ann Intern Med. 2000;133:275–9. doi: 10.7326/0003-4819-133-4-200008150-00011. [DOI] [PubMed] [Google Scholar]
- 79.Miklos DB, Kim HT, Zorn E, Hochberg EP, Guo L, Mattes-Ritz A, et al. Antibody response to DBY minor histocompatibility antigen is induced after allogeneic stem cell transplantation and in healthy female donors. Blood. 2004;103:353–9. doi: 10.1182/blood-2003-03-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cutler C, Miklos D, Kim HT, Treister N, Woo SB, Bienfang D, et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood. 2006;108:756–62. doi: 10.1182/blood-2006-01-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kharfan-Dabaja MA, Mhaskar AR, Djulbegovic B, Cutler C, Mohty M, Kumar A. Efficacy of rituximab in the setting of steroid-refractory chronic graft-versus-host disease: a systematic review and meta-analysis. Biol Blood Marrow Transplant. 2009;15:1005–13. doi: 10.1016/j.bbmt.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 82.Okamoto M, Okano A, Akamatsu S, Ashihara E, Inaba T, Takenaka H, et al. Rituximab is effective for steroid-refractory sclerodermatous chronic graft-versus-host disease. Leukemia. 2006;20:172–3. doi: 10.1038/sj.leu.2403996. [DOI] [PubMed] [Google Scholar]
- 83.Sahaf B, Arai S, Otani J, Schoenrock K, Logan A, Miklos DB. Rituximab provides steroid-sparing therapy in new-onset chronic graft-versus-host disease. BMT Tandem Meeting Abstracts. 2013;19:S140. [Google Scholar]
- 84.Arai S, Sahaf B, Narasimhan B, Chen GL, Jones CD, Lowsky R, et al. Prophylactic rituximab after allogeneic transplantation decreases B-cell alloimmunity with low chronic GVHD incidence. Blood. 2012;119:6145–54. doi: 10.1182/blood-2011-12-395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kohrt HE, Turnbull BB, Heydari K, Shizuru JA, Laport GG, Miklos DB, et al. TLI and ATG conditioning with low risk of graft-versus-host disease retains antitumor reactions after allogeneic hematopoietic cell transplantation from related and unrelated donors. Blood. 2009;114:1099–109. doi: 10.1182/blood-2009-03-211441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Glass B, Hasenkamp J, Gorlitz A, Dreger P, Schubert J, Wulf G, et al. Rituximab for graft-versus-host-disease prophylaxis after allogeneic stem cell transplantation given as treatment of high risk relapse of aggressive lymphoma: results of a randomized phase II study. ASH Annual Meeting Abstracts. 2008;112:1974. [Google Scholar]
- 87.Dominietto A, Tedone E, Soracco M, Bruno B, Raiola AM, Van Lint MT, et al. In vivo B-cell depletion with rituximab for alternative donor hemopoietic SCT. Bone Marrow Transplant. 2012;47:101–6. doi: 10.1038/bmt.2011.28. [DOI] [PubMed] [Google Scholar]