

Abstract
Type 1 diabetes (T1D) is a devastating disease precipitated by an autoimmune response directed at the insulin-producing beta-cells of the pancreas for which no cure exists. Stem cell-derived beta-cells show great promise for a cure as they have the potential to supply unlimited numbers of cells that could be derived from a patient's own cells, thus eliminating the need for immunosuppression. Current in vitro protocols for the differentiation of stem cell-derived beta-cells can successfully generate pancreatic endoderm cells. In diabetic rodents, such cells can differentiate further along the beta-cell lineage until they are eventually capable of restoring normoglycemia. While these observations demonstrate that stem cell-derived pancreatic endoderm has the potential to differentiate into mature, glucose-responsive beta-cells, the signals that direct differentiation and maturation from pancreatic endoderm onwards remain poorly understood. In this review, we analyze the sequence of events that culminates in the formation of beta-cells during embryonic development. and summarize how current protocols to generate beta-cells have sought to capitalize on this ontogenic template. We place particular emphasis on the current challenges and opportunities which occur in the later stages of beta-cell differentiation and maturation of transplantable stem cell-derived beta-cells. Another focus is on the question how the use of recently identified maturation markers such as urocortin 3 can be instrumental in guiding these efforts.
Keywords: type 1 diabetes, insulin, stem cell, pancreatic beta-cell, pancreas development, transplantation, urocortin 3, UCN3, CRF receptor, CRH receptor
Abbreviations: ARX - aristaless-related homeobox; Brn4 - brain 4; CHGB - chromogranin-B; CRF - corticotropin releasing factor; CRFR2 - corticotropin-releasing factor receptor 2; CRH - corticotropin releasing hormone; CX36 - connexin36; CXCR4 - chemokine (C-X-C motif) receptor 4; DAPI - 4',6-diamidino-2-phenylindole; DE - definitive endoderm; E - embryonic day; EMT - epithelial-to-mesenchymal transition; ESC - embryonic stem cell; FACS - fluorescence-activated cell sorting; FGF4 - fibroblast growth factor 4; FOXA2 - forkhead box A2; GCG - glucagon; GLP1 - glucagon-like peptid 1; GLUT1, 2 - glucose transporters 1, 2; GSIS - glucose stimulated-insulin secretion; GSK3-beta - glycogen synthase kinase 3 beta; HEPES - N-(2-Hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid); hESC - human embryonic stem cell; HLXB9 - homeobox HB9; HNF1B - hepatocyte nuclear factor 1 beta; IDE - inducer of definitive endoderm; iPS - induced pluripotent stem; Irx - Iroquois homeobox transcription factor; ISLET1 - insulin gene enhancer protein 1 (aka ISL1); KIR6.2 - inwardly rectifying potassium channel 6.2; MAFB - v-maf musculoaponeurotic fibrosarcoma oncogene homolog A; MMTV - mouse mammary tumor virus; NEUROD1 - neuronal differentiation 1; NGN3 - neurogenin 3; NKX2.2 - NK2 homeobox 2; NKX6.1 - NK6 homeobox 1; Oct-4 - octamer binding transcription factor 4; PAX4 - paired box 4; PC1/3 - proprotein convertase 1; PCR - polymerase chain reaction; PCSK1, 2 - proprotein convertase subtilisin/kexin types 1, 2; PDX1 - pancreatic and duodenal homeobox 1; PE - pancreatic endoderm; POU - Pit-Oct-Unc; POU3F4 - POU class 3 homeobox 4; PP - pancreatic polypeptide; PTF1A - pancreas-specific transcription factor 1a; Sox17 - sex-determining region Y (Sry) box 17; SST - somatostatin; SUR1 - sulfonylurea receptor 1; T1D - type 1 diabetes; TDGF1 - teratocarcinoma-derived growth factor 1; TGF-beta - transforming growth factor beta; TPB - ((2S,5S)-(E,E)-8-(5-(4-(trifluoromethyl) phenyl)-2,4-pentadienoylamino) benzolactam; UCN3 - urocortin 3; Wnt3a - wingless-type MMTV integration site family member 3a; ZNT8 - zinc transporter 8
1. Introduction
Type 1 diabetes (T1D) affects an estimated 3 million people in the US alone. The incidence of the disease is on the rise, while the average age of onset is decreasing [1, 2]. T1D was invariably lethal until the landmark discovery of insulin by Banting and Best [3]. From the early twenties, insulin was widely available to T1D patients, which starkly improved clinical outcomes. Despite considerable progress in the treatment and management of the disease in the decades that followed, insulin remains the standard of care for T1D as a cure has proven elusive.
Strategies designed to cure T1D often involve a two-pronged approach in which the ongoing immune attack is suppressed, followed by the restoration of beta-cell mass. A diverse array of strategies to restore functional beta-cell mass have been investigated. These include the replication of existing beta-cells, the activation of beta-cell neogenesis from endogenous pancreatic progenitors, the transplantation of islet or pancreas tissue of either human or animal origin, and the regeneration and transplantation of beta-cells from stem cells. Human pancreatic beta-cell replication peters out after the neonatal expansion of beta-cell mass [4, 5], and unlike in mice, does not increase under obese or pregnant conditions [6, 7].
To date, the difficulties in promoting human beta-cell proliferation have prevented its use as a feasible strategy to restore beta-cell mass in T1D patients, although recent work shows promise in promoting human beta-cell proliferation by targeting cyclins [8]. Several non-beta-cell types within the pancreas (duct, acinar, and alpha cells) and outside the pancreas (hepatocytes) have been demonstrated to possess the capacity to differentiate into beta-cells under different experimental conditions and/or following genetic manipulations in mice [9]. While these strategies hold promise, the translation of neogenesis into the clinic has not been made [10, 11]. With respect to transplantation, a clinical trial by Living Cell TechnologiesTM using encapsulated porcine islets in human subjects is underway [12]. These islets have already shown longevity potential in humans. Human pancreas [13] and islet transplantations [14] result in varying degrees of insulin independence, with 5-yr independence rates >50% [15, 16]. Nevertheless, the application of human tissue transplantation approaches to a large majority of T1D patients is hampered by the limiting supply of donor tissue [17]. In addition, in the absence of perfect histocompatibility, life-long immune suppression of islet transplant recipients is necessary.
Stem cell-based therapy has gained momentum as a viable alternative, with the potential to overcome or circumvent many of the drawbacks associated with transplantation, and other approaches that aim to harness any endogenous regenerative potential of the pancreas. Potentially, stem cells derived from induced pluripotent stem (iPS) cells obtained from the individual patient, could supply unlimited numbers of cells for transplantation. For these reasons, the generation of stem cell-derived beta-cells has been the subject of intense study in the last decade.
Efforts to generate beta-cells from stem cells have come a long way since the first reports of insulin-positive stem cell-derived cells in the early 2000s [18-21]. These initial attempts employed a variety of approaches for the differentiation of beta-cells. However, after publication of a landmark study by D'Amour and co-workers [22], the focus shifted toward systematically recapitulating in the dish, the developmental sequence of events necessary to generate beta-cells in vivo, by means of a regimen of successive treatments with different media and growth factors. Beta-cell precursor cells generated according to such protocols can be transplanted, proceed to mature in vivo, and cure diabetes in pre-clinical T1D models. While these results are promising, the challenge of generating glucose-responsive, functional beta-cells in vitro capable of maintaining glucose homeostasis without the requirement for exogenous insulin administration has not yet been met.
Here, we review the key sequence of events required for proper pancreas formation during embryonic development. We emphasize the gene expression patterns marking different stages in development. Then, using this ontogenic template, we discuss the significant progress that the stem cell field has made towards the generation of functional beta-cells in a dish since the first reports of in vitro-derived insulin-positive cells. Finally, we discuss some of the major challenges to be overcome before we can generate potentially unlimited pools of mature beta-cells, with the goal of curing T1D.
2. Pancreas ontogeny
The pancreas is a glandular organ whose endocrine cells are organized in small clusters, the islets. The two main endocrine cell types that make up the islets are the insulin-producing beta-cells and the glucagon-producing alpha-cells, whose tightly coordinated and opposing actions are essential for the maintenance of normoglycemia. The islets contain several smaller populations of endocrine cells, including somatostatin-producing delta-cells, ghrelin-producing epsilon-cells, and pancreatic polypeptide (PP) cells. The islets of Langerhans are located amidst a large mass of exocrine cells, grouped in terminal acini that secrete digestive enzymes into a branched pancreatic ductal tree opening onto the duodenum.
The pancreatic anlage first appears around E9, and is derived principally from definitive endoderm, which arises through the actions of nodal. The latter is a transforming growth factor beta (TGF-beta) family member, and wingless-type mouse mammary tumor virus (MMTV) integration site family member 3a (WNT3a) via a mesoderm intermediate [23-25]. Definitive endoderm (DE) is marked by the combined expression of chemokine (C-X-C motif) receptor 4 (CXCR4) [26], forkhead box A2 (FOXA2) [27], SRY (sex determining region Y)-box 17 (SOX17), and the absence of SOX7 [28]. DE next forms the primitive gut tube. The pancreas forms from Cerberus [29], hepatocyte nuclear factor 1 beta (HNF1B), and HNF4-expressing foregut endoderm [30, 31]. Intermediate levels of fibroblast growth factor 4 (FGF4) signaling effectively induce pancreas specification in the posterior foregut, as defined by the expression of pancreatic and duodenal homeobox 1 (PDX1) [32], which is the earliest marker of the dorsal and ventral anlagen of the pancreas in the posterior foregut [33]. While PDX1 is essential for pancreas development [34, 35], some early hormone-positive cells do develop, and the initial budding out of the two pancreatic anlagen still occurs in the absence of PDX1 [35].
Subsequent development of the pancreas is often characterized as consisting of three transitions in which the pancreas undergoes rapid changes in gross and ultrastructural morphology, with changes in enzyme and hormone production [36, 37]. During primary transition (~E8.5-E10.5 in the mouse), the pancreas is defined as an organ, and several pancreas-specific proteins are present in low amounts at the end of the transition. The first wave of endocrine cells is formed and consists of glucagon-positive cells, followed by some early insulin-positive cells. The secondary transition (~E13.5-E16.5) is marked by the generation of cells with high levels of pancreas-specific proteins and little proliferation [38]. The third transition encompasses the perinatal period when islets form and endocrine cells further mature into adult type cells that release hormones to regulate blood glucose in response to appropriate metabolic cues [37].
2.1 Primary transition
Following the expression of PDX1 in the pancreatic anlage, the basic helix-loop-helix protein pancreas-specific transcription factor 1a (PTF1A) becomes expressed. While it was discovered as an activator of acinar cell-specific genes [39], at this stage it helps define exocrine, endocrine, and ductal cell types within the pancreas by binding to and activating the PDX1 promoter [40]. Shortly after the specification of the pancreatic buds, further differentiation into pancreatic endoderm (PE) and subsequent endocrine cells is initiated by the expression of NK2 homeobox 2 (NKX2.2) [41] and NK6 homeobox 1 (NKX6.1) [42]. An early function of NKX6.1 is to specify endocrine identity by repressing the acinar cell specifying activity of PTF1A [43].
At this time of PE development, the basic helix-loop-helix transcription factor neurogenin 3 (NGN3) becomes expressed. Its expression defines the endocrine niche within the PE (reviewed by Rukstalis and Habener [44]), concluded by the observation that NGN3-deficient mice completely lack endocrine cells [45]. NGN3 induces the expression of several transcription factors that locate to the islet cells within the pancreas. These transcription factors are important for development of some or even all lineages of endocrine cells. Downstream targets of NGN3, important for proper endocrine cell formation, include insulin gene enhancer protein (ISLET1) (all islet cells) [46], paired box 4 (PAX4) (beta- and delta-cells) [47], neuronal differentiation 1 (NEUROD1) (beta-cells) [48], paired box 6 (PAX6) (alpha-cells) [49], and aristaless-related homeobox (ARX) (alpha-cells) [45, 50].
2.2 Development of alpha-cells
Towards the end of the primary transition, NGN3 expression is temporarily reduced [51]. The preceding early wave of NGN3 expression mostly results in alpha-cell differentiation, notwithstanding the generation of a few insulin-positive cells [52]. The early glucagon-positive alpha-cells can be seen in the dorsal bud at E9.5 and slightly later in the ventral bud; they are the first endocrine cells to develop. In addition to their signature hormone glucagon, alpha-cells express ARX, FOXA1, FOXA2, PAX6, POU class 3 homeobox 4 (POU3F4 or BRN4), ISLET1, V-Maf musculoaponeurotic fibrosarcoma oncogene homolog B (MAFB), and proprotein convertase subtilisin/kexin type 1 (PCSK2) (reviewed by Bramswig and Kaestner [53]). ARX maintains the apha-cell phenotype, and drives beta-cells towards an alpha-cell fate, if misexpressed in beta-cells [54]. As the alpha-cells mature, they lose PDX1, NKX6.1, and PCSK1 expression [55-59], which continue to mark beta-cells in the adult (Figure 1).
Figure 1. UCN3 expression as a late stage maturation marker for human alpha- and beta-cells.
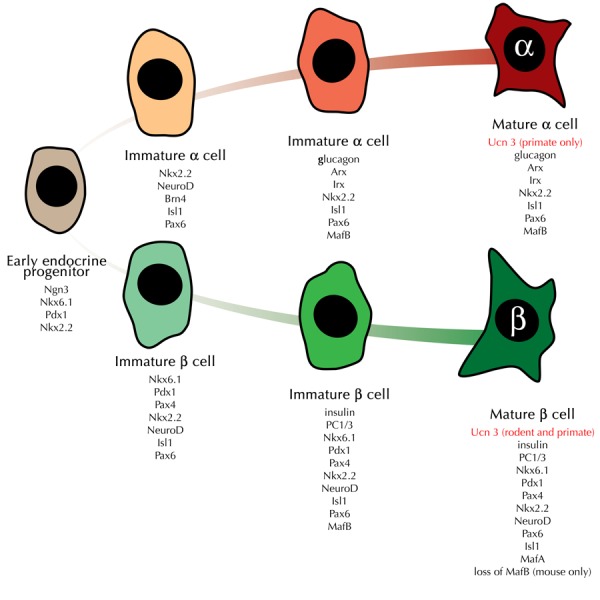
The figure provides a general overview of the (dis)appearance of notable markers that are expressed at key intermediate stages from early endocrine progenitor cells towards the alpha- and beta-cell lineage. UCN3 expression in beta-cells appears later than any of the other markers listed here, with the possible exception of the loss of MafB expression in mouse beta-cells. Note that the appearance of UCN3 in mature alpha-cells is unique for primate alpha-cells and does not occur in rodents islets.
2.3 Development of insulin- and glucagon-co-expressing cells
In mice, early insulin-positive cells appear around E10.5 [60-62]. These cells often co-express glucagon [38, 61]. Also, they may be found in the developing human pancreas, where more than one quarter of the total insulin- and glucagon-positive cell population co-expresses both hormones early in development [63]. These double-positive cells share some of the developmental pathways for both alpha- and beta-cells. They are absent in NKX2.2-deficient [41], NGN3-deficient [45], and MAFB-deficient mice [64]. They lack PDX1 and NKX6.1 expression, which are classical beta-cell markers [58, 59, 63], and indeed persist in PDX1-deficient [58] and NKX6.1-deficient mice [42].
It has been proposed that the double-hormone-positive cells do not contribute to mature islet cells [65]. However, while insulin- and glucagon-double-positive cells disappear as such after E14.5 [55, 61], the possibility that they contribute to the adult alpha-cell pool after losing insulin expression, as suggested by stem cell differentiation and transplantation experiments, cannot be excluded [66, 67].
2.4 Secondary transition
At E12.5, the dorsal and ventral pancreatic primordia fuse to become a single organ. During the secondary transition (E13.5-E16.5), the pancreas undergoes rapid growth and branching. A second wave of NGN3-positive cells occurs, which differentiates into a second wave of endocrine cells [44]. NGN3-positive cells that appear towards the end of the first wave (E10.5), and during the second wave increasingly acquire the competence to generate beta-cells and PP cells, in addition to alpha-cells. NGN3-positive cells can also differentiate into somatostatin-producing delta-cells which develop even later, after E12.5 [52]. The contribution of NGN3-positive cells towards the alpha-cell lineage is greatly reduced in cells appearing after E14.5.
2.5 Development of insulin-single-positive beta-cells
Beta-cells, generated from the second wave of NGN3-positive cells, populate the mature pancreas, and in common with alpha-cells express NKX2.2, ISLET1, PAX6, and MAFB. The expression of homeobox HB9 (HLXB9), PDX1, and NKX6.1 is retained in beta-cells, while PAX4, NEUROD, and ISLET1 are expressed in beta-cells downstream of NGN3 [42, 45, 55, 68-70] (Figure 1). In fact, PAX4 and the alpha-cell transcription factor ARX repress each other’s expression, suggesting they help to establish stable beta- vs. alpha-cell identity [50].
At this stage, NKX6.1 plays a key role as it is both necessary and sufficient to specify insulin-producing beta-cells by repressing alternative endocrine cell fates after the onset of NGN3 expression [71]. The expression of the basic-leucine zipper transcription factor MAFB in beta-cells precedes a surge in PDX1 expression [72], which in turn precedes insulin expression [55, 70]. Following the onset of insulin expression, beta-cells initiate the expression of the basic-leucine zipper transcription factor MAFA [72]. Both MAFA and MAFB induce transcription of insulin. In MAFB-deficient mice, insulin expression is greatly reduced and delayed until the onset of MAFA expression [64].
2.6 Tertiary transition - late prenatal period (E16.5-parturition)
By the end of the secondary transition, the major pancreatic islet hormone-containing cells are present as single hormone-positive cells. During the tertiary transition, typical islet organization is achieved, with the individual endocrine cells forming discrete clusters within the exocrine cell mass. In mice, islets consist of a core of insulin-containing beta-cells surrounded by a mantle of mostly glucagon-containing alpha-cells, in combination with somatostatin-containing delta-cells and PP cells (Figure 2). In humans, the different cell types are more intermingled throughout the islet (Figure 2).
Figure 2. Expression of UCN3 in mouse and human islets.
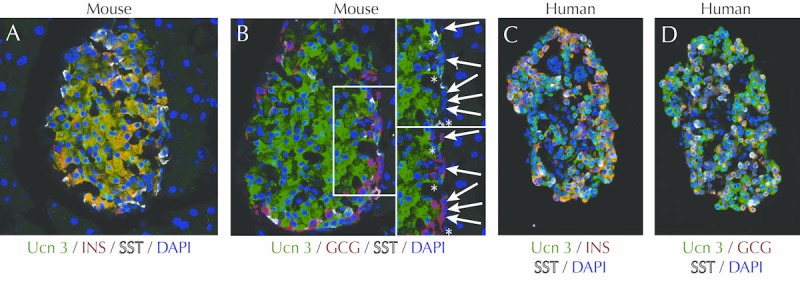
In mouse islets, UCN3 (labeled in green) is expressed exclusively in beta-cells labeled in red for insulin (A). UCN3 is not expressed in mouse alpha-cells (arrows) or delta-cells (asterisks), labeled for glucagon in red and somatostatin in white, respectively (B). In contrast, UCN3 in human islets is expressed by both beta- and alpha-cells, as indicated by its co-localization with insulin (C) and glucagon (D), both in red. These panels were reprinted from [56] under the terms of the Creative Commons Attribution License (CCAL), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The tertiary transition entails the maturation of pancreatic cells to achieve regulated synthesis and secretion of pancreas-specific proteins in response to feeding [37]. While insulin is the sine qua non of the beta-cell, insulin expression alone does not suffice to convey mature beta cell identity. Additional traits are required to transform a mere insulin-expressing cell into a mature, functional beta-cell capable of responding appropriately to changes in ambient glucose levels by starting or arresting insulin exocytosis. These traits include the ability to engage in the following actions:
1. Glucose-sensing (requiring glucokinase and glucose transporters (GLUT2 in mice, GLUT1 in humans))
2. Cell excitability (sulfonylurea receptor 1 (SUR1), inwardly rectifying potassium channel 6.2 (KIR6.2), and others)
3. Beta-cell coordination (e.g. the gap junction protein connexin36 (CX36))
4. Insulin processing (PCSK1 and PCSK2)
5. Packaging (zinc transporter 8 (ZNT8))
6. Secretion (chromogranin-B (CHGB), urocortin 3 (UCN3))
A network of transcription factors underlie the regulation of a number of genes required for these functional traits, including NEUROD, ISLET1, NKX6.1, PAX4, and MAFA, which are all expressed in the beta-cell lineage. These factors have been demonstrated as necessary for beta-cell development and/or function [42, 46-48, 73-75]. The genes listed here provide merely a small sample. Substantially more proteins are required for, or contribute to, these functional traits that collectively define mature beta-cell identity [76]. Expression of many of these genes, and the proteins they encode, starts well before birth to prepare the beta-cells for the independent regulation of glucose homeostasis that follows parturition (Figure 3).
Figure 3. A beta-cell-centric view of the onset of expression of key genes involved in beta-cell development and maturation.
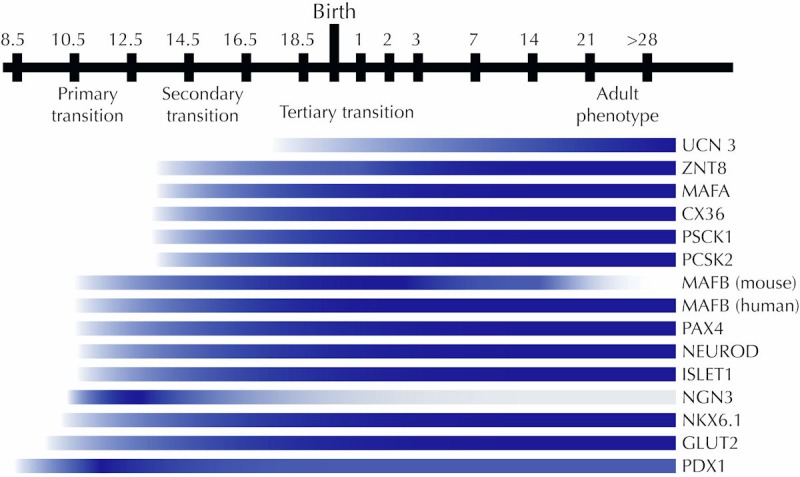
Many of the genes required for normal beta-cell function are already expressed prior to the tertiary transition in which beta-cells mature and acquire glucose responsiveness. Initial expression for UCN3 [56, 77], ZNT8 [126], MAFA [73], Cx36 [127], PCSK1 and PCSK2 [128], MAFB [129], PAX4 [47], NEUROD [48], Islet1 [46], NGN3 [52], NKX6.1 [42], GLUT2 [130], and Pdx1 [58] is shown.
2.7 Postnatal period
The postnatal maturation period is characterized by substantial physiological transitions that shift demand on glucose metabolism and consequently the regulation of beta-cell output. Newborns are no longer able to rely on maternal regulation of blood glucose. The newborn beta-cells have to adapt to maintaining glucose homeostasis from the moment of parturition onwards. Meal quality and pattern also shift over the course of the first weeks of life, with initially frequent intake of mother’s milk, and subsequently gradual supplementation with increasing amounts of solid food at more discrete diurnal intervals. These changes probably necessitate gradual maturation of beta-cell function until glucose sensitivity and insulin output, required for the maintenance of normoglycemia in the adult, have been achieved. These changes include an elevation of the glucose threshold required for full glucose-stimulated insulin secretion [77], and thus an elevation of blood glucose levels around birth [77-79]. In mice, the gradual perinatal increase in blood glucose indeed correlates with a drop in blood insulin levels [79]. Therefore, beta-cells can be considered mature when they are able to maintain blood glucose at normal levels in response to intermittent feeding and fasting, a state which is gradually acquired and only fully achieved after weaning [76, 77].
While it is evident that a full complement of genes is required to direct proper maturation of the beta-cell [76], our understanding of the sequence of events that takes place in the initial days and weeks following birth is incomplete. The expression of the transcription factors MAFA and MAFB changes markedly during the early postnatal period: MAFA expression is activated in most beta-cells perinatally, followed by the gradual loss of MAFB expression in the first postnatal weeks [80]. This shift from MAFB to MAFA expression is considered a defining transition for beta-cell maturation in mice [80], as MAFA is an important transcription factor for the generation and maintenance of mature beta-cell phenotype and normal islet architecture [74, 75]. Enhanced postnatal expression of MAFA plays a crucial role in the acquisition of glucose-responsive insulin secretion [75]. In an interesting example of specie differences between mouse and human islets, MAFB expression is not lost during human beta-cell maturation [81]. While expression of many genes combined is required for maturity, individual genes whose expression marks mature beta-cells can facilitate their recognition, both in vivo and in vitro. One prime example of such a gene is UCN3, which was recently described as marking mature rodent and human primary and embryonic stem (ES) cell-derived beta-cells [56, 77].
2.8 UCN3 as an islet cell maturation marker
UCN3 is a peptide hormone of the corticotropin-releasing hormone family. It is expressed abundantly and exclusively in mouse beta-cells [82] (Figure 2). Recently, it was reported to be expressed relatively late in the beta-cell lineage during mouse development [55, 77]. In an effort to identify traits that distinguish mature beta-cells from their immature counterparts in early postnatal life, the Melton group conducted a microarray experiment comparing gene expression in islets isolated at p10 and p1 [77]. The select number of genes was significantly increased in expression in the more mature (p10) islets included UCN3 [77]. Also, we have recently reported on the expression of UCN3 during mouse embryonic development; we found sporadic UCN3 in beta-cells from E17.5 onwards. Nevertheless, it takes two weeks post parturition before all beta-cells have acquired UCN3 expression [56]. UCN3 expression follows the onset of MAFA expression in most beta-cells, and precedes the postnatal loss of MAFB in mouse beta-cells (Figure 4).
Figure 4. UCN3 expression follows the onset of MafA and MafB expression in mouse development.
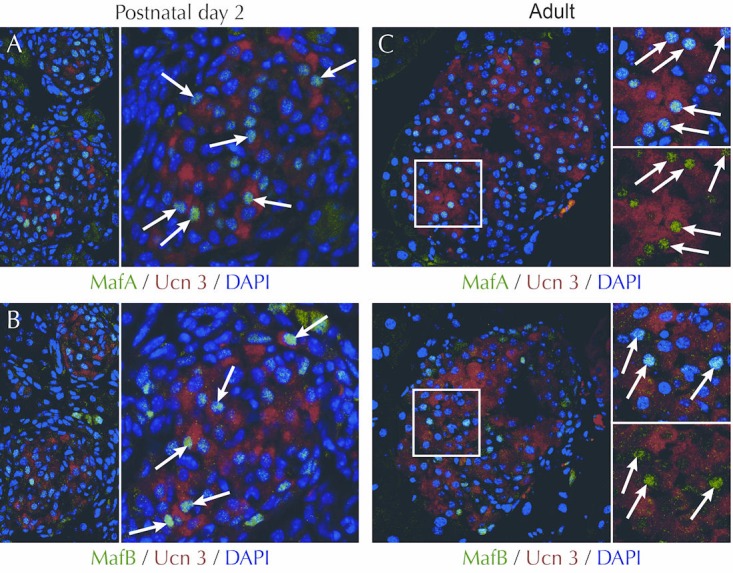
UCN3 expression overlaps with the expression of MafA (A) and MafB (B) at postnatal day 2 (indicated by arrows), when mouse beta-cells express both MafA and MafB. Note that MafB is also expressed in a number of UCN3-negative cells located at the periphery of the islet, indicated by asterisks. These cells are possibly alpha-cells, which retain MafB expression. In adult mouse islets, UCN3 and MafA expression in beta-cells continue to overlap (C, arrows), while MafB expression has been lost from beta-cells, and now labels only a UCN3-negative population of presumptive alpha-cells (D, asterisks). UCN3 was stained by a guinea pig anti-UCN3 antiserum (#44) developed in house. MafA and MafB were stained by rabbit anti-MafA and rabbit anti-MafB (both Bethyl Labs), using standard antigen retrieval, as described in detail in [56].
These observations further establish UCN3 as a late marker for beta-cell maturation, and suggest that UCN3 can be used to monitor beta-cell maturation from human embryonic stem cells (hESCs). Indeed, the onset of UCN3 expression in human pancreas occurs towards the end of the first trimester, and appears notably later than the expression of insulin and glucagon (Figure 5). Interestingly, the expression pattern of UCN3 differs considerably between rodent and primate islets. While mouse UCN3 is restricted to beta-cells [56, 82], its expression in macaque (Macaca nemestrina) and human islets extends to the alpha-cell [56] (Figures 1 and 2).
Figure 5. UCN3 expression follows the onset of insulin and glucagon expression in human pancreas development.

At 55 days post coitus (dpc) numerous cells positive for insulin (red) and glucagon (white) can be readily observed, while there is only faint UCN3 immunoreactivity (A, green). Note the insulin/glucagon co-positive bi-hormonal cell in the circle. At 76 dpc (B) and 94 pdc (C), UCN3 is evidently expressed, and co-localizes with beta-cells (arrows) labeled for insulin (red) and with alpha-cells (asterisks) labeled for glucagon (white). While beta-cells, which do not yet co-express UCN3, can be observed at each of these three developmental stages, their frequency appears to go down as development progresses. Antisera and methods are as previously described in detail in [56]. Human fetal pancreas tissue sections were generously provided by Dr. Ruiyu Xie of the University of California, San Diego, who obtained them from the Birth Defects Research Laboratory of the University of Washington. The use of this material was certified as exempt from IRB approval by UCSD.
2.9 The physiological role of UCN3 in the islet
The physiological function of UCN3, and how it correlates to beta-cell maturity, is not completely understood. UCN3 secretion from MIN6 insulinoma cells is stimulated by glucose, and is blocked by the potassium channel activator diazoxide [82]. This suggests that the mechanism of UCN3 release is similar to that of insulin, and that it requires the closure of Kir6.2 potassium channels to enable the opening of calcium channels triggering exocytosis. Indeed, increasing concentrations of blocking antiserum or peptide antagonist to the cognate receptor of UCN3 in primary rodent islets suppresses insulin secretion stimulated by high glucose or the glucagon-like peptide receptor 1 (GLP-1 receptor) agonist exendin-4. This suggests that UCN3 is required for full glucose- and incretin-stimulated insulin secretion [82, 83]. The fact that blockade of UCN3 actions in vitro in isolated islets inhibits insulin secretion suggests that UCN3 acts in an islet-autonomous fashion. Indeed, we demonstrated the expression of the corticotropin-releasing factor receptor 2 (CRFR2, the cognate receptor for UCN3) in MIN6 insulinoma cells, and in both mouse and human islets [84]. Despite the contribution of endogenous UCN3 to insulin secretion, mice null for UCN3 are normal and healthy. They even appear to retain glucose tolerance in high-fat diet and aging paradigms of insulin resistance [83]. Islets of UCN3-null mice appear normal upon gross histological observation beyond the lack of UCN3 expression [56]. UCN3-null islets display normal basal insulin secretion, and are deficient in glucose-stimulated insulin secretion following stimulation with high (16.8 mM), but not intermediate (8.4 mM), glucose concentrations. This is in line with the effects of acute blockade of endogenous UCN3 actions [83].
Collectively, these observations indicate that UCN3 promotes glucose-stimulated insulin secretion in an islet-autonomous fashion. Also, they suggest that the constitutive absence of UCN3 does not prevent the development of (or can be compensated for to achieve) relatively normal beta-cell function across a range of low to intermediate glucose concentrations. It will be interesting to investigate the physiological contribution of UCN3 to the endocrine output of human islets, where the expression of UCN3 in alpha- and beta-cells may foreshadow a somewhat different physiological role for UCN3 in the regulation of insulin secretion.
3. Stem cells: unlimited potential and practical limitations
Following the overview of beta-cell generation during embryonic development, we now review efforts to regenerate mature beta-cells from stem cells, and how current protocols attempt to recapitulate the ontogenic template described above and summarized in Figure 1. We will discuss some of the challenges towards the ultimate goal of generating mature, glucose-responsive beta-cells in a dish. Over the last decade, stem cells as a source of transplantable beta-cells have become a major focus of research efforts oriented at a cure for diabetes. Stem cells could potentially supply unlimited numbers of cells for transplantation, thereby overcoming the shortage of donor material. iPS cells hold the promise of regenerating beta-cells from a patient's own cells, thus eliminating potential histocompatibility issues.
Despite their potential, stem cell-derived islet cells currently present several limitations. Traditionally, embryonic stem cells are derived from the inner cell mass of surplus embryos generated for in vitro fertilization purposes. Their use continues to be associated with ethical concerns. Moreover, transplantation of cells or tissues derived from ES cells would still require immune suppression similar to immunosuppression required following donor islet or pancreas transplantations. Alternatively, parthenogenetic stem cells, taken from blastocysts that result from activated but unfertilized oocytes [85], carry fewer of the immunogenic epitopes that regular stem cells, thereby decreasing the chance that they are histo-incompatible to transplant recipients.
A patient’s own somatic cells (e.g. skin cells) can be used for the induction of pluripotency prior to their differentiation into beta-cells. Such iPS cells would circumvent host vs. graft rejection of the grafted beta-cells. A major drawback of early protocols to induce pluripotency is that they rely on viral injection to deliver the Yamanaka factors (octamer-binding protein 3/4 (Oct3/4), Sox2, kruppel like factor 4 (Klf4), and myelocytomatosis viral oncogene homolog (c-Myc)) [86]. This permanently integrates the necessary DNA elements in the genome of their target cells, and could lead to undesirable side effects such as increased risk of tumor formation. Several approaches are being considered to induce pluripotency without introducing novel DNA elements, including the use of non-integrating protein treatments to induce the pluripotent state [87, 88].
iPS approaches avoid the histocompatibility issues potentially associated with the use of hES cells. However, the introduction of cells and tissues derived from a pluripotent stem cell population (ES or iPS) is associated with the risk that undifferentiated cells remain at some frequency in the transplanted material. Upon transplantation, such undifferentiated stem cells could potentially give rise to all three germ layers and their derivatives, resulting in teratoma formation. The general strategy for stem cells of either ES or iPS origin is to coach these pluripotent and undifferentiated cells past numerous fate decisions along the beta-cell lineage to generate mature, functional beta-cells.
In essence, one aims to recapitulate the incremental process responsible for successful beta-cell formation during development in a dish. There is a lot of commonality in the plethora of protocols now in use across the scientific community, especially for the initial steps of differentiation leading up to PDX1-positive posterior foregut endoderm [89]. In the following sections, we discuss some of the recent advances the field has made since the initial report of in vitro generated, stem cell-derived hormone-positive cells that made use of the embryonic template [22]. Also, we take a look at the potential for further improvements in current protocols.
3.1 Generation of definitive endoderm
The generation of in vitro stem cell-derived insulin-positive cells by D’Amour and colleagues [22] heralded the potential for success of stepwise differentiation protocols that mimic embryonic development for the generation of pancreatic beta-cells. Many groups have now developed such protocols [22, 90-95] for a variety of starting stem cell populations that include human ES cells [22, 90], human iPS cells [95], and human parthenogenetic stem cells (generation of DE only) [96]. In these protocols, the first step is to differentiate the stem cells into DE. As noted above, DE is characterized by the combined expression of the general endoderm markers CXCR4, FOXA2, and SOX17, but not SOX7, which marks extra-embryonic endoderm.
In DE formation in the embryo, TGF-beta family signaling by nodal and WNT3A plays an important role. Most stem cell protocols make use of the TGF-beta family member activin A instead of nodal. Frequently, this is in combination with WNT3a, under low or no serum conditions, to generate DE that can be successfully differentiated into insulin-positive cells in vitro and glucose-responsive insulin-positive cells in vivo (reviewed by Hosaya et al. [89]). WNT3A signaling is important for the generation of mesendoderm, and is used only on the first day of differentiation. Activin A treatment continues for two more days to generate DE. Nodal and activin A activate very similar pathways, but they respond in opposite ways to teratocarcinoma-derived growth factor 1 (TDGF1 or cripto) [97], which is present in pre-gastrulation embryos [98] and is enriched in ES cells [99]. This suggests that the choice for either activin A or nodal is perhaps not without consequences for the successful induction of DE and the development of beta-cells downstream of that. Indeed, a recent report demonstrates that the use of nodal instead of activin A results in a greater frequency of mouse ES cell-derived beta-cells, despite only modest initial differences in global gene expression at the DE stage [100].
3.2 Generation of primitive gut and posterior foregut tissue
Intermediate fibroblast growth factor (FGF) signaling in the embryo inhibits sonic hedgehog signaling in DE, allowing pancreas specification. From stem cell-derived DE, the use of fibroblast growth factor proteins and cyclopamine is a common strategy to generate primitive gut-like tissue. These factors continue to be applied for further differentiation into posterior foregut-like tissue. Cyclopamine is a plant-derived alkaloid that inactivates the sonic hedgehog signaling pathway by binding and inactivating Smoothened [101]. Also, it is a general component of stem cell differentiation protocols towards a posterior foregut phenotype (reviewed by Hosoya et al. [89]). Once the cultures have acquired primitive gut tube markers such as HNF1b, the addition of retinoic acid is used to initiate PDX1 expression (reviewed by Hosoya et al. [89]), thereby defining the pancreatic region of the posterior foregut. In addition, FGF10 continues to be applied for proliferation of PDX1-positive cells [102, 103]. These steps are reminiscent of the developmental steps necessary for the dorsal, but not necessarily the ventral pancreas.
The induction of PDX1 expression is a hallmark of both the dorsal and ventral pancreatic bud formation. However, the inductive pathways leading up to the PDX1 expression are disparate and may result in slightly different outcomes. This is similar to the induction of DE in vitro by activin A versus nodal [100]. Indeed, during early development, the ventral pancreas exhibits a predominance of PP cells and peptide YY-positive cells over other endocrine cell types [38]. Also, there appears to be a regional diversification of the mature human pancreas, where islets in the posterior part of the splenic head (which originates from the ventral pancreas) contain up to 80% PP cells [63, 104] that are ultrastructurally distinct from the 1% PP cells observed elsewhere in the pancreas [105]. Following the inductive pathways for dorsal pancreas may prevent the development of PP cells, and favor differentiation along the beta-cell lineage.
One way to enhance PDX1 expression without adding new growth factors or other stimuli is to re-seed ES cell-derived DE cells at low densities before the application of retinoic acid. This triplicates the relative number of PDX1-expressing cells [90]. It suggests that fewer cell-to-cell contacts are beneficial for the induction of PDX1. This consideration is in line with the preferential generation of PDX1-positive cells in the extreme periphery of embryoid bodies, where cells lack neighbors on one side [106].
3.3 Generation of pancreatic endoderm
Next, pancreatic endoderm (PE) is generated from posterior foregut endoderm. PE continues to express PDX1, and additionally expresses NKX2.2, NKX6.1, NGN3, and PAX4. From this point forward in the differentiation protocols, there is little consensus on the question what factors are important for the differentiation of PE- and hormone-positive cells downstream of PE. At this stage too, subtle variations introduced earlier in the differentiation protocol begin to show their full effects. For example, the use of nodal versus activin A results in highly similar DE with respect to general gene expression profile. However, it leads to a higher frequency of cells with high levels of PDX1, which in turn have a greater propensity to become insulin-positive cells [100]. PDX1-high cells derived via either method do not appear to differ in their ability to generate beta-cells in vivo, but the fraction of PDX1 cells that differentiate along the beta-cell lineage varies, based on the prior use of nodal vs. activin A for DE induction [100]. In another example, the addition of retinoic acid was not necessary for PDX1 induction per se, but without retinoic acid, no appreciable NGN3 was observed in PE, nor was insulin or glucagon gene expression activated upon further differentiation [22].
3.4 Generation of hormone-positive cells
Early in vitro protocols to differentiate stem cells into beta-cells yielded low numbers of insulin-positive cells (4-7.3%) [22, 91]. More recently, these numbers have been increased to 12% by increased NKX6.1 and sustained NEUROD expression [107]. In vitro-derived insulin-positive cells often co-express other hormones, like glucagon and somatostatin [22, 91, 107, 108], and lack important transcription factors like NKX6.1 [22]. The lack of sustained NKX6.1 expression in ES cell-derived beta-cells may explain the expression of additional hormones, as NKX6.1 suppresses the expression of non-beta-cell hormones [71]. NKX6.1 expression in PE cultures is augmented by omission of HEPES, a common pH-buffering agent, from the culture medium. However, expression of NKX6.1 alone is insufficient to completely suppress non-beta-cell hormone expression, as the insulin-positive cells that were obtained in these cultures frequently still co-expressed glucagon [107]. Interestingly, when cultures enriched for insulin and glucagon-co-positive cells are transplanted, the grafts principally develop into glucagon-single-positive cells [56, 66, 67] that are mature, as suggested by expression of UCN3 [56] and indicated by secretion of fully processed, biologically active glucagon in response to physiological stimuli [66].
These findings suggest that these polyhormonal cells can give rise to mature alpha-cells, but that other populations contained within the PE are more likely to be mature beta-cell precursors [67]. These double hormone-positive cells are reminiscent of the first wave of immature insulin-positive endocrine cells that occur in development. It has been suggested that these double-hormone-positive cells may facilitate the development of mature beta-cells by generating and secreting GLP-1 through processing of proglucagon by PCSK1 [57]. In fact, in conditions of beta-cell stress, mature alpha-cells produce GLP-1 as well. It is thought that this response serves to protect the remaining beta-cells (reviewed in [109]). Indeed, the addition of GLP-1 to stem cell cultures has been reported to improve beta-cell development from human iPS cell-derived PDX1-positive precursors in vitro to the extent that some glucose responsiveness is generated [110].
4. Challenges for the immediate future
The generation of beta-cells in a dish has come a long way in a relatively short period of time. By recapitulating steps necessary to generate beta-cells in vivo, it is now possible to generate insulin-positive cells with insulin secretion in response to several stimuli. Nevertheless, several challenges remain:
- Maturation of stem cells into functionally mature glucose-responsive beta-cells before transplantation.
- Low overall efficiency of differentiation which increases the risk of teratoma formation.
- High cost of growth factors used in the differentiation.
- Undefined factors associated with animal sera and feeder cells.
- Lack of a master protocol that applies to all stem cell lines equally well.
4.1 Islet cell maturation
To date, in vitro protocols have not succeeded in generating mature, glucose-responsive beta-cells. Indeed, the expression of the maturation marker UCN3 at several key intermediate stages towards the differentiation of beta-cells is marginal as determined by quantitative polymerase chain reaction (PCR), and only a handful of faintly UCN3-positive cells exist in the differentiating cultures or in cultures containing insulin-positive cells [56, 77]. This suggests that we have not fully deciphered the cocktail of signals derived from soluble factors within the islet microenvironment that supports beta-cell maturation.
Nevertheless, transplantation and in vivo maturation of ES cell-derived pancreatic endoderm cells that co-express PDX1, FOXA2, HNF6, and NKX6.1 has successfully cured diabetic mice from their disease [93]. Stem cell-derived pancreatic endoderm cells were also transplanted into diabetic mice and allowed to mature over time, while the mice were treated with exogenous insulin, until the grafts were able to successfully take over regulation of blood glucose [107]. When such transplants were analyzed, they were found to contain beta-cells that co-express PDX1, NKX6.1, MAFA, and UCN3 with insulin [56, 77, 93, 107].
These findings are encouraging as they illustrate that stem cell-derived PE can differentiate into mature, glucose-responsive beta-cells fully capable of curing diabetes. They also correlate the acquisition of UCN3 expression with the ability of grafted hESC-derived pancreatic endoderm to restore normoglycemia to diabetic mice [93, 107], and provide support for the idea that UCN3 expression is correlated with beta-cell maturation. Closer inspection of these UCN3-positive cells revealed that they fall into one of two populations of endocrine cells. On the one hand, UCN3 is expressed in mature beta-cells, as the expression of UCN3 trails the onset of expression of both insulin and PCSK1, and is expressed in insulin-positive cells that retain expression of NKX6.1 and PDX1. Also, there is a population of UCN3-positive cells that does not express any of these established beta-cell markers, and instead co-expresses UCN3 with glucagon [56].
Nevertheless, despite the fact that UCN3 in human islets and human ES cell-derived populations is not restricted to beta-cells, UCN3 has great utility as a marker to instruct our efforts towards the differentiation of mature, functional beta-cells in a dish, provided that other markers for alpha- and beta-cell identity (i.e. insulin and glucagon) are incorporated in the experimental design. When glucose-responsive grafts of PE following in vivo maturation are analyzed for the expression of UCN3, only a subset of insulin-positive cells has acquired this maturation marker [56]. If only these UCN3 and insulin-co-positive cells are responsible for normalizing hyperglycemia and curing diabetes in these pre-clinical models, it follows that substantial improvements can still be made regarding the fraction of PE that will go on to differentiate into mature beta-cells, as suggested by the acquisition of the maturation marker UCN3. More efficient induction of beta-cell maturity, prior to or following grafting, would reduce the number of cells or cell-containing devices required to cure a T1D patient.
A major challenge going forward will be to identify the nature of the signal(s) that can promote beta-cell maturation, and to subsequently harness their potential in vitro in beta-cell differentiation and maturation protocols. As shown in several models of beta-cell differentiation, ambient glucose levels are important. For example, fetal rat islets only demonstrate a mature biphasic pattern of GSIS when cultured under high glucose conditions [111]. In addition, while glucose did not alter the differentiation frequency of either PDX1-positive cells or NGN3-positive endocrine precursor cells, in cultured E13.5 rat pancreas, NeuroD expression as well as the insulin and glucagon cell masses were increased under high glucose conditions [112]. A postnatal increase in MAFA expression is a crucial aspect of beta-cell maturation [75], and MAFA expression in differentiating cultures could potentially be increased by glucose [113] to enhance the maturation process.
Current protocols often use high glucose media throughout differentiation. It may be necessary to start at lower glucose at stages preceding the generation of hormone-positive cells to allow for an increase in ambient glucose to potentially stimulate beta-cell differentiation and maturation. An exciting recent finding implicates thyroid hormone signaling in the in vivo upregulation of MAFA expression during beta-cell maturation. Thus, thyroid hormone could constitute a potential new component of differentiation protocols [114]. With the finding that beta-cell maturation can be monitored by following expression of UCN3, combined with insulin expression, we now have a valuable tool to further refine culture conditions in order to obtain mature beta-cells in a dish [56, 77].
4.2 Low overall efficiency of differentiation
Ideally, differentiation protocols should be optimized to yield pure populations of exclusively differentiated cells. This requires perfection in the composition of inducing factors, the timing of their addition to the cultures, and the culture conditions per se. Unfortunately, such uniform differentiation is far from being achieved across any of the various differentiation steps currently employed to differentiate ES cells along the beta-cell lineage. In an attempt to mechanically enhance DE differentiation and purification, a culturing system was designed to mimic the developmental process of migration through the primitive streak, where cells migrate through pores of the membrane on which they are cultured. Treatment of the culture induced an epithelial-to-mesenchymal transition (EMT), which allowed cells to migrate through the pores, away from unresponsive stationary cells. They next differentiate into highly pure DE, with no contamination of undifferentiated OCT4-expressing cells [115].
FACS purification based on lineage-specific cell surface markers has been suggested as an alternatively strategy to purify only appropriately differentiated cells for subsequent use in downstream differentiation of transplantation steps, and has been applied at several steps in current protocols. For example, the DE cell surface marker CXCR4 has been used to enrich DE to near homogeneity by FACS from ES cell-derived DE cultures [90, 116] prior to re-plating and differentiating these cultures further with improved efficiency [90]. Enrichment of PE cells, a later stage in the development of beta-cells, can be obtained by FACS purifying for CD142 (the cell surface glycoprotein coagulation factor III) [67] or discoidin domain receptor tyrosine kinase 1 [117].
CD142 is a marker for PDX1-positive, NKX6.1-positive, and chromogranin A-negative PE cells. These sorted cells are able to generate all major cell types of the pancreas upon transplantation [67]. While FACS purification incurs some losses in cell recovery, the transplanted CD142-enriched PE cells did not show teratoma formation in the seven mice with surviving grafts. This supports the notion that enrichment of differentiated stem cells may reduce the risk of teratomas [67]. Expression of the maturation marker UCN3 can help to guide efforts to differentiate insulin-positive cells into mature beta-cells. However, its intracellular localization precludes its use for live cell sorting in an effort to purify mature beta-cells from a mixed population of cells that are differentiating along the beta-cell lineage. The slow replication rate of beta-cells, compared to their progenitors earlier in the lineage, discourages any attempts to collect mature beta-cells to further expand their number.
Nevertheless, collection of mature stem cell-derived beta-cells could be a desirable strategy to purify mature beta-cells prior to grafting. Cell surface markers or marker combinations for beta-cells exist, but they will also label immature insulin-positive cells early in development [117-119]. At the moment, there are no cell surface markers that specifically label mature beta-cells.
4.3 High cost and undefined chemical composition of animal sera and growth factors
The use of animal serum in differentiation protocols introduces an unknown variation, as serum is not chemically defined, and growth factor content may vary from batch to batch. Also, stem cells are often grown on mouse feeder cells which secrete unknown factors that influence cell behavior. Chemically defined growth and differentiation media that allow stem cells to grow without the use of other cells decreases the risk of cross-species transmission of infectious agents.
The high cost of these chemically defined media derives in large part from the different growth factors added. The use of small molecules could reduce the cost. Also, small molecules may have a more narrowly defined effect on specific signaling pathways, allowing for a more precise tuning of effects. Finally, small molecules are easily adaptable to large scale screens for the optimization of protocols. Several groups have found small molecules that are able to replace growth factors in their protocol, and to generate correctly specified differentiation intermediates. Sometimes, their efficiency surpasses the replaced growth factor. For example, two compounds, termed IDE1 (2-((6-carboxy-hexanoyl)-hydrazonomethyl)-benzoic acid) and IDE2 (7-(2-cyclopentylidenehydrazino)-7-oxohepta-noic acid), which activate the TGF-beta signaling pathway through an unknown mechanism, had the ability to differentiate stem cells into DE [120]. WNT3a activity could be partially mimicked by a small molecule inhibitor of glycogen synthase kinase 3 beta (GSK3-beta) called CHIR99021. Treating stem cells with activin A and CHIR99021 enhances the co-expression of SOX17 and FOXA2 in hiPS cell-derived DE to levels well above of those seen for the combined treatment of activin A and WNT3A [121].
For the induction of PDX1 expression in posterior foregut, retinoic acid is used. One of the ways in which retinoic acid affects cells is by activating protein kinase C, substituting its natural phospholipid cofactor for phosphatidylserine [122]. Addition of small molecule protein kinase C activators like (-)-Indolactam V or ((2S,5S)-(E,E)-8-(5-(4-(trifluoromethyl) phenyl)-2,4-pentadienoylamino) benzolactam (TPB) to cultures of posterior foregut endoderm after retinoic acid treatment increases the frequency of PDX1-expressing cells [107, 110, 123]. Even without retinoic acid treatment, these compounds were able to induce PDX1 expression in primitive gut endoderm, but combined treatment with retinoic acid yielded optimal results [123]. Thus, small molecules are already able to replace growth factors in several of the steps of differentiation. It is hoped that small molecules can contribute more to reproducible and affordable protocols for beta-cell differentiation [124].
4.4 The lack of a master protocol
A final issue worth mentioning is the difficulty associated with the application of a single protocol to different stem cell lines yielding similar results [125]. Even the application of the same protocol, in the same lab, to different cell lines reveals significant variation with respect to the induction of the different stages of differentiation towards beta-cell identity [22]. These difficulties are likely to be the result of as yet unspecified intrinsic differences between cell lines that may originate from chromatin structure, rather than the genetic variation between cell lines. Even iPS clones derived from the same donor vary considerably in their differentiation capacity [110]. This implies that protocols have not been consistently applied across different cell lines or between different laboratories, which hampers a direct comparison of the efficiency of differentiation between different studies and labs. This is particularly important when iPS cells are used as starting material for the generation of specific beta-cell lines to each individual patient. The prospect of empirically optimizing the differentiation protocol for each new iPS cell line would not be appealing. Therefore, the challenge is to achieve a similar state of pluripotency across different iPS cell lines, a state considered optimal for pancreas differentiation, such that differentiation can be started across many different iPS cell lines using essentially the same protocol for beta-cell differentiation.
5. Concluding remarks
In the past decade, we have observed the generation of stem cell-derived beta-cells, and we have witnessed their ability to cure diabetes in preclinical models. Current protocols are able to differentiate a larger fraction of early progenitors towards an endocrine fate compared to normal embryonic development. However, our ability to generate mature functional beta-cells from stem cells is wanting. We have only a rudimentary understanding of the factors that may promote beta-cell maturation from insulin-positive but immature precursors.
The recent discovery of the mature beta-cell marker UCN3 can aid in the development of protocols that generate mature beta-cells in a dish. Transplantation of these mature cells will reduce the number of cells necessary to maintain normoglycemia. Also, transplantation of fully differentiated beta-cells further reduces the risk of teratoma formation. Because stem cells have the potential to generate unlimited numbers of cells, one could imagine periodical replacement of a stem cell-derived graft when the graft function declines over time, similar as in many patients following islet transplantation. Nevertheless, while it is clear that challenges remain, considering the relatively short period of time in which all of these advances have been achieved, there is reason to be cautiously optimistic about the prospects of generating transplantable, mature, glucose-responsive beta-cells as a cure for T1D.
Disclosures: The authors report no conflict of interests.
Acknowledgments
This work is supported by award P01 DK026741 from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and by the Juvenile Diabetes Research Foundation (JDRF), the Clayton Medical Research Foundation, Inc., the Salk Center for Nutritional Genomics, and the Leona M. and Harry B. Helmsley Charitable Trust. The authors would like to thank Dr. Ruiyu Xie who generously provided the human fetal pancreas tissue sections.
References
- 1.JDRF General Diabetes Facts. jdrf.org/about-jdrf/fact-sheets/general-diabetes-facts/ [Google Scholar]
- 2.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91(1):79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 3.Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12:141–146. [PMC free article] [PubMed] [Google Scholar]
- 4.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57(6):1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, Kirby M, Pechhold S, Liu EH, Harlan DM, Tisdale JF. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab. 2010;95(10):E234–E239. doi: 10.1210/jc.2010-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53(10):2167–2176. doi: 10.1007/s00125-010-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 8.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, Troxell R, Wills R, Tanwir M, Casinelli G, Cox AE, Takane KK, Srinivas H. et al. Cytoplasmic-Nuclear Trafficking of G1/S Cell Cycle Molecules and Adult Human beta-Cell Replication: A Revised Model of Human beta-Cell G1/S Control. Diabetes. 2013;62(7):2460–2470. doi: 10.2337/db12-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juhl K, Bonner-Weir S, Sharma A. Regenerating pancreatic beta-cells: plasticity of adult pancreatic cells and the feasibility of in-vivo neogenesis. Curr Opin Organ Transplant. 2010;15(1):79–85. doi: 10.1097/MOT.0b013e3283344932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonner-Weir S, Guo L, Li WC, Ouziel-Yahalom L, Lysy PA, Weir GC, Sharma A. Islet neogenesis: a possible pathway for beta-cell replenishment. Rev Diabet Stud. 2012;9(4):407–416. doi: 10.1900/RDS.2012.9.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lysy PA, Weir GC, Bonner-Weir S. Concise review: pancreas regeneration: recent advances and perspectives. Stem Cells Transl Med. 2012;1(2):150–159. doi: 10.5966/sctm.2011-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott RB, Escobar L, Tan PL, Muzina M, Zwain S, Buchanan C. Live encapsulated porcine islets from a type 1 diabetic patient 9.5 yr after xenotransplantation. Xenotransplantation. 2007;14(2):157–161. doi: 10.1111/j.1399-3089.2007.00384.x. [DOI] [PubMed] [Google Scholar]
- 13.Rogers J, Farney AC, Al-Geizawi S, Iskandar SS, Doares W, Gautreaux MD, Hart L, Kaczmorski S, Reeves-Daniel A, Winfrey S. et al. Pancreas transplantation: lessons learned from a decade of experience at Wake Forest Baptist Medical Center. Rev Diabet Stud. 2011;8(1):17–27. doi: 10.1900/RDS.2011.8.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343(4):230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 15.Vrochides D, Paraskevas S, Papanikolaou V. Transplantation for type 1 diabetes mellitus. Whole organ or islets? Hippokratia. 2009;13(1):6–8. [PMC free article] [PubMed] [Google Scholar]
- 16.Shapiro AM. Islet transplantation in type 1 diabetes: ongoing challenges, refined procedures, and long-term outcome. Rev Diabet Stud. 2012;9(4):385–406. doi: 10.1900/RDS.2012.9.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCall M, Shapiro AM. Update on islet transplantation. Cold Spring Harb Perspect Med. 2012;2(7):a007823. doi: 10.1101/cshperspect.a007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL, Tzukerman M. Insulin production by human embryonic stem cells. Diabetes. 2001;50(8):1691–1697. doi: 10.2337/diabetes.50.8.1691. [DOI] [PubMed] [Google Scholar]
- 19.Hori Y, Rulifson IC, Tsai BC, Heit JJ, Cahoy JD, Kim SK. Growth inhibitors promote differentiation of insulin-producing tissue from embryonic stem cells. Proc Natl Acad Sci U S A. 2002;99(25):16105–16110. doi: 10.1073/pnas.252618999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science. 2001;292(5520):1389–1394. doi: 10.1126/science.1058866. [DOI] [PubMed] [Google Scholar]
- 21.Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes. 2000;49(2):157–162. doi: 10.2337/diabetes.49.2.157. [DOI] [PubMed] [Google Scholar]
- 22.D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24(11):1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 23.Conlon FL, Lyons KM, Takaesu N, Barth KS, Kispert A, Herrmann B, Robertson EJ. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development. 1994;120(7):1919–1928. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- 24.Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22(4):361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- 25.McCracken KW, Wells JM. Molecular pathways controlling pancreas induction. Semin Cell Dev Biol. 2012;23(6):656–662. doi: 10.1016/j.semcdb.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGrath KE, Koniski AD, Maltby KM, McGann JK, Palis J. Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev Biol. 1999;213(2):442–456. doi: 10.1006/dbio.1999.9405. [DOI] [PubMed] [Google Scholar]
- 27.Ang SL, Wierda A, Wong D, Stevens KA, Cascio S, Rossant J, Zaret KS. The formation and maintenance of the definitive endoderm lineage in the mouse: involvement of HNF3/forkhead proteins. Development. 1993;119(4):1301–1315. doi: 10.1242/dev.119.4.1301. [DOI] [PubMed] [Google Scholar]
- 28.Kanai-Azuma M, Kanai Y, Gad JM, Tajima Y, Taya C, Kurohmaru M, Sanai Y, Yonekawa H, Yazaki K, Tam PP, Hayashi Y. Depletion of definitive gut endoderm in Sox17-null mutant mice. Development. 2002;129(10):2367–2379. doi: 10.1242/dev.129.10.2367. [DOI] [PubMed] [Google Scholar]
- 29.Bouwmeester T, Kim S, Sasai Y, Lu B, De Robertis EM. Cerberus is a head-inducing secreted factor expressed in the anterior endoderm of Spemann's organizer. Nature. 1996;382(6592):595–601. doi: 10.1038/382595a0. [DOI] [PubMed] [Google Scholar]
- 30.Coffinier C, Barra J, Babinet C, Yaniv M. Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev. 1999;89(1-2):211–213. doi: 10.1016/s0925-4773(99)00221-x. [DOI] [PubMed] [Google Scholar]
- 31.Coffinier C, Thepot D, Babinet C, Yaniv M, Barra J. Essential role for the homeoprotein vHNF1/HNF1beta in visceral endoderm differentiation. Development. 1999;126(21):4785–4794. doi: 10.1242/dev.126.21.4785. [DOI] [PubMed] [Google Scholar]
- 32.Dessimoz J, Opoka R, Kordich JJ, Grapin-Botton A, Wells JM. FGF signaling is necessary for establishing gut tube domains along the anterior-posterior axis in vivo. Mech Dev. 2006;123(1):42–55. doi: 10.1016/j.mod.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121(1):11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- 34.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 35.Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 36.Pictet RL, Clark WR, Williams RH, Rutter WJ. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. 1972;29(4):436–467. doi: 10.1016/0012-1606(72)90083-8. [DOI] [PubMed] [Google Scholar]
- 37.Rutter WJ, Kemp JD, Bradshaw WS, Clark WR, Ronzio RA, Sanders TG. Regulation of specific protein synthesis in cytodifferentiation. J Cell Physiol. 1968;72(2):1–18. doi: 10.1002/jcp.1040720403. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen MC, Ahnfelt-Ronne J, Hald J, Madsen OD, Serup P, Hecksher-Sorensen J. An illustrated review of early pancreas development in the mouse. Endocr Rev. 2007;28(6):685–705. doi: 10.1210/er.2007-0016. [DOI] [PubMed] [Google Scholar]
- 39.Rose SD, Kruse F, Swift GH, MacDonald RJ, Hammer RE. A single element of the elastase I enhancer is sufficient to direct transcription selectively to the pancreas and gut. Mol Cell Biol. 1994;14(3):2048–2057. doi: 10.1128/mcb.14.3.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiebe PO, Kormish JD, Roper VT, Fujitani Y, Alston NI, Zaret KS, Wright CV, Stein RW, Gannon M. Ptf1a binds to and activates area III, a highly conserved region of the Pdx1 promoter that mediates early pancreas-wide Pdx1 expression. Mol Cell Biol. 2007;27(11):4093–4104. doi: 10.1128/MCB.01978-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sussel L, Kalamaras J, Hartigan-O'Connor DJ, Meneses JJ, Pedersen RA, Rubenstein JL, German MS. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. 1998;125(12):2213–2221. doi: 10.1242/dev.125.12.2213. [DOI] [PubMed] [Google Scholar]
- 42.Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000;127(24):5533–5540. doi: 10.1242/dev.127.24.5533. [DOI] [PubMed] [Google Scholar]
- 43.Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell. 2010;18(6):1022–1029. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rukstalis JM, Habener JF. Neurogenin3: a master regulator of pancreatic islet differentiation and regeneration. Islets. 2009;1(3):177–184. doi: 10.4161/isl.1.3.9877. [DOI] [PubMed] [Google Scholar]
- 45.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385(6613):257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- 47.Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386(6623):399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- 48.Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11(18):2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387(6631):406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- 50.Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17(20):2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villasenor A, Chong DC, Cleaver O. Biphasic Ngn3 expression in the developing pancreas. Dev Dyn. 2008;237(11):3270–3279. doi: 10.1002/dvdy.21740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, Grapin-Botton A. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell. 2007;12(3):457–465. doi: 10.1016/j.devcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Bramswig NC, Kaestner KH. Transcriptional regulation of alpha-cell differentiation. Diabetes Obes Metab. 2011;13(Suppl 1):13–20. doi: 10.1111/j.1463-1326.2011.01440.x. [DOI] [PubMed] [Google Scholar]
- 54.Collombat P, Hecksher-Sorensen J, Krull J, Berger J, Riedel D, Herrera PL, Serup P, Mansouri A. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest. 2007;117(4):961–970. doi: 10.1172/JCI29115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jensen J, Heller RS, Funder-Nielsen T, Pedersen EE, Lindsell C, Weinmaster G, Madsen OD, Serup P. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49(2):163–176. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 56.van der Meulen T, Xie R, Kelly OG, Vale WW, Sander M, Huising MO. Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic alpha and beta cells. PLoS One. 2012;7(12):e52181. doi: 10.1371/journal.pone.0052181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson ME, Kalamaras JA, German MS. Expression pattern of IAPP and prohormone convertase 1/3 reveals a distinctive set of endocrine cells in the embryonic pancreas. Mech Dev. 2002;115(1-2):171–176. doi: 10.1016/s0925-4773(02)00118-1. [DOI] [PubMed] [Google Scholar]
- 58.Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. 1996;122(5):1409–1416. doi: 10.1242/dev.122.5.1409. [DOI] [PubMed] [Google Scholar]
- 59.Oster A, Jensen J, Serup P, Galante P, Madsen OD, Larsson LI. Rat endocrine pancreatic development in relation to two homeobox gene products (Pdx-1 and Nkx 6.1) J Histochem Cytochem. 1998;46(6):707–715. doi: 10.1177/002215549804600602. [DOI] [PubMed] [Google Scholar]
- 60.Herrera PL, Huarte J, Sanvito F, Meda P, Orci L, Vassalli JD. Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development. 1991;113(4):1257–1265. doi: 10.1242/dev.113.4.1257. [DOI] [PubMed] [Google Scholar]
- 61.Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D. Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development. 1993;118(4):1031–1039. doi: 10.1242/dev.118.4.1031. [DOI] [PubMed] [Google Scholar]
- 62.Upchurch BH, Aponte GW, Leiter AB. Expression of peptide YY in all four islet cell types in the developing mouse pancreas suggests a common peptide YY-producing progenitor. Development. 1994;120(2):245–252. doi: 10.1242/dev.120.2.245. [DOI] [PubMed] [Google Scholar]
- 63.Riedel MJ, Asadi A, Wang R, Ao Z, Warnock GL, Kieffer TJ. Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia. 2012;55(2):372–381. doi: 10.1007/s00125-011-2344-9. [DOI] [PubMed] [Google Scholar]
- 64.Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, Sieweke M, Stein R. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104(10):3853–3858. doi: 10.1073/pnas.0700013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127(11):2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- 66.Rezania A, Riedel MJ, Wideman RD, Karanu F, Ao Z, Warnock GL, Kieffer TJ. Production of functional glucagon-secreting alpha-cells from human embryonic stem cells. Diabetes. 2011;60(1):239–247. doi: 10.2337/db10-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly OG, Chan MY, Martinson LA, Kadoya K, Ostertag TM, Ross KG, Richardson M, Carpenter MK, D'Amour KA, Kroon E. et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat Biotechnol. 2011;29(8):750–756. doi: 10.1038/nbt.1931. [DOI] [PubMed] [Google Scholar]
- 68.Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet. 1999;23(1):71–75. doi: 10.1038/12674. [DOI] [PubMed] [Google Scholar]
- 69.Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23(1):67–70. doi: 10.1038/12669. [DOI] [PubMed] [Google Scholar]
- 70.Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993;12(11):4251–4259. doi: 10.1002/j.1460-2075.1993.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schaffer AE, Taylor BL, Benthuysen JR, Liu J, Thorel F, Yuan W, Jiao Y, Kaestner KH, Herrera PL, Magnuson MA. et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. Plos Genet. 2013;9(1):e1003274. doi: 10.1371/journal.pgen.1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, Sharma A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev Biol. 2006;293(2):526–539. doi: 10.1016/j.ydbio.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci U S A. 2004;101(9):2930–2933. doi: 10.1073/pnas.0306233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K. et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25(12):4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, Bonner-Weir S. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia. 2011;54(3):583–593. doi: 10.1007/s00125-010-2026-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, Weir GC, Sgroi D, Bonner-Weir S. Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia. 2011;54(3):594–604. doi: 10.1007/s00125-010-2036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30(3):261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hawdon JM, Ward Platt MP, Aynsley-Green A. Patterns of metabolic adaptation for preterm and term infants in the first neonatal week. Arch Dis Child. 1992;67(4 Spec No):357–365. doi: 10.1136/adc.67.4_spec_no.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang SB, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- 80.Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59(10):2530–2539. doi: 10.2337/db10-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dai C, Brissova M, Hang Y, Thompson C, Poffenberger G, Shostak A, Chen Z, Stein R, Powers AC. Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia. 2012;55(3):707–718. doi: 10.1007/s00125-011-2369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li C, Chen P, Vaughan J, Blount A, Chen A, Jamieson PM, Rivier J, Smith MS, Vale W. Urocortin III is expressed in pancreatic beta-cells and stimulates insulin and glucagon secretion. Endocrinology. 2003;144(7):3216–3224. doi: 10.1210/en.2002-0087. [DOI] [PubMed] [Google Scholar]
- 83.Li C, Chen P, Vaughan J, Lee KF, Vale W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc Natl Acad Sci U S A. 2007;104(10):4206–4211. doi: 10.1073/pnas.0611641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huising MO, Pilbrow AP, Matsumoto M, van der Meulen T, Park H, Vaughan JM, Lee S, Vale WW. Glucocorticoids differentially regulate the expression of CRFR1 and CRFR2alpha in MIN6 insulinoma cells and rodent islets. Endocrinology. 2011;152(1):138–150. doi: 10.1210/en.2010-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Revazova ES, Turovets NA, Kochetkova OD, Kindarova LB, Kuzmichev LN, Janus JD, Pryzhkova MV. Patient-specific stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells. 2007;9(3):432–449. doi: 10.1089/clo.2007.0033. [DOI] [PubMed] [Google Scholar]
- 86.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 87.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4(6):472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y. et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4(5):381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hosoya M, Kunisada Y, Kurisaki A, Asashima M. Induction of differentiation of undifferentiated cells into pancreatic beta cells in vertebrates. Int J Dev Biol. 2012;56(5):313–323. doi: 10.1387/ijdb.123522mh. [DOI] [PubMed] [Google Scholar]
- 90.Cai J, Yu C, Liu Y, Chen S, Guo Y, Yong J, Lu W, Ding M, Deng H. Generation of homogeneous PDX1(+) pancreatic progenitors from human ES cell-derived endoderm cells. J Mol Cell Biol. 2010;2(1):50–60. doi: 10.1093/jmcb/mjp037. [DOI] [PubMed] [Google Scholar]
- 91.Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G, Majumdar AS. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells. 2007;25(8):1940–1953. doi: 10.1634/stemcells.2006-0761. [DOI] [PubMed] [Google Scholar]
- 92.Johannesson M, Stahlberg A, Ameri J, Sand FW, Norrman K, Semb H. FGF4 and retinoic acid direct differentiation of hESCs into PDX1-expressing foregut endoderm in a time- and concentration-dependent manner. PLoS One. 2009;4(3):e4794. doi: 10.1371/journal.pone.0004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J. et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26(4):443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 94.Shim JH, Kim SE, Woo DH, Kim SK, Oh CH, McKay R, Kim JH. Directed differentiation of human embryonic stem cells towards a pancreatic cell fate. Diabetologia. 2007;50(6):1228–1238. doi: 10.1007/s00125-007-0634-z. [DOI] [PubMed] [Google Scholar]
- 95.Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009;19(4):429–438. doi: 10.1038/cr.2009.28. [DOI] [PubMed] [Google Scholar]
- 96.Turovets N, D'Amour KA, Agapov V, Turovets I, Kochetkova O, Janus J, Semechkin A, Moorman MA, Agapova L. Human parthenogenetic stem cells produce enriched populations of definitive endoderm cells after trichostatin A pretreatment. Differentiation. 2011;81(5):292–298. doi: 10.1016/j.diff.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 97.Gray PC, Harrison CA, Vale W. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc Natl Acad Sci U S A. 2003;100(9):5193–5198. doi: 10.1073/pnas.0531290100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ding J, Yang L, Yan YT, Chen A, Desai N, Wynshaw-Boris A, Shen MM. Cripto is required for correct orientation of the anterior-posterior axis in the mouse embryo. Nature. 1998;395(6703):702–707. doi: 10.1038/27215. [DOI] [PubMed] [Google Scholar]
- 99.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113(5):631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 100.Chen AE, Borowiak M, Sherwood RI, Kweudjeu A, Melton DA. Functional evaluation of ES cell-derived endodermal populations reveals differences between Nodal and Activin A-guided differentiation. Development. 2013;140(3):675–686. doi: 10.1242/dev.085431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16(21):2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bhushan A, Itoh N, Kato S, Thiery JP, Czernichow P, Bellusci S, Scharfmann R. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development. 2001;128(24):5109–5117. doi: 10.1242/dev.128.24.5109. [DOI] [PubMed] [Google Scholar]
- 103.Hart A, Papadopoulou S, Edlund H. Fgf10 maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev Dyn. 2003;228(2):185–193. doi: 10.1002/dvdy.10368. [DOI] [PubMed] [Google Scholar]
- 104.Malaisse-Lagae F, Stefan Y, Cox J, Perrelet A, Orci L. Identification of a lobe in the adult human pancreas rich in pancreatic polypeptide. Diabetologia. 1979;17(6):361–365. doi: 10.1007/BF01236270. [DOI] [PubMed] [Google Scholar]
- 105.Fiocca R, Sessa F, Tenti P, Usellini L, Capella C, O'Hare MM, Solcia E. Pancreatic polypeptide (PP) cells in the PP-rich lobe of the human pancreas are identified ultrastructurally and immunocytochemically as F cells. Histochemistry. 1983;77(4):511–523. doi: 10.1007/BF00495805. [DOI] [PubMed] [Google Scholar]
- 106.Phillips BW, Hentze H, Rust WL, Chen QP, Chipperfield H, Tan EK, Abraham S, Sadasivam A, Soong PL, Wang ST. et al. Directed differentiation of human embryonic stem cells into the pancreatic endocrine lineage. Stem Cells Dev. 2007;16(4):561–578. doi: 10.1089/scd.2007.0029. [DOI] [PubMed] [Google Scholar]
- 107.Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, Gauvin R, Narayan K, Karanu F, O'Neil JJ. et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61(8):2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nostro MC, Sarangi F, Ogawa S, Holtzinger A, Corneo B, Li X, Micallef SJ, Park IH, Basford C, Wheeler MB. et al. Stage-specific signaling through TGFbeta family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development. 2011;138(5):861–871. doi: 10.1242/dev.055236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Habener JF, Stanojevic V. Alpha cells come of age. Trends Endocrinol Metab. 2012;24(3):153–163. doi: 10.1016/j.tem.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 110.Thatava T, Nelson TJ, Edukulla R, Sakuma T, Ohmine S, Tonne JM, Yamada S, Kudva Y, Terzic A, Ikeda Y. Indolactam V/GLP-1-mediated differentiation of human iPS cells into glucose-responsive insulin-secreting progeny. Gene Ther. 2011;18(3):283–293. doi: 10.1038/gt.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Freinkel N, Lewis NJ, Johnson R, Swenne I, Bone A, Hellerstrom C. Differential effects of age versus glycemic stimulation on the maturation of insulin stimulus-secretion coupling during culture of fetal rat islets. Diabetes. 1984;33(11):1028–1038. doi: 10.2337/diab.33.11.1028. [DOI] [PubMed] [Google Scholar]
- 112.Guillemain G, Filhoulaud G, Da Silva-Xavier G, Rutter GA, Scharfmann R. Glucose is necessary for embryonic pancreatic endocrine cell differentiation. J Biol Chem. 2007;282(20):15228–15237. doi: 10.1074/jbc.M610986200. [DOI] [PubMed] [Google Scholar]
- 113.Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J Biol Chem. 2002;277(51):49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- 114.Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, Weir GC, Sharma A, Larsen PR, Bonner-Weir S. Thyroid Hormone Promotes Postnatal Rat Pancreatic beta-Cell Development and Glucose-Responsive Insulin Secretion Through MAFA. Diabetes. 2013;62(5):1569–1580. doi: 10.2337/db12-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turovets N, Fair J, West R, Ostrowska A, Semechkin R, Janus J, Cui L, Agapov V, Turovets I, Semechkin A. et al. Derivation of high-purity definitive endoderm from human parthenogenetic stem cells using an in vitro analog of the primitive streak. Cell Transplant. 2012;21(1):217–234. doi: 10.3727/096368911X582723. [DOI] [PubMed] [Google Scholar]
- 116.D'Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005;23(12):1534–1541. doi: 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- 117.Hald J, Galbo T, Rescan C, Radzikowski L, Sprinkel AE, Heimberg H, Ahnfelt-Ronne J, Jensen J, Scharfmann R, Gradwohl G. et al. Pancreatic islet and progenitor cell surface markers with cell sorting potential. Diabetologia. 2012;55(1):154–165. doi: 10.1007/s00125-011-2295-1. [DOI] [PubMed] [Google Scholar]
- 118.Dorrell C, Abraham SL, Lanxon-Cookson KM, Canaday PS, Streeter PR, Grompe M. Isolation of major pancreatic cell types and long-term culture-initiating cells using novel human surface markers. Stem Cell Res. 2008;1(3):183–194. doi: 10.1016/j.scr.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 119.Flamez D, Roland I, Berton A, Kutlu B, Dufrane D, Beckers MC, De Waele E, Rooman I, Bouwens L, Clark A. et al. A genomic-based approach identifies FXYD domain containing ion transport regulator 2 (FXYD2)gammaa as a pancreatic beta cell-specific biomarker. Diabetologia. 2010;53(7):1372–1383. doi: 10.1007/s00125-010-1714-z. [DOI] [PubMed] [Google Scholar]
- 120.Borowiak M, Maehr R, Chen S, Chen AE, Tang W, Fox JL, Schreiber SL, Melton DA. Small molecules efficiently direct endodermal differentiation of mouse and human embryonic stem cells. Cell Stem Cell. 2009;4(4):348–358. doi: 10.1016/j.stem.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kunisada Y, Tsubooka-Yamazoe N, Shoji M, Hosoya M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem Cell Res. 2012;8(2):274–284. doi: 10.1016/j.scr.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 122.Isakov N. Regulation of T-cell-derived protein kinase C activity by vitamin A derivatives. Cell Immunol. 1988;115(2):288–298. doi: 10.1016/0008-8749(88)90182-7. [DOI] [PubMed] [Google Scholar]
- 123.Chen S, Borowiak M, Fox JL, Maehr R, Osafune K, Davidow L, Lam K, Peng LF, Schreiber SL, Rubin LL, Melton D. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol. 2009;5(4):258–265. doi: 10.1038/nchembio.154. [DOI] [PubMed] [Google Scholar]
- 124.Hosoya M. Preparation of pancreatic beta-cells from human iPS cells with small molecules. Islets. 2012;4(3):249–252. doi: 10.4161/isl.20856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mfopou JK, Chen B, Mateizel I, Sermon K, Bouwens L. Noggin, retinoids, and fibroblast growth factor regulate hepatic or pancreatic fate of human embryonic stem cells. Gastroenterology. 2010;138(7):2233–2245. doi: 10.1053/j.gastro.2010.02.056. [DOI] [PubMed] [Google Scholar]
- 126.Tamaki M, Fujitani Y, Uchida T, Hirose T, Kawamori R, Watada H. Downregulation of ZnT8 expression in pancreatic beta-cells of diabetic mice. Islets. 2009;1(2):124–128. doi: 10.4161/isl.1.2.9433. [DOI] [PubMed] [Google Scholar]
- 127.Perez-Armendariz EM, Cruz-Miguel L, Coronel-Cruz C, Esparza-Aguilar M, Pinzon-Estrada E, Rancano-Camacho E, Zacarias-Climaco G, Olivares PF, Espinosa AM, Becker I. et al. Connexin 36 is expressed in beta and connexins 26 and 32 in acinar cells at the end of the secondary transition of mouse pancreatic development and increase during fetal and perinatal life. Anat Rec (Hoboken) 2012;295(6):980–990. doi: 10.1002/ar.22473. [DOI] [PubMed] [Google Scholar]
- 128.Marcinkiewicz M, Ramla D, Seidah NG, Chretien M. Developmental expression of the prohormone convertases PC1 and PC2 in mouse pancreatic islets. Endocrinology. 1994;135(4):1651–1660. doi: 10.1210/endo.135.4.7925129. [DOI] [PubMed] [Google Scholar]
- 129.Artner I, Le Lay J, Hang Y, Elghazi L, Schisler JC, Henderson E, Sosa-Pineda B, Stein R. MafB: an activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes. 2006;55(2):297–304. doi: 10.2337/diabetes.55.02.06.db05-0946. [DOI] [PubMed] [Google Scholar]
- 130.Pang K, Mukonoweshuro C, Wong GG. Beta cells arise from glucose transporter type 2 (Glut2)-expressing epithelial cells of the developing rat pancreas. Proc Natl Acad Sci U S A. 1994;91(20):9559–9563. doi: 10.1073/pnas.91.20.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]