

Abstract
Glycosaminoglycans (GAGs) interact with many proteins to regulate processes such as hemostasis, cell adhesion, growth and differentiation and viral infection. Yet, majority of these interactions remain poorly understood at a molecular level. A major reason for this state is the phenomenal structural diversity of GAGs, which has precluded analysis of specificity of their interactions. We had earlier presented a computational protocol for predicting “high-specificity” GAG sequences based on combinatorial virtual library screening (CVLS) technology. In this work, we expand the robustness of this technology through rigorous studies of parameters affecting GAG recognition of proteins, especially antithrombin and thrombin. The CVLS approach involves automated construction of a virtual library of all possible oligosaccharide sequences (di- to octasaccharide) followed by a two-step selection strategy consisting of “affinity” (GOLD score) and “specificity” (consistency of binding) filters. We find that “specificity” features are optimally evaluated using 100 genetic algorithm experiments, 100,000 evolutions and variable docking radius from 10 Å (disaccharide) to 14 Å (hexasaccharide). The results highlight critical interactions in H/HS oligosaccharides that govern specificity. Application of CVLS technology to the antithrombin–heparin system indicates that the minimal “specificity” element is the GlcAp(1 → 4)GlcNp2S3S disaccharide of heparin. The CVLS technology affords a simple, intuitive framework for the design of longer GAG sequences that can exhibit high “specificity” without resorting to exhaustive screening of millions of theoretical sequences.
Keywords: glycosaminoglycans, heparin/heparan sulfate, molecular docking, specificity, virtual screening
Introduction
Glycosaminoglycans (GAGs) interact with numerous proteins to regulate various physiological and pathological processes such as hemostasis, cell adhesion, growth factor signaling, coagulation, viral pathogenesis and protease regulation (Capila and Linhardt 2002; Gandhi and Mancera 2008). Heparin (H) and heparan sulfate (HS), members of the GAG superfamily, are composed of alternating 1 → 4-linked glucosamine (GlcNp) and uronic acid (UAp) residues [either glucuronic acid (GlcAp) or iduronic acid (IdoAp)] that are incompletely modified through sulfation, acetylation and epimerization reactions (Shriver et al. 2012). These modifications can produce 48 distinct disaccharides, of which 23 have been found in nature to date (Esko and Selleck 2002). Further, the IdoAp residue can exist in multiple conformations, especially 1C4 and 2SO (Mulloy and Forster 2000), that can interconvert easily in solution to enhance structural possibilities. Thus, combinatorial arrangements of the several configurational and conformational variations possible at the monosaccharide level generate millions of distinct H/HS sequences. Although this massive library of natural H/HS sequences offers a major advantage for enhancing the probability of protein recognition, it also presents difficulties in deciphering detailed structure–function relationships for these interesting biopolymers.
A majority of GAG sequences bind to proteins through non-specific interactions because practically any collection of positive charges on a protein surface tends to recognize a sulfated GAG chain. Interactions that rely only on electrostatics, e.g. Coulombic, operate over longer distance than those that rely on hydrogen bond or van der Waals forces [Coulomb forces have a r−1 relationship (r = distance between two ions), whereas van der Waals forces have a r−3 to r−6 dependence]. This implies that unless a GAG sequence forms multiple hydrogen-bonding or equivalent interactions, the protein–GAG system will exhibit poor specificity. In fact, not many GAG sequences exhibit characteristics of high specificity. The prototypic high affinity, high-specificity GAG sequence is the heparin pentasaccharide sequence DEFGH (Figure 1A), which contains several key sulfate groups including a 3-O-sulfate on the central GlcNp residue, that recognizes antithrombin (AT) (Jin et al. 1997; Desai et al. 1998). Other sequences likely to exhibit high specificity include a heparin octasaccharide binding to glycoprotein gD of herpes simplex virus-1 (Copeland et al. 2008) and a dermatan sulfate hexasaccharide binding to heparin cofactor II (Maimone and Tollefsen 1990; Raghuraman et al. 2010). Several other GAG sequences may exhibit specificity of protein recognition (Capila and Linhardt 2002; Gandhi and Mancera 2008) and are awaiting rigorous characterization.
Fig. 1.

(A) Structure of natural pentasaccharide DEFGH, where D, E, F, G and H labels refer to historical assignment of residue labels (Desai et al. 1998). Residue D forms the non-reducing end, while H is at the reducing end of the polysaccharide chain. Groups highlighted in blue are critical for high-affinity interaction with antithrombin. R1, R2, R3 and R4 groups are variable groups. (B) The UANRE library of oligosaccharides (di- to octasaccharide) has a GlcAp or IdoAp residue at the non-reducing end and ends with a GlcNp residue at the reducing end. (C) The GlcNNRE library of oligosaccharides has a GlcNp residue at the non-reducing end and either a GlcAp or IdoAp residue at the reducing end. R1, R2, R3 and R4 variations, UAp epimerization variation and conformational variations (1C4 or 2SO) for IdoAp residue generate 38 disaccharide building blocks.
Biochemical and/or biological studies based on gene knockouts of biosynthetic enzymes support the contention that should be many specific, or selective, GAG sequences within the millions present naturally to induce the biological changes in a spatiotemporal manner (Esko and Selleck 2002). Yet, obtaining a library of thousands of homogenous, synthetic GAG sequences is difficult, which precludes rigorous identification of sequences that exhibit high specificity. Likewise, a large library (e.g. >10,000) of homogenous sequences using biosynthetic enzymes has not been developed as yet and microarray-based identification of specific sequences from the mixture of GAG isolates from nature requires much further development.
Under these conditions, computational library screening approaches (Raghuraman et al. 2006, 2010; Agostino et al. 2014) offer considerable promise for identifying sequences that exhibit high specificity. The rapid increase in computational power has enabled simultaneous screening of GAG sequences so as to afford detailed understanding of structure–function relationships. Yet, modeling GAGs is also fraught with challenges. The computational power is still not sufficient to address the entire theoretical conformational search space of GAGs. For example, a simple, unsulfated H/HS disaccharide (GlcAp(1 → 4)GlcNp) possesses 11 rotatable bonds, which may require a study of up to 1017 bond rotations (Assuming that conformational energetics are calculated every 10°, 36 rotations will have to be analyzed for every rotatable bond. For the unsulfated disaccharide containing 11 rotatable bonds, 3611 = 1.32 × 1017 rotations must be analyzed to understand the entire conformational space.) to understand conformational energetics. For sulfated disaccharides (and higher oligomers), the conformational search space is much larger. Another fundamental challenge is the difficulty of computationally parsing the rare specific GAG interactions from the horde of non-specific interactions. The primary contributory factor to this state is the surface exposure of GAG-binding sites on proteins that induces a primarily electrostatic recognition (Desai 2013).
Despite these difficulties, several computational approaches have been presented in the literature. For example, standard molecular dynamics and docking were used to deduce the pentasaccharide binding site on antithrombin (Grootenhuis and van Boeckel 1991; Bitomsky and Wade 1999). More recently, Agostini et al. (2014) have presented a study of different docking protocols in mapping the binding geometry of GAGs on several proteins. Likewise, we have presented an algorithm that attempted to identify “high affinity, high-specificity” GAG sequences based on two tandem, logical filters including (1) the GOLD score (the “affinity” filter) and (2) consistency of binding (the “specificity” filter) (Raghuraman et al. 2006, 2010). The dual-filter strategy rapidly sorted a small library of H/HS hexasaccharide sequences binding to AT into “specific” and “non-specific” sequences. Yet, this first-generation approach was limited in scope. Herein, we expand the robustness of our GOLD-based combinatorial virtual library screening (CVLS) algorithm by establishing a set of parameters necessary for identification of “high-specificity” sequences, if any, from a haystack of more than 100,000 GAG sequences. Our modified CVLS approach can be readily applied to all possible H/HS sequences ranging from disaccharide to hexasaccharide, irrespective of the number of rotatable bonds. Interestingly, our new CVLS protocol identifies that the minimal sequence capable of recognizing AT with high-specificity is the GlcAp(1 → 4)GlcNp2S3S disaccharide (or alternatively the “EF” disaccharide), which is much smaller than that identified through synthetically prepared tri- and pentasaccharide variants (Desai et al. 1998). Finally, the new CVLS algorithm affords for the first time a rational framework for designing longer H/HS sequences that bind AT with high specificity without resorting to exhaustive screening of all possible sequences.
Methods
Software
SYBYLX 1.3 (Tripos Associates, St. Louis, MO) was used for molecular visualization, minimization and for preparation of protein structures from the Protein Data Bank. GOLD, v5.1 (Jones et al. 1997) was used for molecular docking experiments. GAG sequences were built combinatorially in an automated manner using in-house SPL (SYBYL Programming Language) scripts.
GAG library generation
The first step in the CVLS approach is the generation of two libraries (Figure 2). Starting from either a UAp or a GlcNp at the non-reducing end (NRE), so as to give either a UANRE or GlcNNRE library, respectively, appropriate number of GlcAp (in 4C1 ring pucker), IdoAp (in either 1C4 or 2SO) and GlcNp (in 4C1) residues were added in a combinatorial manner to generate each library of desired chain length (di-, tetra-, hexa- or octasaccharide). The co-ordinates for the two libraries were generated in an automated fashion with a series of SPL scripts and a set of 38 naturally occurring, disaccharide building blocks belonging to each of the UAp-GlcNp (Figure 1B) and GlcNp-UAp (Figure 1C) series (Esko and Selleck 2002). Herein, the different monosaccharide units are substituted with N-acetyl, N-sulfate or O-sulfate groups (Mulloy and Forster 2000), which gives rise to unique sequences. To name each unique H/HS sequence, the symbolic representation employed in the GLYCAM (Kirschner et al. 2008) designation was used. Briefly, the letter “Z” was used for GlcAp, “u” for IdoAp and “Y” for GlcNp. Similarly, ring conformations were encoded as “a” for 1C4, “b” for 4C1 and “c” for 2SO conformations (Cremer and Pople 1975; Forster and Mulloy 1993; Rao et al. 1998). Substituents on rings were represented as “H” (for unsubstituted 2-position), “C” (for N-acetyl), “2” (for N- or O-sulfate), “3” (for 3-O-sulfate) and “6” (for 6-O-sulfate). Anomeric carbon configuration was encoded as “A” for α and “B” for β. This monosaccharide nomenclature is also shown in Table I. Analysis of the available crystal structures showed that the interglycosidic torsions φH (O5–C1–O1–C4′) and ψH (C1–O1–C4′–C5′) fall within a relatively narrow range and are essentially invariant irrespective of the substitution pattern (Jin et al. 1997; McCoy et al. 2003; Johnson et al. 2006; Pol-Fachin and Verli 2008). Thus, average bond torsions, shown in Table II, were used for interglycosidic linkages. The disaccharide building blocks were then used to build the desired library using an SPL script following which each sequence was minimized, in an automated manner. Thus, the two UANRE and GlcNNRE libraries contained a total of 2 × (38 × 38 × 38) = 109,744 unique H/HS hexasaccharide sequences. Likewise, the di- and tetrasaccharide libraries consisted of 76 and 2888 unique sequences, respectively.
Fig. 2.
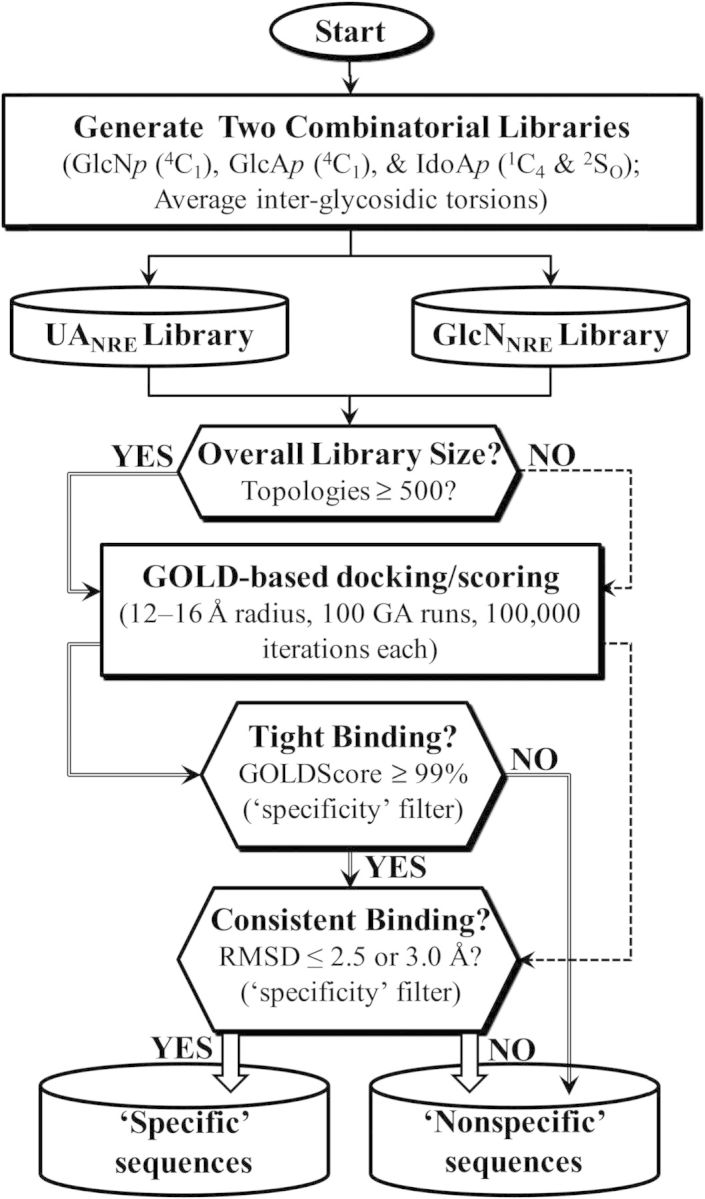
Combinatorial virtual library screening (CVLS) protocol used to study the AT– H/HS interaction. The CVLS protocol assessed the interaction of two H/HS libraries (UANRE and GlcNNRE) using a dual-filter strategy that relied on the geometric convergence filter (RMSD) to assess specificity of binding.
Table I.
Naming convention for the H/HS monosaccharides and the 38 disaccharide building blocks derived from them
| Namea | Name | Conf.b | Anomer | Disaccharide building blocks |
||
|---|---|---|---|---|---|---|
| uaA | IdoAp | 1C4 | α- | ZbB-YbCA | ua2A-YbC6A | ua2A-YbH3A |
| ua2A | IdoAp2S | 1C4 | α- | ZbB-YbC6A | uc2A-YbC6A | uc2A-YbH3A |
| ucA | IdoAp | 2SO | α- | ZbB-Yb2A | uaA-Yb2A | ua2A-YbH36A |
| uc2A | IdoAp2S | 2SO | α- | Zb2B-Yb2A | ua2A-Yb2A | uc2A-YbH36A |
| Yb2A | GlcNp2S | 4C1 | α- | ZbB-Yb26A | uc2A-Yb2A | ucA-Yb2A |
| Yb23A | GlcNp2S3S | 4C1 | α- | Zb2B-Yb26A | uaA-Yb26A | uc2A-Yb23A |
| Yb26A | GlcNp2S6S | 4C1 | α- | ZbB-Yb23A | ucA-Yb26A | |
| Yb236A | GlcNp2S3S6S | 4C1 | α- | Zb2B-Yb23A | ua2A-Yb26A | |
| Yb26A | GlcNp2S6S | 4C1 | α- | ZbB-Yb236A | uc2A-Yb26A | |
| YbCA | GlcNp2Ac | 4C1 | α- | ZbB-YbHA | uaA-Yb23A | |
| YbC6A | GlcNp2Ac6S | 4C1 | α- | uaA-YbCA | ucA-Yb23A | |
| YbHA | GlcNp | 4C1 | α- | ucA-YbCA | ua2A-Yb23A | |
| YbH3A | GlcNp3S | 4C1 | α- | ua2A-YbCA | uaA-Yb236A | |
| YbH36A | GlcNp3S6S | 4C1 | α- | uc2A-YbCA | ucA-Yb236A | |
| ZbB | GlcAp | 4C1 | β- | uaA-YbC6A | ua2A-Yb236Ac | |
| Zb2B | GlcAp2S | 4C1 | β- | ucA-YbC6A | uc2A-Yb236Ac | |
aSymbols: Z = D-GlcAp, u = L-IdoAp, Y = D-GlcNp. Ring conformations: a = 1C4; b = 4C1; c = 2SO. Substituents: H = No substitution at position 2; Ac = N-acetyl, S = sulfate; anomer configuration: A = α, B = β.
bConformation.
cDisaccharides modeled in addition to those presented by Esko and Selleck (2002).
Table II.
Average torsion across the 1 → 4 interglycosidic bonds used in this CVLS study
| Disaccharide building block | Φ (O5–C1–O1–C4′) | Ψ (C1–O1–C4′–C5′) |
|---|---|---|
| GlcAp(1 → 4)GlcNp | −81.8 | −114.0 |
| IdoAp(1 → 4)GlcNp | −87.7 | −128.3 |
| GlcNp(1 → 4)GlcAp | 91.1 | −151.6 |
| GlcNp(1 → 4)IdoAp | 87.4 | −132.3 |
Preparation of AT and GAG sequences for docking
The coordinates for the activated form of AT were extracted from the crystal structure of the ternary AT–pentasaccharide–thrombin complex (Brookhaven Protein Data Bank entry 1TB6) (Li et al. 2004). Likewise, thrombin coordinates were extracted from both the 1TB6 structure and 1XMN structure (chains A and B) (Carter et al. 2005). Hydrogen atoms were added in SYBYL X1.3 and the structure minimized with fixed heavy-atom co-ordinates using the Tripos forcefield for a maximum of 5000 iterations subject to a termination gradient of 0.05 kcal/(mol Å). Energy minimization of the modeled GAG, AT and thrombin structures was performed using the Tripos forcefield with Gasteiger-Hückel charges, a fixed dielectric constant of 80 and a non-bonded cutoff radius of 8 Å.
Docking of the GAG sequences
Molecular docking of the library of sequences onto the activated form of AT was performed using GOLD v.5.1 (Jones et al. 1997). GOLD is a “soft docking” method that implicitly handles local protein flexibility by allowing a small degree of interpenetration, or van der Waals overlap, of ligand and protein atoms. GOLD also optimizes the positions of hydrogen-bond donating atoms on Ser, Thr, Tyr, Lys and Arg residues as part of the docking process. The binding site in AT was defined to cover key heparin-binding residues including Lys11, Arg13, Arg46, Arg47, Trp49, Lys114, Phe121, Lys125, Arg129 and Arg132 (Jin et al. 1997; Desai 2005; Pike et al. 2005). Similarly, the binding site in thrombin was defined to encompass basic residues of exosite 2 including Arg93, Arg101, Arg126, Arg165, Arg233, Lys236 and Lys240. The grid center was defined as the center of the enclosure containing these residues.
For the GAG sequences, the interglycosidic bonds were constrained. In addition to the two libraries for each chain length, docking was also performed for the heparin pentasaccharide sequence (Figure 1A) using the set of optimized parameters. GOLD starts with a population of 100 arbitrarily docked ligand orientations, evaluates them using a scoring function (the GA “fitness” function) and improves their average “fitness” by an iterative optimization procedure that is biased towards high scores. As the initial population is selected at random, several such GA runs are required to more reliably predict correct bound conformations. The optimized parameters included 100 GA runs for each sequence docked onto a binding site of 5–16 Å radius, depending upon chain length and number of rotatable bonds, using a maximum of 100,000 iterations that are continuously evaluated by the GOLD score and/or root-mean-square difference (RMSD) between top-ranked solutions. Collectively, these 100 GA runs form one docking experiment from which the top two solutions were considered for further analysis. Experiments were minimally performed in triplicate, which would yield at least six solutions. To enhance efficiency, the GA was set to preterminate if the top two ranked solutions were within 2.5 Å RMSD. A one or two-step docking protocol was utilized depending on the library size, as described in Figure 2. If the library contained <500 sequences, the analysis relied only on the RMSD between the top six ranked solutions obtained in three independent experiments for every sequence. If the number of sequences was much >500, a two-step protocol involving selection of the most promising sequences based on their GOLD scores (the “affinity” filter) followed by redocking of the selected sequences and evaluation of RMSD between the top-ranked solutions (the “specificity” filter). Docking was driven by equation (HBEXT and VDWEXT are the non-bonded intermolecular hydrogen bond and van der Waals terms, respectively) to prioritize different poses, as reported earlier (Raghuraman et al. 2006).
Results and discussion
Development of a more robust CVLS approach for identifying “high-specificity” sequences
Our previous CVLS protocol could be applied to a small library of H/HS sequences (∼7000 sequences) and screened a limited domain of 3D space to assess “specificity” of binding (Raghuraman et al. 2006). However, the repertoire of nature's sequences is huge, of which the majority are never studied rigorously. Thus, a more robust CVLS protocol that can be optimally applied to a larger H/HS library is desirable. Considering the dramatic increase in conformational and configurational space with the size of the library, it was important to define the optimal parameter set that ensures reproducible and accurate outcome. Hence, we selected a representative group of 65 H/HS hexasaccharide sequences using the following rationale. Fifteen hexasaccharide sequences contained the DEF structure, which is known to be the key high-specificity element of the DEFGH pentasaccharide. This group was identified as GDEF. Likewise, 15 hexasaccharide sequences were selected to contain either the EFG or the FGH structure. These were identified as GEFG and GFGH groups. The GFGH group was expected to be the most non-specific group based on literature reports (Desai et al. 1998). Two additional groups of 10 library members each were constructed to assess specificity elements. The GUSU group contained fewer sulfate groups than the GDEF group, whereas the GSSS contained more sulfate groups (Supplementary data, Table S1).
Because the size of the library in this limited study was small (<500 members), we utilized the one-step CVLS protocol (Figure 2) to assess how well a particular sequence binds to AT in multiple docking runs. A low RMSD (<2.50 Å) between multiple interaction poses in a GA-based (The GA-based approach incorporates domain swaps and mutations so as to screen essentially the entire 3D space for each sequence. In other words, a GA-based run attempts to dock an H/HS sequence onto the binding site with all possible conformations/configurations so as to identify the most optimal fit. This enhances the probability of identifying interactions arising from key structural features on the H/HS sequence) search suggests high consistency of binding. When only a few sequences present in a library display such high consistency of binding, it suggests specific binding with the protein. In contrast, sequences that bind with poor consistency (RMSD > 2.50 Å) possess no structural features that uniquely recognize the target and hence are likely to be non-specific. Thus, based on the literature (Desai et al. 1998; Capila and Linhardt 2002; Raghuraman et al. 2006), the GDEF group should exhibit the highest proportion of sequences that display RMSD < 2.5 Å (high specificity), while the GFGH and GUSU group should exhibit relatively poor specificity. These predictions allowed a rigorous assessment of CVLS parameters that impact the robustness of the protocol including the number of GA runs (10 or 100) and the size of docking radius.
Figure 3 shows CVLS results for the five groups of H/HS hexasaccharide sequences. For each group, the proportion of sequences displaying RMSD < 2.5 Å was found to be highest at 14 Å and 100 GA runs. More importantly, GDEF displayed higher proportion “specific” sequences, as expected. In contrast, the “non-specific” group GFGH displayed much lower proportion mirroring biochemical results (Desai et al. 1998). The GUSU group displayed the least specificity, as one would expect based on the absence of key sulfate groups.
Fig. 3.
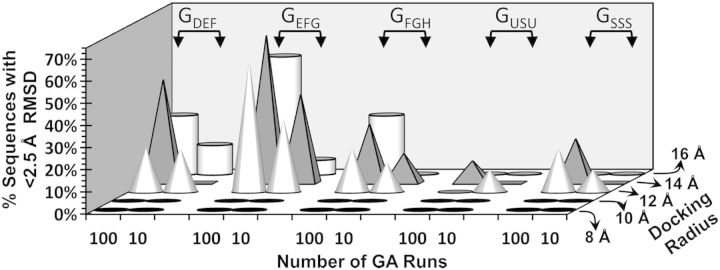
Optimization of GOLD-based CVLS protocol for H/HS hexasaccharide sequences. Parameters including docking radius (8–16 Å), number of GA runs (either 10 or 100) and number of iterations (10,000 or 100,000; not shown here) were evaluated in a rigorous manner for groups of H/HS hexasaccharide sequences including GDEF, GEFG, GFGH, GUSU and GSSS, which refer to 15 sequences containing the DEF structure, 15 sequences containing the EFG structure, 15 sequences containing the FGH structure, 10 sequences containing suboptimal level of sulfation and 10 sequence containing higher level of sulfation, respectively. The structures of the 65 hexasaccharide sequences studied are listed in Supplementary data, Table S1.
Interestingly, a 16 Å radius failed to reliably identify “specific” sequences. This was an unusual result considering that search with 14 Å radius identifies majority of sequences. Most probably, the failure of 16 Å radius search arises from incomplete conformational search imposed by the limitation in the maximum number of GA iterations (100,000) used in this study. It is well established that an increase in docking radius dramatically increases the conformational search space, which in turn requires orders of magnitude increase in the number of GA iterations. At the other extreme, the reason why searches with 8 and 10 Å radii failed is most likely due to the size of the binding site becoming too small for hexasaccharide sequences at these radii. With regard to the number of GA iterations, the results show that 10,000 runs appear to not reliably perform a thorough search of the 3D space. We did not screen an order of magnitude increase in GA iterations, i.e. 1,000,000 runs, because it would be time-wise not feasible. In combination, the results for hexasaccharide sequences show that an arbitrary choice of docking parameters may not yield optimal results within the allocated time frame or make take inexorbitant time to complete. Thus, GAG docking experiments should be approached with measured steps.
Inference of structure on the specificity of recognition
The above study also led to two key advances with regard to H/HS–AT interaction. (1) It is generally assumed that high sulfation level of a GAG chain ensures protein binding. This arises from the non-directional nature of electrostatic forces, which favor recognition of practically any collection of basic residues. In direct contrast to this assumption, the GSSS group, which comprises of sequences that are more sulfated on average than the GDEF group (see Supplementary data, Table S1), displays very few “specific” sequences (Figure 3). In fact, this proportion is even lower than that for GFGH, which is known to be non-specific in solution, and just slightly better than that for GUSU. Clearly, for high-specificity GAG–protein systems hyper sulfation destroys specificity of interaction. (2) The GEFG group has not been studied well in the literature, except for tetrasaccharide EFGH (Petitou et al. 1997; Desai et al. 1998). The CVLS results show that GEFG group displays the highest proportion of sequences with RMSD < 2.5 Å. This proportion is even higher than that for GDEF suggesting that the EFG trisaccharide of the DEFGH sequence may be a better cause of specific recognition of AT. This aspect is further addressed below.
Application of the CVLS protocol to a large H/HS library
Can the above more robust CVLS protocol reliably identify specific sequences from a large library? To address this question, a combinatorial library of H/HS hexasaccharide sequences was generated from all possible monosaccharide residues found in nature (Figure 1B and C, Table I), except for the rare free GlcNp. As in our earlier study, the library considered the conformational flexibility of IdoAp residues in an explicit manner through the inclusion of both the 1C4 and 2SO conformations and utilized the “average backbone” geometry of the inter-glycosidic bonds (Raghuraman et al. 2006). Yet, a significant advance over the earlier study was the explicit consideration of sequences with two different NREs. This led to two libraries, named as UANRE and GlcNNRE libraries, which were treated independently for computational purposes (Figure 2). Considering that AT binding is sequence specific, the UANRE and GlcNNRE libraries may yield different results. Each library contained 54,872 distinct hexasaccharide sequences made combinatorially from 38 disaccharide building blocks, which were generated in a fully automated manner.
The CVLS protocol for the larger library consisted of two steps. The first step involved docking each of the >50,000 hexasaccharides onto AT using the parameters developed above and analyzing the poses using GOLDScore (see Methods). The highest ranking 54 sequences (or top 0.1%) from each library were re-docked in three independent runs and the top two poses of each run were utilized for assessing consistency of binding. The RMSD between the six poses of every sequence was calculated and sequences displaying values <2.5 Å were identified as most promising from the perspective of specificity. This analytical approach eliminates the need for a reference co-crystal structure, which may not be possible for many GAG–protein systems, and thus, greatly expands the applicability of CVLS protocol.
Our CVLS protocol affords deduction of binding specificity from both biological and chemical considerations. Biological specificity refers to a unique mode of interaction in the binding site among many possible modes, whereas chemical specificity refers to a unique ligand sequence among the many sequences available. Because the CVLS protocol operates on a large library and attempts to identify “needles in a haystack”, the final identified GAG sequences capture features of chemical specificity. Likewise, the GA-based identification on one binding mode binding (i.e. low RMSD value) from among many likely captures features of biological specificity. Yet, the CVLS protocol cannot be expected to capture all the chemical and biological specificity features of a GAG–protein system in few sequences because the GAG conformational search space is enormous. However, the dual-filter protocol greatly enhances the probability of rapidly and accurately identifying “specific” GAG sequences.
Of the 54 sequences identified from each of the UANRE and GlcNNRE hexasaccharide libraries following the 1st filter, 10 and 24 sequences, respectively, satisfied the 2nd filter (Table III). These poses were then compared with the pentasaccharide DEFGH geometry of the co-crystal structure (Li et al. 2004) and found to be essentially identical (not shown). Further, the hydrogen-bonding analysis using LIGPLOT (Wallace et al. 1995) showed that the sequences identified by CVLS bound to AT in a manner similar to DEFGH. The 34 sequences represent a significant increase in identification of specific sequences from the 10 such sequences identified in our earlier work from a library of 6859 (Raghuraman et al. 2006). This highlights the enhanced robustness of the new algorithm. More importantly, ua2A-YbCA-ua2A-YbCA-uaA-Yb26A, or alternatively IdoAp2S-GlcNp2Ac-IdoAp2S-GlcNp2Ac-IdoAp-GlcNp2S6S, a false-negative hexasaccharide identified in our first attempt, was effectively eliminated by in this more careful protocol.
Table III.
H/HS hexasaccharide sequences from two libraries of 54,872 sequences each that satisfied the dual-filter CVLS strategy for antithrombin
| No. | Hexasaccharide sequencea | GoldScoreb | No. of H-bondsc |
|---|---|---|---|
| UANRE library | |||
| 1 | ZbB-Yb26A-ZbB-Yb236A-ucA-Yb2A | 137.81 | 13 |
| 2 | uc2A-Yb26A-ZbB-Yb236A-ucA-Yb2A | 136.76 | 13 |
| 3 | ucA-YbC6A-Zb2B-Yb23A-ucA-Yb2A | 133.15 | 14 |
| 4 | ua2A-YbH36A-ZbB-Yb236A-ucA-Yb2A | 132.83 | 12 |
| 5 | ucA-Yb26A-ZbB-Yb23A-ucA-YbCA | 132.66 | 14 |
| 6 | ucA-YbC6A-ZbB-Yb236A-ucA-Yb2A | 132.42 | 14 |
| 7 | ZbB-Yb26A-ZbB-Yb236A-ucA-YbCA | 132.39 | 12 |
| 8 | uaA-Yb23A-ZbB-Yb236A-ucA-Yb2A | 131.63 | 12 |
| 9 | ucA-Yb26A-ZbB-Yb236A-ucA-Yb2A | 131.25 | 14 |
| 10 | ua2A-Yb23A-ZbB-Yb236A-ucA-Yb236A | 129.54 | 14 |
| GlcNNRE library | |||
| 1 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB | 147.76 | 12 |
| 2 | Yb26A-ua2A-Yb26A-ZbB-Yb236A-ZbB | 142.34 | 11 |
| 3 | Yb2A-ua2A-YbC6A-ZbB-Yb236A-ZbB | 141.45 | 11 |
| 4 | Yb23A-uaA-YbC6A-ZbB-Yb23A-ZbB | 139.57 | 14 |
| 5 | Yb2A-ua2A-Yb26A-Zb2B-Yb236A-ZbB | 138.59 | 14 |
| 6 | Yb26A-uaA-YbC6A-ZbB-Yb236A-ZbB | 138.57 | 10 |
| 7 | Yb26A-ua2A-Yb236A-ZbB-Yb236A-ZbB | 136.99 | 10 |
| 8 | Yb236A-ua2A-YbC6A-ZbB-Yb236A-ZbB | 135.44 | 11 |
| 9 | Yb2A-ua2A-Yb26A-Zb2B-Yb23A-ZbB | 134.74 | 10 |
| 10 | Yb26A-uaA-YbC6A-ZbB-Yb236A-uc2A | 134.55 | 13 |
| 11 | Yb2A-uaA-Yb26A-ZbB-Yb236A-ZbB | 134.3 | 12 |
| 12 | Yb26A-ua2A-Yb26A-Zb2B-Yb23A-ZbB | 134.23 | 12 |
| 13 | Yb2A-uaA-Yb236A-ZbB-Yb236A-uc2A | 133.72 | 11 |
| 14 | Yb26A-ua2A-Yb26A-ZbB-Yb23A-ZbB | 133.08 | 12 |
| 15 | Yb23A-ua2A-YbC6A-ZbB-Yb236A-uc2A | 132.82 | 11 |
| 16 | Yb236A-ua2A-YbC6A-ZbB-Yb23A-ZbB | 132.60 | 10 |
| 17 | Yb2A-ua2A-Yb26A-ZbB-Yb236A-ZbB | 132.54 | 12 |
| 18 | Yb26A-uaA-YbC6A-ZbB-Yb23A-uc2A | 132.43 | 11 |
| 19 | Yb236A-ua2A-Yb236A-ZbB-Yb236A-ZbB | 131.50 | 13 |
| 20 | Yb23A-uc2A-Yb26A-Zb2B-Yb236A-ZbB | 131.21 | 13 |
| 21 | YbC6A-ZbB-Yb236A-ucA-Yb2A-ua2A | 130.78 | 16 |
| 22 | Yb23A-uaA-YbC6A-ZbB-Yb23A-uc2A | 130.29 | 9 |
| 23 | Yb23A-ua2A-Yb236A-ZbB-Yb236A-ZbB | 129.75 | 11 |
| 24 | Yb236A-ua2A-Yb26A-Zb2B-Yb236A-uc2A | 129.06 | 13 |
aSee definitions of residue labels and substitution in Table I.
bRefers to modified GoldScore, as defined in Methods.
cNumber of hydrogen bonds calculated using LIGPLOT.
A comparison of the binding geometry of the 10 and 24 sequences from the UANRE and GlcNNRE libraries reveals an interesting insight. Although each sequence is distinct, the binding poses attempt to satisfy a core group of interactions arising from the D, E and F residues. This induces a shift in frame of one residue between UANRE and GlcNNRE sequences (Figure 4). This also explains why more high-specificity sequences were identified from the GlcNNRE library in comparison to the UANRE library. The difference in frame between the two libraries affords interactions with the extended heparin-binding site residues for the GlcNNRE library, which are not realized by sequences of the UANRE library (Figure 4B, Table III). These additional interactions contribute to the specificity of binding.
Fig. 4.
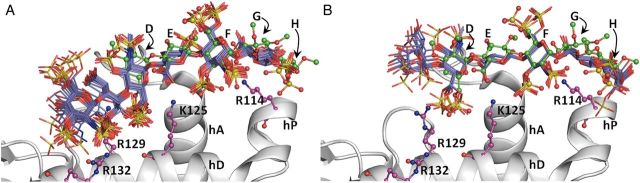
CVLS predicted hexasaccharide sequences from the GlcNNRE (A) and UANRE libraries (B) containing 54,872 sequences each. Shown are overlays of the docked poses of hexasaccharide sequences that bind AT with “high specificity” by satisfying the dual-filter strategy. (A) Twenty-four sequences (blue sticks) from the GlcNNRE library and (B) 24 sequences (blue sticks) from UANRE hexasaccharide library. Helices A (hA), D (hD) and P (hP) of antithrombin are shown in ribbon form and residues Arg132, Arg129, Lys125 and Arg114 are shown in ball and stick display. The crystal structure of DEFGH in green ball and sticks display is shown to highlight correspondence with the CVLS predicted poses.
Importance of appropriate restriction on conformational search space
The results with hexasaccharide sequences indicated that CVLS does not work well with too large a conformational search space, as demonstrated by an optimal docking radius of 14 Å. We reasoned that presumably this would hold true for oligosaccharides with other lengths too. Thus, we docked tetrasaccharide sequences containing the DEF structure onto AT using docking radius ranging from 8 to 16 Å under otherwise identical conditions. Theoretically, each tetrasaccharide sequence should bind with a low RMSD because of the presence of the DEF scaffold. Yet, the results revealed that consistent docking was best obtained with a radius of 12 Å (Figure 5). For disaccharides, we studied all 78 sequences and found that a docking radius of 10 Å was most optimal. Thus, the results highlight the importance of selecting appropriate docking radius, which is surrogate for restricting conformational search space, for identifying “needles in a haystack”. For H/HS sequences, it appears that di-, tetra- and hexasaccharide sequences are best studied by using docking radius of 10, 12 and 14 Å, respectively.
Fig. 5.

Optimization of docking radius to be used in CVLS of H/HS tetrasaccharide sequences. A library of 17 DEF containing tetrasaccharides was assessed for “specificity” of binding using varying docking radius. See Supplementary data, Figure S1 for structural poses of the specific tetrasaccharide sequences.
CVLS predicts the “minimal” H/HS sequence that exhibits high specificity for AT
Although pentasaccharide DEFGH (Figure 1A) is recognized as the minimal AT-specific sequence, detailed biochemical study with pentasaccharide variants led to the conclusion that trisaccharide DEF was minimally needed (Desai et al. 1998). The development of the current more robust CVLS protocol presented an opportunity to assess this computationally using the libraries of tetrasaccharide (2888 sequence), disaccharide (76 sequences) and monosaccharides (15 sequences). The results showed that 14 UANRE and 16 GlcNNRE tetrasaccharide sequences (Supplementary data, Figure S1) and 3 UANRE (and none GlcNNRE) disaccharide sequences satisfied the RMSD filter (Figure 6). Each of the three disaccharide sequences contained the GlcAp(1 → 4)GlcNp2S3S structure (the EF disaccharide, see Supplementary data, Table S3), which was also present in the high-specificity 30 tetrasaccharides (see Supplementary data, Table S2) identified from the library of 2888 sequences. Reducing the size further to monosaccharides eliminated interaction specificity completely. Likewise, neither the GlcNp6S(1 → 4)GlcAp sequence nor the GlcNp2S3S(1 → 4)GlcAp sequence, i.e. neither the DE nor the FE sequence, satisfied the consistency of binding filter (Figure 6B). The results suggested that the exquisite specificity features displayed by the H/HS–AT system arises from the EF disaccharide motif.
Fig. 6.
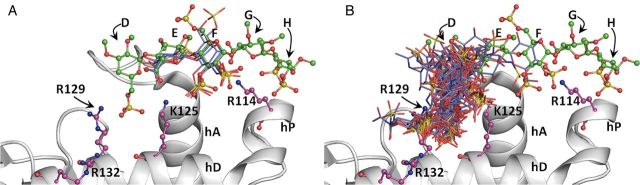
CVLS predicted disaccharide sequences from the UANRE (A) and GlcNNRE libraries (B). Only three disaccharide sequences (blue sticks), each related to EF structure of pentasaccharide DEFGH, were found to satisfy the dual-filter strategy from the library of 78 UANRE sequences (A). None of the 78 possible disaccharides (blue sticks) belonging to the GlcNNRE library bound antithrombin with “high specificity”. Helices A (hA), D (hD) and P (hP) of antithrombin are shown in ribbon form and residues Arg132, Arg129, Lys125 and Arg114 are shown in ball and stick display. The crystal structure of DEFGH in green ball and sticks display is shown to highlight correspondence with the CVLS predicted poses.
The above deduction, in fact, has experimental support. Our detailed studies using stopped flow fluorimetry (Desai et al. 1998) on a group of variants of pentasaccharide DEFGH have shown that removal of residue D (as in tetrasaccharide EFGH) or residues G & H (as in trisaccharide DEF) does not affect specificity of AT recognition. But elimination of residues D and E (as in trisaccharide FGH) resulted in complete loss in specificity. Thus, key elements of specific recognition of antithrombin are actually resident in the disaccharide sequence EF. Thus, these CVLS studies significantly advance understanding on the structural basis of specificity with regard to the antithrombin–heparin system.
Although disaccharide sequence EF may be all that is necessary for specific recognition of AT, this does not imply residues D, G and H of pentasaccharide DEFGH are not important. This work shows that if the EF sequence is not present, then the sequence(s) display(s) multiple modes of binding (i.e. lack biological specificity). The function of D, G and H residues is to contribute binding energy. This is the reason why pentasaccharide DEFGH sequence is a better pharmaceutical agent than tetrasaccharide EFGH or trisaccharide FGH.
A simple CVLS-based algorithm for designing longer H/HS sequences
Considering that many proteins bind longer GAG sequences (Capila and Linhardt 2002; Gandhi and Mancera 2008), we sought to assess the application of our CVLS approach to the design of H/HS sequence(s) longer than hexasaccharide. Yet, designing longer sequences de novo is considerably more challenging. For example, a de novo library of octasaccharide sequences built from the 38 natural disaccharide building blocks would consist of 384, or 2,085,136, unique sequences for each of the two libraries, UANRE and GlcNNRE. Exhaustive screening at this scale is not possible in a reasonable timeframe with current computational power. Hence, we developed a reductionist approach. We reasoned that the 3D space on either side of the most specific hexasaccharide sequence(s) could be explored using our CVLS protocol to derive the most optimal octasaccharide sequence(s).
To test this design algorithm, the docked poses of the five best ranked hexasaccharide sequences from the UANRE library, which satisfied the dual filters, were selected and 38 disaccharide blocks were combinatorially attached to either the NRE or the RE to derive two octasaccharide libraries of 190 sequences each (Figure 7A). Likewise, the same procedure was applied to the five best GlcNNRE hexasaccharide sequences to prepare two libraries of 190 sequences each containing disaccharide extensions at either the NRE or the RE. The octasaccharide sequences were built in an automated manner, their ring conformations and interglycosidic torsions assessed for consistency, and then sequences docked onto AT using the dual-filter CVLS protocol described in Figure 2. Following the application of the first filter, the 10 best ranked sequences of the 190 were selected for the consistency of binding analysis.
Fig. 7.
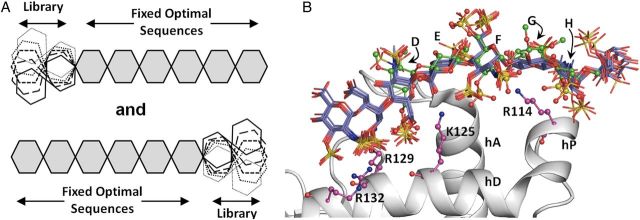
(A) Incremental neighborhood optimization strategy used in the design of octasaccharide sequences. Five hexasaccharide sequences that were found to be most optimal from each of the two libraries (UANRE and GlcNNRE) were selected and then 38 disaccharide sequences of UANRE and GlcNNRE libraries were appended at either the reducing or the non-reducing ends. The total of 5 × 38 × 4 = 760 octasaccharide sequences were then studied using the dual-filter CVLS protocol. (B) Ten octasaccharide sequences from the GlcNNRE library and six sequences from the UANRE library satisfied the CVLS dual-filter criteria. Helices A (hA), D (hD) and P (hP) of antithrombin are shown in ribbon form and residues Arg132, Arg129, Lys125 and Arg114 are shown in ball and stick display. The crystal structure of DEFGH in green ball and sticks display is shown to highlight correspondence with the CVLS predicted poses.
Although each sequence in these four libraries contained the DEFGH sequence, which is theoretically expected to bind to AT with 100% efficiency, the CVLS results suggest a striking preference for the type of library. No octasaccharide sequence satisfied the “specificity” filter for UANRE and GlcNNRE libraries to which disaccharides were added at the RE and NRE, respectively. In contrast, 6 and 10 octasaccharide sequences passed the consistency of binding filter from the UANRE and GlcNNRE libraries possessing disaccharides at the NRE and RE, respectively (Figure 7B, Table IV). The docked poses indicate that longer H/HS sequences possess additional interactions with residues of the extended heparin-binding site of AT, especially Arg132. More importantly, the results reveal for the first time the exact order of residues flanking DEFGH that contribute to AT binding and specificity (see Table IV).
Table IV.
H/HS octasaccharide sequences that satisfied the CVLS strategy for antithrombin
| No. | Octasaccharide sequencea | GoldScoreb | No. of H-bondsc |
|---|---|---|---|
| UANRE library | |||
| 1 | Zb2B-Yb2A-ZbB-Yb26A-ZbB-Yb236A-ucA-Yb2A | 145.53 | 16 |
| 2 | ucA-Yb26A-uc2A-Yb26A-ZbB-Yb236A-ucA-Yb2A | 137.10 | 13 |
| 3 | uc2A-Yb26A-uc2A-Yb26A-ZbB-Yb236A-ucA-Yb2A | 136.19 | 14 |
| 4 | uaA-Yb236A-uc2A-Yb26A-ZbB-Yb236A-ucA-Yb2A | 134.42 | 14 |
| 5 | uaA-Yb2A-uc2A-Yb26A-ZbB-Yb236A-ucA-Yb2A | 131.93 | 13 |
| 6 | uc2A-YbH3A-uc2A-Yb26A-ZbB-Yb236A-ucA-Yb2A | 130.27 | 12 |
| GlcNNRE library | |||
| 1 | Yb2A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-Yb26A-Zb2B | 149.99 | 13 |
| 2 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-YbH3A-ua2A | 149.62 | 13 |
| 3 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-YbCA-uaA | 148.30 | 10 |
| 4 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-Yb26A-ua2A | 147.56 | 14 |
| 5 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-Yb23A-uc2A | 146.40 | 13 |
| 6 | Yb2A-ua2A-YbC6A-ZbB-Yb236A-ZbB-Yb236A-ZbB | 146.09 | 15 |
| 7 | Yb2A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-Yb236A-ua2A | 141.69 | 11 |
| 8 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-Yb23A-ZbB | 141.67 | 12 |
| 9 | Yb23A-ua2A-Yb26A-Zb2B-Yb236A-ZbB-YbCA-ZbB | 138.82 | 11 |
| 10 | Yb2A-ua2A-YbC6A-ZbB-Yb236A-ZbB-Yb26A-ua2A | 136.58 | 9 |
aSee definitions of residue labels and substitution in Table I.
bRefers to modified GoldScore, as defined in Methods.
cNumber of hydrogen bonds calculated using LIGPLOT.
The computational results also provide structural basis for biochemical studies observed earlier by Belzar et al. (2000). In their work, extension of the DEFGH sequence at the RE end was found to induce a frameshift in binding. We find that extension at both the NR and RE ends induces an equivalent shift in the frame of the sequences when compared with pentasaccharide DEFGH (Supplementary data, Figure S2). This frameshift arises to induce better interaction of the extended sequence with AT, which measurably enhances the binding affinity. Also in a recent study, an octasaccharide sequence with two 3-O-sulfated GlcNp residues was reported to induce higher affinity to AT in comparison to pentasaccharide DEFGH (Guerrini et al. 2013). A review of our CVLS results (see Table IV) shows that nine such sequences were identified as the most optimal and specific octasaccharide sequences.
Thus overall, the success of the CVLS algorithm implies that this approach could be used to design longer H/HS sequences one disaccharide block at a time. This is the first simple and intuitive approach to computationally design longer GAG sequences. The approach obviates extensive library screening and may be termed as incremental neighborhood optimization strategy.
Application of the CVLS protocol to identify key heparin sequences binding to thrombin —
To assess the applicability of the CVLS approach to proteins other than AT, we studied the thrombin–heparin system. Thrombin is a key protease of the blood coagulation cascade and is widely recognized as interacting with heparin in a non-specific manner (Olson et al. 1991; Mosier et al. 2012). Heparin binds to thrombin in anion-binding exosite 2 (Carter et al. 2005). A crystal structure of heparin–thrombin complex has been reported (“1XMN”; Carter et al. 2005), which presents two different modes of binding within exosite 2 for the common heparin hexasaccharide, further alluding to lack of specificity presented in the literature.
Each biochemical study performed to date with the thrombin–heparin system has relied either on the most common heparin sequence, i.e. (IdoAp2S-GlcNp2S6S)n, or on a mixture of unfractionated heparin sequences. We reasoned that because our CVLS approach affords a rigorous study of every sequence possible in nature, it would be better suited to assess elements of specificity in heparin binding to thrombin. Thus, the optimized CVLS approach deduced above was applied to thrombin using the library of >50,000 hexasaccharide sequences. Application of the “affinity” filter led to identification of the best 54 sequences (top 0.1%), which were re-docked to assess the consistency of binding. Only one sequence, i.e. IdoAp2S-GlcNp2S6S-IdoAp2S-GlcNp2S-IdoAp-GlcNp2S3S6S, satisfied the second filter (Table V). The pose of this sequence was compared with that of the sequence reported in the heparin–thrombin co-crystal structure and found to be essentially identical (Figure 8A). Thus, the CVLS technology was able to correctly predict the preferred sequence and its binding geometry. It is instructive to note that the GOLDScore for this sequence is only ∼120 (Table V), whereas in comparison, the GOLDScore for DEFGH containing sequences binding to AT is much higher (Table III). This implies that the predicted affinity of the thrombin binding sequence is relatively low.
Table V.
H/HS hexasaccharide sequences that satisfied the CVLS strategy for thrombin
| No. | Hexasaccharide sequencea | GoldScoreb | No. of H-bondsc |
|---|---|---|---|
| 1XMN | |||
| 1 | ua2A-Yb26A-uc2A-Yb2A-uaA-Yb236A | 121.48 | 14 |
| 1TB6 | |||
| 1 | ua2A-Yb23A-uc2A-YbC6A-ua2A-Yb26A | 136.13 | 13 |
| 2 | ua2A-Yb26A-ucA-Yb23A-ua2A-Yb26A | 130.38 | 13 |
| 3 | ua2A-YbC6A-uc2A-YbC6A-ua2A-YbH36A | 129.36 | 12 |
| 4 | ua2A-YbCA-uc2A-Yb26A-ua2A-YbH36A | 128.91 | 14 |
aSee definitions of residue labels and substitution in Table I.
bRefers to modified GoldScore, as defined in Methods.
cNumber of hydrogen bonds calculated using LIGPLOT.
Fig. 8.
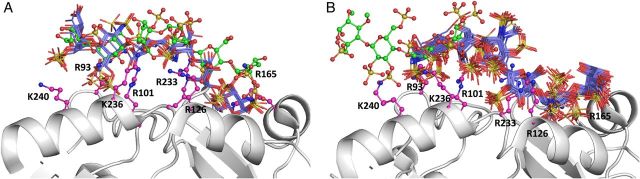
CVLS predicted hexasaccharide sequences that bind with a consistent mode onto exosite 2 of thrombin. (A) Multiple poses of the only sequence (blue sticks) that satisfied the dual-filter CVLS strategy for 1XMN crystal structure of thrombin and (B) poses of the four sequences (blue sticks) identified for the 1TB6 structure. Thrombin surface and key exosite 2 residues are shown in ribbon and ball and stick representations, respectively. Heparin present in the co-crystal structure is shown as green ball and sticks.
To assess these results further, we applied the CVLS technology to the 1TB6 thrombin structure (Li et al. 2004). This structure presents thrombin in a ternary complex with AT and heparin. A key aspect of this structure is that thrombin is bound to a polymeric heparin sequence and therefore presents slightly altered exosite 2 electrostatic surface features (not shown). Once again, the best 54 “affinity” filtered sequences from the >50,000 studied in the first stage were processed for the consistency of binding in multiple docking experiments. Only four sequences were identified in this study (Figure 8B, Table V). All the four sequences bind with relatively high consistency but weaker GOLDScore. The binding pose of these four sequences matches well with that of highly sulfated heparin monomers observed in the 1TB6 structure. The hydrogen-bonding analysis using LIGPLOT (Wallace et al. 1995) suggested favorable interactions of the sequences with key residues of exosite 2 including Arg93, Arg101, Arg126, Arg165, Arg233, Lys236 and Lys240, as expected. Interestingly, all four hexasaccharide sequences identified by CVLS displayed a repeating disaccharide unit of (IdoA –GlcN)3 with variations in the position of sulfate groups. This is essentially identical to the common repeating sequence present in unfractionated heparin. Thus, the heparin–thrombin system turns out to be chemically non-specific, although biologically, a distinct mode of sulfated hexasaccharide binding in exosite 2 is evident.
Significance
Our interest in designing and exploring the huge database of H/HS sequences arose from our previous work (Raghuraman et al. 2006), where we used a limited hexasaccharide library (6859 sequences). Most docking approaches to date focus primarily on the affinity of interaction and minimally on the specificity of interaction (Kitchen et al. 2004). Our genetic algorithm-based approach places major emphasis on the specificity of interaction, which is more challenging to determine for H/HS. Our approach allows screening of considerably large conformational space and attempts to reduce the number of false positives.
This work represents the first, large-scale combinatorial GAG library screening to date. Our work demonstrates that such large library screening is feasible for GAG sequences, especially if a high-resolution crystal structure of the target protein is available. Considering the success achieved for AT and thrombin, two prototypic “highly specific” and “non-specific” GAG-binding proteins, respectively, we expect that CVLS approach may be more generally applicable to other proteins, especially for other serpins such as heparin cofactor II and protein C inhibitor, for which specificity features remain poorly defined. Theoretically, the CVLS technology should be applicable to any GAG–protein system assuming that appropriate validating solution experiments can be performed to assess predictions.
Our CVLS strategy utilized fairly stringent criteria for selection. While the affinity filter selected the upper 0.1% of sequences, the specificity filter was set to select only those sequences that satisfy self-consistency 100% of the time. It was possible to use high filtering stringency because the AT–H/HS system is a biochemically well-studied system. For other less understood systems, such stringent criteria may eliminate potentially useful information, which implies that appropriate relaxation in criteria may be necessary to introduce. One important deduction from our work is that GAG docking onto proteins should be approached with caution and care. We cannot assume that “one-size-fits-all” approach typically used in analyzing protein–ligand interactions will work well for GAG–protein studies as demonstrated by the observation that a higher docking radius does not necessarily result in higher probability of an outcome. This caution becomes even more important for relatively non-specific GAG–protein interactions for which it becomes difficult to estimate the validity of a result.
This work also presents the first algorithm, the incremental neighborhood optimization strategy, to design longer GAG sequences. This approach significantly reduces the computational cost and enhances efficiency over de novo design of a longer GAG sequence. Yet, the approach is expected to work for GAG–protein systems that exhibit high specificity. For non-specific system, it remains to be seen whether the incremental strategy provides meaningful results.
Finally, we expect our CVLS technology to be especially useful in the design of pharmaceutically useful agents. For example, the deduction of minimal “EF” disaccharide sequence as the origin of specificity implies that small GAG mimetics containing the EF domain should function as specific AT activators. Such small GAG mimetics should be possible to design computationally.
Supplementary data
Supplementary data for this article are available online at http://glycob.oxfordjournals.org/.
Funding
The computational facilities are provided by National Center for Research Resources (award Number S10RR027411). The work is supported by a research grant P01 HL107152 provided to URD from National Heart, Lung and Blood Institute.
Conflict of interest statement
None declared.
Abbreviations
AT, antithrombin; GAG, glycosaminoglycan; GlcAp, glucuronic acid; GlcNp, glucosamine; H, heparin; HS, heparan sulfate; IdoAp, iduronic acid;NRE, non-reducing end; RMSD, root-mean-square difference; SPL, SYBYL Programming Language; UAp, uronic acid.
Supplementary Material
Acknowledgements
We thank Drs Aurijit Sarkar and Philip Mosier of VCU for helpful discussions and suggestions.
References
- Agostino M, Gandhi NS, Mancera RL. Development and application of site mapping methods for the design of glycosaminoglycans. Glycobiology. 2014 doi: 10.1093/glycob/cwu045. (in press) [DOI] [PubMed] [Google Scholar]
- Belzar KJ, Dafforn TR, Petitou M, Carrell RW, Huntington JA. The effect of a reducing-end extension on pentasaccharide binding by antithrombin. J Biol Chem. 2000;275:8733–8741. doi: 10.1074/jbc.275.12.8733. [DOI] [PubMed] [Google Scholar]
- Bitomsky W, Wade RC. Docking of glycosaminoglycans to heparin-binding proteins: Validation for aFGF, bFGF, and antithrombin and application to IL-8. J Am Chem Soc. 1999;121:3004–3013. [Google Scholar]
- Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J Biol Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- Copeland R, Balasubramaniam A, Tiwari V, Zhang F, Bridges A, Linhardt RJ, Shukla D, Liu J. Using a 3-O-sulfated heparin octasaccharide to inhibit the entry of herpes simplex virus type 1. Biochemistry. 2008;47:5774–5783. doi: 10.1021/bi800205t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer D, Pople JA. A general definition of ring puckering. J Am Chem Soc. 1975;97:1354–1358. [Google Scholar]
- Desai UR. Antithrombin activation and designing novel heparin mimics. In: Garg HG, Linhardt RJ, Hales CA, editors. Chemistry and Biology of Heparin and Heparan Sulfate. Amsterdam: Elsevier Science; 2005. pp. 483–512. [Google Scholar]
- Desai UR. The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med Chem. 2013;5:1363–1366. doi: 10.4155/fmc.13.117. [DOI] [PubMed] [Google Scholar]
- Desai UR, Petitou M, Bjork I, Olson ST. Mechanism of heparin activation of antithrombin. Role of individual residues of the pentasaccharide activating sequence in the recognition of native and activated states of antithrombin. J Biol Chem. 1998;273:7478–7487. doi: 10.1074/jbc.273.13.7478. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Forster MJ, Mulloy B. Molecular dynamics study of iduronate ring conformations. Biopolymers. 1993;33:575–588. [Google Scholar]
- Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- Grootenhuis PDJ, van Boeckel CAA. Constructing a molecular model of the interaction between antithrombin III and a potent heparin analogue. J Am Chem Soc. 1991;113:2743–2747. [Google Scholar]
- Guerrini M, Elli S, Mourier P, Rudd TR, Gaudesi D, Casu B, Boudier C, Torri G, Viskov C. An unusual antithrombin-binding heparin octasaccharide with an additional 3-O-sulfated glucosamine in the active pentasaccharide sequence. Biochem J. 2013;449:343–351. doi: 10.1042/BJ20121309. [DOI] [PubMed] [Google Scholar]
- Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DJ, Li W, Adams TE, Huntington JA. Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 2006;25:2029–2037. doi: 10.1038/sj.emboj.7601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Disc. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Li W, Johnson DJ, Esmon CT, Huntington JA. Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol. 2004;11:857–862. doi: 10.1038/nsmb811. [DOI] [PubMed] [Google Scholar]
- Maimone MM, Tollefsen DM. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor II with high affinity. J Biol Chem. 1990;265:18263–18271. [PubMed] [Google Scholar]
- McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin's heparin affinity and activity. J Mol Biol. 2003;326:823–833. doi: 10.1016/s0022-2836(02)01382-7. [DOI] [PubMed] [Google Scholar]
- Mosier PD, Krishnasamy C, Kellogg GE, Desai UR. On the specificity of heparin/heparan sulfate binding to proteins. Anion-binding sites on antithrombin and thrombin are fundamentally different. PLoS ONE. 2012;7:e48632. doi: 10.1371/journal.pone.0048632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulloy B, Forster MJ. Conformation and dynamics of heparin and heparan sulfate. Glycobiology. 2000;10:1147–1156. doi: 10.1093/glycob/10.11.1147. [DOI] [PubMed] [Google Scholar]
- Olson ST, Halvorson HR, Björk I. Quantitative characterization of the thrombin-heparin interaction. Discrimination between specific and nonspecific binding models. J Biol Chem. 1991;266:6342–6352. [PubMed] [Google Scholar]
- Petitou M, Barzu T, Herault JP, Herbert JM. A unique trisaccharide sequence in heparin mediates the early step of antithrombin III activation. Glycobiology. 1997;7:323–327. doi: 10.1093/glycob/7.3.323-e. [DOI] [PubMed] [Google Scholar]
- Pike RN, Buckle AM, le Bonniec BF, Church FC. Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. 2005;272:4842–4851. doi: 10.1111/j.1742-4658.2005.04880.x. [DOI] [PubMed] [Google Scholar]
- Pol-Fachin L, Verli H. Depiction of the forces participating in the 2-O-sulfo-alpha-L-iduronic acid conformational preference in heparin sequences in aqueous solutions. Carbohydr Res. 2008;343:1435–1445. doi: 10.1016/j.carres.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Raghuraman A, Mosier PD, Desai UR. Finding a needle in a haystack: Development of a combinatorial virtual screening approach for identifying high specificity heparin/heparan sulfate sequence(s) J Med Chem. 2006;49:3553–3562. doi: 10.1021/jm060092o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman A, Mosier PD, Desai UR. Understanding dermatan sulfate-heparin cofactor II interaction through virtual library screening. ACS Med Chem Lett. 2010;1:281–285. doi: 10.1021/ml100048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver Z, Capila I, Venkataraman G, Sasisekharan R. Heparin and heparan sulfate: Analyzing structure and microheterogeneity. Handb Exp Pharmacol. 2012;207:159–176. doi: 10.1007/978-3-642-23056-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.