

Abstract
Adult T-cell leukemia–lymphoma (ATLL) is an HTLV-1-associated lymphoproliferative malignancy that is frequently fatal. We compared gene expression profiles (GEPs) of leukemic specimens from nine patients with ATLL at the time of diagnosis and immediately after combination therapy with zidovudine (AZT) and interferon α (IFNα). GEPs were also related to genetic aberrations determined by comparative genomic hybridization. We identified several genes anomalously over-expressed in the ATLL leukemic cells at the mRNA level, including LYN, CSPG2, and LMO2, and confirmed LMO2 expression in ATLL cells at the protein level. In vivo AZT–IFNα therapy evoked a marked induction of interferon-induced genes accompanied by repression of cell-cycle regulated genes, including those encoding ribosomal proteins. Remarkably, patients not responding to AZT–IFNα differed most from responding patients in lower expression of these same IFN-responsive genes, as well as components of the antigen processing and presentation apparatus. Demonstration of specific gene expression signatures associated with response to AZT–IFNα therapy may provide novel insights into the mechanisms of action in ATLL.
Keywords: Chemotherapeutic approaches, lymphoid leukemia, prognostication, transcription factor changes
Introduction
Adult T-cell leukemia–lymphoma (ATLL) is a highly aggressive T-cell malignancy etiologically linked to the retrovirus human T-cell leukemia virus type I (HTLV-1) [1]. HTLV-1 is transmitted mainly through breastfeeding, sexually, and via blood transfusions, and is endemic in areas of southern Japan, the Caribbean basin, intertropical Africa, South America, the Middle East, and Papua New Guinea. An estimated 15–20 million people are infected with HTLV-1 worldwide, the majority of whom are asymptomatic carriers [2]. Nevertheless, 2–6% of infected individuals will develop ATLL over a 40–60 year period. This very long latency and the low incidence of ATLL development in carriers suggest that the oncogenic potential of the HTLV-1 virus is quite weak, and that secondary genetic events must somatically arise within infected cells during transformation. Current data support the view that the viral Tax protein is instrumental for oncogenesis, as has been demonstrated in in vitro transformation assays and animal models [3]. The Tax protein affects multiple intracellular processes and signaling pathways, including cell cycle progression, cell survival, mitosis, and genomic stability [4–6]. Surprisingly, however, tax transcripts are detected in only 40% of all primary ATLL tumors [7,8], further pointing to the complexity of the HTLV-1 induced transformation and oncogenesis process, the mechanisms of which remain to be elucidated.
Clinically, ATLL is subdivided into four subtypes: acute, lymphoma-type, chronic, and smoldering [9]. The first two types show aggressive clinical courses with median survival ranging from 7 to 13 months [9]. Although diverse therapeutic approaches have been used, standard or dose-intense chemotherapy regimens result in low response rates, and response durations are typically short. Accordingly, novel therapeutic approaches based on a better understanding of this disease are needed. In this context, several novel approaches include unlabeled and radiolabeled anti-CD25 monoclonal antibodies [10], as well as strategies targeting the nuclear factor NFκB pathway [11,12] and chemokine CCR4 receptor [13]. In addition, promising results have been observed in ATLL patients treated with a combination of zidovudine (AZT) and interferon α (IFNα) [14–17].
To further investigate the deregulated pathways that may contribute to HTLV-1 mediated oncogenesis in ATLL, including the mechanisms that may underlie therapeutic responsiveness, we performed microarray gene expression analyses on ATLL specimens obtained at the time of diagnosis and immediately following combination therapy with AZT and IFNα. Further, correlation of patient cytogenetic profiles with patient gene expression data was performed to determine specific genetic aberrations resulting in over- or under-expression of genes that may contribute to disease pathogenesis. Our studies define a gene expression signature characteristic of ATLL cells, and reveal novel genes associated with response to AZT–IFNα therapy. These data may serve to predict individual responses to these compounds, thus helping to tailor treatment in patients with ATLL.
Materials and methods
Patients and treatment regimen
The diagnosis of acute ATLL was based on characteristic clinical features, immunophenotype, and serological evidence of HTLV-1 antibody by enzyme linked immunosorbent assay (ELISA), which was confirmed by Western blot and/or monoclonal proviral integration. The study group consisted of nine patients with leukemic presentation of HTLV-1 ATLL, all of whom provided informed consent, in accordance with the Declaration of Helsinki. Institutional review board (IRB) approval for these studies was granted by the IRB of the University of Miami.
All patients were treated with a uniform induction regimen combining high dose parenteral AZT (1.5 g administered twice daily) and IFNα (5–10 million units [U] administered twice daily) as induction therapy, a standard approach at our institution for patients with ATLL at the time of this study. Patients who responded to this induction regimen continued to receive outpatient maintenance therapy in the form of oral AZT (600 mg twice daily) and subcutaneous IFNα (5 million U once or twice daily). For patients with prolonged clinical response, maintenance therapy was gradually tapered to twice-daily oral doses of 300 mg AZT and three times weekly subcutaneous injections of 3 million U of IFNα. ‘Clinical response’ was defined as the disappearance of circulating abnormal lymphocytes, hypercalcemia, organomegaly, and measurable disease lasting for at least 1 month.
Isolation of PBMCs from patients with ATLL and cell lines
Peripheral blood mononuclear cells (PBMCs) from nine patients with ATLL at the time of initial presentation and 24 h post initial therapeutic dose (six patients) were obtained and immediately processed for PBMC separation using density gradient centrifugation (Lymphoprep; MediaTech, Herndon, VA). In all the patients, ATLL leukemic cells represented the majority of PBMCs at both time points. After separation, PBMCs were washed in sterile phosphate buffered saline, snap frozen as pellets, and stored at −80°C for later use. The HTLV-1-positive cell line MT2, the CD4-positive lymphoblastic leukemia cell line Jurkat (HTLV1-negative), and the diffuse large B-cell lymphoma cell line SU-DHL6 (HTLV1-negative) were grown in RPMI 1640 medium (Fisher Scientific, Santa Clara, CA) supplemented with 10% fetal calf serum, 2 mM glutamine (GIBCO BRL, Grand Island, NY), and penicillin/streptomycin (GIBCO BRL). MT2 cells were treated with AZT (10 μg/mL) and IFNα (1000 units/mL; Schering-Plough Corporation, Kenil-worth, NJ) in vitro. For this project, the cell lines were given code names (LLAT7A and B for untreated and treated MT2 cells and LLAT8 for Jurkat), and investigators who performed initial gene expression analysis were blinded to the nature of the cell line specimens. For each of six patients (LLAT1 through LLAT6), and one cell line (LLAT7), corresponding sample pairs representing baseline pre-therapy specimens and specimens representing the 24 h time point following therapy with AZT–IFNα were coded as ‘A’ and ‘B,’ respectively.
Immunoblot analyses
Whole-cell extracts for immunoblot analysis were prepared by lysing 5–10 × 106 cells with RIPA buffer (1 × phosphate-buffered saline [PBS], 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 10 mM phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, and 100 mM sodium orthovanadate) on ice for 30 min. After centrifugation at 3000g, the supernatant was assayed for protein concentration by bicinchoninic acid (BCA) assay (Pierce Biotechnology, Rockford, IL). For immunoblotting, 40 μg of whole-cell lysate was separated on 10% SDS–polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes (BioRad Laboratories, Hercules, CA), and probed with anti-LMO2 antibody previously generated by us [18] at 1:1000 dilution and anti-β-actin antibodies at 1:10 000 dilution (Sigma, St Louis, MO) overnight at 4°C. These antibodies were detected using anti-mouse horseradish peroxidase (HRP)-conjugated antibodies at 1:10 000 dilution (Jackson Immuno Research Laboratories, West Grove, PA) and visualized by the Super Signal West Femto Luminol/Enhancer Solution and Super Signal West Pico Luminol/Enhancer Solution (Pierce Biotechnology), respectively.
Gene expression profiling
Total cellular RNA was isolated from the tumor specimens and cell lines using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. Starting from 3–4 μg of total RNA, one round of linear amplification was used to prepare amplified RNA (MessageAmp, Ambion). In each experiment, fluorescent cDNA probes were prepared from an experimental amplified RNA (aRNA) sample (Cy5-labeled) and a reference aRNA sample (Cy3-labeled); the latter used Universal Human Reference RNA (Stratagene), isolated from a pool of 11 cell lines (MCF7, Hs578T, NTERA2, Colo205, OV-CAR-3, UACC-62, MOLT-4, RPMI-8226, NB4+ATRA, SW872, and HepG2). The use of a common reference allows determination of the relative expression of each gene across all of the samples [19]. Fluorescent cDNA probes were then hybridized to microarrays fabricated at the Stanford Functional Genomics Facility (generation SHDW; NCBI GEO GPL3522). The microarrays used included a total of 41 805 features representing 41 409 unique cDNA clones; these clones correspond to approximately 27 319 unique UniGene clusters including at least 14 744 unique named known genes (NCBI UniGene build number 173). Microarrays were hybridized then washed using previously described conditions [19], and then scanned on a GenePix 4000B scanner (Axon Instruments). The data from the microarray experiments is available at the GEO database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE19160.
Statistical methods
Microarray data were deposited into the Stanford Microarray Database, where raw data were normalized using a regression based correlation scheme. Data selection included filtering for well-measured features using the ratio of feature intensity over local background exceeding 2.5-fold for at least 70% of the 18 arrays (n = 9606 features). Variant features (n = 965) were selected by filtering for probes exhibiting a difference 4-fold or more from the median expression level in at least three of 18 (15%) microarrays. Unsupervised analysis was then performed by agglomerative hierarchical clustering using average-linkage that was applied sequentially to both genes and experiments (both median-centered) using the CLUSTER 3.0 program (http://bonsai.ims.u-to-kyo.ac.jp/~mdehoon/software/cluster/software.htm), and the results were analyzed with TREEVIEW (http://rana.lbl.gov/EisenSoftware.htm).
For supervised analyses of gene expression changes upon treatment with AZT–IFNα, Significance Analysis of Microarrays (SAM) [20] was applied to all paired samples with available baseline pre- (‘A’) and post-AZT–IFNα therapy (‘B’) specimens, starting with the data matrix of 9606 features described above. After imputing missing values through K-nearest neighbors (K = 10), the paired metric was applied and a threshold false discovery rate (FDR) of 15% was set using the MeV 4.4.1 software package [21] to identify 150 significant features. For display purposes, paired samples were displayed as the difference in log-ratios between the ‘B’ and ‘A’ samples, and then hierarchically clustered. To assess biological enrichment in response to AZT–IFNα, we applied Gene Set Enrichment Analysis (GSEA v2.0.4, using default parameters) [22] to the pre-ranked list of features as defined above using the paired SAM statistic; we supplemented gene sets within the Molecular Signatures Database (mSIGDB; Broad Institute) [22] with those from the immunological Signatures Database (SIGDB; National Cancer Institute, NIH) [23] as previously described [24]. For supervised analyses of gene expression differences among responding and non-responding patients to AZT–IFNα, SAM was applied to all pre-therapy (‘A’) patient specimens, starting with the data matrix of 9606 features described above. After imputing missing values through K-nearest neighbors (K = 10), the 2-class metric was applied to distinguish responding patients (LLAT1, 2, 3, and 4) versus non-responding patients (LLAT5, 6, 9, 10, and 11). To assess biological processes distinguishing responders from non-responders, we again applied GSEA to the pre-ranked list of features as defined above using the SAM statistic.
Comparative genomic hybridization (array CGH)
High molecular-weight DNA was extracted from 5 million cells with a commercially available kit as described by the manufacturer (QIAamp Tissue Kit; Qiagen, Valencia, CA). To measure DNA copy number changes throughout each tumor genome, standard comparative genomic hybridization was applied to chromosomes (s-CGH) in eight samples, using a reported method [25]. Additionally, two cases were selected and studied in higher detail through array-based CGH using a validated micro-array containing 2.460 BAC and P1 clones spotted in triplicate (UCSF Hum Array 2.0) and representing an average genomic resolution of 1.4 Mb. Relevant experimental methods and analytical procedures for array CGH have previously been described elsewhere in detail [26].
Results
Clinical profile of studied ATLL patients
Nine patients with HTLV-1 associated ATLL with acute leukemia subtype were analyzed. The white blood cell counts at presentation ranged 10–182 × 109/L with absolute leukemic lymphocyte counts ranging 6–153 × 109/L. Flow cytometric analyses demonstrated that leukemic cells from all the patients expressed CD4 and CD25 that stained brightly on the cells. HTLV-1 proviral genome in leukemia cells was detected by Southern blot hybridization or polymerase chain reaction (PCR) analysis. Among the nine patients with ATLL, six also presented with lymphadenopathy. Splenomegaly and hypercalcemia were separately detected in five patients each.
Upon confirmation of the diagnosis, all patients received AZT–IFNα combination therapy. Durable response was observed in four patients (LLAT1, LLAT2, LLAT3, and LLAT4), though the durability of these responses varied (overall survival range of 0.67–8.20 years; patient LLAT1 survived for 6.3 years and only patient LLAT2 remains alive at the last follow-up, 8.20 years since diagnosis). In contrast, all non-responding patients succumbed to complications of their disease and died within 0.25–1.00 years following the diagnosis of ATLL.
Gene expression profiles of HTLV-1+ ATLL
We next explored genome-wide expression profiles obtained from peripheral blood leukemic lymphocytes from diagnostic blood specimens of these nine patients. In six patients (LLAT1, LLAT2, LLAT3, LLAT4, LLAT5, and LLAT6), we also analyzed gene expression profiles (GEPs) of peripheral blood leukemic lymphocytes obtained 24 h after initiation of AZT–IFNα combination therapy. At this time point effects on intracellular signaling can be detected in vitro, while the cells are still not dying, thus allowing comparison of gene expression profiling in the tumor cells. As controls, we also included GEPs of the HTLV-1+ ATLL cell line MT2 (LLAT7) before and after in vitro AZT–IFNα treatment, as well the HTLV-1-negative Jurkat line (LLAT8). Unsupervised hierarchical clustering of specimens based on these genes revealed that distinct GEPs distinguished both transformed leukemic cell lines (i.e. MT2/LLAT7 and Jurkat/LLAT8) from the primary clinical specimens from patients with HTLV-1+ ATLL, as it resulted in their separate clustering (Figure 1). Among GEP features associated with this distinction, we observed differential expression of genes associated with proliferation, whose expression was high in the MT2 cell line, as reflected in a signature comprising many cell-cycle associated genes including PCNA, CCNB1, CCNA2, CDC2, CDK4, CDC25A, and CDCA1. Notably, many of these genes were previously reported as being over-expressed in HTLV-1 infected cell lines [1,27–30].
Figure 1.

Gene expression in ATLL. Gene expression profiles are depicted for nine patients with ATLL, including six pre- (‘A’) and post-AZT–IFNα (‘B’) treatment and two T-cell lines, MT2 (LLAT7A and B) and Jurkat (LLAT8). Each row represents a separate cDNA clone on the microarray and each column a separate mRNA sample. The displayed matrix represents the relative log-base 2 ratio of hybridization of fluorescent cDNA probes prepared from each experimental mRNA sample (compared to the median expression level across samples), as depicted by the corresponding color scale.
In contrast, primary leukemic cells from patients’ peripheral blood demonstrated significantly higher expression of several surface cell markers, such as CD52, CD37, CD28, and CD97. In addition, expression of histocompatibility genes such as HLA-E and HA-1 was higher in primary leukemic samples compared to the cell lines. These observations suggest that despite the fact that the MT2 cell line is commonly used for research of HTLV-1 related ATLL, it may not be representative of primary leukemic cells. Therefore, we excluded the MT2 cell line from subsequent analyses. Overall, distinct clones representing the same gene typically clustered in adjacent rows in this gene-map, indicating that each gene has a distinct pattern of expression and that expression measurements are sufficiently precise to distinguish them.
Expression of several genes that have been previously reported to be induced in Tax-expressing cells was observed to be increased in ATLL cells compared to the Jurkat cells (e.g. IL2Rα, IL2Rβ, and major histocompatibility complex class II genes) [31]. Overall, when focusing on genes exhibiting significant differences between primary diagnostic ATLL specimens in comparison with the Jurkat cell line (i.e. at least 4-fold in at least eight of nine patients with ATLL), 122 arrayed features were reproducibly over-expressed and 50 features had lower expression in ATLL (see ‘Appendix’ supplemental Tables SI and SII).
Multiple genes previously reported as expressed at higher levels in ATLL cells compared to normal CD4 lymphocytes were also found to be expressed at markedly higher levels in our ATLL leukemic cells compared to the Jurkat cell line (e.g. LYN, LYZ, S100A8, S100A9) [30]. Our analysis separately identified several genes highly expressed in the ATLL cells that may have an important role in pathogenesis of this disease. Among these genes, for example, we identified LMO2, which encodes a transcription factor regulating key events in erythropoiesis, angiogenesis, and embryogenesis [32]. Homozygous mice deficient in LMO2 die as a result of defective yolk sac erythropoiesis, whereas heterozygous mice show that LMO2 plays a role in the development of all bone marrow-derived hematopoietic lineages [33]. While LMO2 is not expressed in normal T cells, this gene is of relevance in T-cell lymphoid and myeloid leukemias resulting from the deregulated expression of LMO2 as caused by chromosomal translocations and insertional mutations [34]. Notably, the over-expression of LMO2 mRNA in leukemic cells of patients with ATLL cells observed herein was also reported in a previous study [30].
To further examine whether LMO2 protein is also expressed in ATLL leukemic cases, we performed Western blot analysis (Figure 2). All analyzed primary ATLL cases demonstrated LMO2 protein expression, with significant correlation between LMO2 mRNA and protein expression (data not shown). Consistent with prior reports [35], no expression was observed in the Jurkat cell line. Furthermore, LMO2 was expressed neither at the mRNA nor at the protein level in the MT2 cell line, further confirming differences in gene expression between primary ATLL specimens and this HTLV-1 infected cell line. Our finding of LMO2 expression in the ATLL leukemic T cells suggests that it may have a role in HTLV-1 induced leukemogenesis, similar to its known role in T-cell acute lymphoblastic leukemia.
Figure 2.
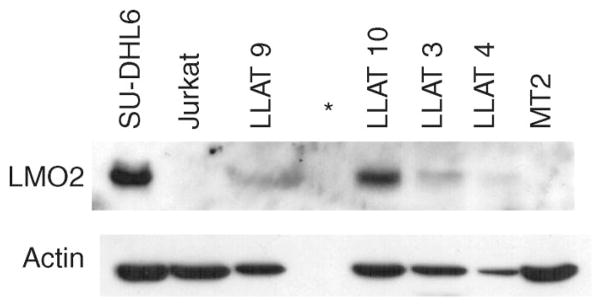
LMO2 protein expression in primary ATLL leukemic cells. Immunoblot analysis using the anti-LMO2 monoclonal antibody shows LMO2 protein expression in primary ATLL leukemic cells, and the diffuse large B-cell lymphoma cell line SU-DHL6, known to express this protein. LMO2 protein is not expressed in the Jurkat and MT2 cells. *Empty lane.
Chondroitin sulfate proteoglycan core protein 2 (CSPG2) expression was also elevated in ATLL leukemic cells as compared to the Jurkat cells. Of note, CSPG2 expression was also previously reported to be higher in ATLL cells compared to CD4 lymphocytes [30]. CSPG2 may play an important role in cancer cell migration and proliferation, and was reported to be elevated in adenomas and carcinomas [36,37]. Interestingly, CSPG2 is positively regulated by β-catenin, which is increased in ATLL cells [38,39].
Among our series of patients with ATLL, in all cases leukemic cells demonstrated at least 2-fold over-expression of CX3CR1 compared to Jurkat cells, similarly to a prior report [30]. CX3CR1 is a typical seven-transmembrane G-protein-coupled receptor involved in adhesion and chemotaxis [40], which binds its ligand, CX3CL1, a membrane bound adhesion molecule. CX3CL1 expression on endothelial cells promotes the metastasis of CX3CR1 expressing tumor cells, as reported for both prostate and breast cancer, and is associated with an increased risk of metastasis to the brain [41,42]. Similar to the expression of CCR7 in T-cell acute lymphoblastic leukemias [43], CX3CR1 over-expression on the ATLL cells may be associated with parenchymal organ infiltration.
Gene expression signatures of response to AZT–IFNα
In unsupervised analyses, comparison of the pre- and post-AZT–IFNα therapy specimens demonstrated that paired specimens derived from individual patients almost invariably clustered together, indicating that intrinsic differences in gene expression among individual patients were more pervasive than the early expression responses induced by AZT–IFNα (Figure 1). Nonetheless, despite baseline variation in expression of responsive genes, comparison of paired specimens from individual patients before and after therapy revealed a concerted signature of transcriptional responses to AZT–IFNα treatment [Figure 3(A)]. Using Gene Set Enrichment Analysis (GSEA), these responses were strikingly enriched for induction of genes known to be responsive to IFN in diverse cell types [Figure 3(B)] and for repression of those associated with ribosomal biogenesis, cell-cycle regulation, and proliferation [Figure 3(C)].
Figure 3.

In vivo gene expression responses of patients with ATLL following AZT–IFNα therapy. (A) Gene expression signature of in vivo response to AZT–IFNα was defined by selecting those exhibiting significant induction or repression in response to AZT–IFNα (n = 150) as determined by SAM, with rows ordered based on the magnitude of the difference. The logarithmic difference between the pre- (‘A’) and post- (‘B’) therapy measurements for each patient is captured in the depicted heat map. Selected gene sets exhibiting significant enrichment by GSEA (p <0.01, FDR <0.05) in response to AZT–IFNα are tabulated, with induced and repressed groups represented in (B) and (C), respectively.
We observed that the inter-individual heterogeneity among these ATLL patients captured gene expression differences relating to their clinical responses to this regimen. Unsupervised analyses revealed that three of the four cases demonstrating a clinical response to AZT–IFNα treatment clustered together on the same dendrogram branch, while the two paired specimens derived from patients that did not exhibit response clustered separately (Figure 1). We applied supervised analyses to explicitly assess differences between signatures of responders to AZT–IFNα and non-responders using GSEA. Notably, despite exhibiting similar response profiles to AZT–IFNα in the magnitude of induction and repression [Figure 3(A)], patients responding to AZT–IFNα differed most from non-responding patients in their baseline relative over-expression of IFN-responsive genes, as well as components of the antigen processing and presentation apparatus (Table I). Conversely, non-responding patients showed relative over-expression of E2F3 and many of its proliferation-associated oncogenic targets (Table I).
Table I.
Gene expression signature of responsiveness to AZT–IFNα treatment. Selected gene sets exhibiting significant enrichment by GSEA (p <0.01, FDR <0.2) in distinguishing patients with clinical responses to AZT–IFNα are tabulated.
| Higher in | Source | Gene set | Annotation | p-Value | FDR | Example enriched gene(s) | Primary author | PMID |
|---|---|---|---|---|---|---|---|---|
| Responders | mSIGDB | HSA04612_ANTIGEN_PROCESSING_AND_PRESENTATION | Genes involved in antigen processing and presentation | <0.001 | <0.005 | HLA-DRB1/B5/B4/DPA, HLA-A, B2M, TAP2, TAPBP, RFX5 | Kanehisa | 18077471 |
| Responders | SIGDB | IS_161_IFN_PMBC_2X_UP | Induced >2-fold in PBMCs by IFNα/IFNβ in vitro (6 h) | <0.001 | <0.05 | ISG20, IRF1, GBP1, STAT1, OAS2, MX1 | Baechler | 12604793 |
| Responders | mSIGDB | DER_IFNA_UP | Induced by IFNα in fibrosarcoma | <0.001 | <0.05 | IFITM1, IRF1, HLA-A, GBP1, PSMB8 | Der | 9861020 |
| Responders | mSIGDB | IFN_GAMMA_UP | Induced by IFNγ in fibrosarcoma | <0.005 | <0.05 | IRF1, ICAM1, STAT1, IFITM1 | Der | 9861020 |
| Responders | mSIGDB | IFN_ALPHA_UP | Induced by IFNα in fibrosarcoma | <0.001 | <0.05 | IFITM1, IRF1, STAT1, OAS2, OAS1 | Der | 9861020 |
| Responders | SIGDB | IS_162_IFN_PMBC_4X_UP | Induced >4-fold in PBMCs by IFNα/IFNβ in vitro (6 h) | <0.001 | <0.05 | IRF7, ISG20, TNFSF10, LY6E, CASP10 | Baechler | 12604793 |
| Non-responders | SIGDB | IS_171_E2F3_OVEREXPRESSION_2X_UP | Induced >2-fold by E2F3 | <0.001 | 0.17 | CDKN1C, FZD1, PTCH1, HEY1, MYB, E2F3 | Bild | 16273092 |
AZT–IFNα, zidovudine–interferon α; mSIGDB, Molecular Signatures Database; SIGDB, immunological Signatures Database; PBMCs, peripheral blood mononuclear cells; FDR, false discovery rate; PMID, PubMed Identifier.
Correlation of genomic and gene expression data
HTLV-1 harboring ATLL tumors frequently carry chromosomal aberrations that may or may not be evident on routine metaphase karyotypes. Consequently, the tumor genomic profiles were examined, and DNA copy number changes were correlated with gene expression profiles. Genomic analysis was performed on eight tumor samples prior to therapy using comparative genomic hybridization (genomic DNA was not available for one patient). Overall, all but one case harbored clonal genomic imbalances. The pattern of genomic aberrations in HTLV-1+ ATLL specimens consisted of 49 genomic imbalances (mean per tumor: 6.125, range: 0–16) and included 15 genomic gains (mean per tumor: 1.875, range: 0–6) and 34 losses (mean per tumor: 4.25, range: 0–15).
The most common chromosomal changes included gains of regions 1q, 3p, and 3q and losses of chromosomal regions 4p, 6q, 9p, 10p, 10q, 16p, 16q, and 17p [Figure 4(A)]. Using array-based CGH, these genomic aberrations were then characterized at higher genomic resolution for two cases [Figure 4(B)]. Despite the anomalous expression of LMO2 in our patients with ATLL, no aberrations were detected at the corresponding cytogenetic interval locus 11p13, suggesting that LMO2 expression in ATLL cells is not due to a genomic amplification.
Figure 4.

Chromosomal aberrations in patients with ATLL. (A) Cumulative gains and losses of chromosomal regions in eight patients with ATLL; (B) representation of the genomic profiling of case LLAT9 using high-resolution BAC array-CGH. Bottom: A number of DNA copy number changes were observed, including the gain of 1q, trisomy of chromosome 3, deletion of 6q, and deletion of 10p. Inserts: details of the genomic gain involving chromosome 1q22–q32 and the loss of chromosome 6q14–q21. The presence of only one chromosome X corresponds to males’ samples.
We next searched for genes within amplified or deleted genomic loci that were correspondingly over-or under-expressed at least 1.5-fold compared to the average expression of the series. This analysis revealed 110 genes located in areas of genomic imbalances (Table II), though only three of the genes (MET2A, IFIT1, and IL2RA) exhibited relative down-regulation in at least two cases with similar chromosomal losses. Of note, expression of MET2A and IFIT1 was also relatively down-regulated in some cases without chromosomal aberrations, suggesting alternative mechanisms. Separately, despite our observation of over-expression of E2F3 and its targets among non-responding patients, we did not observe recurrent amplifications involving the corresponding genomic locus for E2F3 (i.e. 6p22.3) by comparative genomic hybridization of patients with ATLL [Figure 4(A)]. Prior study demonstrated amplification of E2F3 and over-expression of its target genes during progression from the chronic to the acute phase of ATL [44]. The differences between our and previous study may suggest that such lesions may have been occult among our series using conventional CGH, based on limitations in its resolution, or reflect different pathogenetic events in cases progressing from the chronic form compared to the de novo leukemic type of ATLL. Separately, given the number of chromosomal anomalies observed, the small number of analyzed cases limited a more extensive analysis of association between ATLL genotypes, gene expression profiles, and clinical outcomes.
Table II.
Differentially expressed genes* within chromosomal aberrations of adult T-cell leukemia–lymphoma.
| Lesion/chromosome | Differentially expressed genes |
|---|---|
| (−) 1p | RPL27A |
| (−) 1q | UAP1, DAF, FJL10052, FLJ21841, HDGF, ILF2, NUCKS, S100A10, MGC21854, FCER1G |
| (−) 2p | OLD35, MTHFD2 |
| (+) 2q | IL18R1, GPR55, ITM2C, MGAT4A, TTN, CTLA4, STK17B, PSCDBP |
| (+) 3p | AXUD1, SNRK, CISH, LRIG1, PCAF |
| (−) 3p | SATB1 |
| (+) 3q | CCNL1, IFRG28, PLSCR1, TAZ |
| (−) 5q | CSPG2 |
| (+) 7p | NT5C3 |
| (+) 7q | KIAA1718, TES, UBE2H, LR8, PEG10, FLJ20653 |
| (−) 7q | HIMAP4 |
| (+) 8p | PLEKHA2 |
| (+) 8q | MCM4 |
| (−) 9q | NPDC1, ANXA1, UCK1 |
| (−) 10p | IL2RA, FLJ13267 |
| (−) 10q | IFIT1, ABLIM1, ADD3, MRPS16, SCD, IFIT2 |
| (−) 11p | RPL27A, IRF7, H19 |
| (+) 11q | SWAP70, SERPING1 |
| (−) 12p | NINJ2, LTBR, SLC2A3, LLT1, TNFRSF7 |
| (−) 12q | OSBPL8 |
| (−) 13q | NY-REN-34, ALOX5AP, EBI2, KIAA0853, UBL3 |
| (−) 15q | MYO5A, ISG20, B2M, IL16, GLCE |
| (−) 16p | NME4, NK4 |
| (−) 16q | MET2A, MET1L, MET1F, COG8, TUBB4, PNAS-108, FLJ20186, SLC7A5 |
| (−) 17p | HS3ST3B1, ITGAE |
| (+) 18q | MGC10200 |
| (+) 19p | EDG6, MYO1F, JAK3, CTL2, MGC15716 |
| (+) 19q | PIK3AP, LTBP4 |
| (−) 19q | PIK3AP, ZNF134 |
| (+) Xp | KIAA1280 |
Genes with at least 1.5-fold difference in relative expression as compared to the average expression across all specimens were considered. In cases with genomic gain/amplification (denoted as ‘+’), only over-expressed genes were considered, while in cases with genomic deletions (denoted as ‘−’), only genes with decreased expression are shown. In bold are genes whose expression changed in two cases.
Discussion
ATLL is a distinct clinical entity associated with HTLV-1 retroviral infection. Regardless of the extensive progress in virology, immunology, and molecular biology of ATLL and HTLV-1, the prognosis of patients with ATLL remains poor. Recently, several studies suggested that some patients with ATLL may respond to the combination of AZT–IFNα [14–17]; however, factors associated with responsiveness to the treatment are currently unknown. Our objective was to identify genetic parameters which might be associated with responsiveness to AZT–IFNα combination treatment. In addition, application of global gene expression studies to diagnostic specimens might elucidate the pathogenesis of ATLL.
Our analysis of GEPs of diagnostic leukemic ATLL cells revealed changes in gene expression of about 1000 genes, many of which (e.g. IL2Rα, IL2Rβ, and major histocompatibility complex class II genes) were previously reported to be up-regulated in HTLV-1 infected cell lines and tumors [27,30,31]. Moreover, multiple genes deregulated in our cases were also recently reported to be either over-expressed or down-regulated in primary ATLL specimens studied by Pise-Masison et al. [30]. Among the prominent genes similarly expressed in our cases as well as cases reported by Pise-Masison et al. [30], we observed high levels of expression of LYN, LYZ, S100A8, S100A9, LMO2, CSPG2, and CX3CR1. These observations support the validity of our gene expression measurements and analyses, performed on leukemic cells derived from representative patients with ATLL with characteristic clinical manifestations and clinical course. Anomalous expression of the LMO2 protein in ATLL—a transcription factor not normally expressed in normal T cells but implicated in the pathogenesis of T-cell acute lymphoblastic leukemias—may suggest that it could have a similar role in the pathogenesis of ATLL. The mechanism underlying LMO2 expression in ATLL is unknown. We did not detect aberrations at the corresponding cytogenetic interval locus 11p13 that might point to genomic amplification. It is also highly unlikely that HTLV-1 virus specifically integrates into host DNA in the proximity of the LMO2 gene, since previous studies demonstrated random integration sites of the virus [45,46]. Further studies to elucidate the mechanism of LMO2 expression and its possible pathogenic role in ATLL are in progress.
While our results corroborate some of the findings observed by Pise-Masison et al. [30], some studies analyzing gene expression of HTLV-1 immortalized T cells reported different observations with regard to some of the genes. For example, down-regulation of the expression of protein tyrosine kinases of the Src and Syk families (e.g. LCK, HCK, LYN) has been reported in response to HTLV-1 [29], though we did not observe a similar profile among our HTLV-1+ ATLL cases. The latter discrepancy may stem from differences in gene expression between primary tumors and HTLV-1 infected cell lines. Indeed, gene expression analyses presented herein demonstrated marked differences in gene expression profiles between the MT2 HTLV-1-positive cell line and patients’ primary ATLL cells.
Expression of genes associated with cell proliferation was among major differences distinguishing MT2 from primary ATLL patient samples. Similarly, previous studies demonstrated that in vitro HTLV-1 infection induces genes associated with proliferation and cell cycling attributed to expression of the Tax protein [28,29]. However, while the Tax oncogene, encoded by HTLV-1 virus, is expressed in many HTLV-1 infected cell lines, its expression is lost in many primary ATLL cells and may contribute to the observed differences. The differences in gene expression between primary tumors and HTLV-1-positive cell lines suggest that although the latter represent an accessible tool for study of HTLV-1 infection and lymphomagenesis, aberrations observed in these lines may not completely represent changes observed in primary tumor cells.
The primary leukemic cells analyzed in this study harbored genetic aberrations similar to aberrations previously reported in the literature and included frequent chromosomal gains of chromosomal regions 3p, 7q, 1q, 2q, 3q, 8p, 8q, 9q, 18q, and 19p, in addition to losses at chromosomal regions 6q, 2q, 4q, 5q, 10p, 10q, 12p, 18p, and chromosome 13 [47,48]. Analyses of correlation between chromosomal changes and alteration in gene expression of chromosomes located in the chromosomal aberration regions demonstrated relatively poor correlation, similar to our observations in transformed follicular lymphoma specimens [49].
Gene expression profiling demonstrated a difference in gene expression between ATLL cells responsive and unresponsive to AZT–IFNα combination therapy. Noticeably, leukemic cells from non-responsive patients had high expression of E2F3 and many of its proliferation-associated oncogenic target genes, likely reflecting the more aggressive nature of these tumors. Of interest, ATLL specimens exhibiting response to AZT–IFNα therapy displayed high expression of interferon-responsive genes prior to treatment initiation, and a further increase in their expression upon treatment; however, induction of some of these genes was also observed in unresponsive cases. The association between treatment and induction of interferon-inducible genes upon treatment was highly statistically significant. Previous studies demonstrated that HTLV-1 may blunt signaling by IFNα [50], and the extent of this effect may vary between different tumors. Indeed, over-expression of IRF4, a regulatory transcription factor negatively regulating IFN responses, has been reported as a determinant of resistance in ATLL [12]. Interferon γ expressing mice exhibit decreased HTLV-1 Tax protein tumorigenesis compared to animals deficient in interferon γ expression, suggesting that the endogenously active interferon signaling pathway may ameliorate the aggressiveness of these tumors [3]. However, the precise mechanism underlying the baseline differences in interferon target genes among patients with ATLL, and its association with clinical response to the therapy, are currently unknown. A recent in vitro study of ATLL-derived HTLV-1 infected CD4+ T-cell lines treated with arsenic trioxide and IFNα also demonstrated up-regulation of similar interferon-induced genes in the treated cells undergoing apoptosis and death following treatment [11]. Similar to our in vivo findings that demonstrated changes of gene expression at 24 h after AZT–IFNα combination treatment, induction of interferon target genes upon in vitro treatment with arsenic trioxide and IFNα was already detectable at 12 h and was sustained up to 48 h [11].
The NFκB pathway is known to play an integral role in the pathogenesis of HTLV-1 infection and lymphomagenesis [12]. Suppression of NFκB activation by AZT–IFNα combination therapy might lead to apoptosis of ATLL cells, as was recently reported for the combination of arsenic trioxide and IFNα [11], while expression of the C-Rel protein, a component of NFκB, imparts resistance to AZT–IFNα therapy [12]. However, the exact mechanism of the AZT–IFNα induced ATLL cell death is currently unknown. Demonstration of specific gene expression signatures associated with response to therapy may help to further elucidate the exact mechanism of AZT–IFNα induced suppression of NFκB, and needs further evaluation in future studies.
Supplementary Material
Acknowledgments
We would like to thank Joan Climent for help with CGH studies.
Footnotes
Declaration of interest: This study was supported by R01 CA109335 and R01 CA122105 from the United States Public Health Service—National Institutes of Health, Bankhead-Coley, and the Dwoskin Family Foundations (I.S.L.).
References
- 1.Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer. 2007;7:270–280. doi: 10.1038/nrc2111. [DOI] [PubMed] [Google Scholar]
- 2.Franchini G, Nicot C, Johnson JM. Seizing of T cells by human T-cell leukemia/lymphoma virus type 1. Adv Cancer Res. 2003;89:69–132. doi: 10.1016/s0065-230x(03)01003-0. [DOI] [PubMed] [Google Scholar]
- 3.Mitra-Kaushik S, Harding J, Hess J, Schreiber R, Ratner L. Enhanced tumorigenesis in HTLV-1 tax-transgenic mice deficient in interferon-gamma. Blood. 2004;104:3305–3311. doi: 10.1182/blood-2004-01-0266. [DOI] [PubMed] [Google Scholar]
- 4.Felber BK, Paskalis H, Kleinman-Ewing C, Wong-Staal F, Pavlakis GN. The pX protein of HTLV-I is a transcriptional activator of its long terminal repeats. Science. 1985;229:675–679. doi: 10.1126/science.2992082. [DOI] [PubMed] [Google Scholar]
- 5.Sodroski JG, Rosen CA, Haseltine WA. Trans-acting transcriptional activation of the long terminal repeat of human T lymphotropic viruses in infected cells. Science. 1984;225:381–385. doi: 10.1126/science.6330891. [DOI] [PubMed] [Google Scholar]
- 6.Twizere JC, Springael JY, Boxus M, et al. Human T-cell leukemia virus type-1 Tax oncoprotein regulates G-protein signaling. Blood. 2007;109:1051–1060. doi: 10.1182/blood-2006-06-026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda S, Maeda M, Morikawa S, et al. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer. 2004;109:559–567. doi: 10.1002/ijc.20007. [DOI] [PubMed] [Google Scholar]
- 8.Furukawa Y, Kubota R, Tara M, Izumo S, Osame M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood. 2001;97:987–993. doi: 10.1182/blood.v97.4.987. [DOI] [PubMed] [Google Scholar]
- 9.Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87) Br J Haematol. 1991;79:428–437. doi: 10.1111/j.1365-2141.1991.tb08051.x. [DOI] [PubMed] [Google Scholar]
- 10.Waldmann TA, Goldman CK, Bongiovanni KF, et al. Therapy of patients with human T-cell lymphotrophic virus I-induced adult T-cell leukemia with anti-Tac, a monoclonal antibody to the receptor for interleukin-2. Blood. 1988;72:1805–1816. [PubMed] [Google Scholar]
- 11.Nasr R, Rosenwald A, El-Sabban ME, et al. Arsenic/interferon specifically reverses 2 distinct gene networks critical for the survival of HTLV-1-infected leukemic cells. Blood. 2003;101:4576–4582. doi: 10.1182/blood-2002-09-2986. [DOI] [PubMed] [Google Scholar]
- 12.Ramos JC, Ruiz P, Jr, Ratner L, et al. IRF-4 and c-Rel expression in antiviral-resistant adult T-cell leukemia/lymphoma. Blood. 2007;109:3060–3068. doi: 10.1182/blood-2006-07-036368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishida T, Iida S, Akatsuka Y, et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T-cell leukemia/lymphoma. Clin Cancer Res. 2004;10:7529–7539. doi: 10.1158/1078-0432.CCR-04-0983. [DOI] [PubMed] [Google Scholar]
- 14.Matutes E, Taylor GP, Cavenagh J, et al. Interferon alpha and zidovudine therapy in adult T-cell leukaemia lymphoma: response and outcome in 15 patients. Br J Haematol. 2001;113:779–784. doi: 10.1046/j.1365-2141.2001.02794.x. [DOI] [PubMed] [Google Scholar]
- 15.White JD, Wharfe G, Stewart DM, et al. The combination of zidovudine and interferon alpha-2B in the treatment of adult T-cell leukemia/lymphoma. Leuk Lymphoma. 2001;40:287–294. doi: 10.3109/10428190109057927. [DOI] [PubMed] [Google Scholar]
- 16.Gill PS, Harrington W, Jr, Kaplan MH, et al. Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med. 1995;332:1744–1748. doi: 10.1056/NEJM199506293322603. [DOI] [PubMed] [Google Scholar]
- 17.Hermine O, Bouscary D, Gessain A, et al. Brief report: treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med. 1995;332:1749–1751. doi: 10.1056/NEJM199506293322604. [DOI] [PubMed] [Google Scholar]
- 18.Natkunam Y, Zhao S, Mason DY, et al. The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood. 2007;109:1636–1642. doi: 10.1182/blood-2006-08-039024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 20.Tusher V, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saeed AI, Bhagabati NK, Braisted JC, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaffer A, Wright G, Yang L, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev. 2006;210:67–85. doi: 10.1111/j.0105-2896.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- 24.Gentles A, Alizadeh A, Lee S, et al. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood. 2009;114:3158–3166. doi: 10.1182/blood-2009-02-202465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 26.Rubio-Moscardo F, Blesa D, Mestre C, et al. Characterization of 8p21. 3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005;106:3214–3222. doi: 10.1182/blood-2005-05-2013. [DOI] [PubMed] [Google Scholar]
- 27.Pise-Masison CA, Radonovich M, Mahieux R, et al. Transcription profile of cells infected with human T-cell leukemia virus type I compared with activated lymphocytes. Cancer Res. 2002;62:3562–3571. [PubMed] [Google Scholar]
- 28.de La Fuente C, Deng L, Santiago F, Arce L, Wang L, Kashanchi F. Gene expression array of HTLV type 1-infected T cells: up-regulation of transcription factors and cell cycle genes. AIDS Res Hum Retroviruses. 2000;16:1695–1700. doi: 10.1089/08892220050193164. [DOI] [PubMed] [Google Scholar]
- 29.Harhaj EW, Good L, Xiao G, Sun SC. Gene expression profiles in HTLV-I-immortalized T cells: deregulated expression of genes involved in apoptosis regulation. Oncogene. 1999;18:1341–1349. doi: 10.1038/sj.onc.1202405. [DOI] [PubMed] [Google Scholar]
- 30.Pise-Masison CA, Radonovich M, Dohoney K, et al. Gene expression profiling of ATL patients: compilation of disease-related genes and evidence for TCF4 involvement in BIRC5 gene expression and cell viability. Blood. 2009;113:4016–4026. doi: 10.1182/blood-2008-08-175901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue J, Seiki M, Taniguchi T, Tsuru S, Yoshida M. Induction of interleukin 2 receptor gene expression by p40x encoded by human T-cell leukemia virus type 1. EMBO J. 1986;5:2883–2888. doi: 10.1002/j.1460-2075.1986.tb04583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warren AJ, Colledge WH, Carlton MB, Evans MJ, Smith AJ, Rabbitts TH. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell. 1994;78:45–57. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 33.Yamada Y, Warren AJ, Dobson C, Forster A, Pannell R, Rabbitts TH. The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc Natl Acad Sci USA. 1998;95:3890–3895. doi: 10.1073/pnas.95.7.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boehm T, Foroni L, Kaneko Y, Perutz MF, Rabbitts TH. The rhombotin family of cysteine-rich LIM-domain oncogenes: distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. Proc Natl Acad Sci USA. 1991;88:4367–4371. doi: 10.1073/pnas.88.10.4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong WF, Billia F, Atkins HL, Iscove NN, Minden MD. Expression of rhombotin 2 in normal and leukaemic haemopoietic cells. Br J Haematol. 1996;93:280–286. doi: 10.1046/j.1365-2141.1996.5001044.x. [DOI] [PubMed] [Google Scholar]
- 36.Creighton CJ, Bromberg-White JL, Misek DE, et al. Analysis of tumor-host interactions by gene expression profiling of lung adenocarcinoma xenografts identifies genes involved in tumor formation. Mol Cancer Res. 2005;3:119–129. doi: 10.1158/1541-7786.MCR-04-0189. [DOI] [PubMed] [Google Scholar]
- 37.Mauri P, Scarpa A, Nascimbeni AC, et al. Identification of proteins released by pancreatic cancer cells by multidimensional protein identification technology: a strategy for identification of novel cancer markers. FASEB J. 2005;19:1125–1127. doi: 10.1096/fj.04-3000fje. [DOI] [PubMed] [Google Scholar]
- 38.Chung EJ, Hwang SG, Nguyen P, et al. Regulation of leukemic cell adhesion, proliferation, and survival by beta-catenin. Blood. 2002;100:982–990. doi: 10.1182/blood.v100.3.982. [DOI] [PubMed] [Google Scholar]
- 39.Nakayama K, Kamihira S. Survivin an important determinant for prognosis in adult T-cell leukemia: a novel biomarker in practical hemato-oncology. Leuk Lymphoma. 2002;43:2249–2255. doi: 10.1080/1042819021000039956. [DOI] [PubMed] [Google Scholar]
- 40.Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 41.Andre F, Cabioglu N, Assi H, et al. Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann Oncol. 2006;17:945–951. doi: 10.1093/annonc/mdl053. [DOI] [PubMed] [Google Scholar]
- 42.Shulby SA, Dolloff NG, Stearns ME, Meucci O, Fatatis A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004;64:4693–4698. doi: 10.1158/0008-5472.CAN-03-3437. [DOI] [PubMed] [Google Scholar]
- 43.Buonamici S, Trimarchi T, Ruocco MG, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature. 2009;459:1000–1004. doi: 10.1038/nature08020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsukasaki K, Tanosaki S, DeVos S, et al. Identifying progression-associated genes in adult T-cell leukemia/lymphoma by using oligonucleotide microarrays. Int J Cancer. 2004;109:875–881. doi: 10.1002/ijc.20028. [DOI] [PubMed] [Google Scholar]
- 45.Hanai S, Nitta T, Shoda M, et al. Integration of human T-cell leukemia virus type 1 in genes of leukemia cells of patients with adult T-cell leukemia. Cancer Sci. 2004;95:306–310. doi: 10.1111/j.1349-7006.2004.tb03207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozawa T, Itoyama T, Sadamori N, et al. Rapid isolation of viral integration site reveals frequent integration of HTLV-1 into expressed loci. J Hum Genet. 2004;49:154–165. doi: 10.1007/s10038-004-0126-7. [DOI] [PubMed] [Google Scholar]
- 47.Tsukasaki K, Krebs J, Nagai K, et al. Comparative genomic hybridization analysis in adult T-cell leukemia/lymphoma: correlation with clinical course. Blood. 2001;97:3875–3881. doi: 10.1182/blood.v97.12.3875. [DOI] [PubMed] [Google Scholar]
- 48.Tsukasaki K. Genetic instability of adult T-cell leukemia/lymphoma by comparative genomic hybridization analysis. J Clin Immunol. 2002;22:57–63. doi: 10.1023/a:1014471500757. [DOI] [PubMed] [Google Scholar]
- 49.Martinez-Climent JA, Alizadeh AA, Segraves R, et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood. 2003;101:3109–3117. doi: 10.1182/blood-2002-07-2119. [DOI] [PubMed] [Google Scholar]
- 50.Feng X, Ratner L. Human T-cell leukemia virus type 1 blunts signaling by interferon alpha. Virology. 2008;374:210–216. doi: 10.1016/j.virol.2007.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.