

Abstract
DNA aptamer oligonucleotides and their protein conjugates show promise as therapeutics in animal models of diseases such as multiple sclerosis. These molecules are large and highly charged, raising questions about their biodistribution and pharmacokinetics in mammals. Here we exploit the power of quantitative polymerase chain reaction to accurately quantitate the tissue distribution of 40-nucleotide DNA aptamers and their streptavidin conjugates after intraperitoneal injection in mice. We show remarkably rapid distribution to peripheral tissues including the central nervous system. Modeling of tissue distribution data reveals the importance of DNA aptamer sequence, 3′ modification, and protein conjugation in enhancing tissue exposure. These data help to interpret the previously observed effectiveness of aptamer conjugates, as opposed to free aptamers, in stimulating central nervous system remyelination in a mouse model of multiple sclerosis.
Introduction
Nucleic acid aptamers are highly charged macromolecules with the potential to bind and modulate the activities of circulating and cell-surface targets in tissues and organisms [1–7]. As for all drugs, biodistribution, pharmacokinetics, and bioavailability are crucial considerations in the therapeutic application of nucleic acid aptamers. Unlike antibodies [8], nucleic acid aptamers are unnatural macromolecules with unpredictable biostability and biodistribution. Complicating the question of aptamer pharmacokinetics is the potential effect of aptamer modifications, conjugations, and formulations that could influence uptake into circulation, tissue accumulation, metabolism, and clearance.
We have been interested in the potential of DNA aptamers to enhance therapeutic remyelination in animal models of multiple sclerosis (MS). Our prior studies showed that a particular guanosine-rich 40-nucleotide DNA sequence (3064), derived from a longer aptamer selected in vitro for affinity to a crude myelin preparation, displayed central nervous system remyelinating activity after intraperitoneal (i.p.) injection in a Theiler's murine encephalomyelitis virus (TMEV) mouse model of MS [9]. The active formulation was a 3′-biotinylated conjugate of this aptamer in tetrameric complex with streptavidin protein. This result suggested the possibility that, despite a predicted mass of greater than 100 kD for the tetrameric aptamer-protein formulation, the conjugate was gaining access to the central nervous system [9].
Some prior studies with natural and modified oligonucleotides as antisense agents have explored pharmacokinetics using traditional approaches involving radiolabeling [10–20] or other conventional detection methods [21]. These approaches successfully detect and quantitate nucleic acid concentration over a few orders of magnitude. However, these techniques make it difficult to assess low concentrations of nucleic acids in small amounts of tissue early and late after administration of the agent and often cannot distinguish products of drug metabolism. We reasoned that a tremendous advantage of nucleic acid aptamers as drugs is the potential to exploit the exquisite sensitivity of quantitative polymerase chain reaction (qPCR) technology to dramatically improve analysis of DNA aptamer pharmacokinetics in mice. qPCR has the ability to quantitate recovered aptamers over seven orders of magnitude of concentration. Our initial nonquantitative PCR studies of aptamer 3064 confirmed detection in multiple tissues including central nervous system [9], suggesting the potential application of qPCR to this problem.
Here we demonstrate for the first time the extreme sensitivity of qPCR in the analysis of DNA aptamer pharmacokinetics after i.p. administration of a single bolus dose in mice. Extensive buffer perfusion was employed to remove blood contamination from tissues. We report remarkably rapid aptamer uptake from the peritoneum into blood, and dissemination into organs including spinal cord and brain, peaking within minutes of i.p. administration. Following a rapid decrease over a few hours, aptamer concentration then displays a prolonged period of first-order exponential decay. These effects are strongly dependent on tissue, aptamer sequence, and formulation. Maximum tissue exposure was obtained for aptamer 3064 (capable of folding into an intramolecular guanosine quaduplex structure) formulated as a 3′ biotin derivative conjugated with tetrameric streptavidin.
Materials and Methods
Aptamer preparation
HPLC-purified aptamers 3064 and 4971 were purchased at 1 μmol scale from Tri-Link or IDT. Quantitation was by ultraviolet spectrophotometry with calculated molar extinction coefficients. In some cases 3064 was synthesized as a 3′ biotin derivative using biotinTEG control pore glass support (Glen Research 20-2955). Sufficient material for final 1 μM aptamer stocks was made up in 19.5 mL phosphate-buffered saline containing 1 mM MgCl2. This sample was heated to 90°C for 5 min in a water bath, and then snap cooled on ice for 15 min. In cases where biotinylated aptamer stocks were conjugated to streptavidin (Abcam), an appropriate amount of tetrameric streptavidin was added to the 1 μM biotinylated aptamer stock to provide 0.25 μM streptavidin tetramer, and the mixture was incubated at 37°C for 1 h to allow formation of aptamer conjugates. Aptamer solutions were then passed through 0.45-μm filters and stored at 4°C or at −20°C.
Mice and TMEV infection
All studies conformed to Mayo Clinic and National Institutes of Health animal use guidelines and were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee. Where indicated, 8-week-old female SJL/J mice (Jackson Laboratories) received a single intracerebral injection of 2×105 plaque-forming units of the Daniel's strain of Theiler's Murine Encephalomyelitis Virus (TMEV) in Dulbecco's phosphate-buffered saline (10 μL). Infected animals used in this work were chronically demyelinated for 6 months post infection with clear neurologic signs and deficits.
Aptamer administration and tissue preparation
Six- to seven-week-old female CD-1 IGS outbred mice (strain code: 022, Charles River Laboratories International, Inc.) were used for these experiments. Five mice were included for each time-point treatment group, with one mouse left untreated as a negative control for background calculation. Mice received 500 μL of sterile saline containing 1 μM of the indicated aptamer formulation intraperitoneally. Treatment times were staggered to allow accuracy in short time points. At the time of sacrifice, mice were deeply anesthetized with pentobarbital (2 min). Animals were then positioned ventrally on a fixed platform. Disposable absorbent padding was replaced after each animal, and fresh gloves, needles, apparatus, and instruments were used for each animal to minimize cross-contamination. An incision was made in the abdominal wall at the level of the xiphoid process. Using blunt scissors the rib cage was reflected up to expose the pleural cavity and heart. A 1 mL insulin syringe was used to collect 600–800 μL blood from the right ventricle. A 27-gauge needle attached to a primed pressure perfusion apparatus was then inserted into the left ventricle. Fifty mL of normal saline, at a pressure of 120 mm Hg, was perfused through the circulatory system, with the initiation of perfusion defining the assay time point. Variability from aptamer injection time to perfusion initiation time was not greater than 2 min. All tissues appeared white after perfusion, indicating the removal of blood. The brain, heart, liver, pancreas, spleen, kidney, and spinal cord were dissected and frozen on dry ice. All mice showed complete vascular washout, indicated by clearing of the liver and intercostal veins, before tissue collection.
Aptamer extraction
Aptamer extraction was performed using a plasmid maxiprep kit (Qiagen) in a separate facility with filter-tip pipettes and reagents that had been irradiated by ultraviolet light to minimize cross-contamination. Frozen tissue samples (less than 200 mg) were homogenized in 300 μL plasmid preparation buffer P1 in a 1.5-mL microcentrifuge tube using a disposable pestle (Kimble-Kontes No. 749521-1590). Three hundred microliters of plasmid preparation lysis buffer P2 was added and samples mixed gently by inversion, followed by incubation at room temperature for 5 min. 300 μL plasmid preparation buffer P3 was added to precipitate protein and high molecular weight DNA. Samples were gently mixed by inversion and placed on ice for 5 min. After centrifugation at 13,300 rpm for 5 min in a microcentrifuge, the supernatant was recovered. Blood samples (30 μL) were processed similarly after dilution with 195 μL water and 75 μL 2% sodium dodecyl sulfate giving a final sodium dodecyl sulfate concentration of 0.5%. All samples were then treated with proteinase K (30 μL; 10 mg/mL) at 55°C overnight before extraction with 500 μL phenol:chloroform:isoamyl alcohol (25:24:1) and precipitation of the aqueous phase from 700 μL isopropanol, followed by a wash with 250 μL cold 70% ethanol. The purified sample was then resuspended in 1 μL water for each mg of original tissue, with the exception of purified blood samples, which were resuspended in 2 μL water per 1 μL blood.
Quantitative PCR
Aptamer 3064 (Fig. 1; 40 nucleotides) was quantitated by PCR using primers 4556 (5′-CTAGACTAGA2GCTGAGCTGCTAGACTAGA2GCTGAGCTGG3TCG2CG3TG3) and 4557 (5′-ACGT2ACGT2ATGACATGACACGT2ACGT2ATGACATGACAC3AGAGA-CA2GAC2AC), where the underlined regions are shared with 3064. Aptamer 4971 (Fig. 1; 40 nucleotides) was quantitated by PCR using primers 4972 (5′-CTAGACTAGA2G-CTGAGCTGCTAGACTAGA2GCTGAGCTGGCGCTA2TCT2GAC2) and 4973 (5′-ACGT2ACGT2ATGACATGACACGT2ACGT2ATGACATGACAC2CAG2AG2AGCG2AGA), where the underlined regions are shared with 4971.
FIG. 1.

DNA aptamers, conjugates, and quantitative polymerase chain reaction (qPCR) quantitation. (A) Sequences of DNA aptamers 3064 (anti-myelin) and 4971 (control), showing PCR amplification by 55- to 58-nt primers to produce 123-bp PCR products detected and quantitated by qPCR using the indicated primers. (B) Deduced folded structure of 3064 under physiological conditions as reported in (28). (C) Schematic tetramer of 3′-biotinylated 3064 (strands) conjugated to streptavidin (circles).
Quantitative PCR was performed on an iCycler instrument (Life Technologies) with Sybr green master mix according to the manufacturer's instructions. Twenty-five-microliter reactions included 4 μL of tissue extract and 200 nM PCR primers. PCR was performed as follows: 50°C for 2 min; 95°C for 8 min; 40 cycles [95°C for 15 s; 60°C for 60 s]. Raw fluorescence data were fit with equation 1 [22] to estimate the number of PCR cycles for half-maximal amplification (C1/2):
 |
where Fc is the observed fluorescence signal, Fmax is the maximum fluorescence signal, C is the cycle number, C1/2 is the cycle number at half maximal amplification, k is the slope, and Fb is the baseline fluorescence value. These C1/2 values were converted to concentrations (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat), normalized, and outliers removed according to Grubbs test to arrive at the final concentration data sets (Supplementaty Tables S1–S5).
Pharmacokinetic modeling
Aptamer concentration data (C) as a function of time after injection (t) were plotted and then fit [23] with equation 2 reflecting the sum of two gamma variates [24]:
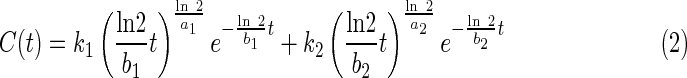 |
where k, a, and b are fitting parameters related to concentration maxima, increase rates, and decrease rates, respectively, for the two components of the function (Supplementary Fig. S2). Estimates of uncertainty in the fitted parameters were obtained by fitting 10,000 simulated data sets (Gaussian noise was added based on the observed variability of experimental concentration data from multiple animals at each time point). Total tissue exposure to aptamer was estimated as the area under the fitted curve with uncertainty again estimated by the Monte Carlo procedure. Derivations and other theoretical considerations are provided in the supplementary materials.
Results
Experimental design
We determined the pharmacokinetics of a 40-nucleotide DNA aptamer (3064) previously shown to induce remeyelination in a TMEV mouse model of MS [9]. This agent is representative of a broad class of potential therapeutics based on DNA oligonucleotides, particularly oligonucleotides capable of folding into intramolecular G-quadruplex structures [16,25–28]. Our previous studies indicated that a formulation of this aptamer as a 3′-biotinylated conjugate in tetrameric complex with streptavidin protein induced statistically significant remyelination of spinal cord axons, while other formulations of the same aptamer (lacking biotin), or other aptamers, did not [9]. We therefore designed the qPCR strategy depicted in Fig. 1A and demonstrated the extreme sensitivity of this qPCR approach to aptamer quantitation (Supplementary Fig. S1). This validation was important because of the demonstrated ability of 3064 to fold into intramolecular G-quadruplex structures, which might be expected to confound the amplification of this construct via PCR [28] (Fig. 1B). Representative PCR products amplified from tissue were also cloned and sequenced to confirm that they originated from the injected aptamers and not from genomic DNA sequences (data not shown).
For convenience and to reflect the conditions previously found for aptamer biological activity in the central nervous system, aptamers and conjugates (Fig. 1C) were injected into the peritoneal cavities of mice, and tissues were recovered and analyzed for aptamer levels at various subsequent times. All tissues were extensively perfused by flushing the entire mouse circulatory system with a large excess of buffer (50 mL). After perfusion, all organs appeared white, indicating the absence of residual blood. This procedure minimizes the tissue contribution of aptamer present in blood vessels. To efficiently extract aptamers and aptamer–streptavidin conjugates from liver, spleen, kidney, blood, spinal cord, and brain, we adapted a DNA preparation protocol normally used to recover bacterial plasmids from bacterial cell contents and genomic DNA, adding a protease digestion step to mimic the classic Hirt lysis method for purification of extrachromosomal DNA [29].
Effects of aptamer formulation on pharmacokinetics
Pharmacokinetic data and fits with a gamma variate model [24,30] (online methods and Supplementary Fig. S2) are shown in Fig. 2. Modeled recovered aptamer concentration is presented as molecules per mg weight of extracted tissue on a logarithmic scale as a function of time. Insets show individual data points graphed against time on a logarithmic scale for increased clarity. The extreme sensitivity of qPCR makes it crucial to measure the experimental background signal. Uninjected control animals harvested at various times throughout this work were analyzed for background PCR signal and the results were pooled. The median background signal is shown by the horizontal dashed line, with the standard deviation of this background estimate shown in the grey region. This depiction is intended to give a sense of the range of experimental background signals encountered in these experiments. Cases of qPCR aptamer concentration data points shown in the grey region imply that background levels for those particular experiments were lower than average.
FIG. 2.
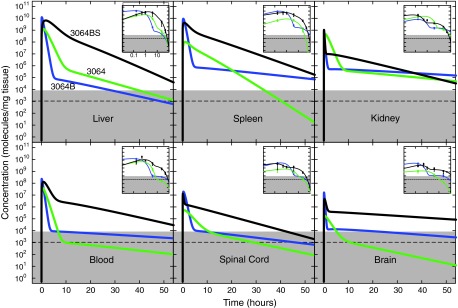
DNA aptamer 3064 pharmacokinetics in healthy mice as a function of formulation. Gamma variate pharmacokinetic fits of recovered aptamer (molecules per mg tissue) from the indicated tissues is plotted on a logarithmic scale as a function of time after intraperitoneal injection of 500 μL of 1 μM solution (500 pmol, ∼1014 molecules) into healthy mice. Mean and standard deviation for at least two determinations are shown in insets with time in hours on a logarithmic scale for clarity. Black: biotinylated 3064 conjugated to streptavidin (3064BS); blue: biotinylated 3064 (3064B); green: unmodified 3064. Horizontal dashed line indicates median background signal from negative control qPCR quantitation with error bounds (gray region). Background in individual qPCR experiments varied.
Kinetic data display a rapid spike in aptamer concentration within minutes after i.p. injection, followed by a rapid decrease and then a long period of first order exponential decay lasting more than 48 h. These features are evident in Fig. 2, and their comparison is facilitated in the derived fitting parameters listed in Table 1 (see also Supplementary Fig. S3). Several trends are evident. For a mouse of average weight (∼20 g), equal distribution of the aptamer dose across all tissues would theoretically give ∼1010 molecules/mg tissue. Independent of aptamer formulation, blood and the abdominal organs (kidney, liver, and spleen) display the highest aptamer concentrations, with peak levels approaching the ∼1010 molecules/mg expected if there had been homogenous distribution of the entire dose, but generally 1–2 orders of magnitude lower (see also Supplementary Fig. S4). Assuming efficient aptamer extraction, this suggests that 1%–10% (maximum observed ∼80%) of the total i.p. aptamer dose is rapidly absorbed by abdominal organs, with the remainder of the aptamer presumably lost by excretion. Central nervous system tissues show peak aptamer concentrations about 100-fold lower than abdominal tissues, but still three orders of magnitude above background. Concentrations of DNA aptamer 3064 in complex with streptavidin remain orders of magnitude above background in all tissues for 48 h. This is a striking result given the large mass and charge of the 3064 aptamer complex.
Table 1.
Aptamer Distribution Parameters from Fitting with Gamma Variate Summation Model
| Tissue | k1 (molecules/mg tissue) | k2 (molecules/mg tissue) | a1 (dimensionless) | a2 (dimensionless) | b1 (min) | b2 (min) | A∞ (min-molecules/mg tissue) |
|---|---|---|---|---|---|---|---|
| 3064BS | |||||||
| Liver | 1.7±0.5×1010 | 1.0±0.5×109 | 0.61±0.15 | 0.77±0.23 | 61±26 | 180±50 | 1.9±0.6×1012 |
| Spleen | 8.3±3.0×109 | 1.3±0.5×109 | 1.2±1.0 | 3.1±2.5 | 46±29 | 240±40 | 9.4±2.3×1011 |
| Kidney | 2.5±1.5×109 | 1.7±0.4×107 | 1.3±0.6 | 3.4±1.8 | 4.5±2.2 | 340±40 | 2.2±0.7×1010 |
| Blood | 3.5±1.4×108 | 6.9±3.1×106 | 0.69±0.15 | 1.5±0.4 | 33±8 | 350±90 | 2.1±1.4×1010 |
| Spinal cord | 2.9±1.4×107 | 2.0±0.5×106 | 1.0±0.4 | 16±6 | 6.3±3.6 | 320±40 | 1.1±0.2×109 |
| Brain | 1.2±0.3×107 | 4.6±2.8×105 | 0.69±0.06 | 18±4 | 11±3 | 1,300±700 | 1.1±0.1×109 |
| 3064B | |||||||
| Liver | 2.6±0.8×1010 | 1.6±0.5×105 | 1.9±0.9 | 3.6±1.5 | 14±2 | 380±100 | 4.6±1.3×1011 |
| Spleen | 2.4±0.6×1010 | 1.2±0.4×106 | 0.89±0.41 | 5.0±2.6 | 14±3 | 770±220 | 4.2±1.4×1011 |
| Kidney | 2.0±0.4×108 | 7.5±1.4×105 | 0.91±0.31 | 5.6±3.9 | 8.3±6.1 | 1,300±200 | 3.7±0.8×109 |
| Blood | 4.4±1.8×108 | 1.1±0.3×104 | 2.4±1.5 | 13±6 | 12±3 | 1,400±600 | 6.2±2.1×109 |
| Spinal cord | 5.1±1.7×107 | 1.6±1.1×104 | 0.87±0.31 | 8.4±5.0 | 16±3 | 700±210 | 1.1±0.4×109 |
| Brain | 4.1±1.3×107 | 2.1±0.5×104 | 1.1±0.6 | 4.3±1.2 | 5.3±1.2 | 1,000±200 | 3.2±1.0×108 |
| 3064 | |||||||
| Liver | 3.4±1.0×109 | 1.7±0.5×106 | 0.74±0.08 | 6.4±4.9 | 34±7 | 300±80 | 1.6±0.4×1011 |
| Spleen | 2.1±0.5×108 | 1.0±0.6×108 | 0.71±0.14 | 1.0±0.5 | 39±28 | 130±30 | 3.0±0.7×1010 |
| Kidney | 1.2±0.2×109 | 6.3±1.9×105 | 1.0±0.1 | 4.7±1.0 | 31±3 | 830±220 | 4.9±0.8×1010 |
| Blood | 1.0±0.4×108 | 1.8±0.6×103 | 0.94±0.33 | 3.7±2.2 | 25±4 | 720±210 | 3.4±1.2×109 |
| Spinal cord | 1.2±0.4×106 | 2.3±0.8×104 | 1.6±0.4 | 21±9 | 63±11 | 410±90 | 1.0±0.2×108 |
| Brain | 3.3±1.6×105 | 2.8±0.9×103 | 2.6±1.6 | 20±9 | 51±14 | 410±70 | 2.4±0.6×107 |
| 3064BS (TMEV) | |||||||
| Liver | 9.5±1.6×108 | 1.1±0.5×105 | 0.76±0.2 | 11±2 | 16±2 | 2,600±300 | 2.1±0.3×1010 |
| Spleen | 5.6±1.3×109 | 4.6±2.1×106 | 0.54±0.07 | 1.2±0.2 | 7.9±1.0 | 280±40 | 7.6±1.4×1010 |
| Kidney | 2.3±0.3×109 | 2.7±1.4×107 | 0.69±0.11 | 1.4±0.3 | 18±3 | 340±60 | 7.1±2.4×1010 |
| Blood | 2.3±1.5×107 | 2.2±0.6×105 | 1.7±0.3 | 8.1±2.3 | 67±19 | 1,900±300 | 2.5±0.4×109 |
| Spinal cord | 6.0±1.7×107 | 1.6±0.1×105 | 0.69±0.04 | 19±2 | 7.7±0.8 | 190±10 | 7.2±1.9×108 |
| Brain | 5.4±1.7×108 | 1.8±0.9×106 | 0.69±0.16 | 14±11 | 9.2±1.9 | 460±210 | 8.9±1.9×109 |
| 4971 | |||||||
| Liver | 2.4±0.3×106 | 7.3±1.7×102 | 1.5±0.4 | 8.5±4.2 | 22±4 | 570±210 | 6.5±1.2×107 |
| Spleen | 2.2±0.2×106 | 9.1±2.3×102 | 1.2±0.4 | 11±7 | 12±3 | 1100±400 | 3.5±0.6×107 |
| Kidney | 2.3±0.8×106 | 5.8±1.0×103 | 2.0±1.2 | 5.2±2.6 | 11±4 | 400±40 | 3.2±1.7×107 |
| Blood | 1.2±0.6×104 | 7.1±0.9×103 | 1.5±0.4 | 5.7±2.7 | 11±2 | 1400±600 | 1.3±0.3×107 |
| Spinal cord | 5.6±2.9×102 | 6.7±2.3×100 | 1.1±0.3 | 3.7±2.4 | 6.5±1.4 | 880±290 | 1.3±0.4×104 |
| Brain | 1.3±0.4×105 | 1.5±0.8×101 | 0.86±0.15 | 2.0±2.1 | 18±4 | 380±230 | 3.1±0.9×106 |
A∞, area under the curve.
Three 3064 formulations are compared in Fig. 2, revealing important effects. The 3′-biotinylated conjugate in tetrameric complex with streptavidin (Fig. 2, black) displays the highest exposure in all tissues, most strikingly in liver and blood; 3′ biotinylation in the absence of streptavidin (Fig. 2, blue) achieves a comparable initial peak concentration, but with a much more rapid decline to the first-order decay phase, where aptamer half-life is greater than the streptavidin conjugate in all tissues (Table 1). The same aptamer formulated without 3′ biotinylation or streptavidin (Fig. 2, green) is similarly reduced in exposure, with significantly lower delivery relative to the other two formulations in brain and blood (Fig. 2, Table 1). Together, these results suggest a profound enhancement of tissue biodistribution for DNA aptamer 3064 in tetrameric complex with streptavidin.
Pharmacokinetic effects of aptamer sequence
The results shown in Fig. 2 reveal a strong role of aptamer formulation in biodistribution and pharmacokinetics. We addressed whether aptamer nucleotide sequence influenced these parameters. In particular, 3064 contains guanosine-rich sequences shown previously to cause folding into compact intramolecular guanosine-quadruplex structures [28] that could affect penetration through endothelial barriers and interactions with transport systems. We therefore synthesized control 4971 (Fig. 1A) to be the same length as 3064, but with no potential for guanosine-quadruplex formation. A qPCR quantitation approach was developed for 4971. Pharmacokinetic results are shown in Fig. 3 and Table 1.
FIG. 3.
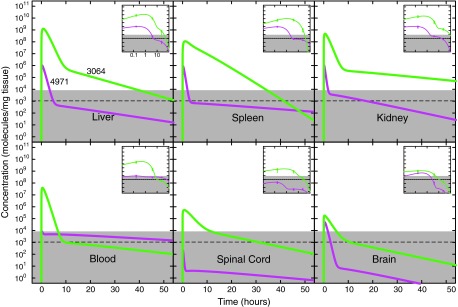
Effect of unmodified DNA aptamer sequence on pharmacokinetics in healthy mice. Depiction is as in Fig. 1, except results are compared for unmodified 3064 (green) and unmodified 4971 (magenta).
These results are striking, showing a strong sequence dependence in pharmacokinetics with the unstructured 4971 control aptamer (Fig. 3, magenta) achieving ∼1000-fold lower maximum concentration in most tissues and a more rapid decline from this peak to the subsequent first-order decay phase. This result indicates that some aspect of the guanosine-rich 3064, presumably related to its ability to adopt intramolecular guanosine-quadruplex structures, improves its pharmacokinetic profile. Folded aptamer structure might also affect tissue uptake or enhance nuclease resistance.
Viral infection effects on aptamer pharmacokinetics
Our original studies with 3064 conjugated to streptavidin showed stimulation of remyelination in animals chronically infected with TMEV[9]. Such animals experience profound inflammatory responses with some perturbation of the blood–brain barrier. We therefore applied qPCR methodology to compare 3064 streptavidin conjugate pharmacokinetics in TMEV-infected and healthy mice. The results are shown in Fig. 4 and Table 1.
FIG. 4.
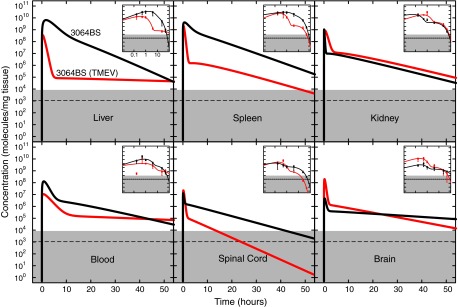
DNA aptamer 3064 streptavidin conjugate pharmacokinetics in healthy and Theiler's murine encephalomyelitis virus–infected mice. Depiction is as in Fig. 1, except results are compared for biotinylated 3064 conjugated to streptavidin injected into healthy mice (black) or Theiler's murine encephalomyelitis virus–infected mice (red).
Tissue distribution and kinetics of the 3064 conjugate with streptavidin are similar in the kidney of healthy and TMEV-infected mice and are reversed between blood and brain. Interestingly, aptamer accumulation in liver, spleen, and spinal cord were substantially reduced in TMEV-infected animals relative to controls, but still much higher than background levels. The basis for this difference is unknown, but it is notable that the stimulation of remyelination previously observed with the 3064 conjugate with streptavidin took place in the spinal cords of TMEV-infected mice. The adequacy of 3064 conjugate dosing for spinal cord remyelination was observed in animals experiencing chronic TMEV infection where demyelination is limited to the spinal cord.
Summary
This work exploits qPCR to provide unprecedented sensitivity in the analysis of the pharmacokinetics of formulations of a guanosine-rich DNA aptamer previously shown to induce remyelination in a mouse model of MS. The results show clear evidence of broad and rapid tissue distribution of the DNA aptamer, including accumulation in the central nervous systems of both healthy and TMEV-infected animals. Fitting with a gamma variate model yields the parameters shown in Table 1. Of particular interest is the value of A(∞), the total tissue exposure to drug after a single i.p. injection of ∼3×1014 molecules. These values are shown in Fig. 5 (see also Supplementary Fig. S4). It is observed that for the active streptavidin conjugate of 3064, this total tissue exposure is highest for liver and spleen, is about 100-fold lower for kidney and blood and about 1,000-fold lower for brain and spinal cord. For comparison, the area under the curve for 48 h based on median background signal is the equivalent of ∼3x106 molecules/mg, so central nervous system exposure to DNA aptamer 3064 conjugated to streptavidin is still more than 300-fold above this background. Aptamer 3064 conjugation to biotin or to biotin and streptavidin both make important favorable contributions to total tissue exposure, and these effects are significant in most tissues (Fig. 5). Importantly, control DNA aptamer 4971, lacking the potential to form guanosine quadruplex structures, showed tissue exposures typically 100- to 1,000-fold lower than the corresponding unmodified 3064. This result points to an important effect of DNA aptamer sequence on pharmacokinetics after i.p. injection in mice.
FIG. 5.

Total tissue exposure to aptamers. Area under the curve (A∞; pharmacokinetic model) for the indicated aptamer formulations and tissues (Table 1). Three vertical arrows highlight similar exposure of spinal cord to 3064 aptamer formulations.
Discussion
Because of the potential applicability of nucleic acids to in vivo diagnosis and therapy [2–6,27], some prior pharmacokinetic experimentation has been reported in animals [10–16,18,21,31]. Studies of radiolabeled phosphorothioate oligonucleotides injected intravenously (i.v.) into mice showed peak concentrations in plasma within minutes, dropping ∼100-fold within 2 h, with liver accumulation dominant over 24 h [14]. In another prior study, pharmacokinetics of an isotopically-labeled guanosine-rich DNA 15-mer targeting thrombin were analyzed after i.v. injection in rats [16]. Here, kinetic studies over 6 h showed extremely rapid tissue distribution, with highest retention in kidney, liver, and blood, and less than 1% of the dose in heart, spleen, lungs, and brain. These results are similar to what was observed for unmodified 3064 tissue distribution at 10 h, comparing brain and spinal cord to, for example, liver (Fig. 2). The present study involved i.p. injection of DNA aptamer. Another previous report suggests that a phosphorothioate DNA oligonucleotide had improved biodistribution after i.p. vs. i.v. injection in mice [11].
Previous studies have suggested that persistence of oligonucleotides in circulation can be promoted by modifications such as conjugation with polyethylene glycol to increase molecular weight [19,21]. We noted the enhancement of aptamer 3064 exposure to most tissues when 3′-biotinylated and conjugated with streptavidin protein to form tetramers (Fig. 2). The effect is least evident in kidney. Using liver as the tissue example, comparison of area-under-the-curve exposure data (Table 1, Fig. 5, and Supplementary Fig. S5) shows the total aptamer exposure is 4-fold and 12-fold higher for the tetrameric streptavidin conjugate than for free biotinylated or unbiotinylated 3064, respectively. In these experiments, 3′ aptamer biotinylation was employed consistently to facilitate oligonucleotide synthesis. Therefore, this work does not allow us to determine whether there would be advantages to 3′ versus 5′ biotinylation with respect to aptamer stability in the presence of serum exonucleases. It is possible that aptamer formulations that enhance molecular weight reduce early urinary elimination.
Of further interest, we compared the pharmacokinetics of DNA aptamer 3064 formulations to those previously reported for immunoglobin M (IgM) antibodies (molecular weights ∼106) determined by an isotopic detection method after i.p. injection [32]. Whereas the DNA aptamer distribution was rapid, peaking within minutes, radiolabel from IgM antibodies peaked at about 24 h and then declined more gradually over the 168-h study period. In contrast to the current aptamer, pharmacokinetic results where aptamer uptake was reduced in liver, spinal cord, and spleen of TMEV-infected mice, TMEV infection status was not observed to affect IgM pharmacokinetics [32].
Essentially all prior studies of DNA oligonucleotide and aptamer pharmacokinetics involved quantitation by isotopic detection or similar methods, often leaving uncertain the extent of oligonucleotide degradation and metabolism of the radiolabel. Because aptamers 3064 and 4971 are amenable to direct PCR amplification, our work quantitated aptamer levels by qPCR, a technique that gives a linear signal over seven orders of magnitude of aptamer concentration. This extreme sensitivity far exceeds conventional pharmacological detection limits and provides unprecedented insight into DNA aptamer pharmacokinetics. The qPCR technique also has the advantage that it detects only aptamers with sufficient integrity for primer annealing and DNA polymerase extension. A similar approach has recently been proposed for nucleic acid quantitation across whole-animal sections [33].
The extreme sensitivity of qPCR methodology necessitates extreme caution during sample preparation and thoughtful treatment of the issue of experimental background signal. Even after implementing extensive measures to avoid sample cross-contamination during animal handling, tissue extraction, and PCR, a variable (extremely low) aptamer signal could be detected even in negative control samples after many amplification cycles. Because this background was variable, it is represented in the data by reporting the median and standard deviation reflecting pooled values from tissue samples representing ∼10 uninjected animals evaluated over ∼10 months (grey shaded regions in Figs. 2–4). Because of the variability of this sample background, true aptamer concentration measurements for some experiments fell within the range sometimes observed for background signal. Such measurements remain valid for those experiments, but they are depicted on the grey background region to emphasize that background signals sometimes exceed these levels.
Previous pharmacokinetic studies of DNA aptamer biodistribution have suggested that the blood–brain barrier limits nucleic acid delivery from circulation to the central nervous system [16,19]. The present experiments allow a more sensitive consideration of this question through the use of qPCR. This advantage permits the conclusion that, while central nervous system exposure to a biotinylated 3064 conjugate with streptavidin is indeed three orders of magnitude lower than, for example, liver, significant central nervous system delivery is observed with an overall exposure more than 300-fold higher than a cautious background estimate. Interestingly, classic antibody studies have shown penetration of the blood-brain barrier [34], as has a recent nucleic acid selection strategy [35]. Our results show that, without special selection, formulations of a 40-nucleotide DNA aptamer reach central nervous system tissues in intact form at concentrations hundreds of fold above background. The mechanism of this delivery is of obvious interest, and may be similar for antibodies.
Table 1 includes area-under-the-curve estimates of total aptamer exposure for each tissue. These results are summarized in Fig. 5 for comparison (see also Supplementary Fig. S5). Our previous work [9] demonstrated that remyelination was stimulated by biotinylated 3064 when complexed into a tetrameric complex with streptavidin, but not by nonbiotinylated 3064 mixed with streptavidin, or by biotinylated 3064 alone (not shown). The data in Fig. 5 suggest that central nervous system tissues (particularly spinal cord, the site of remyelination; vertical arrows in Fig. 5) experience similar total exposure 3064 whether conjugated to streptavidin or not. The observed remyelinating activity of only the streptavidin conjugate therefore suggests that it is some aspect of the multivalent character of this formulation that is responsible for biological activity, not its pharmacokinetics.
Supplementary Material
Acknowledgments
This work was supported by the Mayo Foundation, Mayo Graduate School (J.A.S., M.M.S.), a Collaborative MS Center Grant from the National MS Society, and by grants from the Mayo Clinic Park Endowment (L.J.M. and M.R.) and 5T32GM065841 (J.A.S.), R25GM075148 (M.M.S.), R01NS24180 (M.R.), and R01NS32129 (M.R.) from the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Lee JF, Stovall GM. and Ellington AD. (2006). Aptamer therapeutics advance. Curr Opin Chem Biol 10:282–289 [DOI] [PubMed] [Google Scholar]
- 2.Famulok M, Hartig JS. and Mayer G. (2007). Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem Rev 107:3715–3743 [DOI] [PubMed] [Google Scholar]
- 3.Keefe AD, Pai S. and Ellington A. (2010). Aptamers as therapeutics. Nat Rev Drug Discov 9:537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer C, Hahn U. and Rentmeister A. (2011). Cell-specific aptamers as emerging therapeutics. J Nucleic Acids 2011:904750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong HY. and Byun J. (2013). Nucleic acid aptamers: new methods for selection, stabilization, and application in biomedical science. Biomol Ther (Seoul) 21:423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santosh B. and Yadava PK. (2014). Nucleic acid aptamers: research tools in disease diagnostics and therapeutics. BioMed Res Int 2014:540451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J. and Rossi JJ. (2014). Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol Ther Nucleic Acids 3:e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck A, Wurch T, Bailly C. and Corvaia N. (2010). Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 10:345–352 [DOI] [PubMed] [Google Scholar]
- 9.Nastasijevic B, Wright BR, Smestad J, Warrington AE, Rodriguez M. and Maher LJ. (2012). Remyelination induced by a DNA aptamer in a mouse model of Multiple Sclerosis. PLoS ONE 7:e39595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrawal S, Temsamani and Tang JY. (1991). Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc Natl Acad Sci (USA) 88:7595–7599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saijo Y, Perlaky L, Wang H. and Busch H. (1994). Pharmacokinetics, tissue distribution, and stability of antisense oligodeoxynucleotide phosphorothioate ISIS 3466 in mice. Oncol Res 6:243–249 [PubMed] [Google Scholar]
- 12.Zhang R, Lu Z, Zhao H, Zhang X, Diasio RB, Habus I, Jiang Z, Iyer RP, Yu D. and Agrawal S. (1995). In vivo stability, disposition and metabolism of a “hybrid” oligonucleotide phosphorothioate in rats. Biochem Pharmacol 50:545–556 [DOI] [PubMed] [Google Scholar]
- 13.Cammilleri S, Sangrajrang S, Perdereau B, Brixy F, Calvo F, Bazin H. and Magdelenat H. (1996). Biodistribution of iodine-125 tyramine transforming growth factor alpha antisense oligonucleotide in athymic mice with a human mammary tumour xenograft following intratumoral injection. Eur J Nucl Med 23:448–452 [DOI] [PubMed] [Google Scholar]
- 14.Crooke ST, Graham MJ, Zuckerman JE, Brooks D, Conklin BS, Cummins LL, Greig MJ, Guinosso CJ, Kornbrust D, Manoharan M, et al. (1996). Pharmacokinetic properties of several novel oligonucleotide analogs in mice. J Pharmacol Exp Ther 277:923–937 [PubMed] [Google Scholar]
- 15.Nakada Y, Fattal E, Foulquier M. and Couvreur P. (1996). Pharmacokinetics and biodistribution of oligonucleotide adsorbed onto poly(isobutylcyanoacrylate) nanoparticles after intravenous administration in mice. Pharm Res 13:38–43 [DOI] [PubMed] [Google Scholar]
- 16.Reyderman L. and Stavchansky S. (1998). Pharmacokinetics and biodistribution of a nucleotide-based thrombin inhibitor in rats. Pharm Res 15:904–910 [DOI] [PubMed] [Google Scholar]
- 17.Dougan H, Lyster DM, Vo CV, Stafford A, Weitz JI. and Hobbs JB. (2000). Extending the lifetime of anticoagulant oligodeoxynucleotide aptamers in blood. Nucl Med Biol 27:289–297 [DOI] [PubMed] [Google Scholar]
- 18.Yu RZ, Geary RS, Leeds JM, Watanabe T, Moore M, Fitchett J, Matson J, Burckin T, Templin MV. and Levin AA. (2001). Comparison of pharmacokinetics and tissue disposition of an antisense phosphorothioate oligonucleotide targeting human Ha-ras mRNA in mouse and monkey. J Pharm Sci 90:182–193 [DOI] [PubMed] [Google Scholar]
- 19.Boomer RM, Lewis SD, Healy JM, Kurz M, Wilson C, and McCauley TG. (2005). Conjugation to polyethylene glycol polymer promotes aptamer biodistribution to healthy and inflamed tissues. Oligonucleotides 15:183–195 [DOI] [PubMed] [Google Scholar]
- 20.Pieve CD, Perkins AC, and Missailidis S. (2009). Anti-MUC1 aptamers: radiolabelling with (99m)Tc and biodistribution in MCF-7 tumour-bearing mice. Nucl Med Biol 36:703–710 [DOI] [PubMed] [Google Scholar]
- 21.Healy JM, Lewis SD, Kurz M, Boomer RM, Thompson KM, Wilson C. and McCauley TG. (2004). Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res 21:2234–2246 [DOI] [PubMed] [Google Scholar]
- 22.Rutledge RG. (2004). Sigmoidal curve-fitting redefines quantitative real-time PCR with the prospective of developing automated high-throughput applications. Nucleic Acids Res 32:e178–e178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Offord C. and Bajzer E. (2001). A hybrid global optimization algorithm involving simplex and inductive search. Lect Notes Comput Sc 2074:680–688 [Google Scholar]
- 24.Pfeffer M. (1988). Gamma variate fits to pharmacokinetic data. Eur J Drug Metab Pharmacokinet 13:155–159 [DOI] [PubMed] [Google Scholar]
- 25.Dapic V, Bates PJ, Trent TO, Rodger A, Thomas SD, and Miller DM. (2002). Antiproliferative activity of G-quartet-forming oligonucleotides with backbone and sugar modifications. Biochemistry 41:3676–3685 [DOI] [PubMed] [Google Scholar]
- 26.Dapic V, Abdomerovic V, Marrington R, Peberdy J, Rodger A, Trent JO. and Bates PJ. (2003). Biophysical and biological properties of quadruplex oligodeoxyribonucleotides. Nucleic Acids Res 31:2097–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gatto B, Palumbo M, and Sissi C. (2009). Nucleic acid aptamers based on the G-quadruplex structure: therapeutic and diagnostic potential. Curr Med Chem 16:1248–1265 [DOI] [PubMed] [Google Scholar]
- 28.Smestad J. and Maher LJ., 3rd (2013). Ion-dependent conformational switching by a DNA aptamer that induces remyelination in a mouse model of multiple sclerosis. Nucleic Acids Res 41:1329–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirt B. (1967). Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369 [DOI] [PubMed] [Google Scholar]
- 30.Davenport R. (1983). The derivation of the gamma-variate relationship for tracer dilution curves. J Nuc Med 24:945–948 [PubMed] [Google Scholar]
- 31.Uhlmann E, Peyman A, Ryte A, Schmidt A, and Buddecke E. (2000). Use of minimally modified antisense oligonucleotides for specific inhibition of gene expression. Methods Enzymol 313:268–284 [DOI] [PubMed] [Google Scholar]
- 32.Hunter SF, Miller DJ. and Rodriguez M. (1997). Monoclonal remyelination-promoting natural autoantibody SCH 94.03: pharmacokinetics and in vivo targets within demyelinated spinal cord in a mouse model of multiple sclerosis. J Neurol Sci 150:103–113 [DOI] [PubMed] [Google Scholar]
- 33.Boos JA, Kirk DW, Piccolotto ML, Zuercher W, Gfeller S, Neuner P, Dattler A, Wishart WL, Von Arx F, Beverly M, et al. (2013). Whole-body scanning PCR; a highly sensitive method to study the biodistribution of mRNAs, noncoding RNAs and therapeutic oligonucleotides. Nucleic Acids Res 41:e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friden PM, Walus LR, Musso GF, Taylor MA, Malfroy B. and Starzyk RM, RM. (1991). Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc Natl Acad Sci (USA) 88:4771–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng C, Chen YH, Lennox KA, Behlke MA. and Davidson BL. (2013). In vivo SELEX for Identification of Brain-penetrating Aptamers. Mol Ther Nucleic Acids 2:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.