

Abstract
The DnaK/Hsp70 chaperone system and ClpB/Hsp104 collaboratively disaggregate protein aggregates and reactivate inactive proteins. The teamwork is specific: E. coli DnaK interacts with E. coli ClpB and yeast Hsp70, Ssa1, interacts with yeast Hsp104. This interaction is between the M-domains of hexameric ClpB/Hsp104 and the DnaK/Hsp70 nucleotide-binding domain (NBD). To identify the site on E. coli DnaK that interacts with ClpB, we substituted amino acid residues throughout the DnaK NBD. We found that several variants with substitutions in subdomain IB and IIB of the DnaK NBD were defective in ClpB interaction in vivo in a bacterial two-hybrid assay and in vitro in a fluorescence anisotropy assay. The DnaK subdomain IIB mutants were also defective in the ability to disaggregate protein aggregates with ClpB, DnaJ and GrpE, although they retained some ability to reactivate proteins with DnaJ and GrpE in the absence of ClpB. We observed that GrpE, which also interacts with subdomains IB and IIB, inhibited the interaction between ClpB and DnaK in vitro, suggesting competition between ClpB and GrpE for binding DnaK. Computational modeling of the DnaK-ClpB hexamer complex indicated that one DnaK monomer contacts two adjacent ClpB protomers simultaneously. The model and the experiments support a common and mutually exclusive GrpE and ClpB interaction region on DnaK. Additionally, homologous substitutions in subdomains IB and IIB of Ssa1 caused defects in collaboration between Ssa1 and Hsp104. Altogether, these results provide insight into the molecular mechanism of collaboration between the DnaK/Hsp70 system and ClpB/Hsp104 for protein disaggregation.
Keywords: GrpE, nucleotide exchange factor, Hsp40, protein disaggregation, molecular chaperone
Introduction
The Hsp70 family of ATP-dependent molecular chaperones are involved in a multitude of protein activation and remodeling reactions including protein folding, protein transport and protein disaggregation 1–3. Hsp70 proteins are able to perform this variety of activities because they can collaborate with many additional chaperones and co-chaperones. Escherichia coli Hsp70, DnaK, collaborates with an Hsp40 protein, DnaJ, and a nucleotide exchange factor (NEF), GrpE 1–3. DnaJ recruits DnaK to the substrate and stimulates ATP hydrolysis by DnaK, which promotes stable substrate binding 4. GrpE stimulates ADP and substrate release, priming DnaK for another round of substrate binding 5. DnaK has also been shown to collaborate with additional ATP dependent chaperones, including E. coli Hsp90 and ClpB 6–8.
DnaK is comprised of two domains, the N-terminal nucleotide-binding domain (NBD) and the C-terminal substrate-binding domain (SBD) (Fig. 1A) 9, 10. The NBD is defined by four subdomains: IA, IIA, IB and IIB (Fig. 1A and B). Co-chaperones DnaJ and GrpE have been shown to directly interact with the DnaK NBD (Fig. 1B) 2, 3, 11–13 to regulate nucleotide binding, hydrolysis and exchange, processes which in turn are coupled to substrate binding and release 2, 3. Large conformational changes in DnaK accompany the binding and hydrolysis of ATP by DnaK 2, 3, 14–20. The ADP-bound closed conformation promotes stable interactions with substrate, while the ATP-bound open conformation binds weakly to substrates 14–20.
Fig. 1.

Models of the structure of E. coli DnaK and T. thermophilus ClpB. A. Solution NMR structure of DnaK in the ADP bound conformation (pdb:2KHO) 18. DnaK is comprised of an N-terminal nucleotide-binding domain (NBD) (blue), containing of four subdomains, IA, IB, IIA and IIB and a substrate-binding domain (SBD) (grey) that contains the β-sheet substrate-binding site and α-helical lid region. B. Model of the NBD of DnaK in the ADP bound conformation showing the amino acids identified as interacting with GrpE (cyan) (pdb:2KHO for structure; pdb:1DKG for GrpE binding residues) 12, 18. C. Protomer structure of T. thermophilus ClpB (PDB ID code 1qvr – chain B) 43, with the N-domain, NBD-1, NBD-2 and M-domain shown. Motif 1 and motif 2 of the M-domain identified (ovals) and the four helices of the M-domain are shown in color: H1 (orange), H2 (purple), H3 (blue) and H4 (green). ATP (black) is shown as a CPK model. D. Model of the NBD of DnaK in the ADP bound conformation showing the residues mutated in the present study (pdb:2KHO) 18. Images in A–D were made using PYMOL (www.pymol.org) 57.
The DnaK system has been shown to collaborate both in vivo and in vitro with ClpB for disaggregation and reactivation of insoluble protein aggregates 7, 8, 21. This collaboration is specific: E. coli DnaK (DnaKEc) collaborates with E. coli ClpB (ClpBEc), yeast Hsp70 collaborates with yeast Hsp104 and Thermus thermophilus DnaK (DnaKTth) functions with T. thermophilus ClpB (ClpBTth) 21–23. A recent in vitro study using multiple nuclear magnetic resonance (NMR) spectroscopy techniques, reported an atomic-resolution model for the ClpB-DnaK complex from T. thermophilus 24. To identify residues involved in the intermolecular interaction, monomers and monomeric fragments of both ClpBTth and DnaKTth were utilized 24. Results were consistent with previously described experiments using ClpBEc and Hsp104, and localized the DnaKTth interaction site on ClpBTth to residues in helices 2 and 3 of the M-domain (Fig. 1C) 24–27. The ClpBTth interaction site on DnaKTth was mapped to subdomains IB and IIB of the DnaKTth NBD, and included residues in an extended loop of IIB 24. Specific residues in DnaKTth that could potentially form H-bonds with ClpB M-domain residues were identified (R55 and K285 in subdomain IB and the IIB loop respectively; homologous to E. coli R56 and T291) and were in a region previously shown by x-ray crystallography to interact with GrpE (Fig. 1B and D) 12, 24. Additional functional and NMR based experiments suggested that GrpE and ClpB compete for binding to the DnaK nucleotide-binding domain 24. In another report, the N-terminal domain of human Hsp70 (HSPA1A) was shown to interact with the M-domain of Saccharomyces cerevisiae Hsp104, but the N-terminal domain alone was insufficient to stimulate disaggregation activity in collaboration with Hsp104 27.
A recent study by Reidy et al. showed that E. coli ClpB, DnaK and GrpE support prion propagation and thermotolerance in S. cerevisiae, when expressed from plasmids, thus replacing the yeast chaperones, Hsp104 and Ssa1 for these activities 28. These results suggested that ClpB is able to replace Hsp104 for most cellular activities as long as the species-specific Hsp70 protein, DnaK, is present. Surprisingly, GrpE, which is known to function with DnaK but not Ssa1 29, 30, was also required for these reactions pointing to an essential role for GrpE during protein disaggregation and reactivation with ClpB and DnaK. In contrast E. coli DnaJ, which can function with Ssa1 and other Hsp70s 21, 23, was not essential 28.
Here, we examined collaboration between the E. coli chaperones and co-chaperones involved in protein disaggregation. We identified a region of DnaKEc that is important for interaction and collaboration with ClpBEc. This region includes residues in the IB and IIB subdomains of the DnaKEc NBD, including residues in the GrpE binding site. Additionally, modeling studies support a mutually exclusive GrpE and ClpBEc binding site on DnaKEc and indicate that DnaK interacts with two adjacent ClpB protomers within a hexamer.
Results
Recent in vitro studies from Rosenzweig et al. 24, using T. thermophilus chaperones showed that ClpBTth interacts with residues in the GrpE binding site of DnaKTth. In addition the data suggested that GrpE inhibits disaggregation by ClpB, DnaK and DnaJ by competing with ClpB for binding to the DnaK nucleotide-binding domain (NBD) 24. From these results they proposed that GrpE is not directly involved in protein disaggregation by DnaK, DnaJ and ClpB 24. These results are in apparent contradiction with the studies of Reidy et al., showing that E. coli GrpE, as well as DnaK and ClpB, is required for prion propagation and thermotolerance in yeast cells deleted for Hsp104 28. Because GrpE is specific for DnaKEc and does not act with Ssa1 29, 30, this work implied that GrpE is directly involved in disaggregation and prion propagation with ClpB and DnaK. Therefore, we investigated whether the region in the NBD of DnaKEc that is involved in GrpE binding is important for collaboration with ClpBEc in protein remodeling and whether GrpE is involved in protein disaggregation with ClpB, DnaK and DnaJ.
Residues of the E. coli DnaK NBD subdomain IIB are important for functional collaboration between E. coli DnaK and ClpB
We first tested whether residues in the GrpE binding region of the E. coli DnaK NBD are involved in functional interaction and collaboration with E. coli ClpB, as they are in T. thermophilus DnaK. Several variants were constructed containing amino acid substitutions in the NBD of DnaKEc in residues shown by X-ray crystallography to be involved in GrpE binding (Table 1 and Fig. 1B and D) 12. Some were single substitutions and others were double or triple substitutions, denoted by an (*) after the first residue (Table 1). The DnaK mutants were then tested for their ability to reactivate heat-denatured GFP (hdGFP) with DnaJ and GrpE or with ClpB, DnaJ and GrpE. As previously seen, wild type DnaK with DnaJ and GrpE reactivated hdGFP slowly; the addition of ClpB stimulated the rate of reactivation by ~3-fold (Fig. 2A and Supplemental Fig. S1A) 31. When DnaK mutants with substitutions in subdomain IIB, including DnaKL257A, DnaKR261C and DnaKN282A*, were tested, we observed that they were able to reactivate hdGFP, although the rates of reactivation were slower than for wild type (Fig. 2A and B and Supplemental Fig. S1A). Importantly, reactivation was not appreciably stimulated by the addition of ClpB, suggesting that these residues are important for the collaboration between DnaK and ClpB. In contrast, DnaK mutants with substitutions in the subdomain IIB loop, DnaKA288C and DnaKA290C, had activity similar to DnaKwt either with DnaJ and GrpE or with ClpB, DnaJ and GrpE, indicating these substitutions do not affect the collaboration with ClpB for the reactivation of hdGFP (Fig. 2A and Supplemental Fig. S1A). DnaK with substitutions in residues in subdomain IB DnaKL49A, DnaKR56A and DnaKQ57A*, were partially defective in protein reactivation in collaboration with DnaJ and GrpE, but ClpB stimulated the rate of disaggregation between ~5 and 15-fold (Fig. 2C and Supplemental Fig. S1A). The results suggest that the subdomain IB residues that we characterized are important for DnaK, DnaJ and GrpE reactivation of hdGFP. However, they are not important for reactivation of hdGFP in collaboration with ClpB. In additional experiments, DnaK mutants with substitutions in other regions of the DnaK NBD were tested for the ability to reactivate hdGFP (Table 1 and Supplemental Fig. S1B and S1C). Many of these DnaK mutants were defective in hdGFP reactivation with DnaJ and GrpE, however ClpB stimulated hdGFP reactivation by all mutants at least ~3-fold (Supplemental Fig. S1B and S1C). Together these results show that residues in the GrpE binding region of E. coli DnaK are important for DnaK collaboration with E. coli ClpB.
Table 1.
Substitutions in the nucleotide-binding domain of DnaK
| Name of Mutanta | Region of NBD | Substitution |
|---|---|---|
| K106A* | No known interactions | K106A, G107A, Q108A, K109A |
| K121E* | K121E, K122E, K124E, K125E | |
| R159A* | R159A, I160A, E164A | |
| E310K* | E310K, D311K, N314K | |
| K351A* | K351A, K352A, A354G, E355A | |
| Y145A* | DnaJ binding region | Y145A, N147A, D148A |
| K166A* | K166A, R167A, I169A | |
| N187A* | N187A, T189A | |
| E217A* | E217A, V218A | |
| L49A | GrpE binding region | L49A |
| R56A | R56A | |
| Q57A* | Q57A, V59A, T60A | |
| L257A | L257A | |
| R261C | R261C | |
| N282A* | N282A, P284A, Y285A | |
| A288C | A288C | |
| A290C | A290C |
* indicates multiple amino acid substitutions
Fig. 2.

Substitution mutants in DnaK NBD subdomain IIB are defective for collaboration with ClpB in GFP reactivation. A–C. Reactivation of heat-denatured GFP was measured in reactions containing DnaK wild-type or mutant. DnaK wild-type or mutant protein was incubated with DnaJ, GrpE, ClpB and ATP as indicated and the increase in fluorescence intensity was monitored over time as described in methods. Subdomain IIB loop mutants, subdomain IIB mutants and subdomain IB mutants are shown in A, B and C, respectively. In A–C, the initial fluorescence was set equal to 0 and a representative experiment is shown (A–C) of three or more replicates.
Based on our previous observation that the DnaK system and ClpB act synergistically to hydrolyze ATP in the presence of substrate 31, we next tested if the DnaK mutants were defective in this activity. We monitored ATPase activity by the mutant DnaK proteins both with and without ClpB in the presence of DnaJ, GrpE and heat-denatured malate dehydrogenase (hdMDH). As previously seen, the ATPase activity of ClpB in combination with the DnaK system in the presence of hdMDH was ~2.5-fold higher than the additive rates of the DnaK system and ClpB separately in the presence of hdMDH (Fig. 3A) 31. In contrast, when the subdomain IIB mutants, DnaKL257A, DnaKR261C and DnaKN282A*, were incubated with ClpB, DnaJ, GrpE and hdMDH, ATP hydrolysis rates were not stimulated above the additive rates of the DnaK mutants with cochaperones and ClpB separately (Fig. 3A). The ATPase activity exhibited by subdomain IB mutant, DnaKR56A, with ClpB, DnaJ, GrpE and hdMDH was approximately additive and similar to the subdomain IIB mutants, suggesting a defect in collaboration between ClpB and this mutant (Fig. 3B). The ATPase activity of other subdomain IB mutants, DnaKQ57A* and DnaKL49A and subdomain IIB loop mutants, DnaKA288C and DnaKA290C was stimulated ~1.6–2.3-fold more than additive in the presence of ClpB, DnaJ, GrpE and hdMDH, however, the stimulation was less than the ~2.5-fold stimulation observed for DnaKwt (Fig. 3B). For other E. coli DnaK mutant proteins with substitutions in the NBD, DnaKY145A* and DnaKN187A*, the ATPase activity in the presence of DnaJ, GrpE, ClpB and hdMDH was between ~1.7–2.1-fold more than additive, again similar to DnaKwt (Supplemental Fig. S1D). Together, these results show that residues in DnaKEc subdomains IB and IIB are important for functional collaboration between ClpB and DnaK.
Fig. 3.
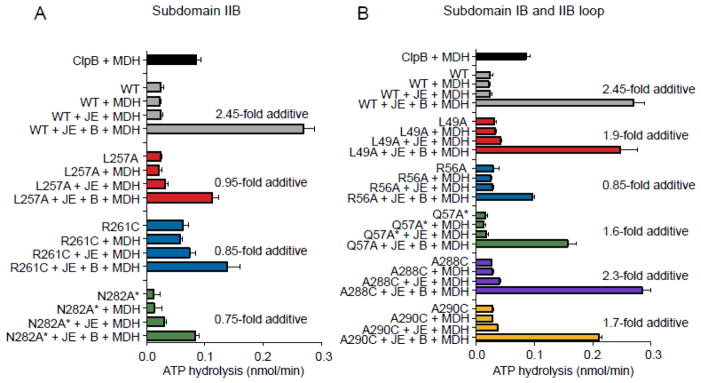
DnaK mutants with substitutions in subdomain IB and IIB are defective in synergistic ATPase activity with ClpB. A, B. Steady-state ATP hydrolysis was measured in reactions containing DnaK wild-type or mutant, DnaJ, GrpE, ClpB and heat-denatured malate dehydrogenase (MDH) and ATP as indicated and described in Methods. The fold stimulation above additive is calculated by dividing the rate of ATP hydrolysis for DnaK, J, E, B and MDH by the sum of the rates for DnaK, J, E, and MDH plus the rate for ClpB and MDH. DnaK subdomain IIB mutants are shown in A and subdomain IB and IIB loop mutants are shown in B. For A and B data are means ± SEM (n=3).
E. coli DnaK variants with substitutions in subdomains IB and IIB of the NBD are defective for in vivo and in vitro interactions with ClpB
To determine if variants that were defective in functional collaboration with ClpB were defective in intermolecular interaction with ClpB, we tested our mutants using a bacterial two-hybrid assay 32. We had previously used this assay to demonstrate an interaction between DnaK and ClpB in vivo 25. DnaK wild type or mutant was fused to the C-terminus of one fragment of Bordetella pertussis adenylate cyclase, T25, while ClpB was fused to the C-terminus of the other adenylate cyclase fragment, T18. If ClpB and the DnaK mutant being tested interact, the T18 and T25 fragments will form an active adenylate cyclase and a cAMP dependent reporter gene, β-galactosidase, is expressed. As previously observed, coexpression of T18-ClpB and T25-DnaKwt resulted in higher levels of β-galactosidase activity than coexpression of T18-ClpB and the T25 vector, suggesting that ClpB and DnaK interact in vivo (Fig. 4A) 25. When the T25-subdomain IIB mutant fusions, T25-DnaKL257A, T25-DnaKR261C or T25-DnaKN282A*, were each coexpressed with T18-ClpB, we observed less β-galactosidase activity than seen with T25-DnaKwt, indicating that the mutants were partially defective in ClpB interaction in vivo (Fig. 4A). The subdomain IB mutants, DnaKR56A and DnaKQ57A*, were also partially defective in ClpB interaction in vivo (Fig. 4A). In control experiments all of the mutant proteins were produced at levels similar to T25-DnaKwt (Supplemental Fig. S2).
Fig. 4.

Interaction between DnaK and ClpB in vivo and in vitro. A. A bacterial two-hybrid system was used to detect interactions between DnaK mutants and ClpB in vivo, as described in Methods. Fusion proteins were constructed between DnaK wild-type or mutant and one domain of B. pertussis adenylate cyclase, T25. ClpB was fused to the other domain, T18. The fusion proteins were coexpressed in BTH101 cells and the interaction between ClpB and DnaK was monitored by the expression of a reporter gene, β-galactosidase in liquid assays. Data are means ± SEM (n=9–12) B–C. Interaction of DnaK (0.075 μM) with ClpBE279A,E678A (ClpBtrap) (5 μM) in the presence of ATP was measured by fluorescence anisotropy using pyrene-maleimide labeled DnaKK106C (DnaKpy(wt)) or DnaKpy with additional substitutions (DnaKpy(L257A), DnaKpy(N282A*), DnaKpy(R56A) and DnaKpy(Q57A*)) as described in Methods. Data in B are presented as the fold change in signal upon addition of ClpBtrap to the reaction. In C, either GrpE or DnaKwt (5 μM) was added to the DnaKpy(wt) (0.075 μM) and ClpBtrap (5 μM) reaction as indicated and the change in fluorescence anisotropy was measured. Data in C are presented as the change in anisotropy signal (arbitrary units) upon addition of ClpBtrap to the reaction. Data in B–C are means ± SEM (n=3).
We tested DnaK mutants for interaction with ClpB in vitro by measuring the fluorescence anisotropy signal of pyrene-labeled DnaK in the presence and absence of ClpB. To do this, a K106C substitution was introduced into DnaKwt and the DnaK mutants and then the purified proteins were labeled with pyrene-maleimide, referred to as DnaKpy-wt, DnaKpy-L257A, DnaKpy-N282A*, DnaKpy-R56A, and DnaKpy-Q57A*. A ClpB mutant that contains mutations in each NBD (E279A and E678A) 33, ClpBtrap, was used to promote the ClpB ATP bound conformation and stabilize the DnaK-ClpB interaction. We observed that the addition of ClpBtrap and ATP to DnaKpy-wt stimulated the anisotropy signal ~2.2-fold above the DnaKpy-wt signal alone, indicating an interaction between the two proteins (Fig. 4B). The anisotropy signals of DnaKpy-L257A, DnaKpy-N282A*, DnaKpy-R56A and DnaKpy-Q57A* also increased upon addition of ClpB, however the increase was only ~1.2–1.3-fold above the signal for the DnaK mutant alone (Fig. 4B). Taken together, the results suggest that the decrease in the functional collaboration in ATPase and protein remodeling activities between E. coli ClpB and the DnaK mutants in subdomain IB and IIB is likely caused by a weaker interaction between the ClpB M-domain and DnaK.
GrpE competes with ClpB for a binding site on DnaK
Since the region on E. coli DnaK involved in collaboration and interaction with ClpB (Fig. 2, 3 and 4A and B) is also involved in interactions with GrpE 12, we tested whether GrpE competes with ClpB for DnaK interaction in vitro. Fluorescence anisotropy was carried out using pyrene labeled wild type DnaK as above. We observed that the fluorescence signal seen by the combination of DnaKpy-wt, ClpBtrap and ATP decreased by ~33% with the addition of GrpE at a concentration equimolar to ClpB (Fig. 4C). These data suggest that GrpE interferes with the interaction between ClpB and DnaK. As a control, excess unlabeled DnaKwt caused a decrease in signal of ~30% compared to DnaKpy-wt and ClpBtrap alone (Fig. 4C), indicating that unlabeled DnaKwt competes with DnaKpy-wt for binding to ClpB.
E. coli GrpE and ClpB can both collaborate with DnaK during the process of protein remodeling
We next wanted to explore the proposal by Rosenzweig et al. that GrpE does not aid ClpB, DnaK and DnaJ in extracting polypeptides from aggregates, but rather participates in a separate later reaction with DnaK and DnaJ to refold the unfolded polypeptides released from ClpB 24. To eliminate the effects of the DnaK system in protein refolding following release from ClpB, we utilized RepA as a substrate. RepA is activated for DNA binding by conversion of inactive dimers to active monomers using the combined activity of ClpB, DnaK and DnaJ 31. RepA does not require chaperone assisted refolding of monomers following dissociation of dimers into monomers 34. Our lab has also shown that with some conditions DnaK and DnaJ alone activate RepA 31 and with other conditions activation requires GrpE in addition to DnaK and DnaJ 35. For example, DnaK and DnaJ efficiently remodel RepA dimers into monomers in the absence of GrpE when the concentration of DnaK is ~1.0 μM (Fig. 5A). However, at lower concentrations of DnaK (~0.5 μM), GrpE is required for RepA remodeling (Fig. 5A). We observed that RepA activation was stimulated by either a low (0.03 μM) or a high concentration of GrpE (2 μM), but the lower concentration was more effective (Fig. 5A). Moreover, as we increased the concentration of GrpE in the presence of DnaK (0.4 μM) and DnaJ, RepA activation was stimulated and then inhibited (Fig. 5B). RepA activation decreased at ratios of 1:0.5 or lower DnaK to GrpE. Previously, inhibition of DnaK chaperone activity by excess GrpE was observed 36–39 and it was suggested that a balance between ATP binding and ATP hydrolysis must be maintained for optimal chaperone activity. Thus, the presence of excess GrpE likely shifts the balance towards ATP-bound DnaK, which has low affinity for substrate, and would lead to less RepA being bound and activated by DnaK.
Fig. 5.

The effect of GrpE on protein activation and remodeling by DnaK, DnaJ and ClpB. A, C. RepA was incubated with ATP, DnaJ and increasing concentrations of DnaKwt in the presence of 0 μM, 0.03 μM or 2 μM GrpE without ClpB (A) or with 180 nM ClpB (C), as indicated. RepA activation was measured as described in Methods. In A and C, gray dashed lines at 0.2 and 0.4 μM DnaK are present to aid the eye. B, D. RepA was incubated with 0.4 μM DnaKwt (B) or 0.1 μM DnaKwt (D), DnaJ and ATP in the presence of increasing GrpE concentrations without (B) or with 20 nM ClpB (D) as indicated. RepA activation was measured as described in Methods. E. DnaKwt (1.4 μM) and DnaJ (0.25 μM) were incubated with hdGFP and ATP in the absence or presence of ClpB (0.25 μM) and GrpE as indicated. The increase in fluorescence intensity was monitored over time. Initial rates of reactivation of hdGFP were determined as described in Methods. Data in A–E are means ± SEM (n=3). Some error bars are hidden by symbols.
We next investigated whether GrpE stimulated or inhibited RepA activation by the combination of DnaK, DnaJ and ClpB. A lower concentration of DnaKwt (0.2 μM) was sufficient for RepA activation in the presence of ClpB compared to in the absence (Fig. 5C) 31. The addition of GrpE (0.03 μM) to the ClpB, DnaK and DnaJ reaction did not alter the requirement for DnaK, neither stimulating nor inhibiting the reaction (Fig. 5C). However, at a higher GrpE concentration (2 μM), a higher concentration of DnaKwt (0.5 μM) was required for RepA activation, suggesting that GrpE may interfere with the collaboration between ClpB and DnaKwt for the monomerization of RepA (Fig. 5C). To better understand the effect of GrpE on the activation of RepA by DnaK, DnaJ and ClpB, we varied the GrpE concentration in the reaction (Fig. 5D). Addition of GrpE up to ~0.05 μM slightly stimulated RepA activation by DnaK, DnaJ and ClpB; at higher GrpE concentrations, chaperone activity was inhibited (Fig. 5D). Similar to our results in the absence of ClpB (Fig. 5B), RepA activation decreased at ratios of 1:0.5 or lower DnaK (0.1 μM) to GrpE in the presence of ClpB. These results indicate that GrpE is essential for the activation of RepA at low concentrations of DnaK in the absence of ClpB. But when ClpB is present, GrpE is not required for RepA monomerization and GrpE inhibits the reaction at high concentrations. GrpE may inhibit RepA activation by ClpB, DnaK and DnaJ by competing with ClpB for binding to DnaK (Fig. 5D). However, the decrease in RepA activation observed for the DnaK and DnaJ reaction with high GrpE concentrations makes it difficult to interpret the results (Fig. 5B).
The effect of GrpE on the remodeling of another substrate, hdGFP, by the DnaK system and ClpB was also tested. Rosenzweig et al., used this substrate and found that GrpE inhibited the disaggregation of GFP when a 10:2:1 molar ratio of T. thermophilus DnaK:DnaJ:GrpE was used in the presence of ClpBTth 24. We monitored hdGFP reactivation by E. coli DnaK and DnaJ or DnaK, DnaJ and ClpB in the absence or presence of GrpE. In the presence of a 10:2:1 ratio of DnaK:DnaJ:GrpE, we observed that the initial rate of hdGFP reactivation was two-fold higher in the presence than in the absence of GrpE (Fig. 5E). Moreover, the 2-fold GrpE stimulation was also seen when ClpB was present (Fig. 5E). A similar stimulation by GrpE was observed using a molar ratio of 10:2:10 DnaK:DnaJ:GrpE either without or with ClpB (Fig. 5E). These results suggest that GrpE aids in the reactivation of hdGFP by DnaK and DnaJ, and under the conditions tested it does not interfere with the stimulation observed upon addition of ClpB. Taken together, the data suggest that the roles ClpB and GrpE play during collaboration with DnaK may be overlapping, but they are not identical, despite sharing a binding site on DnaK.
Subdomains IB and IIB of eukaryotic Hsp70 are important for collaboration with Hsp104
Since yeast Hsp104 and Hsp70, like E. coli ClpB and DnaK, interact in a species-specific manner 21, 25–27, 40, 41, we tested if the same region of yeast Hsp70, Ssa1, was important for collaboration with Hsp104. We constructed mutants in subdomains IB, IIB and the IIB loop of the Ssa1 NBD (Table 2). The Ssa1 mutants were tested for the ability to collaborate with Hsp104 in reactivating heat-denatured luciferase (hdLuc) (Fig. 6). Ssa1wt reactivated hdLuc in combination with yeast Hsp40, Ydj1, and the addition of Hsp104 stimulated the rate of the reaction ~2-fold, as previously observed (Fig. 6A) 25, 42. Ssa1S283A reactivated hdLuc as well as Ssa1wt and was further stimulated by Hsp104 (Fig. 6A). Ssa1D282A also reactivated hdLuc similarly to Ssa1wt, but the addition of Hsp104 did not stimulate the reaction, suggesting that this residue is likely important for collaboration with Hsp104 (Fig. 6A). At the same concentration as Ssa1wt, Ssa1R255A, Ssa1R259A, Ssa1G287A and Ssa1D289A had limited activity with Ydj1. When these mutant proteins were added at a 3-fold higher concentration, reactivation of hdLuc was observed, but Hsp104 did not stimulate reactivation further (Fig. 6B). The subdomain IB mutants, Ssa1L48A and Ssa1Q56A, had the ability to act with Ydj1 similar to Ssa1wt (Fig. 6C). However, Ssa1L48A had a slight defect in collaboration with Hsp104, and Ssa1Q56A was unable to collaborate with Hsp104 (Fig. 6C). Together these results suggest that the region on DnaK and Ssa1 that interacts with ClpB/Hsp104 is similar, but not identical (Fig. 6A–C).
Table 2.
Substitutions in the nucleotide-binding domain of Ssa1 or hHsp70
| Substitution | Hsp70 | Homologous residue in E. coli DnaK |
|---|---|---|
| Ssa1 | ||
|
| ||
| L48A | L49 | |
|
| ||
| Q56A | Q57 | |
|
| ||
| R255A | L257 | |
|
| ||
| R259A | R261 | |
|
| ||
| D282A | P284 | |
|
| ||
| S283A | Y285 | |
|
| ||
| G287A | D289 | |
|
| ||
| D289A | H295 | |
|
| ||
| Human Hsp70 | ||
| R262A | R261 | |
Fig. 6.

Substitutions in subdomains IB and IIB of the yeast Hsp70, Ssa1, cause defects in collaboration with Hsp104. A–C. Reactivation of heat-denatured luciferase (hdLuc) was measured over time as described in Methods. hdLuc was incubated with Ssa1wt (2 μM) (A–C) or Ssa1 mutant (2 μM in A and C and 6 μM in B), yeast Hsp40, Ydj1, (0.5 μM when 2 μM Ssa1 was used and 1.5 μM when 6 μM Ssa1 was used) and ATP in the presence or absence of 0.4 μM Hsp104, as indicated. Data in A–C are means ± SEM (n=3). Some error bars are hidden by symbols.
A substitution mutant in subdomain IIB of human Hsp70, hHsp70R262A (homologous to DnaKR261 and Ssa1R259), was also tested for the ability to collaborate with Hsp104 and Ydj1 for the reactivation of hdLuc. This mutant was defective in collaboration with Hsp104 in agreement with the Ssa1 and DnaK results (Supplemental Fig. S3). Together, these results suggest that NBD subdomains IB and IIB of eukaryotic homologues of DnaK are important for specific collaboration with Hsp104 in protein reactivation, implying that this region of Hsp70 is conserved among species as a site of ClpB/Hsp104 interaction.
Computational modeling of the DnaK-ClpB complex indicates multivalent binding of DnaK to ClpB
Our experimental data suggest that the collaboration between ClpB and DnaK in reactivation reactions is inhibited by high concentrations of GrpE. Moreover, the mutants that we found to be defective in interaction with ClpB were in residues known to interact directly with GrpE. These results imply that ClpB and GrpE interact with DnaK in a mutually exclusive manner. To gain a better understanding of the interaction of DnaK with ClpB, we employed a computational approach. We utilized the available solution NMR structure of E. coli DnaK in the ADP bound conformation 18 and we constructed both trimer and hexamer models of E. coli ClpB. The trimer model was based on the crystal structure of trimeric T. thermophilus ClpB, which presented three distinct subunit conformations 43. In the ClpBEc hexamer model we accounted for these distinct subunit conformations and addressed asymmetry by satisfying restraints provided by the recently reported asymmetric EM map of E. coli ClpB (EMD-2558) 47 as described in Methods. Binding models of the DnaK-ClpB complex were obtained by rigid body docking 44 of DnaK with the ClpB trimer and hexamer models (see Methods) 44. The resulting decoys were ranked based on cluster size and then compared with experimental data. The top ranking DnaK-ClpB decoy generated using the ClpB hexamer model is discussed below. The decoy generated using the DnaK-ClpB trimer model was very similar and is described in Supplemental Information (Supplemental Table S2 and Supplemental Figure S4).
Analysis of the top docking DnaK-ClpB hexamer decoy indicated a binding interface on DnaK involving subdomains IB and IIB of the NBD. The decoy further showed DnaK interacting with the M-domain of one ClpB protomer and with the M-domain and NBD-1 of an adjacent ClpB protomer (Fig. 7). The majority of the DnaK residues shown to be involved in the collaboration between DnaK and ClpB by protein remodeling assays were in contact with the ClpB M-domains (Table 3 and Supplemental Table S1). These include subdomain IB residues R56 and V59 and subdomain IIB residues R261, P284 and Y285. DnaK interacted with one ClpB subunit (chain E) in motif 2 of the M-domain (Fig. 1C) between amino acid 490 and 518, forming 14 contacts within a cutoff of 6 Å (Fig. 7 and Supplemental Table S1). DnaK made 16 additional contacts with motif 1 of the M-domain of the adjacent ClpB protomer (chain F) between amino acids 423 and 456 (Fig. 7 and Supplemental Table S1). In addition, there were seven contacts between DnaK and the NBD-1 of ClpB (chain E) (Fig. 7 and Supplemental Table S1). An interaction between DnaK and NBD-1 of ClpB has not been experimentally identified, and further studies are necessary to corroborate these predictions. The model corresponds well with recent studies, which show that residues between amino acids 435–448 in motif 1 of the M-domain of Hsp104 (corresponding to residues 433–444 of ClpBEc) are involved in collaboration and interaction with Hsp70 25–27, 40, 41, 45. Additionally, homologous substitution mutants in E. coli ClpB are defective for collaboration with DnaK in protein remodeling 25–27, 40, 41. Altogether the protein-protein interactions identified in the model of the DnaK-ClpB hexamer complex support the functional and direct interactions identified in biochemical studies. Furthermore, they suggest a mechanism for collaboration between DnaK and ClpB for protein disaggregation.
Fig. 7.

Computational model of DnaK and ClpB indicating that one DnaK interacts with two ClpB protomers. A, B. Model of full-length ClpB hexamer shown as a top view (A) or a bottom view (B) bound to full-length DnaK (gray). The two ClpB protomers that interact with DnaK are shown in blue (chain E) and green (chain F). ClpB M-domain residues identified from modeling performed in this study as interacting with DnaK are shown in cyan (chain E) and yellow (chain F). ClpB NBD-1 residues identified as interacting with DnaK are shown in purple (chain E). DnaK residues identified as interacting with the ClpB chain E M-domain are shown in red and with ClpB chain F M-domain are in magenta (Supplemental Table S1). DnaK residues interacting with ClpB chain E NBD-1 are shown in orange. The black boxes indicate the region of interest that is shown larger in panels C and D. C, D. The region of interaction between ClpB M-domains and the DnaK NBD highlighted by the black boxes in A and B are enlarged, with colors as indicated in A and B. Models were obtained as described in Methods and images in A–D were made using PYMOL (www.pymol.org) 57.
Table 3.
Residues involved in DnaK-ClpB interaction
| Residue in DnaK | SASAa | DnaK | DnaK-ClpB hexamer complex | DnaK-GrpE complex |
|---|---|---|---|---|
| R56 | Absolute SASA (Å)2 | 199.0 | 21.7 | 10.7 |
| Relativeb SASA | 0.8 | 0.1 | 0 | |
| V59 | Absolute SASA (Å)2 | 84.2 | 58.1 | 13.8 |
| Relative SASA | 0.5 | 0.4 | 0.1 | |
| T60 | Absolute SASA (Å)2 | 108.3 | 87.4 | 25.6 |
| Relative SASA | 0.7 | 0.6 | 0.2 | |
| L257 | Absolute SASA (Å)2 | 89.1 | 65.7 | 8.6 |
| Relative SASA | 0.5 | 0.4 | 0 | |
| R261 | Absolute SASA (Å)2 | 66.2 | 3.4 | 0 |
| Relative SASA | 0.3 | 0.0 | 0 | |
| N282 | Absolute SASA (Å)2 | 90.9 | 95.2 | 35.4 |
| Relative SASA | 0.6 | 0.6 | 0.2 | |
| P284 | Absolute SASA (Å)2 | 80.2 | 50.7 | 0.3 |
| Relative SASA | 0.6 | 0.35 | 0 | |
| Y285 | Absolute SASA (Å)2 | 160.9 | 37.7 | 70.1 |
| Relative SASA | 0.7 | 0.2 | 0.3 |
SASA = solvent accessible surface area (see Methods)
Relative SASA values of ~1 suggest the residue is located on the surface of the protein
Next, we compared the DnaK-ClpB binding interface identified in our modeling study to the DnaK-GrpE binding interface as determined by crystal structure 12 (see Methods). We used the DnaK-GrpE crystal structure to evaluate the change in SASA of the experimentally identified DnaK residues that are important for ClpB binding, including DnaK subdomain IB and IIB residues R56, V59, T60, L257, R261, N282, P284, and Y285 (Table 3). The SASA of these residues in DnaK-GrpE complexes was greatly reduced compared to DnaK alone, showing that these residues are buried (R56, L257, R261, and P284) or partially buried (V59, T60, N282 and Y285) when DnaK interacts with GrpE (Table 3 and Supplemental Fig. S5). Additionally, each of these DnaK residues made 1–3 contacts with GrpE. The results of this analysis are consistent with the experimental results suggesting that ClpB and GrpE compete for the same interaction site on DnaK, and bind in a mutually exclusive fashion.
Collectively, the results presented here indicate that the interaction between DnaK and ClpB comprises one step in the protein disaggregation pathway, and suggest that GrpE is not present on ClpB-bound DnaK. However, they do not rule out a role for GrpE in the disaggregation process.
Discussion
Our results show that residues within subdomains IB and IIB of the NBD of E. coli DnaK are important for interaction and collaboration with E. coli ClpB, consistent with recent results using T. thermophilus homologues 24. Since many of the residues identified as part of the ClpB binding region of DnaKEc and DnaKTth were shown previously to interact with GrpE 12, 24, the observations presented here and recently 24 suggest that ClpB and GrpE of E. coli and T. thermophilus utilize the same DnaK binding region in a mutually exclusive fashion. We also found that yeast Hsp104 interacts with subdomain IB and IIB of Ssa1 through residues homologous to those in DnaK that interact with ClpB. However, it remains to be explored whether yeast NEF and Hsp104 utilize a single binding site on Ssa1.
The results further indicate that there are some differences in the interaction and collaboration between DnaK and ClpB in different species. For example, we observed that collaboration between DnaKEc and ClpBEc for hdGFP reactivation was unperturbed at a 1:1 molar ratio of DnaKEc to GrpEEc with or without ClpB (Fig. 5E). In contrast, GrpETth inhibited the collaboration between ClpBTth and DnaKTth for hdGFP disaggregation, even at substoichiometric levels (10:1 DnaKTth to GrpETth) 24. Some, but not all of the DnaKTth residues shown by NMR structural analysis to interact with ClpBTth 24 were identified as DnaKEc residues involved in collaboration with ClpBEc. This could explain the differences observed for GrpE competition with ClpB for binding on DnaKEc versus what has been observed for the T. thermophilus chaperones 24. The loop in subdomain IIB appears to be important for interaction with ClpBTth 24, but substitutions in two loop residues in DnaKEc did not alter either intrinsic or collaborative activity of DnaKEc with ClpBEc. Interestingly, substitutions in the IIB loop of Ssa1 cause major defects in chaperone activity as well as a loss in collaboration with Hsp104. The DnaKTth loop is two amino acids longer while the Ssa1 loop is four residues shorter than the ~12 residue DnaKEc loop, possibly accounting for differences in the collaboration in protein reactivation with the corresponding disaggregase.
Our results from the characterization of DnaK mutants are in agreement with several recently published studies: 1) an NMR structural study of the DnaKTth-ClpBTth interaction 24, 2) an HD exchange study showing that some residues in the ClpB M-domain helix 2 of motif 1 (Fig. 1C) are solvent exposed in hexameric ClpBEc 46, and 3) crosslinking studies demonstrating that residues in M-domain helices 2 and 3 of motif 2 (Fig. 1C) directly interact with DnaKEc 26. Our modeling study of the DnaK-ClpB hexamer (shown in Fig. 7) predicts that DnaK interacts simultaneously with the M-domains from two adjacent ClpB protomers, a result with potential functional implications. In this model residues in M-domain helices 1 and 2 (motif 1) of one ClpB protomer (chain F) and residues in M-domain helices 2, 3 and 4 (motif 2) of the neighboring ClpB protomer (chain E) contribute to the binding of a single DnaK.
The structural predictions of our model and that of Rosenzweig et al. 24 differ. The Rosenzweig et al. model was generated using the NBD fragment of DnaK and a fragment of a ClpB protomer containing the NBD-1 and M-domain, but lacking the N-domain and NBD-2 24. Due to the use of the truncated structures and the absence of the oligomeric assembly of ClpB, this model could only predict complexes that involve binding sites from a single ClpB subunit. Additionally, possible steric clashes may occur with structural overlapping of the docked DnaK-ClpB fragments with full-length DnaK and oligomeric ClpB. In contrast, the model presented here used full-length DnaK and the ClpB hexamer model generated in this study (Fig. 7). Although our modeling did not preclude interactions of DnaK with a single ClpB protomer as proposed by the Rosenzweig et al. study 24, our results suggested binding of DnaK to two neighboring ClpB protomers as the most favorable interaction for complex formation.
Our modeling data indicating multivalent binding of DnaK to the ClpB hexamer through the M-domains of adjacent ClpB protomers is consistent with the recent EM study of Carroni et al. 47. The EM data suggests that the ClpB M-domains adopt various positions in a single ClpB hexamer 47, as does the crystal structure of the T. thermophilus ClpB trimer 43. Studies of a DnaK-independent mutant with a substitution in motif 2 showed that the M-domains were tilted upward, while the M-domains of a motif 1 mutant deficient in DnaK interaction were horizontal. The results suggest that when adjacent M-domains are horizontal, the M-domains form head-to-tail contacts preventing DnaK interaction and repressing ClpB activity. The model presented by Carroni et al. proposes that when an M-domain transiently tilts upward, contacts between it and the adjacent M-domains are broken, DnaK binds to motif 2 of the M-domain and ClpB disaggregase activity is activated 47. In addition, our model also predicts that DnaK contacts two ClpB protomers simultaneously. While the two models differ in how ClpB interacts with DnaK, both are consistent with the idea that the binding of one or more DnaK to a ClpB hexamer promotes a functionally active conformation of ClpB, poised for protein disaggregation. We expect that future studies will continue to reveal information regarding the DnaK-ClpB hexamer interaction and its functional implications.
In summary, the complexity surrounding protein disaggregation and refolding is apparent. Not only are DnaK and ClpB or Ssa1 and Hsp104 major players in this process, but also co-chaperones, specifically the nucleotide-exchange factors, add another layer of regulation to the process. Whether the competition between ClpB and GrpE for binding to DnaK plays a role in protein disaggregation or other DnaK processes in the cell has yet to be addressed.
Materials and Methods
Strains and Plasmids
Plasmids p18link-ClpB and p25link-DnaK (Supplemental Table S3) were transformed into BTH101 cells for expression 25, 48. The plasmids for expression of DnaK mutants (Table 1) were constructed using the QuikChange site-directed mutagenesis kit from Agilent Technologies (Santa Clara, CA) and plasmid pET24b-dnak or p25link-DnaK (Supplemental Table S3) as template. Plasmid for the expression of the human Hsp70 (HSPA1A) was cloned into pET-23c and was a gift from Len Neckers (NIH). The plasmid was transformed into BL21(DE3)pLysS cells. Human Hsp70 mutants and yeast Ssa1 mutants (Table 2) were constructed using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) and plasmids pET23c-HSPA1A (Supplemental Table S3) and pET24b-SSA1 respectively as templates.
Proteins
P1 RepA 49, GFP 50, wild type and mutant ClpB 51, Hsp104 25, wild type and mutant DnaK 35, DnaJ 35, GrpE 35, wild type and mutant human Hsp70 25, Ydj1 52 and [3H]oriP1 DNA 49 (4475 cpm/fmol) were prepared essentially as described. Wild type and mutant Ssa1 were purified as previously described for DnaK 35 with some modifications. MDH was purchased from Roche (Indianapolis, IN). Luciferase was purchased from Promega (Madison, WI). Protein concentrations given are for monomeric DnaK, Ssa1, human Hsp70, luciferase and GFP, dimeric DnaJ, Ydj1, GrpE and MDH, and hexameric ClpB and Hsp104.
ATPase activity
Reaction mixtures (50 μL) contained HKE buffer [25 mM Hepes, pH 7.5, 50 mM KCl, 0.1 mM EDTA], 0.005% Triton X-100 (vol/vol), 2 mM DTT, 4 mM ATP, 0.1 μCi of [γ-33P]ATP (>3000 Ci/mM; GE Healthcare), 10 mM MgCl2, 1.3 μM DnaK wild-type or mutant, 0.25 μM DnaJ, 0.13 μM GrpE, 0.4 μM ClpB, 0.6 μM heat-denatured MDH (heated at 47 °C for 33 min in Buffer B [50 mM Tris, pH 7.5, 150 mM KCl, 20 mM MgCl2] at 2 μM) as indicated. Reactions were initiated by the addition of ATP, incubated 60 min at 25°C and analyzed as described 53.
GFP disaggregation
GFP reactivation assays (100 μL) contained HKE buffer, 5 mM DTT, 4 mM ATP, an ATP regenerating system (10 mM creatine phosphate and 0.3 μg/ml creatine kinase), 20 mM MgCl2, 10 μL heat-aggregated GFP (heated 10 min at 80 °C at 4.5 μM) and, where indicated, 1.4 μM wild-type or mutant DnaK, 0.25 μM DnaJ, 0.13 μM or 1.3 μM GrpE and 0.25 μM ClpB. Reactivation was monitored over time at 23 °C using a Tecan Infinite M200Pro plate reader. Excitation and emission wavelengths were 395 nm and 510 nm, respectively. Refolding rates were determined from the initial linear increase in fluorescence intensities of GFP.
RepA activation
Reaction mixtures (20 μL) contained HKE buffer, 2 mM DTT, 4 mM ATP, 10 mM MgCl2, 50 μg/ml BSA, 0.005% Triton X-100 (vol/vol) and 7 nM RepA. For DnaK titrations, DnaK concentration varied between 0 and 2 μM protein as indicated, with 15 nM DnaJ, 180 nM ClpB as indicated, and either 30 nM or 2 μM GrpE as indicated. For GrpE titrations, GrpE concentration varied between 0 and 2 μM protein as indicated, with 15 nM DnaJ, 100 nM or 400 nM DnaK as indicated, and 20 nM ClpB as indicated. For MgCl2 titrations, reaction mixtures contained HK buffer [25 mM Hepes, pH 7.5, 50 mM KCl], 0.01 to 15 mM MgCl2, 0.3 mM EDTA, 0.6 μM DnaK, 25 nM DnaJ, 30 nM GrpE and 180 nM ClpB as indicated. After 10 min at 23 °C the reactions were chilled to 0 °C. Calf thymus DNA (1 μg) and 13 fmol of [3H]oriP1 plasmid DNA were added. After 5 min at 0 °C, the mixtures were filtered through nitrocellulose filters and retained radioactivity was measured.
Heat-denatured luciferase reactivation
Luciferase (40 nM) was denatured in HKE buffer (55 μl) supplemented with 0.05 mg/ml BSA, 2 mM DTT and 10 mM MgCl2 for 7 min at 45 °C followed by 1 min at 4 °C in a thermocycler. Denatured luciferase was added to reaction mixtures (final total volume of 75 μL) containing HKE buffer, 3 mM ATP, an ATP regenerating system (10 mM creatine phosphate and 0.3 μg/ml creatine kinase), 15 mM MgCl2, 2 μM Ssa1 wild type or mutants as indicated, 1 μM Ydj1, and 0.4 μM Hsp104 as indicated. Reactions were incubated at 23 °C for 30 min and aliquots (5 μL) were removed at the times indicated. Luciferase activity was determined at 23 °C by injecting 120 μL of 25 mM Hepes, pH 7.5, 0.5 mM ATP 10 mM MgCl2 and 200 μM luciferin (Roche) and measuring light output in a Tecan Infinite M200Pro plate reader. Reactivation was determined compared to a non-denatured luciferase control.
Fluorescence anisotropy
Reaction mixtures (150 μL) contained HKE buffer with 0.5 mM TCEP-HCl from Thermo Scientific (Rockford, IL), 10 mM MgCl2, 4 mM ATP, 0.075 μM DnaKK106C single or double mutant labeled with N-(1-pyrene)maleimide as per included instructions from Invitrogen (Carlsbad, CA) (degree of labeling was between 0.1 and 0.5 for all mutants) and 5 μM ClpBE279A,E678A. 5 μM DnaK wild type or 5 μM GrpE were added as indicated. Fluorescence anisotropy measurements were performed on a Perkin Elmer LS55 spectrofluorimeter with excitation and emission wavelengths set at 338 nm and 380 nm, respectively with 10 nm slits, and with polarizers in place. Data was collected for 15 min at a frequency of ~1 point every 10 sec and the average anisotropy signal between 400 to 900 sec was determined.
Bacterial two-hybrid system
A bacterial two-hybrid system utilizing B. pertussis adenylate cyclase was used as previously described 25, 32. The clpB coding region was fused in frame to the 3′ end of the T18 fragment (pEB355) and dnaK wild type or mutant was fused to the 3′ end of the T25 fragment (pEB354) of B. pertussis adenylate cyclase (see Supplemental Table S3). Plasmid pEB354 carrying dnaK wild type or mutant, p25link-DnaK (see Supplemental Table S3), was co-transformed with p18link-ClpB into BTH101 (E. coli Δcya) cells. Cells were plated on LB plates containing ampicillin (100 μg/ml) and kanamycin (50 μg/ml) and incubated at 30°C. Single colonies were selected and used to inoculate LB selective media and cultures grown at 30 °C. β-galactosidase reporter activity was measured as previously described 54.
Modeling of protein-protein interactions
A. Modeling of E. coli ClpB (ClpBEc) monomeric and oligomeric structures
Full-length monomeric structures of ClpBEc subunits were obtained through the threading approach available in the I-TASSER structure prediction software (http://zhanglab.ccmb.med.umich.edu/I-TASSER/)55. Templates were chosen from the T. thermophilus ClpB (ClpBTth) counterpart (Protein Data Bank (PDB) entry 1QVR) 43, which has 56% sequence similarity with ClpBEc. Although the functionally active state of ClpB is a hexamer, the only well-resolved crystal structure of oligomeric ClpB is a trimer. While all ClpBTth subunits have the same sequence, each protomer possesses a distinct orientation of the N-terminal domain (residues 1-157) and the M-domain (residues 401–512). This structural heterogeneity is reflected in relatively small (~2–3Å) root-mean-square deviations (RMSDs) between subunit pairs of ClpBTth ΔN and large RMSDs of ~2.5–10.5Å between wild-type subunit pairs. To account for the intra-ring asymmetry, our first step was to obtain models of ClpBEc using individual ClpBTth subunits (A, B, and C) as templates.
The ClpBEc hexamer model was generated to satisfy restraints provided by the asymmetric electron microscopy map of E. coli ClpB (EMD-2558) 47. In addition, we maintained consistency with the subunit ordering found in the ClpBTth trimer. The map restraints were generated as follows: Fragments of NBD-1-M (residues 161-550) from I-TASSER generated ClpBEc subunits A, B, and C were fitted into the density map using Chimera 56. Dimers of ClpBEc NBD-2 were then fit into the electron density map. The NBD-2 dimers were built using a structural alignment in PyMol 57 to minimize the RMSD between NBD-2 fragments of individual ClpB subunits (residues 601-858) and the ClpX ring (PDB ID 3HWS) 58. Next, full-length individual subunits generated by I-TASSER were fit into the density map, with subunit sequence ABCABC (counterclockwise orientation viewed along the pore axis from NBD-1) that preserved the asymmetric subunit ordering found in the ClpBTh trimer. This structure was then minimized to satisfy map restraints by imposing best-fit positional restraints for NBD-1, NBD-2 and M-domain atoms. To this end, we performed 20000 steps of steepest descent and 1000 steps of adapted basis Newton-Raphson minimization using CHARMM 59. Overall, our final model illustrates the assembly of full-length ClpBEc subunits into a hexamer structure compatible with the asymmetric map restraints.
B. Modeling full-length ADP-bound DnaK and the DnaK/GrpE complex
The crystal structure of the DnaK/GrpE complex (PDB: 1DKG)12 is not fully resolved, lacking linker and peptide-binding domains of DnaK (residues 384-638). We modeled missing DnaK amino acids 384-605 by using the well-resolved isolated ADP-bound DnaK structure (PDB:2KHO) 18. To this end, we first used the CHARMM 59 molecular modeling program to build missing amino acids 1-3 and 604-605, as well as hydrogen atoms in the 2KHO structure. The missing regions in the DnaK/GrpE complex were then modeled by superimposing the isolated ADP-bound structure of DnaK onto the apo-DnaK in the DnaK/GrpE complex. Next, we minimized the RMSD of the ATPase domain fragment between the two structures (~6.5Å).
C. Protein-Protein docking and selection of decoys of docking complexes by using experimental information on binding interfaces
Protein-protein docking models of the ClpBEc/DnaK complexes were obtained using the ClusPro server (http://cluspro.bu.edu/login.php) 44, 60–62. Our approach incorporated experimental data, which showed that a subset of residues within the M-domains of ClpB (residues 409-530) and subdomains IB and IIB of DnaK (residues 38-119 and 228-310 respectively) participate in binding between these complexes. In ClusPro, all of the residues in these regions were defined as attractive restraints. Although these favorable interactions bias the binding towards these regions of ClpB and DnaK complex, they do not preclude binding of other regions. ClusPro pre-processing of the isolated ClpB and DnaK structures included removal of steric clashes by using 300 steps of minimization of the van der Waals energy with fixed backbone restraints using CHARMM 59. To remove remaining steric clashes within the docking decoys, we performed an additional 1000 steps of adapted basis Newton-Raphson and 1000 steps of steepest descent minimization in the absence of restraints using CHARMM59. The top 30 decoys were ranked based on cluster size and the highest-ranking decoy was chosen. The top 30 decoys from docking were analyzed for protein interaction sites using the SPPIDER webserver (http://sppider.cchmc.org/) 63. This server implements a neural network to determine the probability of forming binding interfaces, while accounting for structural and sequential features of the proteins. Using these predicted binding sites, the models were then matched for overlaps with experimentally identified residues on ClpB and DnaK 12, 24, 26.
Accessible surface area per residue:
We computed the accessible surface area per residue using the formula:
Where as 64 is the accessible surface area of a specific amino acid and a0 65 is the reference value given by Gly – X – Gly accessible surface area. Values of ~1 suggest the residue is located on the surface of the protein.
Contacts:
Contacts were determined by calculating the distances between the center of geometry of residue pairs in DnaK and ClpB or GrpE using a cutoff distance of 6Å (Supplemental Table S1).
Supplementary Material
Highlights.
ClpB and DnaK collaborate in protein disaggregation via specific interactions
Residues in DnaK subdomains IB and IIB are important for cooperation with ClpB
Homologous residues in yeast Ssa1 are required for collaboration with Hsp104
Modeling indicates multivalent binding of DnaK to adjacent ClpB protomers
ClpB and GrpE both function with DnaK in protein remodeling, but not simultaneously
Acknowledgments
This research was supported by the National Science Foundation CAREER grant no. MCB-0952082 (to G.S.) and the Intramural Research Program of the NIH, NCI, Center for Cancer Research.
Abbreviations
- NBD
nucleotide-binding domain
- SBD
substrate-binding domain
- NEF
nucleotide exchange factor
- M-domain
middle domain
- N-domain
N-terminal domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–84. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–55. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 3.Zuiderweg ER, et al. Allostery in the Hsp70 chaperone proteins. Top Curr Chem. 2013;328:99–153. doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–92. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison C. GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperones. 2003;8:218–24. doi: 10.1379/1466-1268(2003)008<0218:ganeff>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genest O, Hoskins JR, Camberg JL, Doyle SM, Wickner S. Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc Natl Acad Sci U S A. 2011;108:8206–11. doi: 10.1073/pnas.1104703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goloubinoff P, Mogk A, Zvi AP, Tomoyasu T, Bukau B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc Natl Acad Sci U S A. 1999;96:13732–7. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zolkiewski M. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J Biol Chem. 1999;274:28083–6. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 9.Flaherty KM, DeLuca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346:623–8. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X, et al. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–14. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gassler CS, et al. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A. 1998;95:15229–34. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–5. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- 13.Suh WC, et al. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci U S A. 1998;95:15223–8. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48:863–74. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 15.Qi R, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol. 2013;20:900–7. doi: 10.1038/nsmb.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhuravleva A, Clerico EM, Gierasch LM. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell. 2012;151:1296–307. doi: 10.1016/j.cell.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci U S A. 2011;108:6987–92. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–6. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlecht R, Erbse AH, Bukau B, Mayer MP. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol. 2011;18:345–51. doi: 10.1038/nsmb.2006. [DOI] [PubMed] [Google Scholar]
- 20.Schweizer RS, Aponte RA, Zimmermann S, Weber A, Reinstein J. Fine tuning of a biological machine: DnaK gains improved chaperone activity by altered allosteric communication and substrate binding. Chembiochem. 2011;12:1559–73. doi: 10.1002/cbic.201000786. [DOI] [PubMed] [Google Scholar]
- 21.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 22.Doyle SM, Genest O, Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol. 2013;14:617–29. doi: 10.1038/nrm3660. [DOI] [PubMed] [Google Scholar]
- 23.Krzewska J, Langer T, Liberek K. Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 2001;489:92–6. doi: 10.1016/s0014-5793(00)02423-6. [DOI] [PubMed] [Google Scholar]
- 24.Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR, Kay LE. Unraveling the Mechanism of Protein Disaggregation Through a ClpB-DnaK Interaction. Science. 2013 doi: 10.1126/science.1233066. [DOI] [PubMed] [Google Scholar]
- 25.Miot M, et al. Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc Natl Acad Sci U S A. 2011;108:6915–20. doi: 10.1073/pnas.1102828108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seyffer F, et al. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol. 2012;19:1347–55. doi: 10.1038/nsmb.2442. [DOI] [PubMed] [Google Scholar]
- 27.Lee J, et al. Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc Natl Acad Sci U S A. 2013;110:8513–8. doi: 10.1073/pnas.1217988110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reidy M, Miot M, Masison DC. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics. 2012;192:185–93. doi: 10.1534/genetics.112.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabani M, Beckerich JM, Brodsky JL. Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol Cell Biol. 2002;22:4677–89. doi: 10.1128/MCB.22.13.4677-4689.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levy EJ, McCarty J, Bukau B, Chirico WJ. Conserved ATPase and luciferase refolding activities between bacteria and yeast Hsp70 chaperones and modulators. FEBS Lett. 1995;368:435–40. doi: 10.1016/0014-5793(95)00704-d. [DOI] [PubMed] [Google Scholar]
- 31.Doyle SM, Hoskins JR, Wickner S. Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proc Natl Acad Sci U S A. 2007;104:11138–44. doi: 10.1073/pnas.0703980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A. 1998;95:5752–6. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weibezahn J, Schlieker C, Bukau B, Mogk A. Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem. 2003;278:32608–17. doi: 10.1074/jbc.M303653200. [DOI] [PubMed] [Google Scholar]
- 34.Wickner S, et al. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc Natl Acad Sci U S A. 1994;91:12218–22. doi: 10.1073/pnas.91.25.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skowyra D, Wickner S. The interplay of the GrpE heat shock protein and Mg2+ in RepA monomerization by DnaJ and DnaK. J Biol Chem. 1993;268:25296–301. [PubMed] [Google Scholar]
- 36.Sugimoto S, Saruwatari K, Higashi C, Sonomoto K. The proper ratio of GrpE to DnaK is important for protein quality control by the DnaK-DnaJ-GrpE chaperone system and for cell division. Microbiology. 2008;154:1876–85. doi: 10.1099/mic.0.2008/017376-0. [DOI] [PubMed] [Google Scholar]
- 37.Grimshaw JP, et al. The heat-sensitive Escherichia coli grpE280 phenotype: impaired interaction of GrpE(G122D) with DnaK. J Mol Biol. 2005;353:888–96. doi: 10.1016/j.jmb.2005.08.069. [DOI] [PubMed] [Google Scholar]
- 38.Packschies L, et al. GrpE accelerates nucleotide exchange of the molecular chaperone DnaK with an associative displacement mechanism. Biochemistry. 1997;36:3417–22. doi: 10.1021/bi962835l. [DOI] [PubMed] [Google Scholar]
- 39.Brehmer D, Gassler C, Rist W, Mayer MP, Bukau B. Influence of GrpE on DnaK-substrate interactions. J Biol Chem. 2004;279:27957–64. doi: 10.1074/jbc.M403558200. [DOI] [PubMed] [Google Scholar]
- 40.Sielaff B, Tsai FT. The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J Mol Biol. 2010;402:30–7. doi: 10.1016/j.jmb.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeSantis ME, et al. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell. 2012;151:778–93. doi: 10.1016/j.cell.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haslbeck M, Miess A, Stromer T, Walter S, Buchner J. Disassembling protein aggregates in the yeast cytosol. The cooperation of Hsp26 with Ssa1 and Hsp104. J Biol Chem. 2005;280:23861–8. doi: 10.1074/jbc.M502697200. [DOI] [PubMed] [Google Scholar]
- 43.Lee S, et al. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–40. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 44.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 45.Desantis ME, et al. Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide-binding domain 2 and enable Hsp70-dependent protein disaggregation. J Biol Chem. 2014;289:848–67. doi: 10.1074/jbc.M113.520759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oguchi Y, et al. A tightly regulated molecular toggle controls AAA+ disaggregase. Nat Struct Mol Biol. 2012;19:1338–46. doi: 10.1038/nsmb.2441. [DOI] [PubMed] [Google Scholar]
- 47.Carroni M, et al. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. Elife. 2014;3:e02481. doi: 10.7554/eLife.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Battesti A, Bouveret E. Acyl carrier protein/SpoT interaction, the switch linking SpoT-dependent stress response to fatty acid metabolism. Mol Microbiol. 2006;62:1048–63. doi: 10.1111/j.1365-2958.2006.05442.x. [DOI] [PubMed] [Google Scholar]
- 49.Wickner S, Hoskins J, McKenney K. Monomerization of RepA dimers by heat shock proteins activates binding to DNA replication origin. Proc Natl Acad Sci U S A. 1991;88:7903–7. doi: 10.1073/pnas.88.18.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoskins JR, Singh SK, Maurizi MR, Wickner S. Protein binding and unfolding by the chaperone ClpA and degradation by the protease ClpAP. Proc Natl Acad Sci U S A. 2000;97:8892–7. doi: 10.1073/pnas.97.16.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnett ME, Zolkiewski M. Site-directed mutagenesis of conserved charged amino acid residues in ClpB from Escherichia coli. Biochemistry. 2002;41:11277–83. doi: 10.1021/bi026161s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cyr DM, Lu X, Douglas MG. Regulation of Hsp70 function by a eukaryotic DnaJ homolog. J Biol Chem. 1992;267:20927–31. [PubMed] [Google Scholar]
- 53.Shacter E, Chock PB, Stadtman ER. Energy consumption in a cyclic phosphorylation/dephosphorylation cascade. J Biol Chem. 1984;259:12260–4. [PubMed] [Google Scholar]
- 54.Zubay G, Morse DE, Schrenk WJ, Miller JH. Detection and isolation of the repressor protein for the tryptophan operon of Escherichia coli. Proc Natl Acad Sci U S A. 1972;69:1100–3. doi: 10.1073/pnas.69.5.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 57.PyMOL. The PyMOL Molecular Graphics System. Schrodinger, LLC; 2006. [Google Scholar]
- 58.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–56. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks BR, et al. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217d. [Google Scholar]
- 60.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–9. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kozakov D, et al. How good is automated protein docking? Proteins. 2013;81:2159–66. doi: 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 63.Porollo A, Meller J. Prediction-based fingerprints of protein-protein interactions. Proteins. 2007;66:630–45. doi: 10.1002/prot.21248. [DOI] [PubMed] [Google Scholar]
- 64.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 65.Miller S, Janin J, Lesk AM, Chothia C. Interior and surface of monomeric proteins. J Mol Biol. 1987;196:641–56. doi: 10.1016/0022-2836(87)90038-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.