

Abstract
A network of mechanisms operates to maintain tolerance in the gut mucosa. Although CD103 marks many lymphoid cells within the gut, its direct functional role in intestinal tolerance is poorly understood. CD103 may be part of a redundant pathway as CD103−/− mice do not exhibit autoimmunity. To reduce such redundancy, CD103−/− mice were crossed to mice (designated Y3) whose T cells expressed a mutant IL-2Rβ chain that lowers IL-2R signaling. Unlike overtly healthy Y3 mice, all Y3/CD103−/− mice rapidly developed severe colitis. The large intestine of these mice contained an increase in CD4+ Th1 and Th17 effector cells and a reduced ratio of regulatory T cells (Tregs). Importantly, colitis was effectively prevented by the transfer of wild type (WT) Tregs into Y3/CD103−/− mice. Impaired intestinal tolerance was not attributed to an obvious lack of CD103-dependent gene regulation or intestinal homing/retention by Tregs nor a lack of functional activities typically associated with CD103+ dendritic cells (DCs), such as peripheral induced Treg (pTreg) development or imprinting CCR9 and α4β7 homing molecules on Treg and T effector cells. Transcriptome analysis of Tregs was consistent with altered homeostasis due to impaired IL-2Rβ-dependent signaling with minimal dysregulation added by the absence of CD103. Rather the absence of CD103 functioned to alter the localization of the cells within the gut microenvironment that may alter Treg homeostasis. Thus, IL-2Rβ-dependent signaling and CD103 normally cooperate through distinctive processes to promote Treg homeostasis and immune tolerance.
Introduction
Foxp3+ Tregs are the major cell type that dominantly functions in mucosal tolerance by suppressing responses to food antigens and commensal bacteria (1). Treg-mediated suppression in the intestine is mainly dependent on IL-10, TGF-β and IL-35 (2-6). However, gut homeostasis also depends on other regulatory mechanisms, including tolerogenic CD103+ DCs, which support the development of pTregs, IL-10-producing type1 regulatory T cells (Tr1s), and secretion of inhibitory cytokines, such as TGF-β, by epithelial cells (7). A defect in a single regulatory component, even one crucial to maintain mucosal tolerance, such as IL-10, does not immediately tip the balance to inflammatory bowel disease (IBD). Rapid develop of IBD in the absence of IL-10 depends on agents that trigger strong inflammatory responses, such as infection by H.hepaticus (8).
Establishing the function of an individual component within mucosal regulatory circuits is sometimes difficult. αE-integrin (CD103) represents one such molecule that likely directly contributes to mucosal tolerance but its function is poorly understood. CD103 is a marker that is found on many cells within the gut mucosa, including T effector cells, Tregs, and DCs. Cells bearing CD103, paired with β7 integrin, interacts with E-cadherin on gut epithelial cells and some DCs and this interaction has been hypothesized to mediate long-term retention of Intr-aepithelial lymphocytes (IEL) (9). With respect to CD103 expression by Tregs, evidence is limited and contradictory concerning a direct functional role for CD103 to effectively mediate tolerance. For example, expression of CD103 was reported to be required for Treg retention in the skin to limit inflammation during L. major infection (10). In contrast, CD103−/− Tregs readily suppressed the T cell-transfer model of colitis (11). Furthermore, CD103−/− mice do not exhibit pathological abnormalities, including those related to IBD (12). These latter two findings suggest that CD103 expression by Tregs is not required for mucosal tolerance. Thus, CD103 represents an important marker found on mucosal cells but redundant and co-operative mechanisms may obscure its functional activity for intestinal homeostasis.
Proper IL-2R signaling represents another activity essential for tolerance in the gut mucosa (13). Polymorphisms in IL-2, IL-2Rα, and IL-2Rβ are genetic risks for several autoimmune diseases, including IBD (14, 15). We have developed a mouse model that permits the evaluation of the outcome of impaired IL-2R signaling on Treg function and the risk for autoimmune disease (16). IL-2RβY3 are transgenic mice on the IL-2Rβ−/− genetic background (referred to as Y3 in this report) where all T cells express a transgenic IL-2Rβ chain whose cytoplasmic tail contains three tyrosine to phenylalanine mutations that impairs IL-2-dependent PI3K and STAT5 activation. A low level of STAT5 activation occurs upon IL-2 binding to this IL-2Rβ mutant molecule and this amount of signaling readily supports outwardly normal thymic Treg development and peripheral homeostasis. Nevertheless, some IL-2-dependent functions remain impaired in these Tregs. Y3 mice do not develop severe autoimmunity associated with parental IL-2Rβ−/− mice and are long-lived, but upon aging (> 16 weeks) some exhibit immune activation and mild to moderate lymphocytic infiltrates in several tissues, principally the lung and salivary gland, with less frequent involvement of the intestine. The representation of Tregs in the lamina propria (LP) of the small intestine (SI-LP) are normal, but their homeostasis is altered and characterized by enhanced proliferation that is balanced by greater turnover (17).
In this study, we take a closer look at the role of CD103 in maintaining mucosal tolerance in the context of autoimmune-prone mice by crossing CD103−/− mice to Y3 mice. We predicted that generally lowering Treg activity through impaired IL-2R signaling may help to reveal the potential contribution of CD103 to mucosal tolerance. Unlike Y3 mice that lack severe autoimmune-related symptoms, all Y3/CD103−/− mice spontaneously develop severe colitis. We show distinct roles for IL-2 and CD103 in regulation of Treg homeostasis in the gut mucosa with a role for IL-2 in the SI-LP but with a cooperative role for IL-2 and CD103 in the LP of the large intestine (LI-LP). A breakdown in the latter mechanism leads to colitis.
Materials and Methods
Mice
C57BL/6, B6.129S2(C)-Itgaetm1Cmp/J (CD103−/−) mice were originally obtained from The Jackson Laboratory. Foxp3/RFP mice were kindly provided by R.A. Flavell (18). Blimp-1/GFP mice were kindly provided by S.L. Nutt (19) and further crossed to Foxp3/RFP mice. Y3 mice were generated in our lab as previously described (16). Y3 mice were crossed to CD103−/− mice to generate Y3/CD103−/− mice. To perform adoptive transfer experiments and microarray assays, CD103−/−, Y3 and Y3/CD103−/− mice were each crossed to Foxp3/RFP reporter mice. All mice were maintained in the same animal room and CD103−/−, Y3, and Y3/CD103−/− mice were typically co-housed. OT-II and OT-II/Foxp3/RFP transgenic mice were kindly provided by Dr. E.R. Podack. All mice were on C57BL/6 background and maintained in the animal facility at the University of Miami. Animal experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee at the University of Miami.
Cell preparation
LP cells were prepared as previously described with minor modifications (17). Briefly, small intestine was cut 0.5 cm below the stomach and 1 cm above the cecum. Large intestine was cut from the cecum to anus. After removing the fat and connective tissue, the intestine was flushed with Ca2+-Mg2+ free HBSS containing 5% FBS and Peyer's Patches (PPs) were dissected. The intestine was cut longitudinally and sliced into 0.2-0.5 cm pieces, followed by incubating in 20 ml Ca2+-Mg2+ free HBSS containing 10% FBS, 15mM HEPES, 5mM EDTA at 37° C on 200 rpm shaker for 20 min to collect IELs. The Intestine fragments were further digested in 20 ml RPMI 1640 containing 5% FBS, collagenase VIII (0.24 mg/ml) (Sigma Aldrich) and trypsin inhibitor (0.24 mg/ml) (Sigma Aldrich) at 37 °C on 200 rpm shaker for 60 min. Samples were passed through 70 μm cell strainer and LP cells were collected. IEL and LP cells were then purified on a 44%/67% Percoll (GE Healthcare) gradient by centrifugation (800g, 20 min at room temperature).
Antibodies and flow cytometry
The following reagents were used for FACS staining or confocal microscopy: CD4 (Gk1.5), CD8α (53-6.7), CD44 (pgp-1), Ly6G (1A8) were prepared in our laboratory; CD4 (Gk1.5), CD62L (MEL-14), CD69 (H1.2F3), CD103 (2E7), CCR5 (HM-CCR5), CCR4 (2G12), CCR6 (29-2L17), CTLA4 (UC10-4B9), Helios (22F6), IL-17 (TC11-18H10.1), IFN-γ (XMG1.2), TCR γδ (GL3), PE Streptavidin were obtained from BioLegend; Klrg1 (2F1), CD127 (A7R34), ICOS (15F9), CD39 (24DMS1), CD73 (eBioTY/11.8), Thy1.1 (HIS51), CCR9 (eBioCW-1.2), α4β7 (DATK32), CXCR3 (CXCR3-173), Nrp-1 (3DS304M), Foxp3 (FJK-16s), CD8β (H35.17.2), IL-2 (JES6-5H4), TNF-α (MP6-XT22), Streptavidin PE-Cy7, Streptavidin PerCP-eFluor 710 were obtained from eBioscience; CD25 (PC61), Thy1.2 (53-2.1), TCR Vα2 (B20.1), TCR Vβ5 (MR9-4), Ki67 (B56), E-Cadherin (36/E-Cadherin) were obtained from BD Biosciences; Alexa Fluor 568 Streptavidin was obtained from Invitrogen. Intracellular staining of Foxp3, Ki67, CTLA4, and Helios was performed using Foxp3/Transcription Factor Staining Buffer Set (eBioscience) according to manufacturer's instruction. FACS data were acquired on LSR II and FACSFortesa (BD) and analyzed with BD-FACSDiva software. In general, 200-300 × 103 total events were collected for spleen and mesenteric lymph node (MLN) samples and roughly 500 × 103 total events were collected for LP samples. Cell sorting was performed on FACSAria II.
Ex vivo cytokine analysis
To measure the intracellular cytokine production, unfractionated cells (2-2.5 × 106) were cultured in 24-well plates in 1 ml RPMI 1640 complete medium (CM) in the presence of Brefeldin A (GolgiPlug; BD Biosciences), PMA and Ionomycin (Sigma Aldrich) at 37 °C incubator for 4 hr. Cells were harvested and subjected to surface staining with antibodies for 15 min. Cells were then fixed, permeabilized with Foxp3/Transcription Factor Staining Buffer Set and stained with mAbs to IL-2, TNF-α, IL-17, IFN-γ and Foxp3 according to manufacturer's instruction. Cells were analyzed by flow cytometry.
Adoptive transfer
In general, lymphocytes from spleen, peripheral lymph node and MLN were collected and CD4+ cells were enriched using anti-CD4 magnetic beads (Miltenyi Biotec). Cells were stained for desired surface antibodies and subjected to cell sorting. CD4+ Tregs or CD4+ Foxp3− T conventional cells were purified based on the Foxp3/RFP reporter. The purity of sorted cells was always ≥ 98%. In particular, the purity of sorted CD4+ T conventional cells to assess pTreg development in vivo was > 99.9%. In some experiments, total CD4+ T cells were MACS-enriched and transferred. Recipient mice received purified cells by i.v. or i.p. injection. Details of each transfer are provided in the Figure legends.
Histopathology
Tissues/organs were removed and fixed with 10% formalin solution. Fixed tissue blocks were paraffin-embedded, sectioned and stained with hematoxylin and eosin for pathological analysis. Slides were coded and scored blindly by a Board Certified Veterinary Pathologist.
Microarray analysis
Tregs from Foxp3/RFP reporter mice were purified by FACS sorting from the SI-LP and LI-LP. Total RNA was prepared and processed to probe Affymetrix Mouse Gene ST2.0 arrays as previously described (17). In brief, 10-20 ng of RNA was used in a single round of linear RNA probe amplification using Ovation Pico WTA System V2 and biotin-labeled using Encore Biotin Module (NuGEN, San Carlos, CA, USA). The resulting data were normalized by the RMA method using software at GeneSifter (Seattle, WA) and expressed as log2 signal intensity. Group comparisons of the transformed data of 3 independent biological replicates were performed using ANOVA, applying the Benjamini Hochberg correction for false positives. Data analysis was limited to annotated mRNAs after setting a lower signal intensity limit of 4 units (one group must pass). Genome-wide expression data in the article have been submitted to the Gene Expression Omnibus data base (http:/www.ncbi.nlm.nihgov/geo/) under accession number GSE56438.
Confocal microscopy and image analysis
The intestine was cut, flushed with PBS and then snap frozen in OCT. Frozen tissues were cut into 6μm thickness and fixed in ice-cold acetone for 10 min. Prior to staining, sections were blocked with 20% BSA containing 0.3% Triton-X 100 for 30 min. Sections were stained with Biotin-Foxp3 at 4 °C overnight, washed with PBS and then stained with Alexa Fluor 568 Streptavidin, Alexa Fluor 647 anti-CD4, and FITC anti-E-Cadherin. Stained sections were mounted with Fluoroshield with DAPI (Sigma Aldrich). Images were acquired on TCS-SP5 II Leica microscope, using 40 × objective lens, NA = 1.25 and processed with ImageJ software.Prior to analysis, images were individually background subtracted. Background was defined as mean pixel intensity of the area that carried no tissue. Localization analysis was performed as follows: regions of interest (ROIs) were selected using the CD4 channel as guidance. Individual CD4+ T cells were enclosed into separate ROIs, leaving an additional ½ cell diameter around the cell. ROIs were then transferred onto the E-cadherin channel images. Mean pixel intensity was calculated for ROIs in the E-cadherin channel, and the value was normalized against mean pixel intensity of E-cadherin in the E-cadherin channel for the current image. These data were evaluated by the, Relative Mean pixel intensity = (MROIs in E-cadherin channel - Mbackground)/(ME-cadherin in E-cadherin channel - Mbackground), as one approach to define the extent of co-localization.
Statistical Analysis
Data were shown as the mean ± SD in most of the figures. Statistical analysis was performed using one-way ANOVA with Turkey's multiple comparison test when comparing groups (> 2) and unpaired Student's t test when comparing 2 groups. Some data (Incidence of disease, Figure 1A and Disease free, Figure 4B) were analyzed using Log-rank test. Statistical significance were considered as * (P < 0.05), **(P < 0.01), ***(P < 0.001) and ****(P < 0.0001).
Figure 1. Y3/CD103−/− mice develop severe colitis.

(A) Incidence of disease. Mice from each genotype were monitored weekly for signs of colitis, and were considered diseased when they exhibit at least one of the following severe diarrhea, rectal prolapse and/or wasting. (B) MLN cellularity. (C) Histology scoring of the large intestine of age-matched 12-24 week-old mice (left). Formalin-fixed and H&E stained colon sections (right) were examined. Typically, 1 = mild inflammatory infiltrates, 2 = moderate, 3 = extensive, with or without necrosis (n ≥ 3 mice/group), (original magnification 12.5 X). (D) Splenic cellularity (top) and the frequency of Ly6G+ granulocytes (bottom) in the spleen were assessed by flow cytometry. (E, F) The proportion and number of total CD4+ (E) and CD4+ Foxp3+ Tregs (F) in the LI-LP and SI-LP were determined by flow cytometry. (B, D-F) Age-matched 12-24 week-old mice were assessed for each genotype. The number of mice/group is indicated within each bar or by individual symbols in scatter graphs.
Figure 4. Adoptive transfer of WT Tregs prevents colitis in Y3/CD103−/− mice.

0.8-1 × 106 FACS-sorted Tregs from Foxp3/RFP mice were injected i.v. into day 12-15 old male and female Y3/CD103−/− mice. The adoptive transfer was performed on 9 and 11 mice for Treg-treated and untreated Y3/CD103−/− mice respectively in 7 independent experiments. Control untreated and adoptively transferred mice were analyzed at 7-34 and 28-34 weeks of age, respectively. (A) Animal weights were monitored weekly. (B) Mice (combined males and females) were monitored for the colitis and were considered disease-free when they did not exhibit diarrhea, rectal prolapse and wasting disease. (C) Representative H&E stained colon sections from Treg-treated and untreated mice (n = 3 mice/group) (original magnification 10 X). Histology scoring of the large intestine was performed as described in the legend to Fig. 1 (left). (D) MLN cellularity. (E) The proportion (top) and number (bottom) of total CD4+ T cells and (F) Ki67 expression by CD4+ Foxp3− T conventional cells in the LI-LP and SI-LP. (G) The frequency and origin of Tregs in the LI-LP and SI-LP, as indicated, from untreated and adoptively transferred mice (n = 5 mice/group). (H) Cytokine production by CD4+ Foxp3− T conventional cells from the LI-LP (top) and SI-LP (bottom), as indicated, after culture with PMA and ionomycin for 4 hr. (D-F, H) The number of mice/group is indicated within each bar or by individual symbols in scatter graphs.
Result
Y3/CD103−/− mice develop colitis
CD103 is expressed on most Tregs in the gut mucosa but is on fewer Tregs in secondary lymphoid tissues (Supplemental Fig. 1A, left). Many fewer CD4+ T conventional cells express CD103, with the highest proportion found in the IEL compartment of the small intestine (Supplemental Fig. 1A, right). In comparison to CD103− Tregs, CD103+ Tregs from the spleen (Supplemental Fig. 1B) and the SI-LP (Supplemental Fig. 1C) express a more activated phenotype (i.e. increased CD44, CD69, ICOS, Klrg1, Blimp-1) and higher levels of some suppressive molecules (i.e. CD39 and CTLA4). These trends were more noticeable for the spleen as it contains many fewer CD103+ Tregs. CD103+ Tregs show increased capacity to suppress T cell proliferation in vitro and colitis induced by CD4+ T conventional cells in lymphopenic recipients (20-22).
The high prevalence of CD103 on mucosal Tregs raises the possibility that CD103 might be required to facilitate Treg-mediated suppression. CD103−/− mice, which lack autoimmunity, were bred to Y3 mice, which exhibit mild inflammation, to assess whether more severe autoimmune disease may develop. Strikingly, all Y3/CD103−/− mice developed severe colitis characterized by diarrhea, rectal prolapse and wasting disease, usually between 12-16 weeks of age (Fig. 1A). These mice exhibited enlarged MLN with increased cellularity (Fig. 1B) and thickening of the wall of the large intestine (data not shown). Histological evaluation revealed extensive lymphoid infiltration and tissue destruction in the large intestine consistent with chronic active colitis (Fig. 1C). Some mice also showed mild inflammation of the pancreas, lung, liver and salivary gland (not shown), similar to that reported for Y3 mice (16). However, there was not histological evidence of inflammation in the small intestine of Y3/CD103−/− mice (not shown). Coincident with colitis, splenic cellularity was slightly, but significantly, increased for Y3/CD103−/− mice that was primarily due to an increase in Ly-6G+ granulocytes (Fig. 1D). The LI-LP from Y3/CD103−/− mice showed a large increase in the proportion and number of CD4+ T cells (Fig. 1E). A decreased frequency (2-fold) of Tregs was found in the LI-LP from Y3 and Y3/CD103−/− mice. However, only Y3/CD103−/− mice showed a substantial increase in the number of Foxp3+ Tregs (Fig. 1F), due to the high CD4+ T cell inflammatory response in the LI-LP. In contrast, conventional CD4+ T cells and Tregs each increased in the SI-LP from Y3 and Y3/CD103−/− (Fig. 1E, F), but the proportion of Tregs to conventional CD4+ T cells was similar to WT and CD103−/− mice (Fig. 1F).
Increased percentages of CD44hi CD62Llo activated CD4+ Foxp3− T cells were detected in the spleen and MLN of Y3 and Y3/CD103−/− mice (Fig. 2A). In comparison to these tissues, more activated CD44hi CD62Llo CD4+ Foxp3− T cells were found in the SI-LP and LI-LP from WT mice. Nevertheless, these levels further increased only in the LI-LP of Y3/CD103−/− mice (Fig. 2A). The LI-LP of Y3/CD103−/− mice also showed increased number of IL-2, IL-17, IFN-γ and TNF-α producing CD4+ T effector cells (Fig. 2B). In marked contrast, the SI-LP of Y3 and Y3/CD103−/− mice showed comparable increases of IL-17-producing CD4 T effector cells (Fig. 2B). These findings demonstrate that a CD103-dependent induction of colitis in the context of altered IL-2Rβ signaling is due to a substantial CD4+ T cell inflammatory response, with a lower ratio of Treg:T conventional cells, in the LI-LP. However, with the exception of increased CD4+ T conventional and Treg cells in the LI-LP of Y3/CD103−/− mice, leading to a low proportion of Tregs, all other significant immunological changes noted were detected in Y3 and Y3/CD103−/− mice, indicating that these were primarily dependent upon the Y3 IL-2Rβ mutation.
Figure 2. Increased T cell activation in Y3/CD103−/− mice as assessed by flow cytometry.

(A) Activated CD44hi CD62Llo CD4+ Foxp3− T conventional cells were enumerated for the indicated tissues. (B) Cytokine production by CD4+ Foxp3− T conventional cells from the LI-LP (top) and SI-LP (bottom) after culture with PMA and ionomycin for 4 hr. Age-matched 12-24 week-old mice were assessed for each genotype. The number of mice/group is indicated within each bar or by individual symbols in scatter graphs.
Altered Treg homeostasis in the gut mucosa of Y3 and Y3/CD103−/− mice
CD4+ T conventional and Treg cells were evaluated for their proliferative activity by Ki67 expression. The increased number of Y3/CD103−/− Tregs and activated CD4+ T cells (Figs. 1F, 2A) in the LI-LP was associated with similar and increased proliferation of both cell populations (Fig. 3A). This effect is due to the absence of CD103 as Tregs in the LI-LP of Y3 mice did not show increase in Ki67+ cells. However, the percentage of Tregs to total CD4+ T cells substantially decreased (Fig. 1F), indicating that this increased proliferation by Tregs is unable to keep up with the CD4+ T inflammatory responses. Both Y3 and Y3/CD103−/− mice showed a similar lower percentage of Tregs in the LI-LP (Fig. 1F), indicating that this effect is primarily due to impaired IL-2R signaling associated with the Y3 IL-2Rβ mutation.
Figure 3. Increased Treg proliferation in Y3/CD103−/− mice.

(A) Representative (left) and averaged (right) Ki67 expression by CD4+ Foxp3− T conventional cells (Tconvs) and CD4+ Foxp3+ Tregs from the LI-LP and SI-LP. Age-matched 12-24 week-old mice were assessed for each genotype. (B-D) 6-10 week-old age-matched mice from each genotype were analyzed for MLN cellularity (B), the proportion of total CD4+ and CD4+ Foxp3+ Tregs in the LI-LP (C) and Ki67 expression by CD4+ Foxp3+ Tregs and CD4+ Foxp3− T conventional cells from the LI-LP (D). The number of mice/group is indicated within each bar or by individual symbols in scatter graphs.
With respect to the SI-LP, CD4+ T conventional and Treg cells from Y3 and Y3/CD103−/− mice each showed increased proliferative Ki67+ cells, indicating that this effect is due to the Y3 IL-2Rβ mutation. Past BrdU uptake and loss studies have shown that the increased proliferation by Tregs in the SI-LP of Y3 mice is due to increased turnover of these cells (17). Thus, an identical mechanism, but with greater turnover, likely accounts for the lower percentage of Tregs in the LI-LP in Y3/CD103−/− mice, because these Tregs showed higher proliferation but a similar lower percentage within the CD4+ T cell compartment when compared to Tregs in the LI-LP of Y3 mice. Overall these data suggest a role for CD103 in normal homeostasis of Tregs in the LI-LP.
The preceding experiments examined CD4+ T conventional and Treg proliferation in the LI-LP of Y3/CD103−/− mice when colitis was evident. The increased proliferation of Tregs, therefore, might reflect an intrinsic abnormality or a response to the proliferating CD4+ inflammatory T cells. To address this point, the CD4+ T cell compartment within the LI-LP of younger colitis-free Y3/CD103−/− mice were examined. At this time extensive lympho-proliferation had not yet developed as assessed by relatively low cellularity of the MLN, which is comparable to that in age-matched Y3 mice (Fig. 3B). An increase in CD4+ T cells was detected in the LI-LP of Y3/CD103−/− mice, including a lower percentage of Tregs (Fig. 3C). However, Y3/CD103−/− Tregs, but not CD4+ T conventional cells, showed increased proliferation (Fig. 3D). Thus, altered proliferation of Tregs precedes changes in proliferation of inflammatory CD4+ T cells. This finding suggests that altered IL-2R signaling and lack of CD103 affect Treg homeostasis which eventually drives colitis.
WT Tregs prevent the development of colitis in Y3/CD103−/− mice
The development of colitis in Y3/CD103−/− mice and the associated immune abnormalities raise the possibility that Tregs were not able to fully maintain tolerance in the intestine. To test this possibility, the capacity of WT Tregs to suppress this form of colitis was evaluated. Y3/CD103−/− mice (12-15 days old) received FACS-purified WT Tregs from Foxp3/RFP reporter mice. At 4 weeks post-transfer, 40% to 70% of total Tregs in the peripheral blood of these recipients were of donor origin (data not shown). Cohorts of sex-matched treated and untreated Y3/CD103−/− mice were monitored for weight change, diarrhea and rectal prolapse. Disease severity as measured by weight loss was more pronounced in untreated female Y3/CD103−/− mice (Fig. 4A). Adoptively transferred WT Tregs prevented this weight loss and other symptoms of colitis (Fig. 4A, B). Accordingly, histological examination of the large intestine showed normal colonic histology (Fig. 4C) and significantly reduced total cellularity for the MLN (Fig. 4D). Further analysis of the LI-LP of Treg-treated mice showed a lower proportion and number of CD4+ T cells (Fig. 4E) with decreased proliferation of CD4+ T conventional cells (Fig. 4F) and an increased frequency of Tregs, which was largely donor derived (75 ± 8.9%) (Fig. 4G). In addition, the number of IL-2+, IL-17+, IFN-γ+ and TNF-α+ CD4+ T effector cells in the LI-LP was substantially decreased (Fig. 4H). These results demonstrate that adoptive transfer of WT Tregs efficiently prevented colitis and the intestinal inflammation in the LI-LP in Y3/CD103−/− mice.
It is worth noting that the increase in proportion, cellularity (Fig. 4E), and proliferation of CD4+ T conventional cells (Fig. 4F) and proportion of Tregs (Fig. 4G) in the SI-LP were not affected by the donor WT Tregs. In addition, increased cytokine production by effector CD4+ T cells in the SI-LP (Fig. 4H) was also unaffected by the WT donor Tregs. This finding suggests that inflammation associated with the SI-LP of Y3/CD103−/− mice is cell intrinsic and independent of altered function of Tregs.
WT Tregs mediate suppression in the absence of CD103
Somewhat surprisingly, the donor WT Tregs in the spleen, MLN, and SI-LP (not shown) and the LI-LP (Fig. 5A) from these long-term colitis-free Y3/CD103−/− mice did not express CD103. These WT donor Tregs, nevertheless, readily expressed Klrg1, which is a marker of highly activated terminally differentiated Tregs. This effect was dependent on the absence of CD103 on recipient Y3/CD103−/− cells because CD103 was readily detected on donor Tregs after adoptive transfer to Y3, but not Y3/CD103−/−, mice 4 weeks post-transfer (Fig. 5B). The CD4+ T cell compartment of both types of recipients contained a substantial fraction of RFP+ donor Tregs and many of these were activated Tregs based on Klrg1 expression (Fig. 5B). This finding suggests that the absence of CD103 on WT Tregs in Y3/CD103−/− recipient is due to a failure to maintain CD103 levels on these Tregs rather than a failure to develop activated Tregs.
Figure 5. Tregs that solely lack CD103 prevent colitis in Y3/CD103−/− mice.

(A) Representative FACS plot of donor and recipient LI-LP Tregs derived from Y3/CD103−/− mice that were adoptively transferred by WT Tregs 26-32 weeks previously (n = 5 mice). (B) FACS purified WT CD4+ RFP+ Tregs (1.5-2.0 × 105) were injected i.v. into the indicated recipient mice. 4 weeks post-transfer, donor Treg engraftment and their activation phenotype (Klg1 and CD103) by donor Tregs in the indicated tissues. Data are pooled from 3 independent experiments (n = 7-11/group). Age-matched 8-12 week-old Y3 and Y3/CD103−/− mice were used as recipients in the experiments, the latter without apparent signs of severe disease. (C) 2 week-old Y3/CD103−/− mice were adoptively transferred with MACS-enriched CD4+ T cells (6 × 106) from CD103−/− mice. Health was assessed 32-33 weeks later in part by evaluation of body weight, MLN cellularity, and activation status of CD4+ Foxp3− T cells in the MLN (n = 3 mice). Controls were autoimmune-free littermate Y3+ or Y3neg IL-2Rβ+/- CD103−/− mice.
To further investigate the requirement for CD103 expression by donor Tregs for suppression of colitis, CD4+ T cells from CD103−/− mice were adoptively transferred into 2 week-old Y3/CD103−/− mice. When examined 32-33 weeks after transfer, these mice were healthy in appearance without signs of colitis, i.e. no diarrhea, rectal prolapse (not shown), wasting disease, lymphoproliferation, or abnormal immune activation, the latter 3 as evidenced by normal body weight, MLN cellularity, and activation of CD4+ T conventional cells (CD44hi and CD62Llo) in the MLN (Fig. 5C). In this setting >80% of the Treg pool in the spleen and MLN was donor-derived. These findings indicate that Tregs, which only lack CD103, exhibit sufficient redundancy to compensate for this deficiency to suppress autoimmunity and maintain mucosal tolerance. This result is not so surprising considering that CD103−/− mice do not exhibit colitis and illustrates the importance for impaired IL-2R signaling as a co-factor to reveal the functional contribution of CD103 to maintain intestinal tolerance.
CD103−/− DCs maintain tolerogenic functions
The preceding experiments raised the possibility that CD103 expression on a non-Treg cell in the setting of impaired IL-2R signaling is also important for mucosal tolerance. CD103 is not only expressed on Tregs, also it is expressed on other cells, such as DCs. CD103+ DCs in the MLN and SI-LP mark a subpopulation of DCs that are tolerogenic due in part to their production of TGF-β and retinoic acid (23). These cytokines are required for pTreg development and expression of molecules, especially CCR9 and α4β7, on T lymphocytes required for homing to the gut mucosa (24-27). To determine whether CCR9 imprinting function was active in Y3/CD103−/− mice, T lymphocytes from the MLN were examined for CCR9 expression. CD4+ T conventional, Treg, and CD8+ T cells from the Y3/CD103−/− mice contained normal proportions of CCR9+ cells when compared to WT, Y3, and CD103−/− mice (Fig. 6A). The latter finding also is consistent with a lack of requirement for expression of CD103 by DCs for CCR9 imprinting (28).
Figure 6. Deficiency of CD103 does not affect homing molecules expression on T cells and pTreg induction.

(A) MLN T cells were analyzed for the expression of CCR9 by flow cytometry. Representative FACS plots (left) or averaged values (right) for CCR9 expression by CD4+ Foxp3− T conventional cells (T convs), CD4+ Foxp3+ Tregs, and CD8+ T cells. Age-matched 8-12 week-old mice were assessed for each genotype. The number of mice/group is indicated within each bar. (B) CD4+ OT-II cells (1 × 106) were i.v. transferred into the indicated recipients and one day later they were fed with OVA (20 mg/ml) in the drinking water for 9 days. Day 10 post-transfer, the percent of OT-II cells (based on the expression of TCR Vα2 Vβ5) among total CD4+ T cells in indicated tissues was determined (left). Representative (middle) and averaged (n = 3 mice/group) (right) expression of α4β7 and CCR9 on donor OT-II cells in MLN was assessed. Similar results were obtained in a second experiment (not shown) for recipients fed with OVA (10 mg/ml) for 14 days. (C) FACS-purified CD4+ Foxp3− Thy1.1+ cells (1 × 106) from Foxp3/RFP mice were injected i.v. into Thy1.2+ Y3 and Y3/CD103−/− recipients. The frequency of CD4+ RFP+ pTregs among total donor cells in the indicated tissues was assessed by flow cytometry. Data are pooled from 2 independent experiments (n = 6-7 mice/group). (D) FACS-purified CD4+ Foxp3− cells (1 × 106) from OT-II/Foxp3/RFP mice were i.v. transferred into Y3 and Y3/CD103−/− recipients. One day later, recipient mice were fed with OVA (5 mg/ml) in the drinking water for 7 days. Day 8 post-transfer, the percent of donor OT-II cells (based on the expression of TCR Vα2 Vβ5) among total CD4+ T cells (left) and the frequency of CD4+ RFP+ pTregs among donor OT-II cells (right) in MLN were assessed by flow cytometry. Data are from one experiment (n = 2-3 mice/group). (B-D) Age-matched 8-12 week-old Y3 and Y3/CD103−/− mice were used as recipients in the experiments, the latter without apparent signs of severe disease.
To more directly assess homing and CCR9 and α4β7 imprinting in the absences of CD103 expression by DCs, we followed the fate and phenotype of OVA-specific class-II restricted TCR transgenic OT-II CD4+ T cells after transfer to Y3 and Y3/CD103−/− mice. OT-II T cells were stimulated by antigen through OVA in the drinking water to induced antigen-specific proliferation and migration to the gut mucosa. Ten days post-transfer, OT-II cells were enumerated in the MLN and SI-LP by their co-expression of the Vα2 and Vβ5 subgroups associated with the OT-II TCR. Comparable proportions of OT-II cells were found in the MLN (1.4% vs. 1.7%) and SI-LP (2.9% vs. 3.1%), respectively, in both recipients (Fig. 6B). Analysis of the phenotype of donor OT-II cells in the MLN revealed that they expressed similar levels of CCR9 and α4β7 homing molecules (Fig. 6B). Collectively, these data are consistent with a CD103-independent role for DC-dependent gut homing and CCR9 and α4β7 expression by T lymphocytes.
To determine whether CD103 is required for induction of pTregs, purified CD4+ Foxp3− T cells were transferred into Y3 and Y3/CD103−/− mice. In this setting pTregs develop in response to host auto or environmental antigens. At 2 weeks post-transfer, pTreg development was assessed for the spleen, MLN, SI-LP and LI-LP (Fig. 6C) and comparable proportions of pTregs were detected in both recipients.
To further examine pTreg development in the absence of CD103 expression by DCs, Treg-depleted OT-II CD4+ T cells from OT-II/Foxp3/RFP reporter mice were transferred into Y3 and Y3/CD103−/− mice followed by feeding OVA in the drinking water in order to induce antigen-specific pTregs in the periphery. Seven days post-transfer, comparable frequencies of donor Vα2+ Vβ5+ OT-II cells were detected in the spleen (data not shown) and MLN (0.25% vs. 0.35%) of Y3 and Y3/CD103−/− mice (Fig. 6D). Notably, RFP expression by OT-II cells, which represents antigen-specific pTregs, was detected in the spleen (data not shown) and MLN at similar proportions in both recipients (Fig. 6D). These data indicate that expression of CD103 by host cells, including DCs, is not required for pTreg development. Collectively, these data indicate that several important aspects of mucosal DCs functioned normally in the absence of CD103.
Normal trafficking/retention of CD103−/− Tregs in the gut mucosa
The effective suppression of inflammation within the colon by CD103+ Tregs might be related to higher expression of suppressive and/or gut homing molecules (29). CD103 might directly contribute to these distinct properties or alternatively may simply mark cells with these activities. CD103 has been reported to promote Treg retention in the skin (10), raising the possibility that CD103 directly regulates Treg trafficking and/or retention within the gut. To test this possibility, the fate of a 1:1 mixture of input Thy1.1+ WT and Thy1.2+ CD103−/− RFP+ Tregs was followed after transfer into lympho-replete Y3 mice. Past studies have shown that adoptively transferred WT Tregs engraft in Y3 recipients (17), which easily permits following the fate of the donor Tregs, in this case based on expression of RFP and Thy1.1(Fig. 7A). Four weeks post-transfer, Tregs of WT and CD103−/− origin were detected in a largely similar proportion in the SI-LP (Fig. 7B). The spleen, MLN and LI-LP also contained similar proportions of Tregs derived from WT and CD103−/− donor cells (Fig. 7C). On average, donor WT Tregs in the spleen, MLN, SI-LP and LI-LP were approximately 40%, 75%, 75% and 50% CD103+, respectively. These results indicate that CD103 expression is not required for Treg homing or retention within the LP of the intestine. Thus, normal Treg homeostasis in the SI-LP and LI-LP and suppression of colitis in Y3/CD103−/− mice by adoptively transferred WT Tregs is not due to CD103-dependent tissue homing and retention in the gut.
Figure 7. CD103 molecule is not required for Treg retention in the gut mucosa.
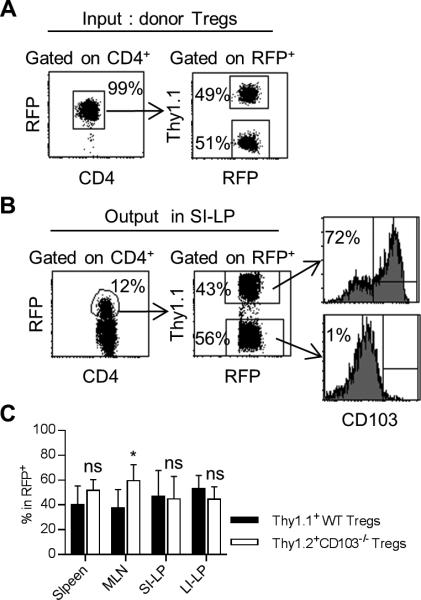
FACS-purified CD4+ RFP+ Thy1.1+ Tregs from Foxp3/RFP mice were mixed with CD4+ RFP+ Thy1.2+ Tregs from CD103−/− Foxp3/RFP mice at 1:1 ratio (4 × 105 total) (input donor Tregs) and injected i.v. into Y3 mice. (A) Representative FACS plots showing the purity and ratio of mixed input Tregs of WT Thy1.1+ and CD103−/− Thy1.2+ origin. Four weeks post-transfer, representative (B) and averaged (C) distribution of Tregs of WT and CD103−/− origin for the indicated tissues. 8-12 week-old Y3 mice were used as recipients. Data are pooled from 2 independent experiments (n = 7 mice/group).
Similar gene programs in Y3 and Y3/CD103−/− Tregs
The inability of Y3/CD103−/− Tregs to maintain tolerance within the gut mucosa might reflect intrinsic defects in these cells when compared to Y3 Tregs. Differences in gene expression between WT and Y3 Tregs provide information concerning properties that predispose Y3 mice to autoimmunity. CD103 might directly or indirectly influence Y3 Treg function by interacting with E-cadherin on DCs and/or epithelial cells or by permitting other molecular interactions due to proximity to these cells. In these situations gene expression is expected to vary between Y3 and Y3/CD103−/− Tregs. Initially to address these points, FACS analysis was performed for SI-LP and LI-LP Tregs from B6, Y3, and Y3/CD103−/− mice to examine several important activation, functional, and homing molecules (Fig. 8A). In comparison to WT Tregs, many of the molecules examined (Foxp3, CD73, CXCR3, CCR6, and CCR5) were similarly expressed by Y3 and Y3/CD103−/− Tregs in both the SI-LP and LI-LP. As expected, CD25 expression, which is upregulated by IL-2R signaling, was lower for Y3 and Y3/CD103−/− Tregs in the SI-LP and LI-LP. However, CD39, Nrp-1 and CTLA4, molecules linked to Treg function, and CCR4 were lower whereas Helios was higher in both Y3 and Y3/CD103−/− Tregs, indicating that their expression was altered due to impaired IL-2Rβ signaling. In each case, however, these differences in expression by Tregs showed localized effects, i.e. Nrp-1, and CCR4 was lower in the SI-LP whereas CTLA4 was lower and Helios was higher only in the LI-LP. As these changes were not unique to Y3/CD103−/− Tregs, they may contribute to mild/moderate inflammation associated with the SI-LP of Y3 and Y3/CD103−/− mice and are likely permissive for colitis associated with Y3/CD103−/− mice.
Figure 8. Similar gene programs in Y3 and Y3/CD103−/− Tregs.

(A) Expression of the indicated markers in Tregs from the SI-LP and LI-LP were assessed by flow cytometry (n ≥ 3 mice/group). (B) Euclidean cluster analysis of all differentially expressed genes (≥ 1.5-fold) for the indicated groups (n = 3 mice/group). (C) Euclidean clustering of genes within the major enrichment groups (EG). EG 1-5 are defined in the text of the Results. Age-matched 8-10 week-old mice were assessed for each genotype.
We have previously reported that Klrg1+ Tregs are IL-2-dependent terminally differentiated short-lived cells that have undergone more than 8 cell divisions (17). The decreased proportion of Klrg1+ Tregs could be interpreted as increased death of Tregs before they differentiate into terminal stage Klrg1+ Tregs due to low IL-2R signaling. As expected, very few Klrg1+ Tregs were found in the SI-LP of Y3 and Y3/CD103−/− mice (Fig. 8A). However, in the LI-LP, Klrg1+ Tregs were reduced only for Y3/CD103−/− mice. This result indicates that the LI-LP contains fewer terminally differentiated Tregs and that the development of the Klrg1+ Tregs may depend on other local signals other than IL-2Rβ, suggesting a contribution of CD103 to Treg homeostasis.
To obtain sufficient number of cells, Tregs were purified from the total LP (both SI-LP and LI-LP) from WT, Y3, and Y3/CD103−/− mice and the RNA was used for genome-wide expression studies to globally address the contribution of impaired IL-2Rβ signaling and the lack of CD103 function for intestinal inflammation and loss of tolerance within the colon. 204 unique annotated mRNAs were differentially expressed by ≥1.5-fold between these 3 groups (Fig. 8B). Very few mRNAs were uniquely up (4) or down (8) regulated in relationship to Y3 and Y3/CD103−/− mucosal Tregs (Fig. 8B, regions 1 & 2). In contrast, 175 mRNAs (88%) showed largely similar expression between Y3 and Y3/CD103−/− Tregs but were up- (139 mRNAs) or down- (36 mRNAs) regulated in comparison to WT Tregs. The former group included 28 mRNAs that showed a somewhat closer expression levels between WT and Y3/CD103−/− Tregs, but were not statistically different from Y3 Tregs due to their variability in expression (Fig. 8B, region 3), perhaps due to colitis. These results indicate that the large majority of differentially expressed genes in this comparison are mainly related to impaired IL-2R signaling by the Y3 mutations rather than the absence of CD103.
Gene enrichment analysis was performed for the differential expressed mRNAs and 4 major enrichment groups (EG), consisting of 75 mRNAs, were identified. Euclidian clustering of these mRNAs represents a microcosm of the entire list of differentially expressed mRNAs (Fig. 8C). The EGs are: EG1, Cytokine-cytokine receptor interaction (p = 2.2 × 10−8) and JAK-STAT signaling pathway (p = 3.2 × 10−6); EG2, regulation of lymphocyte activation (p = 9.7 × 10−9) and proliferation (p = 0.02); EG3, regulation of cell death (p = 2.4 × 10−4) and the Caspase pathway in apoptosis (p = 0.005), and EG4, transcription (p = 0.006). EG5 consists of 8 other genes related to immune function, e.g. Itgae (CD103). Inspection of these mRNAs is consistent with cells more prone to cell death due to decreases in receptors that deliver growth and survival signals (Il2ra, Il7r) and increases in pro-apoptotic molecules (Fas, Apaf1, Birc4, Casp4, Lmnb1, Gzmb). Increases in mRNA were also detected for molecules that promote T-helper activity (Il12rb2, Il12rb1, Ifng, Il21r, Tbx21 (T-bet), Stat1, and Stat3) and aspects of innate immune responses, in particular, the IL-1R pathway (Il1r1, Il1r2, Il18r1, Il18rap). All these genes were similarly expressed in Y3 and Y3/CD103−/− Tregs suggesting that these effects are primarily related to impaired IL-2Rβ signaling. Expression of Foxp3 and most mRNAs linked to Treg function was similarly expressed by all three samples tested (data not shown). One exception was Nrp1, which was reduced, and a trend for lower IL-10, but this was judged not be statistically significant. Overall, these findings are consistent with the known role of IL-2 to regulate cell growth and death and suggest that IL-2Rβ signaling may also normally function in the gut microenvironment to maintain Treg identity by inhibiting responsiveness to inflammatory mediators and expression of molecules that favor T helper activity. These alterations in Tregs, however, on their own, did not lead to colitis.
CD103 is required for proper localization in the gut mucosa
The modest transcriptome changes associated with intestinal Y3/CD103−/− Tregs when compared to Y3 Tregs raise the question how CD103 contributes to Treg homeostasis. Although CD103−/− Tregs were readily found in the gut, their localization within the intestinal microenvironment might be altered. IELs represent an important line of defense to maintain gut homeostasis and are normally abundant in CD103+ cells. Initially, we assessed whether the prevalence of Tregs or other cell populations was altered in the IEL due to the absence of CD103. This analysis revealed decreased CD8αα T cells in the IEL of the small intestine (SI-IEL) and large intestine (LI-IEL) and TCR γδ+ T cells in the SI-IEL from Y3 and Y3/CD103−/− mice (Supplemental Fig. 2). These results are explained by the known requirement of IL-2 and IL-15 signaling, which is impaired by the Y3 transgene, for the development and survival of these T cell subsets (30). Thus, differences in these types of T cells are unlikely to explain colitis that is selectively associated with Y3/CD103−/− mice.
With respect to the function of CD103, the SI-IEL and LI-IEL of CD103−/− mice contained a substantial reduction in the number of total CD4+ T and Tregs cells in comparison to WT B6 mice (Fig. 9A), leading to a relatively normal proportion of Tregs in the CD4+ T cell compartment (Supplemental Fig. 2). This finding demonstrates a role for CD103 in the localization of CD4+ T cells in the IEL of the gut. A trend, albeit not statistically significant, was also noted for fewer conventional CD4+ T and Treg cells within the SI-IEL, but not the LI-IEL, of Y3/CD103−/− mice. This result may reflect the increased T cell cellularity in the intestines of these mice that lead to some contamination of the IEL with LP cells, which are numerically much higher than IELs, especially from Y3/CD103−/− mice.
Figure 9. CD103 is important for proper T cell localization in the small intestine.

(A) The number of total CD4+ T cells and CD4+ Foxp3+ Tregs in the IEL compartment of small and large intestine was determined by flow cytometry. Age-matched 12-24 week-old mice were assessed for each genotype. The number of mice/group is indicated by individual symbols in scatter graphs. (B) Representative confocal images show E-cadherin+ epithelial cells, CD4+ T cells, Foxp3+ Tregs and DAPI stained nucleated cells in the small intestine from age-matched 8-10 week-old mice. Foxp3+ Tregs are marked with yellow arrow head and co-localized CD4+ and E-cadherin+ cells are indicated with yellow arrow. (C) Co-localization of CD4+ T and E-cadherin+ cells was determined by two approaches. Left, total CD4+ T cells and CD4+ T cells that co-localized or closely associated with E-cadherin were counted. The percent of cells that were co-localized or closely associated with E-cadherin+ cells was determined after counting 674 total CD4+ T cells from 26 fields for Y3 and 576 total CD4+ T cells from 27 fields for Y3/CD103−/− mice (PPs were excluded). Right, co-localization was measured by relative mean pixel intensity (Relative Mean) and show graphically. Each symbol represents one relative mean pixel intensity from one field (n ≥ 6 sections per tissue obtained from 3 individual mice for each group).
To directly examine T cell localization within the intestine, confocal imaging was performed after immunofluorescent labeling of frozen intestine sections. We focused this analysis on the small intestine of Y3 and Y3/CD103−/− mice for several reasons: CD103+ cells are more prevalent in the small intestine (Supplemental Fig. 1A); the architecture of the SI-LP with its well-define villi is more favorable to distinguish cells in close association with the epithelial layer; and cells are limited from LI-LP in the B6 and CD103−/− mice and the accumulated T cells in the LI-LP tend to form clusters, even in younger Y3 and Y3/CD103−/− mice, which makes it difficult to detect altered T cell localization in the large intestine.
Localization of CD4+ T conventional and Treg cells with respect to E-cadherin+ epithelial cells was determined for by staining for CD4, Foxp3, E-cadherin and DAPI, the latter to generally reveal nucleated cells. In agreement with FACS analysis, CD4+ T cells were more numerous in the small intestine of Y3 and Y3/CD103−/− mice, which exhibited increased CD4+ T cell cellularity (Figs. 1E, 9B). However, altered T cell localization was noted in the gut mucosa. CD4+ T cells were diffusely localized in the villi of Y3 small intestine (Fig. 9B). More Y3 CD4+ T cells were closely associated to epithelial cells as assessed by co-localization of CD4+ and E-cadherin+ cells (Fig. 9C). In contrast, CD4+ T cells were more centered in the villi of the Y3/CD103−/− small intestine and fewer cells were associated with E-cadherin+ cells (Fig. 9B, C). Although many fewer Foxp3+ Tregs were detected, these trends likely hold true for Tregs because they co-localize with the CD4+ T cells. This distinct localization of CD4+ T cells in the small intestine of Y3 vs. Y3/CD103−/− mice indicates that CD103 is required for close interaction with E-cadherin+ epithelium. Consistent with this notion, DAPI stained nucleated cells in the villi were closer to the epithelial cells in the small intestine from WT and Y3 mice whereas these cells were more clustered within the center of the villi in the small intestine from CD103−/− and Y3/CD103−/− mice. Thus, not only CD4+ T cells, but also other cell types in the intestine require CD103 to closely associate to the E-cadherin+ epithelium.
Collectively, these findings revealed that CD103 is not required for Treg homing or retention (Fig. 7C) in the gut mucosa, but rather is necessary for proper lymphoid cell localization. The disruption of this property in conjunction with the tendency for apoptosis associated with Tregs with impaired IL-2R signaling represents one important condition that leads to colitis.
Discussion
Tregs and DCs marked by expression of CD103 have been associated with maintaining tolerance in the gut mucosa (20, 25), but the direct functional contribution of CD103 in tolerance remains poorly understood. Our study shows that there is a dynamic and important role for CD103 to functionally participate for tolerance in the intestine, primarily through a complex and redundant mechanism that supports the homeostasis of Tregs. This activity of CD103 only became apparent in the context of impaired IL-2R signaling where colitis develops in Y3/CD103−/−, but not Y3 mice, indicating that the Y3 IL-2Rβ mutations on its own does not result in colitis. These effects can be viewed as a risk for colitis, perhaps in an analogous manner by which SNPs in IL-2 or IL-2R subunits are risk for other autoimmune disease, including Crohn's disease (14, 15). CD103 has not been implicated as a genetic risk for colitis. Nevertheless, the Y3/CD103−/− mice represent a new model of colitis and illustrates how an additional genetic lesion, which on its own is not associated with disease, cooperates in a highly penetrant manner with other risk factors to cause autoimmunity.
The importance for CD103 in mucosal tolerance at the level of Tregs is apparent based on the findings that the LI-LP from Y3/CD103−/− mice contained distinctive alterations in Treg homeostasis and WT Tregs readily suppressed immune activation and colitis associated with Y3/CD103−/− mice. If inflammation and autoreactivity in Y3/CD103−/− mice was cell intrinsic or otherwise independent of a defect in Tregs, the high effective reversal of disease was not expected to occur. Nevertheless, Tregs that solely lack CD103 remain competent to suppression colitis in Y3/CD103−/− mice, indicating that the activity supplied by CD103 has an effective undefined redundant counterpart. This redundancy likely explains why the functional contribution of CD103 has been difficult to define, relegating CD103 as primarily an important immunological marker, and has also complicated our ability to more precisely define the function of CD103 in mucosal tolerance.
In contrast to LI-LP, when one considers the inflammatory responses in the SI-LP in Y3/CD103−/− mice, which is closely paralleled in Y3 mice, donor WT Tregs did not reverse the increase in total CD4+ T cell numbers and IL-17 producing cells. These aspects of altered immunity likely represent intrinsic effects at the level of T conventional cells due to impaired IL-2R signaling. This conclusion is in line with the known role of IL-2 to repress Th17 development (31, 32), which also did not function normally by T cells that expressed the Y3 IL-2Rβ mutation.
CD103 has been implicated in other functions that promote immune tolerance, including homing/retention of Tregs within tissue sites, CD103+ DC-mediated imprinting of CCR9 and α4β7 on T cells, and promoting the development of pTregs (10, 25, 33). However, these activities were generally comparable in Y3 and Y3/CD103−/− mice. These findings indicate that CD103 largely serves as a marker on DCs, rather than a functional participant in these tolerogenic activities. Thus, colitis associated with Y3/CD103−/− mice is not accounted for by these activities associated with CD103, further pointing to the importance of CD103-dependent homeostasis of CD103+ Tregs as an important contributing factor for tolerance in the colon. We cannot rule out that CD103 may participate in other functions associated with CD103+ DCs or other cells types (11).
Genome-wide mRNA expression profiling of Tregs from the intestine provides several important clues concerning how impaired IL-2R signaling and the absence of CD103 contribute to the development of colitis. First, very few mRNAs were found differentially expressed between Y3 and Y3/CD103−/− mice and these were not included in the IL-2-dependent gene enrichment groups including those related to T cell activation and proliferation and regulation of cell death. This finding suggests that CD103 does not directly signal to promote the homeostasis of CD103+ Tregs. Second, IL-2-dependent mRNAs included cell death mediators Casp4, Apaf1, and Birc2. This finding indicates that the one important effect of the Y3 mutation is to render Tregs to be more prone to cell death.
Past work and the current study indicate that homeostasis of Y3 and Y3/CD103−/− Tregs from the SI-LP is altered (17). Although Treg number and proportion in the SI-LP were relatively normal, these Tregs showed increased proliferation and turnover, so the increased proliferative rate must be offset by increased cell death to maintain a normal homeostatic level of cells. The SI-LP of Y3 and Y3/CD103−/− mice contain a near 10-fold reduction in Klrg1+ Tregs, consistent with cells that have died before the 8-10 divisions required for becoming terminally differentiated short-lived Klrg1+ Tregs. Some aspects of altered Treg homeostasis related to the Y3 mutation was also noted for cells in the LI-LP, especially a lower ratio of Tregs in CD4+ T cells from Y3 and Y3/CD103−/− mice. This IL-2R signaling defect on its own is not sufficient to cause severe pathology or disease. However, two unique features related to the lack of CD103 expression by Y3/CD103−/− Tregs in the LI-LP is heightened proliferative activity coupled with lower levels of Klrg1+ Tregs. This represents a scenario of CD103-dependent aberrant Treg homeostasis accompanied by increased proliferation and cell death over that associated with Y3 Tregs. This altered homeostasis likely results in a failure in Tregs to effectively suppress autoreactive T cells and represents at least one aspect by which the lack of CD103 leads to colitis.
If CD103 does not directly deliver a survival signal, as suggested by our gene expression profiling data, how does it contribute to Treg survival? The most likely possibility is that CD103 expression is required for the proper localization of Tregs in a niche with other lymphoid cells, and perhaps epithelial cells, and these latter cells provide a survival signal independent of CD103 through secreted molecules or cell-cell interactions. One candidate cytokine is TGF-β, which is highly produced by epithelial cells and CD103+ DCs, and might directly mediate Treg survival or indirectly influence Treg survival by its ability to upregulate CD103 on lymphocytes (34-38). The observation that WT Tregs cannot maintain expression of CD103 in Y3/CD103−/− mice suggests that another CD103+ non-Treg cell is required within the Treg niche to supply TGFβ to maintain expression of CD103. Thus, far we have not been able to define this accessory cell.
Y3 mice showed a clear tendency for increased lymphocyte cellularity in the small and large intestine, yet only Y3/CD103−/− mice developed severe pathology, and only in the large intestine. Why was the small intestine spared from a more severe autoimmune attack? First, even though Foxp3+ Tregs in the SI-LP have impaired gene expression due to the Y3 IL-2Rβ mutation and altered localization due to the absence of CD103, Tr1 cells, which are abundant in the small intestine, and other regulatory mechanisms may help to counteract autoreactive T cells (1, 39, 40). In contrast, tolerance in the colon is more dependent on Tregs because these cells represent the major cell type that produces IL-10. Second, the small intestine contains much less microflora compared to the large intestine (41). The substantially larger bacterial load of the microbiota in the large intestine represents a potential trigger to drive colitis. Thus, tolerance in the large intestine depends more stringently on relatively normal Treg activity. Lastly, a unique mechanism operates within the small intestine to control Th17 cells where SI-LP IL-17+ cells are eliminated via lumen leading to a less colitogenic phenotype (42).
In summary, at this juncture we favor the following model, which provides a framework for future studies. The IL-2Rβ Y3 mutation lowers IL-2 and IL-15-dependent signaling on Treg and T effector cells. Lower IL-2R signaling is primarily responsible for mild subacute inflammation associated with Y3 mice in part due to altered Treg homeostasis with a tendency for increased Treg apoptosis and lower negative regulation of Th17 development. This IL-2-dependent risk, however, does not generally progress to severe autoimmune disease. Other aspects of low IL-2 or IL-15 signaling might interfere and lower T effector responses, memory programming, and memory homeostasis (43), but these effects should not increase self-reactivity, and might counteract lower Treg activity. The absence of CD103, which on its own is not pathogenic, consistently leads to severe colitis in the context of impaired IL-2R signaling imposed by the Y3 mutation. This form of IBD is a result of an inability of these Tregs to maintain tolerance in the gut mucosa. Altered activity of a gut specific niche promotes colitis through a failure of CD103 to properly localize Tregs to receive growth and/or survival signals. In the absence of CD103, the defects associated with the Y3 IL-2Rβ mutation cannot compensate the failure in producing this suppressive niche, leading to tissue-specific severe self-reactivity. The lack of autoimmunity associated with CD103−/− mice, indicates that redundant mechanisms compensate for the lack of CD103 to prevent IBD and explain the ability of CD103−/− Tregs to prevent colitis. It seems highly likely that some of this redundancy depends on molecules that are normally expressed in an IL-2Rβ-dependent fashion.
Supplementary Material
Acknowledgments
We thank the Flow Cytometry Core Facility in the SCCC for FACS analysis and sorting, M. Boulina and the Immaging Core for assistance with confocal microscopy and comments on image analysis, M. Fukata, A. Tomei and M. Najjar for help with preparation of frozen tissue sections and immunofluorescent staining, T. Guettouche and L. Espinoza and Gene Expression Core Facility for the microarray assays. We also thank Louis Gonzalez for critically reading the manuscript.
This work was supported by the NIH (R01 AI055815).
Abbreviations used in this article
- DC
dendritic cell
- EG
enrichment group
- IBD
inflammatory bowel disease
- IEL
intra-epithelial lymphocyte
- LI-IEL
intra-epithelial lymphocytes of the large intestine
- LI-LP
lamina propria of the large intestine
- MLN
mesenteric lymph node
- PP
Peyer's Patch
- pTreg
peripheral induced Treg
- ROIM
region of interest
- SI-IEL
intra-epithelial lymphocytes of the small intestine
- SI-LP
lamina propria of the small intestine
- Tr1
type1 regulatory T cells
- Treg
regulatory T cell
- WT
wild type
Footnotes
Disclosures
The authors have no financial conflicts of interest
References
- 1.Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-β1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr., Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 6.Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-β but not interleukin 4 in the suppression of T helper type 1- mediated colitis by CD45RBlow CD4+ T cells. J. Exp. Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009;27:313–338. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 8.Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, Jankovic D, Sher A. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and γ interferon-dependent mechanism. Infect. Immun. 1998;66:5157–5166. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, Brenner MB. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature. 1994;372:190–193. doi: 10.1038/372190a0. [DOI] [PubMed] [Google Scholar]
- 10.Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J. Immunol. 2005;174:5444–5455. doi: 10.4049/jimmunol.174.9.5444. [DOI] [PubMed] [Google Scholar]
- 11.Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, Agace WW, Parker CM, Powrie F. Essential role for CD103 in the T cell- mediated regulation of experimental colitis. J. Exp. Med. 2005;202:1051–1061. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schon MP, Arya A, Murphy EA, Adams CM, Strauch UG, Agace WW, Marsal J, Donohue JP, Her H, Beier DR, Olson S, Lefrancois L, Brenner MB, Grusby MJ, Parker CM. Mucosal T lymphocyte numbers are selectively reduced in integrin α E (CD103)-deficient mice. J. Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- 13.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 14.Wellcome Trust Case Control, C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204.–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng G, Yuan X, Tsai MS, Podack ER, Yu A, Malek TR. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J. Immunol. 2012;189:1780–1791. doi: 10.4049/jimmunol.1103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. USA. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, Hodgkin PD, Nutt SL. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat. Immunol. 2006;7:466–474. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann J, Huehn J, de la Rosa M, Maszyna F, Kretschmer U, Krenn V, Brunner M, Scheffold A, Hamann A. Expression of the integrin αEβ7 identifies unique subsets of CD25+ as well as CD25− regulatory T cells. Proc. Natl. Acad. Sci. USA. 2002;99:13031–13036. doi: 10.1073/pnas.192162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 22.Banz A, Peixoto A, Pontoux C, Cordier C, Rocha B, Papiernik M. A unique subpopulation of CD4+ regulatory T cells controls wasting disease, IL-10 secretion and T cell homeostasis. Eur. J. Immunol. 2003;33:2419–2428. doi: 10.1002/eji.200324205. [DOI] [PubMed] [Google Scholar]
- 23.del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R. Development and functional specialization of CD103+ dendritic cells. Immunol. Rev. 2010;234:268–281. doi: 10.1111/j.0105-2896.2009.00874.x. [DOI] [PubMed] [Google Scholar]
- 24.Svensson M, Johansson-Lindbom B, Zapata F, Jaensson E, Austenaa LM, Blomhoff R, Agace WW. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal. Immunol. 2008;1:38–48. doi: 10.1038/mi.2007.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J. Exp. Med. 204. 2007:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, Berg PL, Davidsson T, Powrie F, Johansson-Lindbom B, Agace WW. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 2008;205:2139–2149. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. USA. 2010;107:5919–5924. doi: 10.1073/pnas.1002006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porter BO, Malek TR. IL-2Rβ/IL-7Rα doubly deficient mice recapitulate the thymic and intraepithelial lymphocyte (IEL) developmental defects of γc−/− mice: roles for both IL-2 and IL-15 in CD8αα IEL development. J. Immunol. 1999;163:5906–5912. [PubMed] [Google Scholar]
- 31.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea J. J. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O'Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Marquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J. Exp. Med. 2003;198:963–969. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and - independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 35.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-β receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 36.Zhang N, Bevan MJ. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39:687–696. doi: 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, Drachenberg CB, Hadley GA. TGF-β-dependent CD103 expression by CD8+ T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J. Exp. Med. 2005;201:1647–1657. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galan JE, Harhaj E, Flavell RA. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity. 2006;25:941–952. doi: 10.1016/j.immuni.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 40.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat. Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 41.Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu. Rev. Immunol. 2010;28:623–667. doi: 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O'Connor W, Jr., Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro I, Yu A, Dee MJ, Malek TR. The basis of distinctive IL-2- and IL-15- dependent signaling: weak CD122-dependent signaling favors CD8+ T central-memory cell survival but not T effector-memory cell development. J. Immunol. 2011;187:5170–5182. doi: 10.4049/jimmunol.1003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.