

Abstract
Mitochondria are critically important in providing cellular energy ATP as well as their involvement in anti-oxidant defense, fat oxidation, intermediary metabolism and cell death processes. It is well-established that mitochondrial functions are suppressed when living cells or organisms are exposed to potentially toxic agents including alcohol, high fat diets, smoking and certain drugs or in many pathophysiological states through increased levels of oxidative/nitrative stress. Under elevated nitroxidative stress, cellular macromolecules proteins, DNA, and lipids can undergo different oxidative modifications, leading to disruption of their normal, sometimes critical, physiological functions. Recent reports also indicated that many mitochondrial proteins are modified via various post-translation modifications (PTMs) and primarily inactivated. Because of the recently-emerging information, in this review, we specifically focus on the mechanisms and roles of five major PTMs (namely oxidation, nitration, phosphorylation, acetylation, and adduct formation with lipid-peroxides, reactive metabolites, or advanced glycation end products) in experimental models of alcoholic and nonalcoholic fatty liver disease as well as acute hepatic injury caused by toxic compounds. We also highlight the role of the ethanol-inducible cytochrome P450-2E1 (CYP2E1) in some of these PTM changes. Finally, we discuss translational research opportunities with natural and/or synthetic anti-oxidants, which can prevent or delay the onset of mitochondrial dysfunction, fat accumulation and tissue injury.
Abbreviations: AA-AGE, acetaldehyde-derived advanced glycation end product; ACR, acrolein; AFLD, alcoholic fatty liver disease; AGE-albumin, advanced glycation end product-albumin adduct; ALDH2, mitochondrial low-Km aldehyde dehydrogenase 2; AMPK, AMP-activated protein kinase; APAP, acetaminophen; Complex I, NADH-dependent ubiquinone oxidoreductase; Complex III, ubiquinone cytochrome bc1 oxidoreductase; Complex IV, cytochrome c oxidase; Complex V, ATP synthase; CYP2E1, ethanol-inducible cytochrome P450 2E1 isozyme; DILI, drug-induced liver injury; DTT, dithiothreitol; eNOS, endothelial NOS; ER, endoplasmic reticulum; ERK, extracellular signal regulated protein kinase; ETC, electron transport chain; Gpx, glutathione peroxidase; GSH, glutathione; 4-HNE, 4-hydroxynonenal; HIF, hypoxia-inducible factor; ICDH, isocitrate dehydrogenase; I-kB, inhibitor protein of NF-κB; iNOS, inducible nitric oxide synthase; I/R, ischemia–reperfusion; JNK, c-Jun N-terminal protein kinase; Keap1, Kelch-like ECH-associated protein 1; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MDMA, 3,4-methylenedioxymethamphetamine; MGO, methylglyoxal; mito-CP, mitochondria-targeted carboxy-proxyl; mitoQ, mitochondria-targeted ubiquinone; MPT, mitochondrial permeability transition; mtGSH, mitochondrial glutathione; NAC, N-acetylcysteine; NAFLD, nonalcoholic fatty liver disease; NF-κB, nuclear factor-κB; NAPQI, N-acetyl-p-benzoquinone imine; NEL-adduct, N-ethyllysine adduct; nNOS, neuronal NOS; NO, nitric oxide; Nrf2, nuclear factor (erythroid-derived 2)-like 2; 4-ONE, 4-oxonon-2-enal; p38K, p38 protein kinase; PGC1α, peroxisomal proliferator activated receptor gamma coactivator-1α; PKC, protein kinase C; Prx, peroxiredoxin; PTEN, lipid phosphatase and tensin homolog; RAGE, receptor for advanced glycation end product; RNS, reactive nitrogen species; ROS, reactive oxygen species; S-NO-Cys, S-nitrosylated Cys; SAMe, S-adenosyl-methionine; SOD, superoxide dismutase; SREBP, sterol regulated element binding protein; Thiolase, 3-ketoacyl-CoA thiolase; TIMP, tissue inhibitor of metalloproteinase 3; TPP+, triphenylphosphonium cation.
Keywords: Nitroxidative stress, Redox, Post-translational modifications, Mitochondrial proteins, Mitochondrial dysfunction, Tissue injury
Graphical abstract
Highlights
-
•
Hepatotoxic agents including alcohol and high fat elevate nitroxidative stress.
-
•
Increased nitroxidative stress promotes post-translational protein modifications.
-
•
Post-translational protein modifications of many proteins lead to their inactivation.
-
•
Inactivation of mitochondrial proteins contributes to mitochondrial dysfunction.
-
•
Mitochondrial dysfunction contributes to necrotic or apoptotic tissue injury.
Introduction
All living organisms require exogenous foods/nutrients for producing energy in the form of ATP, which is needed for numerous cellular functions. Most of the cellular energy is efficiently produced in specialized organelles mitochondria by oxidative phosphorylation. In addition to energy production, mitochondria play an important role in other cellular functions such as fatty acid oxidation, anti-oxidant defense, intermediary metabolism including ammonia and glutamate detoxification, synthesis of heme and steroids, cell death process, and autophagy [1]. Recent reports showed that mitochondria also undergo constant morphological changes with fusion and fission, following the exposure to toxic agents and/or under pathological conditions [2], [3]. After decreased glucose supply like during fasting or inefficient oxidative phosphorylation in certain disease states, the mitochondrial fat oxidation pathway becomes important in providing an alternative source of energy (e.g., ketone bodies) [1]. Without these energy supply mechanisms due to suppressed mitochondrial function (i.e., mitochondrial dysfunction), living cells would die or become susceptible to cell death processes (necrosis and apoptosis). In fact, it is well-established that mitochondrial function in different tissues is significantly suppressed in numerous medical disorders such as metabolic syndrome including obesity/diabetes, cardiovascular disorders, ischemia reperfusion injury (I/R)1 and cancer as well as many neurological disorders like Alzheimer's disease and Parkinson's disease [4], [5], [6], [7], [8], [9], [10], [11], [12], although etiological causes of each disease are different.
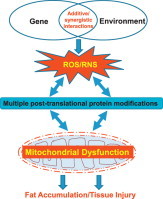
Hepatic mitochondrial abnormalities are observed in both alcoholic fatty liver disease (AFLD) [4], [5], [6], nonalcoholic fatty liver disease (NAFLD) [7], [8], [9] and acute liver injury. As a result, it is conceivable to observe decreased ATP levels and increased fat accumulation (micro-vesicular and macro-vesicular steatosis) in the liver under these conditions. Continued mitochondrial dysfunction with increased oxidative stress also sensitizes the hepatocytes to subsequent necrosis and/or apoptosis of hepatocytes, which likely lead to activation of resident liver macrophage (i.e., Kupffer cells) and recruitment of infiltrating immune cells into the liver with elevated levels of hepatic inflammation (steatohepatitis) and pro-inflammatory cytokines/chemokines [13], [14], [15]. Consequently, hepatic stellate cells can be activated and transformed into myofibroblast-like cells, producing pro-fibrotic cytokines such as transforming growth factor-beta and platelet derived growth factor, leading to hepatic fibrosis/cirrhosis and cancer. These sequential events can be observed in both AFLD [13], [14], [15] and NAFLD [16], [17]. In addition, acute or sub-chronic exposure to various hepatotoxic compounds, including clinically-used drugs, such as acetaminophen (APAP) [18], [19], [20], a major ingredient of Tylenol®, an anti-breast cancer agent tamoxifen [21], an anti-retroviral drug zidovudine (AZT) [22] and antidepressants [23], can cause mitochondrial dysfunction, contributing to liver injury with or without fat accumulation, depending on the injurious agent, as extensively reviewed [24], [25], [26]. These hepatotoxic agents and pathological conditions are known to elevate the levels of reactive oxygen and nitrogen species (ROS/RNS) and nitroxidative stress through the suppression of the mitochondrial electron transport chain (ETC) and induction/activation of NADPH oxidase, cytochrome P450 isozymes including ethanol-inducible P450-2E1 (CYP2E1) and CYP4A, xanthine oxidase, and inducible nitric oxide synthase (iNOS). Although increased nitroxidative stress can oxidatively damage mitochondrial DNA and lipids, the majority of insults can also take place at the protein levels through different forms of post-translational modification (PTM) [26], [27]. Because of the newly-emerging information on redox-related protein modifications, we briefly describe five major forms of PTM and functional consequences of some modified mitochondrial proteins in the experimental models of the AFLD, NAFLD and acute liver injury (Fig. 1). In addition, we highlight potential roles of CYP2E1 in promoting various PTMs.
Fig. 1.
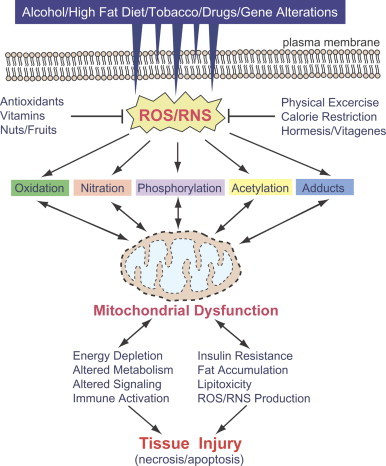
Five major types of PTM contributing to mitochondrial dysfunction and tissue injury in AFLD, NAFLD and acute liver damage. Alcohol, smoking, high fat diet, hepatotoxic compounds and aberrant genetic mutations can lead to increased production of ROS and RNS either individually or synergistically, leading to elevated nitroxidative stress. Under increased nitroxidative stress, many cellular (mitochondrial) proteins can be modified by different forms of PTM and inactivated, leading to mitochondrial dysfunction. Under increased mitochondrial dysfunction, greater amounts of ROS/RNS are produced while energy supply and normal metabolism can be suppressed. Accumulation of fat resulting from disrupted fat oxidation can trigger insulin resistance and lipotoxicity while protein adducts can stimulate immune responses. Sustained mitochondrial dysfunction would lead to the development of vicious cycles of PTMs and energy depletion and lipotoxicity, ultimately promoting tissue injury (necrosis/apoptosis). Many small molecule antioxidants contained in fruits and vegetables as well as exercise and calorie restrictions can be used to prevent or reduce the nitroxidative stress. Uni-directional and bi-directional arrows indicate exclusive and mutual influences (in vicious cycles), respectively.
Post-translational modifications of mitochondrial proteins
Oxidation of mitochondrial proteins
Under normal conditions, transiently elevated ROS is known to be involved in cellular signaling pathways [28], [29] and mitochondrial functions are correctly maintained through proper redox balance. However, chronic and/or binge alcohol, high fat diets, tobacco smoking, or certain hepatotoxic drugs can directly damage the mitochondrial ETC, producing greater amounts of ROS leaked from the ETS [24], [25], [26], [27]. Without proper counter-balance by various cellular anti-oxidants, the persistent imbalance in cellular redox states result in decreased levels of mitochondrial antioxidants including mitochondrial glutathione (mtGSH), which serves as a critical determinant between toxic damage and cellular protection [30]. When the cellular defense system is overwhelmed, greater amounts of ROS and RNS remain elevated, ultimately leading to increased nitroxidative stress.
It is well-established that many amino acids such as cysteine (Cys), methionine (Met), histidine (His), proline (Pro), lysine (Lys), tyrosine (Tyr), phenylalanine (Phe), threonine (Thr) and tryptophan (Trp) in most proteins can also be redox regulated. As recently reviewed [26], [27], Cys residue(s) can be oxidatively modified in many forms [sulfenic acid, disulfide, sulfinic/sulfonic acids, NO- or peroxynitrite-dependent S-nitrosylation, NO-independent ADP-ribosylation, mixed disulfide formation between Cys residues and glutathione (glutathionylation), cysteine (cystathionylation), succinic acid, myristic acid [31] or palmitic acid (Cys-palmitoylation) [32] prior to membrane attachment or cellular trafficking].
Since the sensitive methods to detect redox-regulated Cys residues in cellular proteins were extensively described in our previous reviews [26], [33], we will only highlight the functional consequences of oxidative modification of Cys residues in some proteins. If one of these amino acids serves as the active site or is located near the active site of certain enzymes, it is highly likely that oxidative modifications of these amino acids can result in their inactivation, as shown by the oxidation and/or S-nitrosylation of Cys residues including the active site Cys in the mitochondrial aldehyde dehydrogenase (ALDH2) [34] and 3-keto-acyl CoA-thiolase (thiolase) [34] in binge alcohol-exposed rodents. In fact, mass-spectral analysis revealed that more than 87 mitochondrial and 60 cytosolic proteins were oxidatively-modified in alcohol-exposed rodents [34], [35]. The rates of protein oxidation in CYP2E1-containing E47-HepG2 hepatoma cells [36] exhibited a dose- and time- dependent pattern in response to alcohol as well as the presence of CYP2E1 [37]. Similar numbers of oxidatively-modified mitochondrial proteins were also identified by mass-spectral analysis in mice with I/R injury [38] and rats exposed to 3,4-methylenedioxymethamphetamine (MDMA) [39]. The number of oxidatively-modified mitochondrial proteins we characterized may represent a small fraction of the estimated number of 1100–1400 total mitochondrial proteins [40], [41]. However, we believe that the actual number of oxidized proteins could be significantly higher than the proteins originally reported [34], [38], [39], because of the technical limitations in the identification and purification of oxidized proteins and detection methods, especially for those proteins expressed in low abundance, as previously discussed [26], [33].
In the case of oxidative inactivation of mitochondrial ALDH2 through active site Cys modification [42], immunoblot analysis of the immunoaffinity purified ALDH2 proteins by using the specific antibody against ALDH2 or S-NO-Cys was performed [34]. Protein detection using the anti-ALDH2 antibody revealed that similar levels of ALDH2 (54 kDa) were observed in all 3 mitochondrial samples [i.e., mitochondria from control rats, mitochondria from alcohol-exposed rats, and dithiothreitol (DTT)-treated mitochondria from alcohol-exposed rats]. However, one specific immunoreactive band with the anti-S-NO-Cys antibody was detected only in mitochondria from alcohol-exposed rats. Furthermore, the significantly decreased ALDH2 activity in ethanol-exposed rats was fully restored by DDT treatment [34]. Similar results of S-nitrosylation and inactivation of ALDH2 were also observed in rat hepatoma H4IIE-C3 cells exposed to NO donors such as S-nitrosoglutathione (GSNO), S-nitroso-N-acetylpenicillamine, and 3-morpholino-sydnonimine, respectively [43]. These results indicate that ALDH2 and other ALDH isozymes, which contain the highly conserved active site Cys residue, can be reversibly modulated by S-nitrosylation under increased nitroxidative stress, as reviewed [42]. Other PTMs of Cys and other amino acid residues in ALDH2 protein are summarized (Fig. 2). Oxidative modification and suppression of ALDH2 in alcohol-exposed rodents were consistently observed by other groups [44], [45]. In fact, these phenomena could explain the decreased levels of ALDH2 activity in alcoholic individuals, as discussed [34], [42]. Oxidative modification and inactivation of ALDH2 were also observed after exposure to MDMA [39] or I/R injury [38], resulting in the accumulation of lipid peroxidation products, which are metabolized by ALDH2 [46]. Similar modifications and inactivation of active site Cys residues in thiolase and possibly other enzymes in the β-oxidation pathway of fatty acids are likely to lead to fat accumulation observed in alcohol-exposed rodents [34], in MDMA-exposed rats [39] as well as in mouse liver following I/R injury [38]. Furthermore, oxidative modification of the active site Cys residue of peroxiredoxin 1 (Prx1) and inactivation was observed in alcohol-exposed mice [47]. Since Prx1 was determined to be localized on the cytosolic side of the endoplasmic reticulum (ER) and interacts with CYP2E1 [47], it is likely that this oxidative inactivation of Prx1 contributes to increased ROS production and fatty liver following alcohol exposure. Based on the conserved redox-sensitive Cys residues among the multiple isoforms [47], [48], [49], many Prx enzymes could be also oxidatively-modified and inactivated, contributing to elevated levels of oxidative stress. Furthermore, the activities of NAD+- or NADP+-dependent isocitrate dehydrogenases (ICDH) are known to be oxidatively-modified and inactivated in experimental models exposed to hydrogen peroxide, menadione, nitric oxide, or alcohol [50], [51], [52], [53]. Oxidative inactivation of ICDH enzymes would increase oxidative stress since they are important in the regulation of mitochondrial redox balance and cellular defense through providing NADPH for the regeneration of reduced GSH [50]. Mass-spectral analysis confirmed that Cys305 and Cys387 of NADP+-ICDH were S-nitrosylated, resulting in its inactivation in alcohol-exposed animals [53]. By the same logic, it is expected that many other enzymes that contain active site Cys residue would likely undergo similar types of redox-related modifications and become inactivated, as reviewed [26], [33]. For instance, it was reported that a DNA repair enzyme O6-methylguanine-DNA-methyltransferase which contains redox-sensitive Cys in its active site can be inactivated through S-nitrosylation [54], resulting in increased levels of oxidative DNA modifications, which can contribute to carcinogenesis.
Fig. 2.
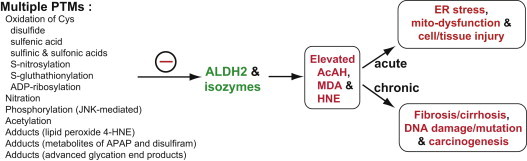
Multiple PTM of ALDH2 protein, contributing to its inactivation. Different types of PTM on ALDH2 are illustrated. Most PTMs led to the suppression of ALDH2 activity, although phosphorylation of ALDH2 by the PKCε isoform was shown to stimulate the ALDH2 activity, as recently reviewed [42]. The suppressed activities of ALDH2 and other isozymes, all of which share the 100%-conserved active site Cys residue, likely contribute to increased levels of toxic and carcinogenic acetaldehyde (AcAH) and lipid peroxides (MDA and 4-HNE) following exposure to ethanol, high fat and other toxic compounds. ER stress, mitochondrial dysfunction and tissue injury can be observed in acute and sub-chronic cases. Long-term chronic suppression of ALDH2 and other isozymes can also contribute to fibrosis, cirrhosis and carcinogenesis. The negative sign represents the inactivation of ALDH2 and other isozymes.
It is also possible that redox regulation can indirectly affect the patterns of gene expression by modulating the conserved Cys residues in proteins involved in the cell signaling pathways like many protein phosphatases (see the Section “Phosphorylation of mitochondrial proteins”) or directly affecting many redox-sensitive transcription factors such as nuclear factor-kappa B (NF-κB), nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and hypoxia-inducible factor-1 (HIF-1), as recently reviewed [55]. The modulation of transcription factors can either be harmful or beneficial. For instance, NF-κB, a critical transcription factor in inflammation and innate immune regulation, can be activated by alcohol [56] and other toxic agents such as lipopolysaccharide (LPS) [57]. NF-κB can be activated through proteasomal degradation of the inhibitor protein (I-κB) of the NF-κB after its phosphorylation by I-κB kinase. Consequently, NF-κB activation leads to the transcriptional initiation of many pro-inflammatory cytokines, chemokines and pro-oxidant proteins such as TNF-α, IL-12, monocyte chemotactic protein-1, macrophage inflammatory protein-2, cyclooxygenase-2 and iNOS [55], [56], [57]. Chronic or acute alcohol exposure and other hepatotoxic agents can also activate HIF-1α in mice and rats [57], [58], [59], [60], [61]. However, activation of HIF-1α, which is co-localized with CYP2E1 in centrilobular areas, seems to contribute to CYP2E1-dependent oxidative stress and liver toxicity, as recently suggested in chronic and acute alcohol exposure [59], [60], [61]. In contrast, a cell-protective transcription factor Nrf2 can be activated under increased oxidative stress in AFLD and NAFLD [62], [63], [64], [65]. Increased oxidative stress activates Nrf2 through oxidative modifications of redox-sensitive Cys residues of the regulatory binding protein Kelch-like ECH-associated protein 1 (Keap1), leading to ubiquitin-dependent degradation of Keap1, promoting Nrf2 translocation into the nucleus for transcriptional activation of its downstream targets, as reviewed [66]. Activation of Nrf2 with its down-stream target proteins, including glutamate cysteine ligase, heme oxygenase-1, quinone reductase, and thioredoxin reductase, provides anti-oxidant defense against increased oxidative stress caused by alcohol exposure [62].
As described thus far, oxidative modifications of Cys residues in many cellular proteins are associated with inactivation or suppression of the activity of modified target proteins. However, redox modification of Cys, especially the mixed disulfide formation, such as S-glutathionylation, can also be considered as a protective mechanism in some cases. For instance, reversible oxidation of Cys residues of mitochondrial NADH-dependent ubiquinone oxidoreductase (complex I), ALDH2, and Prx3 seem to be protective, despite temporary inactivation, against further tissue damage from irreversible hyper-oxidation of Cys to Cys-sulfinic acid (Cys-SO2) and Cys-sulfonic acid (Cys-SO3) followed by proteolytic degradation [67], [68], as in the case of I/R injury [69], [70], [71], [72]. It is known that Cys-glutathionylation of mixed disulfide groups can create a functional cycle consisting of the forward reaction (glutathione S-transferase P) and the reverse reaction (glutaredoxin system) [67], [73], [74]. Furthermore, some Cys residues can be modified through adduct formation with carbonyl compounds, including lipid peroxide 4-hydroxy-2-nonenal (4-HNE) and reactive metabolites of APAP. However, Cys residues in the adducts with reactive metabolites or lipid peroxides cannot be restored even in the presence of a strong reducing agent DTT and thus are considered irreversible, as discussed [26], [33]. The functional implications of some examples of these adducts are described later (see “Section Electrophilic adduct formation of mitochondrial proteins”).
Nitration of many mitochondrial proteins
It is well-established that the physiological levels of NO produced by endothelial NOS (eNOS) and neuronal NOS (nNOS) are relatively small and can act as a signaling molecule within the cell or interact with surrounding cells [27], [75], [76]. However, many toxic agents such as alcohol, APAP and LPS can induce iNOS, which produces greater amounts of NO, likely leading to nitrosative/nitrative stress [27], [75], [76], [77]. In this case, NO can interact with ROS to produce a potently toxic peroxynitrite, which can nitrate Tyr residues to generate 3-nitroTyr (3-NT) or S-nitrosylate Cys residues of many proteins [77]. These two PTMs can lead to alterations of both protein structure and function, contributing to increased cell and tissue injury.
Protein nitration may occur in various cell compartments. Although the concentration of mtGSH is similar to that of cytosolic GSH (5–10 mM), it represents a small fraction of the total cellular GSH [30]. Because mitochondria are a major source of ROS/RNS as previously mentioned, they can be very vulnerable to nitroxidative damage [27]. Indeed, mitochondrial DNA is extremely sensitive to oxidative/nitrative damage due to (A) its location closer to the inner mitochondrial membrane; (B) the relatively low amount of histone, anti-oxidant enzymes, and mtGSH, and (C) the low levels of polyamines and DNA repair enzymes [78], [79]. In support of the view of mitochondrial sensitivity to nitration, it has been reported that the turn-over rates of rat liver mitochondria proteins were markedly decreased from few days to hours as a result of proteolytic degradation following exposure to nitrating conditions [80], [81]. Interestingly, mitochondrial DNA encodes 13 polypeptides all of which are members of the 4 mitochondrial ETC proteins, namely Complexes I, III, IV, and V [82], [83]. Taken together, mitochondrial DNA mutation and/or deletion may lead to abnormal expression or deletion of some of the ETC components, contributing to mitochondrial respiratory dysfunction and more leakage of ROS/RNS. Consequently, mitochondrial damage or dysfunction due to oxidative/nitrative protein modifications can be expected in various pathological conditions or following exposure to some toxic agents such as alcohol, high fat, MDMA, and APAP [18], [19], [20], [21], [22], [23], [24], [84], [85]. Nitration of mitochondrial proteins and their functional roles in fatty liver disease and/or acute liver injury have been extensively reviewed recently [27]. For instance, NO-dependent inhibition of the mitochondrial respiration was monitored in both AFLD and NAFLD [86], [87], [88], [89], [90].
Since protein nitration is one of the emerging research topics in acute tissue injury, we specifically focused on this PTM in promoting mitochondrial dysfunction and acute hepatotoxicty. We used APAP as a well-established model agent for acute drug-induced liver injury (DILI) to study the functional role of nitrated mitochondrial proteins and the mechanisms of mitochondrial dysfunction and hepatotoxicity. APAP was first shown to induce and promote mitochondrial protein nitration and that peroxynitrite can cause mitochondrial DNA damage by Cover et al. [18]. Hepatic necrosis highly correlated with the areas of nitrated proteins [91]. Direct evidence for a critical role of peroxynitrite in APAP-mediating hepatic injury has been provided with the seminal work by Knight et al. [92]. This study showed that the protective effects of GSH were mainly due to the restoration of cellular GSH levels for efficient scavenging of peroxynitrite, thus providing direct evidence of the central role of peroxynitrite in APAP-induced liver injury. CYP2E1, one of the main enzymes involved in APAP metabolism, seems to play a critical role in APAP-induced protein nitration and liver injury [93]. Based on these results, we hypothesized that CYP2E1-dependent nitration of many mitochondrial proteins is important in promoting mitochondrial dysfunction and liver injury. In fact, after immunoaffinity purification followed by mass-spectral analysis, we demonstrated that more than 70 mitochondrial proteins and 30 cytosolic proteins were nitrated in APAP-exposed mice [94]. These results are in line with another recent report [95]. The identified nitrated proteins are involved in cellular energy production, fat metabolism, intermediary metabolism, urea cycle, chaperone activity, and anti-oxidant defense. To further determine the functional roles of protein nitration, we mainly focused on the five selected mitochondrial enzymes, namely SOD2, Gpx, ATP synthase, ALDH2 and thiolase. We specifically studied the presence of nitration and activity change in each of these five enzymes at 2 h after exposure to a toxic dose of APAP in the presence or absence of the co-treatment with an anti-oxidant N-acetyl-cysteine (NAC) [94]. Treatment with NAC completely protected the liver from the APAP-induced necrotic injury measured by increased serum transaminase activities and liver histology. Furthermore, NAC ameliorated the inactivation of the five critical enzymes via preventing Tyr nitration, as evidenced by immunoprecipitation with a specific antibody to each protein followed by immunoblot with anti-3-NT antibody. Thus, protein nitration serves as one of the critical causal factors in APAP-induced acute liver damage usually observed at later time points. Indeed, the suppressed activities of anti-oxidant enzymes such as SOD2 and Gpx would certainly interfere with the cellular anti-oxidant abilities. Suppressed ATP synthase activity would lead to decreased ATP production, interfering with vital cell functions and ultimately causing necrosis. Furthermore, inactivation of thiolase likely leads to the inhibition of the β-oxidation of fatty acids with increased hepatic fat accumulation or inefficient supply of an alternative energy (e.g., ketone bodies produced from fat degradation). Furthermore, suppressed ALDH2 would lead to the accumulation of toxic, reactive aldehydes and lipid peroxidation, as previously mentioned in the oxidation section [42]. Based on our results, we expect similar results of mitochondrial dysfunction and tissue injury with other conditions such as I/R injury [38] and LPS treatment [96], which caused extensive protein nitration. It is important to mention that the inactivation of many nitrated proteins can result from the nitration of Tyr residue in the active site, leading to interference of the protein’s catalytic activity. However, it is still possible that nitration of other Tyr residues may cause a conformational change in the target protein and thus block the accessibility of ATP or substrates for example, as previously discussed [27]. However, it would be prudent to double-check the function of each nitrated protein, since nitration can also increase the activity of some proteins such as glutathions S-transferase (GST), heat shock protein 90 (HSP90), and protein phosphatase type 2 [PP2], or exert no effects at all as reported with transferrin [27].
Although most reported results mainly focused on Tyr nitration, nitration of Trp residues in proteins was also reported [97]. However, there have been fewer reports about Trp nitration and functional consequence than those of Tyr nitration, since Trp is much less abundant than Tyr and that Trp is usually buried inside the protein core with less surface exposure for nitration. Nonetheless, excellent reports discussed the occurrence of Trp nitration that may cause functional alterations in detail [97], [98], [99]. The effects of peroxynitrite on nitration of Tyr or Trp and S-nitrosylation of Cys likely depend on cellular status of antioxidants, various microenvironments including surface exposure, peptide loop structure, local charge groups, and the presence of competing amino acids nearby (e.g., Cys near the potentially-nitrated Trp or Tyr), as suggested [98].
Phosphorylation of mitochondrial proteins
Many cellular proteins can be regulated by reversible cycles of phosphorylation and dephosphorylation by protein kinases and phosphatases, respectively. It is well-established that many exogenous agents such as alcohol, smoking, high fat, and APAP can modulate many different classes of protein kinases including cAMP-dependent protein kinases (PKA), PI3K/AKT/mTOR (PKB), protein kinase C isoforms (PKC), calcium-calmodulin-dependent protein kinases, stress activated mitogen-activated protein kinases (MAPKs) including c-Jun N-terminal protein kinase (JNK) and p38 kinase (p38K), and receptor-mediated tyrosine kinases. These kinases can work together synergistically or antagonize each other’s activities. In fact, crosstalk between different classes of protein kinases becomes complicated, contributing to different outcomes, depending on their inducibility, tissue distribution, subcellular localization or translocation to another compartment, cellular environment, treatment agent, and activity status of phosphoprotein phosphatase, as recently reviewed [100], [101], [102], [103], [104], [105]. For instance, concurrent activation of PKC-delta isoform along with stress activated protein kinases generally enhances cell death [106], [107], [108], although some exceptions exist. In contrast, activation of PI3K/Akt and extracellular signal regulated protein kinase (ERK) is generally known to support cell survival and proliferation. However, we briefly describe the redox-mediated regulation of stress activated MAPKs and AMP-activated protein kinase (AMPK) in this review, based on their involvement in mitochondrial homeostasis, intermediary metabolism for steatosis and liver injury.
Under increased nitroxidative stress, stress-activated MAPKs can be stimulated partly through inhibition of phosphoprotein phosphatases. For instance, the highly-conserved active site Cys residue of phosphoprotein phosphatases including MAPK phosphatases [103], [104], [105] can be oxidatively-modified and inactivated by the mechanisms described earlier (“Section of Oxidation of mitochondrial proteins”). This redox-related activation of MAPKs is similar to that of Akt activation through oxidative inactivation of the essential Cys residue of its binding partner lipid phosphatase and tensin homolog (PTEN) [109]. The inactivation of MAPK-phosphatases leads to activation of various stress-activated JNK and p38K [104], [105]. In fact, acute exposure to many hepatotoxic agents including alcohol [110], [111], APAP [112], [113], [114], [115], [116], arachidonic acid [117], 4-HNE and carbon tetrachloride [118], palmitic acid [119], and troglitazone [120] significantly activated JNK (p-JNK) and/or p38K (p-p38K), contributing to toxicity in a variety of cells. Pretreatment with a specific inhibitor of CYP2E1 blocked metabolic activation of some hepatotoxic agents, which are CYP2E1 substrates [110], [111], [112], [113], [114], [115], [116], [117], [118], and prevented JNK activation. Taken together, CYP2E1 is critically involved in JNK activation and cell death caused by those hepatotoxic CYP2E1 substrates. In addition, activation of JNK was positively related to high fat diet-induced insulin resistance and metabolic syndrome [119], [121], [122], [123]. A recent study also showed that sustained JNK activation led to phosphorylation at Ser-46 of sirtuin 1, NAD+-dependent deacetylase in cytosol (described later), followed by ubiquitin-dependent degradation, contributing to fat accumulation in obese mice [124]. Furthermore, persistent activation of JNK and/or p38K is positively associated with various liver diseases such as steatohepatitis and I/R injury [122], [123], [124], [125], suggesting the clinical importance of JNK and p38K in human disease states, as reviewed [126]. Experiments with gene deletion technology and anti-sense oligonucleotides were conducted to determine the role of JNK1 versus JNK2 in hepatotoxicity. For instance, both JNK1 and JNK2 seem to be important in alcohol-induced fatty liver injury [111], while JNK1 seems critical in hepatotoxicity caused by co-treatment of pyrazole and TNFα [127]. In contrast, JNK2 plays a dominant role in APAP-induced acute liver injury since JNK2-null mice are resistant to acute hepatotoxicity [113], suggesting that this area needs further clarification.
Several reports on the redox-related activation of the JNK and/or p38K or their upstream kinases such as mitogen activated protein kinase kinase (M2K) and mitogen activated protein kinase kinase kinase (M3K), as reviewed [128]. However, the number and identity of the mitochondrial and cytosolic proteins, that are phosphorylated by active p-JNK and/or p-p38K, need to be investigated. Our results showed that pro-apoptotic Bax in the cytosol can be phosphorylated (activated) by p-JNK and/or p-p38K, resulting in its conformational change with exposure of the C-terminal membrane domain. These events were followed by translocation of the activated Bax to mitochondria, leading to changes in mitochondrial permeability transition (MPT) and apoptosis of cultured hepatoma cells [129]. The importance of phosphorylation was further confirmed using site-directed mutagenesis of potential amino acids for phosphorylation followed by functional analysis. Our results revealed that Thr167 of Bax was phosphorylated before it was translocated to mitochondria to stimulate mitochondria-dependent apoptosis [129]. In addition, activated p-JNK is known to translocate to mitochondria to initiate MPT change and damage following exposure to hepatotoxic agents including APAP [115], [116], [130], [131]. Since JNK does not contain the canonical mitochondrial leader sequence [132], it would be interesting to identify the mechanism(s) by which activated p-JNK is transported into mitochondria and then phosphorylates mitochondrial matrix proteins such as ALDH2 in CCl4-exposed rats [133]. One of the potential mechanisms of JNK translocation to mitochondria could be through its interaction with a scaffold protein Sab [116], [134] or Bcl-XL [130] on the mitochondrial outer membrane before it gets transported into mitochondria. When the active p-JNK is translocated to mitochondria, it can phosphorylate many mitochondrial proteins such as ATP synthase β subunit, α-KGDH, PDH E1α and β subunits, which were phosphorylated by the recombinant JNK, as shown in an in vitro study using bioenergetically competent mitochondria [135]. However, the identities and physiological roles of numerous mitochondrial proteins phosphorylated by p-JNK (e.g., JNK-specific phosphoproteomes) need to be determined in animal models and human disease states in the future.
Regulatory mechanism of AMP-dependent protein kinase (AMPK) by alcohol and other non-alcoholic substances, including high fat diet has become an important research topic, since AMPK serves as a key sensor and/regulator of metabolic syndrome and many cellular processes [136]. In fact, the redox sensitive AMPK can be paradoxically regulated by activation or suppression. For instance, AMPK can be activated by hydrogen peroxide and mitochondria-derived ROS since its activation can be prevented by pretreatment with antioxidants such as NAC or GSH-ethyl ester [101]. AMPK activation by hydrogen peroxide was tightly associated with the elevated levels of AMP with concurrent depletion of ATP. Recent results showed that AMPK can be also activated in an AMP-independent manner. Regardless of AMP dependency, AMPK activation under oxidative stress or by chemical activators such as metformin and AICAR can lead to increased cell survival signal by promoting autophagy to remove damaged mitochondria and to stimulate mitochondrial energetics [101]. In contrast, AMPK can be suppressed by alcohol [137] or other agents such as high fat diet [138] and APAP [139]. Inactivation of AMPK is known to be associated with AFLD and NAFLD or acute liver injury, as reviewed [13].
Acetylation of mitochondrial proteins
Reversible protein acetylation and deacetylation, catalyzed by acetyltransferases and deacetylases, respectively, represent another form of PTMs, although functional roles of many acetylated proteins are still unclear and need further investigations. In fact, acetylation on N-ε-Lys residues in proteins becomes an important research area through epigenetic regulation of many genes involved in normal cellular growth and physiology by acetylated histones and other chromatin associated proteins [140]. A large scale proteomics study revealed that approximately 20% of mitochondrial proteins and 44% of NAD+-dependent mitochondrial dehydrogenases are acetylated under physiological conditions [141]. The number of acetylated proteins can be significantly increased in experimental models of pathological conditions or following exposure to hepatotoxic agents, possibly resulting from redox-mediated suppression of deacetylases, as shown in alcohol-exposed [142], [143], [144], [145] or high-fat exposed rodents [146], [147], [148]. For instance, chronic and binge alcohol exposure suppressed the activities or decreased levels of various isoforms of sirtuin, Zn and NAD+-dependent deacetylases, involved in cellular aging, lipid metabolism, and anti-oxidant defense. Because of NAD+-dependence, the activities of sirtuins can be suppressed by the redox change (e.g., a decreased NAD+/NADH ratio) following alcohol metabolism via alcohol dehydrogenase (ADH) and low Km ALDH2, both of which use NAD+ as a cofactor for their activities. Earlier reports also showed that chronic alcohol exposure suppressed the activities and protein amounts of sirtuin 1, 3 and 5 isoforms [142], [143], [144], [145], [149], [150], in a CYP2E1-independent manner [144]. In addition, the activities of sirtuin isoforms can be allosterically suppressed through thiol-specific modification of Cys280 in the Zn binding site with 4-HNE adduct formation, as demonstrated [151]. Consequently, the levels of acetylated proteins were significantly elevated in alcohol-exposed rodents. Some of the acetylated proteins, that are deacetylated by sirtuin isoforms, are several transcription factors involved in the regulation of lipid metabolism and cell apoptosis such as sterol regulated element binding protein (SREBP), peroxisomal proliferator activated receptor gamma coactivator-1 (PGC-1), forkhead box protein O1 (FoxO1), NF-κB and p53 [142], [143], [144], [149]. Acetylation of these transcription factors is a well-established cause for fat accumulation and altered cell proliferation in the liver. For instance, alcohol-related suppression of sirtuin 1 increased the levels of acetylated SREBP-1 and PGC-1α with functional activation and suppression, respectively, resulting in alcoholic fatty liver [142]. By using immunoprecipitation with anti-Ac-Lys antibody followed by mass spectral analysis, 91 acetylated mitochondrial proteins were recently identified in alcohol exposed mice [152]. Some of the acetylated mitochondrial proteins are ALDH2, ATP synthase, Gpx, and thiolase, suggesting that these acetylated mitochondrial proteins likely contribute to their functional alterations and fat accumulation [152]. Another report further indicated that alcohol treatment elevated the number of acetylated proteins as well as propionylated protein in mitochondria and nuclear fractions with little changes in cytosolic and microsomal fractions through suppression of sirtuin activities [153]. However, in this study, alcohol-exposure did not seem to change the protein levels of sirtuin 3, 4, and 5. The reasons for dissimilar results on sirtuin levels, observed in different laboratories, are unknown, but could be due to distinct ethanol dose, time course, environment, or animal strains, and warrants further investigation.
In addition, high fat feeding can suppress the activities of mitochondrial sirtuin 3 and other isoforms, thus increasing the number of acetylated proteins. However, the mechanism(s) by which high fat decreased levels of sirtuin isoforms is poorly understood. The levels of hyper-acetylated proteins are markedly increased in mice deficient of mitochondrial sirtuin 3 [147], [154], [155]. For instance, acetylation of long-chain fatty acyl-CoA dehydrogenase, one of the four enzymes involved in the mitochondrial fat oxidation pathway, causes its inactivation, leading to fat accumulation [154], [155]. In contrast, activation of sirtuins by small molecule polyphenols including resveratrol [156] and green tea extracts [157] can reduce the number of acetylated proteins, including PGC-1α, which contributes to increased fat oxidation and insulin sensitivity with improved outcome against metabolic syndrome. Therefore, sirtuin 3 and its isoforms, involved in fat metabolism and aging process, become important therapeutic targets in managing metabolic syndrome and cellular senescence in the liver and other tissues.
As mentioned earlier, key mitochondrial proteins in regulating the fatty acid metabolism are acetylated and that the number of acetylated proteins can be significantly increased through suppression of the mitochondrial deacetylase sirtuin 3 and other sirtuin isoforms by alcohol [142], [143], [144], [145], [149], [150], [151], [152], [153], high fat [148], and other conditions [154], [155]. Compared to numerous reports on the regulation of deacetylase proteins including mitochondrial sirtuin 3, the expression and properties of the mitochondrial acetyltransferase enzyme(s) have been less characterized although acetylation sites in many mitochondrial proteins have been recently reported [158]. In addition, other studies indicated that protein acetylation can take place nonenzymatically in the presence of low levels of acetyl-CoA [159] or acetyl-phosphate and acetyl-adenylate [160]. These reports indicate that the characteristics of acetyltransferase(s) are poorly understood and need further studies, although the GCN5 family seems to be the prototype of (histone) acetyltransferases [161].
Electrophilic adduct formation of mitochondrial proteins
It is well-established that excessive alcohol intake and many potentially toxic compounds can suppress low Km mitochondrial ALDH2 activity in rodents and humans through various PTMs of the active site Cys residue and other redox-sensitive amino acids, as reviewed [26], [27], [42], resulting in the accumulation of toxic acetaldehyde and other lipid aldehydes. Consequently, cellular macromolecules including mitochondrial proteins [162], [163] and DNA [164] are subjected to modification by these toxic aldehydes, leading to mitochondrial dysfunction and oral and gastrointestinal tract cancers [165]. Chronic alcohol drinking and/or smoking or high fat can also cause mitochondrial damage with increased nitroxidative stress and lipid peroxidation products such as 4-HNE, 4-oxonon-2-enal [4-ONE], malondialdehyde (MDA), and acrolein (ACR) [166], [167], [168]. In addition, many potentially toxic drugs can produce reactive metabolites, which can induce mitochondrial dysfunction and acute liver injury [18], [19], [20], [21], [24]. For instance, APAP metabolism produces N-acetyl-p-benzoquinone imine (NAPQI) while the metabolism of potentially hepatotoxic troglitazone generates reactive quinolone-like metabolites. Other drugs such as halothane, tienilic acid, and dihydralazine can produce reactive metabolites, which promote hepatotoxicity. The underlying mechanisms of these idiosyncratic toxicities by these agents could be due to interactions between highly reactive electrophilic intermediates with various mitochondrial proteins, leading to mitochondrial dysfunction and tissue damage including DILI. They can also damage membrane integrity and alter calcium homeostasis between endoplasmic reticulum and mitochondria, leading to hepatotoxicity. Furthermore, these protein adducts can serve as a neo-antigen of hapten–carrier conjugate proteins to activate immune responses and to facilitate infiltration of neutrophils into the liver to promote inflammation [18], [19], [20], [21], [24], [166], [167], [168].
Recent results showed that highly-reactive lipid peroxides including 4-HNE and 4-ONE, despite their extremely short half-lives, can suppress the activities of many mitochondrial proteins likely through adduct formation with a few amino acids such as Cys, His, Arg, and Lys of target proteins [168]. For instance, earlier reports showed that acetaldehyde and 4-HNE produced by alcohol intake interact with mitochondrial proteins, including cytochrome C oxidase (complex IV), leading to its inactivation, mitochondrial dysfunction and liver injury [169], [170], [171], [172]. By using mass-spectrometry, Petersen and colleagues recently determined the specific amino acids that are modified by 4-HNE. They showed that the active site Cys301 of ALDH2 [173] and Cys280 in the critical Zn binding site of sirtuin 3 [151] were covalently modified with 4-HNE, leading to their inactivation. In addition, many other proteins such as protein disulfide isomerase [174] and Grp78 [175] in the ER, gamma-glutamyl-cysteine synthase (GCS) [176], ERK [177], PTEN [178], or AMPK [179] in cytosol could also be inactivated through interaction with 4-HNE, likely leading to ER stress, GSH depletion and alteration of the cell signaling pathways frequently observed in alcohol-fed models and humans.
Unlike mixed disulfide bonds of Cys such as S-nitrosylation and S-glutathionylation, the protein adducts with reactive carbonyls including 4-HNE, 4-ONE, MGO and ACR cannot be reversed by the presence of reducing agents such as DTT, GSH and ascorbate, as recently reviewed [28]. Therefore, these protein adducts may exist for an extended time period unless they are removed by cellular proteolytic enzymes or phagocytic macrophages. Furthermore, HNE-protein adducts can induce immune cell activation of hepatic macrophage Kupffer cells and/or infiltration of neutrophils into the liver, producing inflammatory cytokines and activation of stellate cells, leading to advanced liver disease, as reported in alcoholic liver injury [166], [167], [168], [180]. In fact, the average levels of MDA- and HNE-protein adducts in 50 alcoholic cirrhosis patients were significantly higher than those in a similar number of patients with nonalcoholic cirrhosis or healthy individuals [180]. In contrast, no differences in the levels of ACR- and MGO-protein adducts were found among different groups. Furthermore, the average levels of MDA and HNE-adducts were higher in 51 alcoholic patients with advanced liver disease with fibrosis/cirrhosis than 23 fatty liver disease or 30 healthy control people, suggesting that MDA- and HNE-protein adducts likely served as antigens or activators of immune responses in advanced alcoholic liver disease. The ethanol-induced CYP2E1 was partly responsible for the production of 37 kD-acetaldehyde protein adducts [181] while 4-HNE protein adduct could be produced in CYP2E1-independent manner, although CYP2E1 seems to at least play a permissive role [182].
In case of high-fat fed rodents, the total number of the reports on HNE-protein adducts in the livers of NAFLD seems relatively small compared to those in AFLD, although the identities of HNE-protein adducts were reported in extra-hepatic tissues such as heart [183], [184]. Mass spectral analysis revealed 39 unique lipoxidation sites in 27 mitochondrial proteins. These lipid peroxide-modified mitochondrial proteins include: pyruvate dehydrogenase, malate dehydrogenase, ICDH, methylmalonate-semialdehyde dehydrogenase, long chain fatty acyl-CoA dehydrogenase, NADH-dependent ubiquinone oxidoreductase (complex I), succinate dehydrogenase (complex II), ubiquinone-cytochrome bc1 oxidoreductase (complex III), cytochrome c oxidase (complex IV), ATP synthase (complex V), and others [184]. Although the activities of each HNE-modified protein were not determined in this study, it is reasonable to assume that the functional activities of some of the modified proteins could be suppressed. We also believe similar types of lipid peroxide adducts present in the liver of high-fat exposed rodents (and humans), due to the elevated levels of oxidative stress and lipid peroxides including 4-HNE [185], as comparable to the alcoholic fatty liver [170], [171], [172]. In fact, ALDH2 activity in the heart was significantly inhibited in high fat-exposed diabetic mice [186]. Further characterization revealed that ALDH2 was inactivated through 4-HNE adduct formation, as evidenced by immunodetection of 4-HNE adduct in the immunoprecipitated ALDH2 with the anti-ALDH2 antibody [186]. The levels of cardiomyocyte hypertrophy and dysfunction in high fat-exposed mice were inversely correlated with the ALDH2 activity, suggesting a causal role of ALDH2 in diabetic cardiomyopathy. These results are consistent with the cardioprotective role of ALDH2 which can be activated by small molecule activators [42], [187] or over-expression by genetic modulation [188], [189].
In addition to lipid peroxide-related protein adducts, recent data suggest that sugar moiety-related protein adduct such as advanced glycation end products (AGEs) can be produced by high fat or excessive alcohol exposure or under pathological states such as diabetes, cardiovascular diseases and Alzheimer's disease, as recently reviewed [190], [191], [192], [193]. Elevated levels of cross-linked AGEs in many tissues can up-regulate the expression of the receptor for AGEs (RAGE), which activates NADPH oxidase, leading to increased oxidative stress, insulin resistance, and inflammation with disruption of normal cell signaling pathways. These pro-oxidative events with accumulated proteins contribute to acute injury and/or persistent chronic disease states in many tissues [190], [191], [192], [193]. For instance, chronic alcohol exposure elevated the levels of acetaldehyde-derived AGE (AA-AGE) and HNE-protein adducts in the livers of rats and humans while their elevated levels disappeared after alcohol abstinence. The immunostaining intensities of AA-AGE and HNE-adducts positively correlated with the severity of alcoholic liver disease [193]. Furthermore, AA-AGE but not N-ethyllysine (NEL) caused apoptosis of rat hepatocytes, suggesting an important role of AA-AGE in alcohol-induced hepatotoxicity and fatty liver injury [193]. Similar results of elevated production of AGE and its plasma levels in cirrhotic patients compared to those in healthy individuals were previously reported [194]. High fat feeding also elevated the levels of AGEs and RAGE in many tissues, including kidneys, hearts and epididymal fat in an aldose reductase-dependent manner [195]. In a mouse model of NASH caused by high fat western diet or choline-deficient diet, AGEs induced TNFα-cleavage enzyme in an NADPH oxidase-dependent manner and stimulated fibrogenic activity via decreasing the levels of sirtuin 1 and tissue inhibitor of metalloproteinase 3 (TIMP3) [196]. The significantly lower levels of sirtuin 1 and TIMP3 were also observed in livers from NASH patients than those of healthy individuals, suggesting a role of AGE adducts in promoting inflammatory responses. Another report recently indicated that several mitochondrial proteins in aged rats were modified with AGEs, thus their activities inactivated. These AGE-modified proteins were: complexes I and V, catalase, ALDH2, medium chain acyl Co-A dehydrogenase, keto-acyl CoA dehydrogenase, and others, leading to decreased ETC, anti-oxidant defense and urea cycle in aged animals [197].
In addition to liver injury, AA-AGE but not NEL stimulated death of neuronal cells [198], suggesting an important role of AA-AGE in neuronal cell death. Consistent with this report, elevated amounts of AGE-albumin adduct, produced by activated microglial cells, were found in alcohol-exposed rat brains and post-mortem brains from human alcoholic individuals compared to those of healthy normal counterparts [199]. Increased oxidative stress can produce AGE-albumin adduct, which elevates the level of RAGE and activates JNK- and p38K-mediated death of neuronal cells. These results are in agreement with the increased expression of RAGE in the brains of alcohol-exposed rats and humans [200]. Consistently, the elevated amounts of AGE-albumin, which was confirmed by mass-spectral analysis [201], were found in the brains of Alzheimer’s disease patients than those of the healthy individuals [202]. Inhibition of AGE-albumin production or blockade of RAGE by a chemical antagonist or a specific antibody to RAGE efficiently prevented alcohol-induced brain damage [199], suggesting an important role of AGE-protein adducts in promoting neurodegeneration. Based on these recent results [199], [200], [201], [202], it would be of interest to study the role of AGE-protein adducts in promoting organ damage in the peripheral tissues such as the liver and heart.
Translational research with mitochondrial-targeted antioxidants
As described above, the increased nitroxidative stress in AFLD and NAFLD or after exposure to hepatotoxic agents can stimulate multiple PTMs, including the hydroxyethyl-adducts observed in alcohol-exposed rats [203] and individuals [204] (Fig. 3). All these PTMs of cellular proteins are likely to contribute to increased ER stress and mitochondrial dysfunction, leading to energy depletion, fat accumulation, altered metabolism, inflammation and necrotic/apoptotic tissue damage (Fig. 3). Since elevated nitroxidative stress is a critical contributing factor for acute and chronic liver diseases, beneficial effects of many anti-oxidants from natural and synthetic origins have been tested in experimental model systems. These anti-oxidant agents include: l-arginine, small molecule metabolites (e.g., GSH-ethyl ester and NAC), natural antioxidants [e.g., vitamins C, E, co-enzyme Q10, alpha-lipoic acid, fish oil containing n-3 fatty acids, betaine and S-adenosyl-methionine (SAMe)], and herbal molecules and polyphenols (curcumin, esculetin, sulforaphane, resveratrol, epigallocatechin-3-gallate, caffeic acid phenethyl ester, and many others) [205], [206], [207], [208], [209], [210], [211]. Some of these antioxidants, contained in many fruits and vegetables, show beneficial effects on obesity, NAFLD and liver injury [205], [206], [207], [208] while other antioxidants are potentially neuroprotective. Recent reports showed that naturally occurring antioxidants such as SAMe and betaine preserved mitochondrial function and proteome in the animal models of AFLD [212], [213], [214] as well as NAFLD [215], [216], [217], partly through blockade of the ROS/RNS production with restoring the physiological levels of SAMe, S-adenosyl-homocysteine and GSH. Nonetheless, the list of these antioxidants with therapeutic potential is exponentially growing and will reduce the oxidative burden in various pathophysiological conditions including fatty liver disease.
Fig. 3.
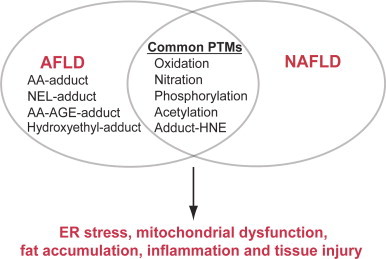
Overlapping PTMs between AFLD and NAFLD. Overlapping PTMs commonly observed in both AFLD and NAFLD are illustrated. However, acetaldehyde–protein adduct (AA–adduct) [171], [181], NEL-adduct [193], AA-AGE-adduct [193], and hydroxyethyl-adduct [203], [204] seem uniquely observed in AFLD. These PTMs observed in AFLD and NAFLD are likely to suppress the functions of the target proteins, contributing to altered cell signaling, ER stress, mitochondrial dysfunction, fat accumulation and inflammatory tissue injury.
One of the main problems with some of the natural antioxidants is to guarantee good manufacturing quality control [e.g., a constant amount of the active component(s) with acceptable levels of impure materials]. Other problems can be low solubility, poor bioavailability and little mitochondrial transport [208], [209], [210], [211]. To overcome these problems, many compounds with better mitochondrial targeting properties have been synthesized to prevent or treat mitochondrial dysfunction in many pathological conditions [211], [218], [219], [220], [221], [222], [223], [224], [225]. For instance, to accomplish better mitochondrial transport, Szeto and Schiller developed a series of small, cell-permeable, mitochondria targeted antioxidant peptides (Szeto–Schiller or SS-tetrapeptide) that protect mitochondria from oxidative damage [220]. These peptide antioxidants represent a novel approach with targeted delivery of antioxidants to the inner mitochondrial membrane. The structural motif of these SS-peptides centers on alternating aromatic residues and basic amino acids (aromatic-cationic peptides) that can scavenge hydrogen peroxide and peroxynitrite while they inhibit lipid peroxidation. Their antioxidant activities can be attributed to the tyrosine or dimethyltyrosine residue with inhibition of mitochondrial permeability transition and cytochrome c release, thus preventing oxidant-induced cell death. Because these peptides can accumulate >1000-fold in the inner mitochondrial membrane, they prevent oxidative cell death with EC50 in the ~nM range. Similarly, the analogs of SOD-catalase mimetics also showed beneficial effects on many tissues including the liver and brain [221].
In contrast to the SS-peptides or peptide-mimetics, a small molecule triphenyl phosphonium (TPP+, a cell permeable lipophilic cation) has been developed to conjugate with various drugs and antioxidants to enhance the transport of target molecules to reduce oxidative stress and damage, as reviewed [222], [223]. The results with mitochondria-targeted ubiquinone (MitoQ) or mitochondria-targeted carboxy-proxyl (Mito-CP) so far have shown promising results in preventing mitochondrial abnormalities and nitroxidative tissue injury in various disease models including Friedreich Ataxia fibroblasts [224], diabetic nephropathy [225], cisplatin-induced renal toxicity [226] and cocaine-induced cardiac dysfunction [227]. In addition, the protective effects of MitoQ and Mito-CP were clearly demonstrated in a mouse model of hepatic I/R injury [228]. In a dose-dependent manner, these mitochondria-targeted compounds blocked the early and delayed nitroxidative stress responses (e.g., HNE/carbonyl adducts, malondialdehyde, 8-OHdG, and 3-nitrotyrosine formation), mitochondrial dysfunction and histopathological signs of liver injury as well as delayed inflammatory cell infiltration and cell death [228]. Consistently, mitoQ was shown to be protective against micro- and macro-vesicular steatosis in AFLD [229] without affecting the levels of CYP2E1 and ALDH2 as well as ethanol-induced mitochondrial respiratory abnormality. The effects of mitoQ were rather mediated through the suppression of ROS/RNS production and their down-stream targets such as protein nitration and HIF-1α stabilization [229]. These promising results from different laboratories suggest that mitochondria-targeted antioxidants are more effective against elevated nitroxidative stress than untargeted natural antioxidants. Because of the recent development of these antioxidant agents and clinical testing [230], [231], we expect approval of some of these beneficial agents in treating mitochondrial dysfunction-related organ damage and hence preventing the health deterioration.
Conclusion and future research opportunities
We have thus far briefly described five major types of PTMs that promote mitochondrial dysfunction, fat accumulation, inflammation and hepatic injury in alcoholic and nonalcoholic substances (Fig. 1). Although several PTMs in many mitochondrial proteins were identified in different disease states or under increased nitroxidative stress, it is quite possible that many proteins can be modified by multiple PTMs simultaneously. Therefore, it seems difficult to clearly demonstrate the deleterious effect of each single PTM and which PTM plays a critical role in causing mitochondrial dysfunction and tissue damage. In addition, it is challenging to identify the modified amino acid(s) and their roles in activity changes. It is also still unknown what other proteins, especially those expressed in low amounts in a particular tissue, are modified by different PTMs following exposure to a potentially toxic agent. Furthermore, mitochondrial functions can be altered by other types of PTM such as methylation, O-linked glycosylation (Ser or Thr), N-linked glycosylation (Asn), sumoylation, and ubiquitin conjugation. The identities of the proteins modified by these PTMs and their roles in mitochondrial dysfunction, fat accumulation and tissue injury need to be studied in the future. Finally, the patterns of PTM and their functional roles in other extra-hepatic tissues such as brain, heart, pancreas, adipose tissue and intestine need to be investigated. For instance, binge alcohol exposure [232], [233], [234], [235], [236], [237], [238], [239] and nonalcoholic substances including fructose [240], [241], [242] are known to promote gut leakiness, contributing to more severe inflammatory liver disease. In the case of binge alcohol-induced gut leakiness, CYP2E1-mediated increased oxidative stress plays an important role, since treatment with an antioxidant NAC or a CYP2E1 inhibitor chlormethiazole significantly prevented gut leakiness and liver injury. In addition, Cyp2e1-null mice were resistant to alcohol-mediated gut leakiness, which was clearly observed in the corresponding wild-type mice [238]. It is of interest whether gut leakiness caused by nonalcoholic substances also depends on CYP2E1-mediated oxidative stress. Despite numerous reports on gut leakiness caused by alcohol and fructose [232], [233], [234], [235], [236], [237], [238], [239], [240], [241], [242], the functional roles of redox-regulated PTMs on many intestinal proteins are still poorly understood. Therefore, characterization of the modified proteins and their functional consequences in intestinal epithelial tissues and other tissues are not only good for basic mechanistic studies in understanding tissue–tissue interactions but also provide future translational and clinical research opportunities.
Acknowledgments
This research was supported by the Intramural Program Fund at the National Institute on Alcohol Abuse and Alcoholism. The authors thank Dr. Klaus Gawrisch for his support. The authors do not have any conflict of interest.
References
- 1.Soboll S. Regulation of energy metabolism in liver. Journal of Bioenergetics and Biomembranes. 1995;27(6):571–582. doi: 10.1007/BF02111655. 8746844 [DOI] [PubMed] [Google Scholar]
- 2.Dimmer K.S., Scorrano L. (De)constructing mitochondria: What for? Physiology (Bethesda, Md.) 2006;21:233–241. doi: 10.1152/physiol.00010.2006. 16868312 [DOI] [PubMed] [Google Scholar]
- 3.Galloway C.A., Yoon Y. Mitochondrial morphology in metabolic diseases. Antioxidants and Redox Signaling. 2013;19(4):415–430. doi: 10.1089/ars.2012.4779. 22793999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoek J.B., Cahill A., Pastorino J.G. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122(7):2049–2063. doi: 10.1053/gast.2002.33613. 12055609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantena S.K., King A.L., Andringa K.K., Eccleston H.B., Bailey S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radical Biology & Medicine. 2008;44(7):1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. 18242193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu D., Cederbaum A.I. Oxidative stress and alcoholic liver disease. Seminars in Liver Disease. 2009;29(2):141–154. doi: 10.1055/s-0029-1214370. 19387914 [DOI] [PubMed] [Google Scholar]
- 7.Sanyal A.J., Campbell-Sargent C., Mirshahi F., Rizzo W.B., Contos M.J., Sterling R.K., Luketic V.A., Shiffman M.L., Clore J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–1192. doi: 10.1053/gast.2001.23256. 11266382 [DOI] [PubMed] [Google Scholar]
- 8.Gambino R., Musso G., Cassader M. Redox balance in the pathogenesis of nonalcoholic fatty liver disease: Mechanisms and therapeutic opportunities. Antioxidants and Redox Signaling. 2011;15(5):1325–1365. doi: 10.1089/ars.2009.3058. 20969475 [DOI] [PubMed] [Google Scholar]
- 9.Blake R., Trounce I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochimica et Biophysica Acta. 2014;1840(4):1404–1412. doi: 10.1016/j.bbagen.2013.11.007. 24246956 [DOI] [PubMed] [Google Scholar]
- 10.Wallace D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. 16285865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaturvedi R.K., Flint Beal M.. Mitochondrial diseases of the brain. Free Radical Biology and Medicine. 2013;63:1–29. doi: 10.1016/j.freeradbiomed.2013.03.018. 23567191 [DOI] [PubMed] [Google Scholar]
- 12.Dexter D.T., Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biology and Medicine. 2013;62:132–144. doi: 10.1016/j.freeradbiomed.2013.01.018. 23380027 [DOI] [PubMed] [Google Scholar]
- 13.Purohit V., Gao B., Song B.J. Molecular mechanisms of alcoholic fatty liver. Alcoholism, Clinical and Experimental Research. 2009;33(2):191–205. doi: 10.1111/j.1530-0277.2008.00827.x. 19032584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao B., Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–1585. doi: 10.1053/j.gastro.2011.09.002. 21920463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes M.A., Roychowdhury S., Nagy L.E. Innate immunity and cell death in alcoholic liver disease: role of cytochrome P4502E1. Redox Biology. 2014;2:929–935. doi: 10.1016/j.redox.2014.07.007. 25180169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell C.L., Kosyk O., Bradford B.U., Parker J.S., Lobenhofer E.K., Denda A., Uematsu F., Nakae D., Rusyn I. Temporal correlation of pathology and DNA damage with gene expression in a choline-deficient model of rat liver injury. Hepatology (Baltimore, Md.) 2005;42(5):1137–1147. doi: 10.1002/hep.20910. 16250055 [DOI] [PubMed] [Google Scholar]
- 17.Baffy G., Brunt E.M., Caldwell S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. Journal of Hepatology. 2012;56(6):1384–1391. doi: 10.1016/j.jhep.2011.10.027. 22326465 [DOI] [PubMed] [Google Scholar]
- 18.Cover C., Mansouri A., Knight T.R., Bajt M.L., Lemasters J.J., Pessayre D., Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. Journal of Pharmacology and Experimental Therapeutics. 2005;315(2):879–887. doi: 10.1124/jpet.105.088898. 16081675 [DOI] [PubMed] [Google Scholar]
- 19.Jaeschke H., McGill M.R., Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metabolism Reviews. 2012;44(1):88–106. doi: 10.3109/03602532.2011.602688. 22229890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han D., Dara L., Win S., Than T.A., Yuan L., Abbasi S.Q., Liu Z.X., Kaplowitz N. Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends in Pharmacological Sciences. 2013;34(4):243–253. doi: 10.1016/j.tips.2013.01.009. 23453390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribeiro M.P., Santos A.E., Custódio J.B. Mitochondria: the gateway for tamoxifen-induced liver injury. Toxicology. 2014;323:10–18. doi: 10.1016/j.tox.2014.05.009. 24881593 [DOI] [PubMed] [Google Scholar]
- 22.Banerjee A., Abdelmegeed M.A., Jang S., Song B.J. Zidovudine (AZT) and hepatic lipid accumulation: implication of inflammation, oxidative and endoplasmic reticulum stress mediators. PloS One. 2013;8(10):e76850. doi: 10.1371/journal.pone.0076850. 24146933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Couch L., Higuchi M., Fang J.L., Guo L. Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicological Sciences: an Official Journal of the Society of Toxicology. 2012;127(2):582–591. doi: 10.1093/toxsci/kfs100. 22387747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pessayre D., Fromenty B., Berson A., Robin M.A., Lettéron P., Moreau R., Mansouri A. Central role of mitochondria in drug-induced liver injury. Drug Metabolism Reviews. 2012;44(1):34–87. doi: 10.3109/03602532.2011.604086. 21892896 [DOI] [PubMed] [Google Scholar]
- 25.Yuan L., Kaplowitz N. Mechanisms of drug-induced liver injury. Clinics in Liver Disease. 2013;17(4):507–518. doi: 10.1016/j.cld.2013.07.002. 24099014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song B.J., Abdelmegeed M.A., Henderson L.E., Yoo S.H., Wan J., Purohit V., Hardwick J.P., Moon K.H. Increased nitroxidative stress promotes mitochondrial dysfunction in alcoholic and nonalcoholic fatty liver disease. Oxidative Medicine and Cellular Longevity. 2013;2013:781050. doi: 10.1155/2013/781050. 23691267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdelmegeed M.A., Song B.J. Functional roles of protein nitration in acute and chronic liver diseases. Oxidative Medicine and Cellular Longevity. 2014;2014:1–21. doi: 10.1155/2014/149627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhee S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science (New York, N.Y.) 2006;312(5782):1882–1883. doi: 10.1126/science.1130481. 16809515 [DOI] [PubMed] [Google Scholar]
- 29.Hamanaka R.B., Chandel N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends in Biochemical Sciences. 2010;35(9):505–513. doi: 10.1016/j.tibs.2010.04.002. 20430626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Von Montfort C., Matias N., Fernandez A., Fucho R., Conde de la Rosa L., Martinez-Chantar M.L., Mato J.M., Machida K., Tsukamoto H., Murphy M.P., Mansouri A., Kaplowitz N., Garcia-Ruiz C., Fernandez-Checa J.C. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. Journal of Hepatology. 2012;57(4):852–859. doi: 10.1016/j.jhep.2012.05.024. 22687340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bastidas A.C., Pierce L.C., Walker R.C., Johnson D.A., Taylor S.S. Influence of N-myristylation and ligand binding on the flexibility of the catalytic subunit of protein kinase A. Biochemistry. 2013;52(37):6368–6379. doi: 10.1021/bi400575k. 24003983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadolski M.J., Linder M.E. Protein lipidation. FEBS Journal. 2007;274(20):5202–5210. doi: 10.1111/j.1742-4658.2007.06056.x. 17892486 [DOI] [PubMed] [Google Scholar]
- 33.Song B.J., Suh S.K., Moon K.H. A simple method to systematically study oxidatively modified proteins in biological samples and its applications. Methods in Enzymology. 2010;473:251–264. doi: 10.1016/S0076-6879(10)73013-5. 20513482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moon K.H., Hood B.L., Kim B.J., Hardwick J.P., Conrads T.P., Veenstra T.D., Song B.J. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology (Baltimore, Md.) 2006;44(5):1218–1230. doi: 10.1002/hep.21372. 17058263 [DOI] [PubMed] [Google Scholar]
- 35.Kim B.J., Hood B.L., Aragon R.A., Hardwick J.P., Conrads T.P., Veenstra T.D., Song B.J. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics. 2006;6(4):1250–1260. doi: 10.1002/pmic.200500447. 16408314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai J., Cederbaum A.I. Adenovirus-mediated overexpression of catalase in the cytosolic or mitochondrial compartment protects against cytochrome P450 2E1-dependent toxicity in HepG2 cells. Journal of Biological Chemistry. 2001;276(6):4315–4321. doi: 10.1074/jbc.M008895200. 11071897 [DOI] [PubMed] [Google Scholar]
- 37.Suh S.K., Hood B.L., Kim B.J., Conrads T.P., Veenstra T.D., Song B.J. Identification of oxidized mitochondrial proteins in alcohol-exposed human hepatoma cells and mouse liver. Proteomics. 2004;4(11):3401–3412. doi: 10.1002/pmic.200400971. 15449375 [DOI] [PubMed] [Google Scholar]
- 38.Moon K.H., Hood B.L., Mukhopadhyay P., Rajesh M., Abdelmegeed M.A., Kwon Y.I., Conrads T.P., Veenstra T.D., Song B.J., Pacher P. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135(4):1344–1357. doi: 10.1053/j.gastro.2008.06.048. 18778711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon K.H., Upreti V.V., Yu L.R., Lee I.J., Ye X., Eddington N.D., Veenstra T.D., Song B.J. Mechanism of 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-mediated mitochondrial dysfunction in rat liver. Proteomics. 2008;8(18):3906–3918. doi: 10.1002/pmic.200800215. 18780394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134(1):112–123. doi: 10.1016/j.cell.2008.06.016. 18614015 1098 proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calvo S.E., Mootha V.K. The mitochondrial proteome and human disease. Annual Review of Genomics and Human Genetics. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. 20690818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song B.J., Abdelmegeed M.A., Yoo S.-H., Kim B.-J., Jo S.A., Jo I., Moon K.-H. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. Journal of Proteomics. 2011;74(12):2691–2702. doi: 10.1016/j.jprot.2011.05.013. 21609791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon K.H., Kim B.J., Song B.J. Inhibition of mitochondrial aldehyde dehydrogenase by nitric oxide-mediated S-nitrosylation. FEBS Letters. 2005;579(27):6115–6120. doi: 10.1016/j.febslet.2005.09.082. 16242127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venkatraman A., Landar A., Davis A.J., Ulasova E., Page G., Murphy M.P., Darley-Usmar V., Bailey S.M. Oxidative modification of hepatic mitochondria protein thiols: effect of chronic alcohol consumption. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2004;286(4):G521–G527. doi: 10.1152/ajpgi.00399.2003. 14670822 [DOI] [PubMed] [Google Scholar]
- 45.Venkatraman A., Landar A., Davis A.J., Chamlee L., Sanderson T., Kim H., Page G., Pompilius M., Ballinger S., Darley-Usmar V., Bailey S.M. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. Journal of Biological Chemistry. 2004;279(21):22092–22101. doi: 10.1074/jbc.M402245200. 15033988 [DOI] [PubMed] [Google Scholar]
- 46.Hartley D.P., Ruth J.A., Petersen D.R. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Archives of Biochemistry and Biophysics. 1995;316(1):197–205. doi: 10.1006/abbi.1995.1028. 7840616 [DOI] [PubMed] [Google Scholar]
- 47.Bae S.H., Sung S.H., Cho E.J., Lee S.K., Lee H.E., Woo H.A., Yu D.Y., Kil I.S., Rhee S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology (Baltimore, Md.) 2011;53(3):945–953. doi: 10.1002/hep.24104. 21319188 [DOI] [PubMed] [Google Scholar]
- 48.Rhee S.G., Woo H.A., Kil I.S., Bae S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. Journal of Biological Chemistry. 2012;287(7):4403–4410. doi: 10.1074/jbc.R111.283432. 22147704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rhee S.G., Woo H.A. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxidants and Redox Signaling. 2011;15(3):781–794. doi: 10.1089/ars.2010.3393. 20919930 [DOI] [PubMed] [Google Scholar]
- 50.Jo S.H., Son M.K., Koh H.J., Lee S.M., Song I.H., Kim Y.O., Lee Y.S., Jeong K.S., Kim W.B., Park J.W., Song B.J., Huh T.L. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. Journal of Biological Chemistry. 2001;276(19):16168–16176. doi: 10.1074/jbc.M010120200. 11278619 [DOI] [PubMed] [Google Scholar]
- 51.Lee S.M., Koh H.J., Park D.C., Song B.J., Huh T.L., Park J.W. Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radical Biology and Medicine. 2002;32(11):1185–1196. doi: 10.1016/s0891-5849(02)00815-8. 12031902 [DOI] [PubMed] [Google Scholar]
- 52.Yang E.S., Richter C., Chun J.S., Huh T.L., Kang S.S., Park J.W. Inactivation of NADP(+)-dependent isocitrate dehydrogenase by nitric oxide. Free Radical Biology and Medicine. 2002;33(7):927–937. doi: 10.1016/s0891-5849(02)00981-4. 12361803 [DOI] [PubMed] [Google Scholar]
- 53.Lee J.H., Yang E.S., Park J.W. Inactivation of NADP+-dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. Journal of Biological Chemistry. 2003;278(51):51360–51371. doi: 10.1074/jbc.M302332200. 14551203 [DOI] [PubMed] [Google Scholar]
- 54.Laval F., Wink D.A. Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA-methyltransferase. Carcinogenesis. 1994;15:443–447. doi: 10.1093/carcin/15.3.443. [DOI] [PubMed] [Google Scholar]
- 55.Marinho H.S., Real C., Cyrne L., Soares H., Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biology. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. 24634836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen J.I., Chen X., Nagy L.E. Redox signaling and the innate immune system in alcoholic liver disease. Antioxidants and Redox Signaling. 2011;15(2):523–534. doi: 10.1089/ars.2010.3746. 21126203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nath B., Szabo G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology (Baltimore, Md.) 2012;55(2):622–633. doi: 10.1002/hep.25497. 22120903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zelickson B.R., Benavides G.A., Johnson M.S., Chacko B.K., Venkatraman A., Landar A., Betancourt A.M., Bailey S.M., Darley-Usmar V.M. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochimica et Biophysica Acta. 2011;1807(12):1573–1582. doi: 10.1016/j.bbabio.2011.09.011. 21971515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X., Wu D., Yang L., Gan L., Cederbaum A.I. Cytochrome P450 2E1 potentiates ethanol induction of hypoxia and HIF-1α in vivo. Free Radical Biology and Medicine. 2013;63:175–186. doi: 10.1016/j.freeradbiomed.2013.05.009. 23669278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou J.Y., Jiang Z.A., Zhao C.Y., Zhen Z., Wang W., Nanji A.A. Long-term binge and escalating ethanol exposure causes necroinflammation and fibrosis in rat liver. Alcoholism, Clinical and Experimental Research. 2013;37(2):213–222. doi: 10.1111/j.1530-0277.2012.01936.x. 23009062 [DOI] [PubMed] [Google Scholar]
- 61.Yun J.W., Son M.J., Abdelmegeed M.A., Banerjee A., Morgan T.R., Yoo S.H., Song B.J. Binge alcohol promotes hypoxic liver injury through CYP2E1-HIF1a-dependent apoptosis pathway in mice and humans. Free Radical Biology and Medicine. 2014;77:183–194. doi: 10.1016/j.freeradbiomed.2014.08.030. (Pubmed: 25236742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gong P., Cederbaum A.I. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology (Baltimore, Md.) 2006;43(1):144–153. doi: 10.1002/hep.21004. 16374848 [DOI] [PubMed] [Google Scholar]
- 63.Wang Z., Dou X., Li S., Zhang X., Sun X., Zhou Z., Song Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology (Baltimore, Md.) 2014;59(4):1381–1392. doi: 10.1002/hep.26912. 24170703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kathirvel E., Chen P., Morgan K., French S.W., Morgan T.R. Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. Journal of Gastroenterology and Hepatology. 2010;25(6):1136–1143. doi: 10.1111/j.1440-1746.2009.06196.x. 20594230 [DOI] [PubMed] [Google Scholar]
- 65.Xu W., Shao L., Zhou C., Wang H., Guo J. Upregulation of Nrf2 expression in non-alcoholic fatty liver and steatohepatitis. Hepato-Gastroenterology. 2011;58(112):2077–2080. doi: 10.5754/hge10501. 22024078 [DOI] [PubMed] [Google Scholar]
- 66.Niture S.K., Khatri R., Jaiswal A.K. Regulation of Nrf2-an update. Free Radical Biology and Medicine. 2014;66:36–44. doi: 10.1016/j.freeradbiomed.2013.02.008. 23434765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grek C.L., Zhang J., Manevich Y., Townsend D.M., Tew K.D. Causes and consequences of cysteine S-glutathionylation. Journal of Biological Chemistry. 2013;288(37):26497–26504. doi: 10.1074/jbc.R113.461368. 23861399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gould N., Doulias P.T., Tenopoulou M., Raju K., Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. Journal of Biological Chemistry. 2013;288(37):26473–26479. doi: 10.1074/jbc.R113.460261. 23861393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun J., Morgan M., Shen R.F., Steenbergen C., Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circulation Research. 2007;101(11):1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. 17916778 [DOI] [PubMed] [Google Scholar]
- 70.Kohr M.J., Sun J., Aponte A., Wang G., Gucek M., Murphy E., Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circulation Research. 2011;108(4):418–426. doi: 10.1161/CIRCRESAHA.110.232173. 21193739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murphy E., Kohr M., Sun J., Nguyen T., Steenbergen C. S-nitrosylation: a radical way to protect the heart. Journal of Molecular and Cellular Cardiology. 2012;52(3):568–577. doi: 10.1016/j.yjmcc.2011.08.021. 21907718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mailloux R.J., Jin X., Willmore W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biology. 2014;2:123–139. doi: 10.1016/j.redox.2013.12.011. 24455476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radical Biology and Medicine. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. 23899494 [DOI] [PubMed] [Google Scholar]
- 74.Pastore A., Piemonte F. S-glutathionylation signaling in cell biology: progress and prospects. European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences. 2012;46(5):279–292. doi: 10.1016/j.ejps.2012.03.010. 22484331 [DOI] [PubMed] [Google Scholar]
- 75.Calcerrada P., Peluffo G., Radi R. Nitric oxide-derived oxidants with a focus on peroxynitrite: molecular targets, cellular responses and therapeutic implications. Current Pharmaceutical Design. 2011;17(35):3905–3932. doi: 10.2174/138161211798357719. 21933142 [DOI] [PubMed] [Google Scholar]
- 76.Brenner C., Galluzzi L., Kepp O., Kroemer G. Decoding cell death signals in liver inflammation. Journal of Hepatology. 2013;59(3):583–594. doi: 10.1016/j.jhep.2013.03.033. 23567086 [DOI] [PubMed] [Google Scholar]
- 77.Radi R., Beckman J.S., Bush K.M., Freeman B.A. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. Journal of Biological Chemistry. 1991;266(7):4244–4250. 1847917 [PubMed] [Google Scholar]
- 78.Wan J., Bae M.A., Song B.J. Acetoaminophen-induced accumulation of 8-oxodeoxyguanosine through reduction of Ogg1 DNA repair enzyme in C6 glioma cells. Experimental and Molecular Medicine. 2004;36(1):71–77. doi: 10.1038/emm.2004.10. 15031674 [DOI] [PubMed] [Google Scholar]
- 79.Begriche K., Igoudjil A., Pessayre D., Fromenty B. Mitochondrial dysfunction in Nash: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1–28. doi: 10.1016/j.mito.2005.10.004. 16406828 [DOI] [PubMed] [Google Scholar]
- 80.Elfering S.L., Haynes V.L., Traaseth N.J., Ettl A., Giulivi C. Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. American Journal of Physiology: Heart and Circulatory Physiology. 2004;286(1):H22–H29. doi: 10.1152/ajpheart.00766.2003. 14527943 [DOI] [PubMed] [Google Scholar]
- 81.Pickering A.M., Davies K.J. Degradation of damaged proteins: the main function of the 20S proteasome. Progress in Molecular Biology and Translational Science. 2012;109:227–248. doi: 10.1016/B978-0-12-397863-9.00006-7. 22727423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallace K.B. Mitochondrial off targets of drug therapy. Trends in Pharmacological Sciences. 2008;29(7):361–366. doi: 10.1016/j.tips.2008.04.001. 18501972 [DOI] [PubMed] [Google Scholar]
- 83.Chandra D., Singh K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochimica et Biophysica Acta. 2011;1807(6):620–625. doi: 10.1016/j.bbabio.2010.10.023. 21074512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fromenty B., Pessayre D. Impaired mitochondrial function in microvesicular steatosis. Effects of drugs, ethanol, hormones and cytokines. Journal of Hepatology. 1997;26(Suppl. 2):43–53. doi: 10.1016/s0168-8278(97)80496-5. 9204409 [DOI] [PubMed] [Google Scholar]
- 85.Song B.J., Moon K.H., Upreti V.V., Eddington N.D., Lee I.J. Mechanisms of MDMA (ecstasy)-induced oxidative stress, mitochondrial dysfunction, and organ damage. Current Pharmaceutical Biotechnology. 2010;11(5):434–443. doi: 10.2174/138920110791591436. 20420575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McKim S.E., Gäbele E., Isayama F., Lambert J.C., Tucker L.M., Wheeler M.D., Connor H.D., Mason R.P., Doll M.A., Hein D.W., Arteel G.E. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125(6):1834–1844. doi: 10.1053/j.gastro.2003.08.030. 14724835 [DOI] [PubMed] [Google Scholar]
- 87.Venkatraman A., Shiva S., Davis A.J., Bailey S.M., Brookes P.S., Darley-Usmar V.M. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology (Baltimore, Md.) 2003;38(1):141–147. doi: 10.1053/jhep.2003.50293. 12829996 [DOI] [PubMed] [Google Scholar]
- 88.Venkatraman A., Shiva S., Wigley A., Ulasova E., Chhieng D., Bailey S.M., Darley-Usmar V.M. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology (Baltimore, Md.) 2004;40(3):565–573. doi: 10.1002/hep.20326. 15349894 [DOI] [PubMed] [Google Scholar]
- 89.Mantena S.K., Vaughn D.P., Andringa K.K., Eccleston H.B., King A.L., Abrams G.A., Doeller J.E., Kraus D.W., Darley-Usmar V.M., Bailey S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochemical Journal. 2009;417(1):183–193. doi: 10.1042/BJ20080868. 18752470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spruss A., Kanuri G., Uebel K., Bischoff S.C., Bergheim I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxidants and Redox Signaling. 2011;14(11):2121–2135. doi: 10.1089/ars.2010.3263. 21083420 [DOI] [PubMed] [Google Scholar]
- 91.Hinson J.A., Pike S.L., Pumford N.R., Mayeux P.R. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chemical Research in Toxicology. 1998;11(6):604–607. doi: 10.1021/tx9800349. 9625727 [DOI] [PubMed] [Google Scholar]
- 92.Knight T.R., Ho Y.S., Farhood A., Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. Journal of Pharmacology and Experimental Therapeutics. 2002;303(2):468–475. doi: 10.1124/jpet.102.038968. 12388625 [DOI] [PubMed] [Google Scholar]
- 93.Abdelmegeed M.A., Moon K.H., Chen C., Gonzalez F.J., Song B.J. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochemical Pharmacology. 2010;79(1):57–66. doi: 10.1016/j.bcp.2009.07.016. 19660437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abdelmegeed M.A., Jang S., Banerjee A., Hardwick J.P., Song B.J. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free Radical Biology and Medicine. 2013;60:211–222. doi: 10.1016/j.freeradbiomed.2013.02.018. 23454065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yun J.W., Lum K., Lei X.G. A novel upregulation of glutathione peroxidase 1 by knockout of liver-regenerating protein Reg3β aggravates acetaminophen-induced hepatic protein nitration. Free Radical Biology and Medicine. 2013;65:291–300. doi: 10.1016/j.freeradbiomed.2013.06.034. 23811004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yoo S.H., Park O., Henderson L.E., Abdelmegeed M.A., Moon K.H., Song B.J. Lack of PPARα exacerbates lipopolysaccharide-induced liver toxicity through STAT1 inflammatory signaling and increased oxidative/nitrosative stress. Toxicology Letters. 2011;202(1):23–29. doi: 10.1016/j.toxlet.2011.01.013. 21262334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nuriel T., Hansler A., Gross S.S. Protein nitrotryptophan: formation, significance and identification. Journal of Proteomics. 2011;74(11):2300–2312. doi: 10.1016/j.jprot.2011.05.032. 21679780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bregere C., Rebrin I., Sohal R.S. Detection and characterization of in vivo nitration and oxidation of tryptophan residues in proteins. Methods in Enzymology. 2008;441:339–349. doi: 10.1016/S0076-6879(08)01219-6. 18554544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yamakura F., Kawasaki H. Post-translational modifications of superoxide dismutase. Biochimica et Biophysica Acta. 2010;1804(2):318–325. doi: 10.1016/j.bbapap.2009.10.010. 19837190 [DOI] [PubMed] [Google Scholar]
- 100.Ron D., Messing R.O. Signaling pathways mediating alcohol effects. Current Topics in Behavioral Neuroscience. 2013;13:87–126. doi: 10.1007/7854_2011_161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sid B., Verrax J., Calderon P.B. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochemical Pharmacology. 2013;86(2):200–209. doi: 10.1016/j.bcp.2013.05.007. 23688501 [DOI] [PubMed] [Google Scholar]
- 102.Son Y., Kim S., Chung H.T., Pae H.O. Reactive oxygen species in the activation of MAP kinases. Methods in Enzymology. 2013;528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1. 23849857 [DOI] [PubMed] [Google Scholar]
- 103.Theodosiou A., Ashworth A. MAP kinase phosphatases. Genome Biology. 2002;3(7) doi: 10.1186/gb-2002-3-7-reviews3009. REVIEWS3009.1-REVIEWS3009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–661. doi: 10.1016/j.cell.2004.12.041. 15766528 [DOI] [PubMed] [Google Scholar]
- 105.Heneberg P., Dráber P. Regulation of cys-based protein tyrosine phosphatases via reactive oxygen and nitrogen species in mast cells and basophils. Current Medicinal Chemistry. 2005;12(16):1859–1871. doi: 10.2174/0929867054546636. 16101506 [DOI] [PubMed] [Google Scholar]
- 106.Geraldes P., Hiraoka-Yamamoto J., Matsumoto M., Clermont A., Leitges M., Marette A., Aiello L.P., Kern T.S., King G.L. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nature Medicine. 2009;15(11):1298–1306. doi: 10.1038/nm.2052. 19881493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ryer E.J., Sakakibara K., Wang C., Sarkar D., Fisher P.B., Faries P.L., Kent K.C., Liu B. Protein kinase C delta induces apoptosis of vascular smooth muscle cells through induction of the tumor suppressor p53 by both p38-dependent and p38-independent mechanisms. Journal of Biological Chemistry. 2005;280(42):35310–35317. doi: 10.1074/jbc.M507187200. 16118209 [DOI] [PubMed] [Google Scholar]
- 108.Gonzalez-Guerrico A.M., Kazanietz M.G. Phorbol ester-induced apoptosis in prostate cancer cells via autocrine activation of the extrinsic apoptotic cascade: a key role for protein kinase C delta. Journal of Biological Chemistry. 2005;280(47):38982–38991. doi: 10.1074/jbc.M506767200. 16183650 [DOI] [PubMed] [Google Scholar]
- 109.Kwon J., Lee S.R., Yang K.S., Ahn Y., Kim Y.J., Stadtman E.R., Rhee S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(47):16419–16424. doi: 10.1073/pnas.0407396101. 15534200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee Y.J., Shukla S.D. Pro- and anti-apoptotic roles of c-Jun N-terminal kinase (JNK) in ethanol and acetaldehyde exposed rat hepatocytes. European Journal of Pharmacology. 2005;508(1–3):31–45. doi: 10.1016/j.ejphar.2004.12.006. 15680252 [DOI] [PubMed] [Google Scholar]
- 111.Yang L., Wu D., Wang X., Cederbaum A.I. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radical Biology and Medicine. 2012;53(5):1170–1180. doi: 10.1016/j.freeradbiomed.2012.06.029. 22749809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bae M.A., Pie J.E., Song B.J. Acetaminophen induces apoptosis of C6 glioma cells by activating the c-Jun NH(2)-terminal protein kinase-related cell death pathway. Molecular Pharmacology. 2001;60(4):847–856. 11562448 [PubMed] [Google Scholar]
- 113.Gunawan B.K., Liu Z.X., Han D., Hanawa N., Gaarde W.A., Kaplowitz N. C-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131(1):165–178. doi: 10.1053/j.gastro.2006.03.045. 16831600 [DOI] [PubMed] [Google Scholar]
- 114.Henderson N.C., Pollock K.J., Frew J., Mackinnon A.C., Flavell R.A., Davis R.J., Sethi T., Simpson K.J. Critical role of c-jun (NH2) terminal kinase in paracetamol-induced acute liver failure. Gut. 2007;56(7):982–990. doi: 10.1136/gut.2006.104372. 17185352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hanawa N., Shinohara M., Saberi B., Gaarde W.A., Han D., Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. Journal of Biological Chemistry. 2008;283(20):13565–13577. doi: 10.1074/jbc.M708916200. 18337250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Win S., Than T.A., Han D., Petrovic L.M., Kaplowitz N. C-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial sab protein expression in mice. Journal of Biological Chemistry. 2011;286(40):35071–35078. doi: 10.1074/jbc.M111.276089. 21844199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu D., Cederbaum A.I. Role of p38 MAPK in CYP2E1-dependent arachidonic acid toxicity. Journal of Biological Chemistry. 2003;278(2):1115–1124. doi: 10.1074/jbc.M207856200. 12403788 [DOI] [PubMed] [Google Scholar]
- 118.Soh Y., Jeong K.S., Lee I.J., Bae M.A., Kim Y.C., Song B.J. Selective activation of the c-Jun N-terminal protein kinase pathway during 4-hydroxynonenal-induced apoptosis of PC12 cells. Molecular Pharmacology. 2000;58(3):535–541. doi: 10.1124/mol.58.3.535. 10953046 [DOI] [PubMed] [Google Scholar]
- 119.Solinas G., Naugler W., Galimi F., Lee M.S., Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16454–16459. doi: 10.1073/pnas.0607626103. 17050683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bae M.A., Song B.J. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Molecular Pharmacology. 2003;63(2):401–408. doi: 10.1124/mol.63.2.401. 12527812 [DOI] [PubMed] [Google Scholar]
- 121.Schattenberg J.M., Wang Y., Singh R., Rigoli R.M., Czaja M.J. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. Journal of Biological Chemistry. 2005;280(11):9887–9894. doi: 10.1074/jbc.M410310200. 15632182 [DOI] [PubMed] [Google Scholar]
- 122.Schattenberg J.M., Singh R., Wang Y., Lefkowitch J.H., Rigoli R.M., Scherer P.E., Czaja M.J. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology (Baltimore, Md.) 2006;43(1):163–172. doi: 10.1002/hep.20999. 16374858 [DOI] [PubMed] [Google Scholar]
- 123.Tuncman G., Hirosumi J., Solinas G., Chang L., Karin M., Hotamisligil G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(28):10741–10746. doi: 10.1073/pnas.0603509103. 16818881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gao Z., Zhang J., Kheterpal I., Kennedy N., Davis R.J., Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. Journal of Biological Chemistry. 2011;286(25):22227–22234. doi: 10.1074/jbc.M111.228874. 21540183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Uehara T., Bennett B., Sakata S.T., Satoh Y., Bilter G.K., Westwick J.K., Brenner D.A. JNK mediates hepatic ischemia reperfusion injury. Journal of Hepatology. 2005;42(6):850–859. doi: 10.1016/j.jhep.2005.01.030. 15885356 [DOI] [PubMed] [Google Scholar]
- 126.Seki E., Brenner D.A., Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143(2):307–320. doi: 10.1053/j.gastro.2012.06.004. 22705006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang X., Wu D., Yang L., Cederbaum A.I. Hepatotoxicity mediated by pyrazole (cytochrome P450 2E1) plus tumor necrosis factor alpha treatment occurs in c-Jun N-terminal kinase 2-/- but not in c-Jun N-terminal kinase 1−/− mice. Hepatology (Baltimore, Md.) 2011;54(5):1753–1766. doi: 10.1002/hep.24540. 21748763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Son Y., Cheong Y.K., Kim N.H., Chung H.T., Kang D.G., Pae H.O. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? Journal of Signal Transduction. 2011;2011:792639. doi: 10.1155/2011/792639. 21637379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim B.J., Ryu S.W., Song B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. Journal of Biological Chemistry. 2006;281(30):21256–21265. doi: 10.1074/jbc.M510644200. 16709574 [DOI] [PubMed] [Google Scholar]
- 130.Kharbanda S., Saxena S., Yoshida K., Pandey P., Kaneki M., Wang Q., Cheng K., Chen Y.N., Campbell A., Sudha T. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. Journal of Biological Chemistry. 2000;275(1):322–327. doi: 10.1074/jbc.275.1.322. 10617621 [DOI] [PubMed] [Google Scholar]
- 131.Xie Y., McGill M.R., Dorko K., Kumer S.C., Schmitt T.M., Forster J., Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicology and Applied Pharmacology. 2014;279(3):266–274. doi: 10.1016/j.taap.2014.05.010. 24905542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ferramosca A., Zara V. Biogenesis of mitochondrial carrier proteins: molecular mechanisms of import into mitochondria. Biochimica et Biophysica Acta. 2013;1833(3):494–502. doi: 10.1016/j.bbamcr.2012.11.014. 23201437 [DOI] [PubMed] [Google Scholar]
- 133.Moon K.H., Lee Y.M., Song B.J. Inhibition of hepatic mitochondrial aldehyde dehydrogenase by carbon tetrachloride through JNK-mediated phosphorylation. Free Radical Biology and Medicine. 2010;48(3):391–398. doi: 10.1016/j.freeradbiomed.2009.11.008. 19922789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Win S., Than T.A., Fernandez-Checa J.C., Kaplowitz N. JNK interaction with sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death and Disease. 2014;5:e989. doi: 10.1038/cddis.2013.522. 24407242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schroeter H., Boyd C.S., Ahmed R., Spencer J.P.E., Duncan R.F., Rice-Evans C., Cadenas E. C-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochemical Journal. 2003;372(2):359–369. doi: 10.1042/BJ20030201. 12614194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hardie D.G. AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annual Review of Nutrition. 2014;34:31–55. doi: 10.1146/annurev-nutr-071812-161148. 24850385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.You M., Matsumoto M., Pacold C.M., Cho W.K., Crabb D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127(6):1798–1808. doi: 10.1053/j.gastro.2004.09.049. 15578517 [DOI] [PubMed] [Google Scholar]
- 138.Singhal S.S., Figarola J., Singhal J., Reddy M.A., Liu X., Berz D., Natarajan R., Awasthi S. RLIP76 protein knockdown attenuates obesity due to a high-fat diet. Journal of Biological Chemistry. 2013;288(32):23394–23406. doi: 10.1074/jbc.M113.480194. 23821548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Saberi B., Ybanez M.D., Johnson H.S., Gaarde W.A., Han D., Kaplowitz N. Protein kinase C (PKC) participates in acetaminophen hepatotoxicity through c-jun-N-terminal kinase (JNK)-dependent and -independent signaling pathways. Hepatology (Baltimore, Md.) 2014;59(4):1543–1554. doi: 10.1002/hep.26625. 23873604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun Z., Feng D., Everett L.J., Bugge A., Lazar M.A. Circadian epigenomic remodeling and hepatic lipogenesis: lessons from HDAC3. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:49–55. doi: 10.1101/sqb.2011.76.011494. 21900149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kim S.C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N.V., White M., Yang X.J., Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Molecular Cell. 2006;23(4):607–618. doi: 10.1016/j.molcel.2006.06.026. 16916647 [DOI] [PubMed] [Google Scholar]
- 142.You M., Liang X., Ajmo J.M., Ness G.C. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2008;294(4):G892–G898. doi: 10.1152/ajpgi.00575.2007. 18239056 [DOI] [PubMed] [Google Scholar]
- 143.Lieber C.S., Leo M.A., Wang X., Decarli L.M. Effect of chronic alcohol consumption on hepatic SIRT1 and PGC-1alpha in rats. Biochemical and Biophysical Research Communications. 2008;370(1):44–48. doi: 10.1016/j.bbrc.2008.03.005. 18342626 [DOI] [PubMed] [Google Scholar]
- 144.Picklo M.J. Sr. Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochemical and Biophysical Research Communications. 2008;376(3):615–619. doi: 10.1016/j.bbrc.2008.09.039. 18804449 [DOI] [PubMed] [Google Scholar]
- 145.Shepard B.D., Tuma D.J., Tuma P.L. Chronic ethanol consumption induces global hepatic protein hyperacetylation. Alcoholism, Clinical and Experimental Research. 2010;34(2):280–291. doi: 10.1111/j.1530-0277.2009.01091.x. 19951295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kendrick A.A., Choudhury M., Rahman S.M., McCurdy C.E., Friederich M., Van Hove J.L., Watson P.A., Birdsey N., Bao J., Gius D., Sack M.N., Jing E., Kahn C.R., Friedman J.E., Jonscher K.R. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochemical Journal. 2011;433(3):505–514. doi: 10.1042/BJ20100791. 21044047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hirschey M.D., Shimazu T., Jing E., Grueter C.A., Collins A.M., Aouizerat B., Stančáková A., Goetzman E., Lam M.M., Schwer B., Stevens R.D., Muehlbauer M.J., Kakar S., Bass N.M., Kuusisto J., Laakso M., Alt F.W., Newgard C.B., Farese R.V., Jr, Kahn C.R., Verdin E. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular Cell. 2011;44(2):177–190. doi: 10.1016/j.molcel.2011.07.019. 21856199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Valdecantos M.P., Pérez-Matute P., González-Muniesa P., Prieto-Hontoria P.L., Moreno-Aliaga M.J., Martínez J.A. Lipoic acid improves mitochondrial function in nonalcoholic steatosis through the stimulation of sirtuin 1 and sirtuin 3. Obesity (Silver Spring, Md.) 2012;20(10):1974–1983. doi: 10.1038/oby.2012.32. 22327056 [DOI] [PubMed] [Google Scholar]
- 149.Lieber C.S., Leo M.A., Wang X., Decarli L.M. Alcohol alters hepatic FoxO1, p53, and mitochondrial SIRT5 deacetylation function. Biochemical and Biophysical Research Communications. 2008;373(2):246–252. doi: 10.1016/j.bbrc.2008.06.006. 18555008 [DOI] [PubMed] [Google Scholar]
- 150.Shulga N., Pastorino J.G. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. Journal of Cell Science. 2010;123(23):4117–4127. doi: 10.1242/jcs.073502. 21062897 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 151.Fritz K.S., Galligan J.J., Smathers R.L., Roede J.R., Shearn C.T., Reigan P., Petersen D.R. 4-hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chemical Research in Toxicology. 2011;24(5):651–662. doi: 10.1021/tx100355a. 21449565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fritz K.S., Galligan J.J., Hirschey M.D., Verdin E., Petersen D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. Journal of Proteome Research. 2012;11(3):1633–1643. doi: 10.1021/pr2008384. 22309199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fritz K.S., Green M.F., Petersen D.R., Hirschey M.D. Ethanol metabolism modifies hepatic protein acylation in mice. PloS One. 2013;8(9):e75868. doi: 10.1371/journal.pone.0075868. 24073283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hirschey M.D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D.B., Grueter C.A., Harris C., Biddinger S., Ilkayeva O.R., Stevens R.D., Li Y., Saha A.K., Ruderman N.B., Bain J.R., Newgard C.B., Farese R.V., Jr, Alt F.W., Kahn C.R., Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–125. doi: 10.1038/nature08778. 20203611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hirschey M.D., Shimazu T., Huang J.Y., Schwer B., Verdin E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:267–277. doi: 10.1101/sqb.2011.76.010850. 22114326 [DOI] [PubMed] [Google Scholar]
- 156.Yang S.J., Lim Y. Resveratrol ameliorates hepatic metaflammation and inhibits NLRP3 inflammasome activation. Metabolism: Clinical and Experimental. 2014;63(5):693–701. doi: 10.1016/j.metabol.2014.02.003. 24629563 [DOI] [PubMed] [Google Scholar]
- 157.Wang S., Moustaid-Moussa N., Chen L., Mo H., Shastri A., Su R., Bapat P., Kwun I., Shen C.L. Novel insights of dietary polyphenols and obesity. Journal of Nutritional Biochemistry. 2014;25(1):1–18. doi: 10.1016/j.jnutbio.2013.09.001. 24314860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Still A.J., Floyd B.J., Hebert A.S., Bingman C.A., Carson J.J., Gunderson D.R., Dolan B.K., Grimsrud P.A., Dittenhafer-Reed K.E., Stapleton D.S., Keller M.P., Westphall M.S., Denu J.M., Attie A.D., Coon J.J., Pagliarini D.J. Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. Journal of Biological Chemistry. 2013;288(36):26209–26219. doi: 10.1074/jbc.M113.483396. 23864654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Paik W.K., Pearson D., Lee H.W., Kim S. Nonenzymatic acetylation of histones with acetyl-CoA. Biochimica et Biophysica Acta. 1970;213(2):513–522. doi: 10.1016/0005-2787(70)90058-4. 5534125 [DOI] [PubMed] [Google Scholar]
- 160.Ramponi G., Manao G., Camici G. Nonenzymatic acetylation of histones with acetyl phosphate and acetyl adenylate. Biochemistry. 1975;14(12):2681–2685. doi: 10.1021/bi00683a018. 238571 [DOI] [PubMed] [Google Scholar]
- 161.Tanner K.G., Trievel R.C., Kuo M.H., Howard R.M., Berger S.L., Allis C.D., Marmorstein R., Denu J.M. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. Journal of Biological Chemistry. 1999;274(26):18157–18160. doi: 10.1074/jbc.274.26.18157. 10373413 [DOI] [PubMed] [Google Scholar]
- 162.French S.W., Wong K., Jui L., Albano E., Hagbjork A.L., Ingelman-Sundberg M. Effect of ethanol on cytochrome P450 2E1 (CYP2E1), lipid peroxidation, and serum protein adduct formation in relation to liver pathology pathogenesis. Experimental and Molecular Pathology. 1993;58(1):61–75. doi: 10.1006/exmp.1993.1006. 8454037 [DOI] [PubMed] [Google Scholar]
- 163.Niemelä O., Parkkila S., Pasanen M., Iimuro Y., Bradford B., Thurman R.G. Early alcoholic liver injury: formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of CYP2E1 and CYP3A. Alcoholism, Clinical and Experimental Research. 1998;22:2118–2124. doi: 10.1111/j.1530-0277.1998.tb05925.x. 9884160 [DOI] [PubMed] [Google Scholar]
- 164.Brooks P.J., Enoch M.A., Goldman D., Li T.K., Yokoyama A. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Medicine. 2009;6(3):e50. doi: 10.1371/journal.pmed.1000050. 19320537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yokoyama A., Muramatsu T., Omori T., Yokoyama T., Matsushita S., Higuchi S., Maruyama K., Ishii H. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis. 2001;22(3):433–439. doi: 10.1093/carcin/22.3.433. 11238183 [DOI] [PubMed] [Google Scholar]
- 166.Jaeschke H., Gores G.J., Cederbaum A.I., Hinson J.A., Pessayre D., Lemasters J.J. Mechanisms of hepatotoxicity. Toxicological Sciences: an Official Journal of the Society of Toxicology. 2002;65(2):166–176. doi: 10.1093/toxsci/65.2.166. 11812920 [DOI] [PubMed] [Google Scholar]
- 167.Galligan J.J., Smathers R.L., Fritz K.S., Epperson L.E., Hunter L.E., Petersen D.R. Protein carbonylation in a murine model for early alcoholic liver disease. Chemical Research in Toxicology. 2012;25(5):1012–1021. doi: 10.1021/tx300002q. 22502949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fritz K.S., Petersen D.R. An overview of the chemistry and biology of reactive aldehydes. Free Radical Biology and Medicine. 2013;59:85–91. doi: 10.1016/j.freeradbiomed.2012.06.025. 22750507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cohn J.A., Tsai L., Friguet B., Szweda L.I. Chemical characterization of a protein-4-hydroxy-2-nonenal cross-link: immunochemical detection in mitochondria exposed to oxidative stress. Archives of Biochemistry and Biophysics. 1996;328(1):158–164. doi: 10.1006/abbi.1996.0156. 8638925 [DOI] [PubMed] [Google Scholar]
- 170.Niemelä O., Parkkila S., Ylä-Herttuala S., Villanueva J., Ruebner B., Halsted C.H. Sequential acetaldehyde production, lipid peroxidation, and fibrogenesis in micropig model of alcohol-induced liver disease. Hepatology (Baltimore, Md.) 1995;22(4 Pt 1):1208–1214. 7557872 [PubMed] [Google Scholar]
- 171.Li C.J., Nanji A.A., Siakotos A.N., Lin R.C. Acetaldehyde-modified and 4-hydroxynonenal-modified proteins in the livers of rats with alcoholic liver disease. Hepatology (Baltimore, Md.) 1997;26(3):650–657. doi: 10.1002/hep.510260317. 9303495 [DOI] [PubMed] [Google Scholar]
- 172.Chen J., Robinson N.C., Schenker S., Frosto T.A., Henderson G.I. Formation of 4-hydroxynonenal adducts with cytochrome c oxidase in rats following short-term ethanol intake. Hepatology (Baltimore, Md.) 1999;29(6):1792–1798. doi: 10.1002/hep.510290611. 10347122 [DOI] [PubMed] [Google Scholar]
- 173.Doorn J.A., Hurley T.D., Petersen D.R. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chemical Research in Toxicology. 2006;19(1):102–110. doi: 10.1021/tx0501839. 16411662 [DOI] [PubMed] [Google Scholar]
- 174.Carbone D.L., Doorn J.A., Kiebler Z., Petersen D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chemical Research in Toxicology. 2005;18(8):1324–1331. doi: 10.1021/tx050078z. 16097806 [DOI] [PubMed] [Google Scholar]
- 175.Galligan J.J., Fritz K.S., Backos D.S., Shearn C.T., Smathers R.L., Jiang H., MacLean K.N., Reigan P.R., Petersen D.R. Oxidative stress-mediated aldehyde adduction of GRP78 in a mouse model of alcoholic liver disease: functional independence of ATPase activity and chaperone function. Free Radical Biology and Medicine. 2014;73:411–420. doi: 10.1016/j.freeradbiomed.2014.06.002. 24924946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Backos D.S., Fritz K.S., Roede J.R., Petersen D.R., Franklin C.C. Posttranslational modification and regulation of glutamate-cysteine ligase by the α,β-unsaturated aldehyde 4-hydroxy-2-nonenal. Free Radical Biology and Medicine. 2011;50(1):14–26. doi: 10.1016/j.freeradbiomed.2010.10.694. 20970495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sampey B.P., Stewart B.J., Petersen D.R. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. Journal of Biological Chemistry. 2007;282(3):1925–1937. doi: 10.1074/jbc.M610602200. 17107949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shearn C.T., Smathers R.L., Backos D.S., Reigan P., Orlicky D.J., Petersen D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radical Biology and Medicine. 2013;65:680–692. doi: 10.1016/j.freeradbiomed.2013.07.011. 23872024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shearn C.T., Backos D.S., Orlicky D.J., Smathers-McCullough R.L., Petersen D.R. Identification of 5′ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. Journal of Biological Chemistry. 2014;289(22):15449–15462. doi: 10.1074/jbc.M113.543942. 24722988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mottaran E., Stewart S.F., Rolla R., Vay D., Cipriani V., Moretti M., Vidali M., Sartori M., Rigamonti C., Day C.P., Albano E. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radical Biology and Medicine. 2002;32(1):38–45. doi: 10.1016/s0891-5849(01)00757-2. 11755315 [DOI] [PubMed] [Google Scholar]
- 181.Jeong K.S., Soh Y., Jeng J., Felder M.R., Hardwick J.P., Song B.J. Cytochrome P450 2E1 (CYP2E1)-dependent production of a 37-kDa acetaldehyde-protein adduct in the rat liver. Archives of Biochemistry and Biophysics. 2000;384(1):81–87. doi: 10.1006/abbi.2000.2119. 11147839 [DOI] [PubMed] [Google Scholar]
- 182.Bardag-Gorce F., Li J., French B.A., French S.W. The effect of ethanol-induced CYP2E1 on proteasome activity: the role of 4-hydroxynonenal. Experimental and Molecular Pathology. 2005;78(2):109–115. doi: 10.1016/j.yexmp.2004.10.005. 15713435 [DOI] [PubMed] [Google Scholar]
- 183.Chen J., Henderson G.I., Freeman G.L. Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. Journal of Molecular and Cellular Cardiology. 2001;33(11):1919–1927. doi: 10.1006/jmcc.2001.1454. 11708837 [DOI] [PubMed] [Google Scholar]
- 184.Chavez J.D., Wu J., Bisson W., Maier C.S. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. Journal of Proteomics. 2011;74(11):2417–2429. doi: 10.1016/j.jprot.2011.03.031. 21513823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Abdelmegeed M.A., Banerjee A., Yoo S.H., Jang S.H., Gonzalez F.J., Song B.J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. Journal of Hepatology. 2012;57(4):860–866. doi: 10.1016/j.jhep.2012.05.019. 22668639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mali V.R., Ning R., Chen J., Yang X.P., Xu J., Palaniyandi S.S. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Experimental Biology and Medicine (Maywood, N.J.) 2014;239(5):610–618. doi: 10.1177/1535370213520109. 24651616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chen C.-H., Budas G.R., Churchill E.N., Disatnik M.-H., Hurley T.D., Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science (New York, N.Y.) 2008;321(5895):1493–1495. doi: 10.1126/science.1158554. 18787169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Doser T.A., Turdi S., Thomas D.P., Epstein P.N., Li S.Y., Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119(14):1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. 19332462 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 189.Zhang Y., Mi S.L., Hu N., Doser T.A., Sun A., Ge J., Ren J. Mitochondrial aldehyde dehydrogenase 2 accentuates aging-induced cardiac remodeling and contractile dysfunction: role of AMPK, Sirt1, and mitochondrial function. Free Radical Biology and Medicine. 2014;71:208–220. doi: 10.1016/j.freeradbiomed.2014.03.018. 24675227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Daffu G., del Pozo C.H., O’Shea K.M., Ananthakrishnan R., Ramasamy R., Schmidt A.M. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. International Journal of Molecular Sciences. 2013;14(10):19891–19910. doi: 10.3390/ijms141019891. 24084731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ott C., Jacobs K., Haucke E., Navarrete Santos A., Grune T., Simm A. Role of advanced glycation end products in cellular signaling. Redox Biology. 2014;2:411–429. doi: 10.1016/j.redox.2013.12.016. 24624331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Niforou K., Cheimonidou C., Trougakos I.P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biology. 2014;2:323–332. doi: 10.1016/j.redox.2014.01.017. 24563850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hayashi N., George J., Takeuchi M., Fukumura A., Toshikuni N., Arisawa T., Tsutsumi M. Acetaldehyde-derived advanced glycation end-products promote alcoholic liver disease. PloS One. 2013;8(7):e70034. doi: 10.1371/journal.pone.0070034. 23922897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ahmed N., Lüthen R., Häussinger D., Sebeková K., Schinzel R., Voelker W., Heidland A., Thornalley P.J. Increased protein glycation in cirrhosis and therapeutic strategies to prevent it. Annals of the New York Academy of Sciences. 2005;1043:718–724. doi: 10.1196/annals.1333.083. 16037298 [DOI] [PubMed] [Google Scholar]
- 195.Baba S.P., Hellmann J., Srivastava S., Bhatnagar A. Aldose reductase (AKR1B3) regulates the accumulation of advanced glycosylation end products (AGEs) and the expression of AGE receptor (RAGE) Chemico-Biological Interactions. 2011;191(1–3):357–363. doi: 10.1016/j.cbi.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jiang J.X., Chen X., Fukada H., Serizawa N., Devaraj S., Török N.J. Advanced glycation endproducts induce fibrogenic activity in nonalcoholic steatohepatitis by modulating TNF-α-converting enzyme activity in mice. Hepatology (Baltimore, Md.) 2013;58(4):1339–1348. doi: 10.1002/hep.26491. 23703665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bakala H., Ladouce R., Baraibar M.A., Friguet B. Differential expression and glycative damage affect specific mitochondrial proteins with aging in rat liver. Biochimica et Biophysica Acta. 2013;1832(12):2057–2067. doi: 10.1016/j.bbadis.2013.07.015. 23906978 [DOI] [PubMed] [Google Scholar]
- 198.Takeuchi M., Saito T. Cytotoxicity of acetaldehyde-derived advanced glycation end-products (AA-AGE) in alcoholic-induced neuronal degeneration. Alcoholism, Clinical and Experimental Research. 2005;29(12 Suppl):220S–224S. doi: 10.1097/01.alc.0000190657.97988.c7. 16385226 [DOI] [PubMed] [Google Scholar]
- 199.Byun K., Bayarsaikhan D., Bayarsaikhan E., Son M., Oh S., Lee J., Son H.I., Won M.H., Kim S.U., Song B.J., Lee B. Microglial AGE-albumin is critical in promoting alcohol-induced neurodegeneration in Rats and humans. PloS One. 2014;9(8):e104699. doi: 10.1371/journal.pone.0104699. 25140518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Vetreno R.P., Qin L., Crews F.T. Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiology of Disease. 2013;59:52–62. doi: 10.1016/j.nbd.2013.07.002. 23867237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ahn S.M., Byun K., Cho K., Kim J.Y., Yoo J.S., Kim D., Paek S.H., Kim S.U., Simpson R.J., Lee B. Human microglial cells synthesize albumin in brain. PloS One. 2008;3(7):e2829. doi: 10.1371/journal.pone.0002829. 18665237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Byun K., Bayarsaikhan E., Kim D., Kim C.Y., Mook-Jung I., Paek S.H., Kim S.U., Yamamoto T., Won M.H., Song B.J., Park Y.M., Lee B. Induction of neuronal death by microglial AGE-albumin: implications for Alzheimer's disease. PloS One. 2012;7(5):e37917. doi: 10.1371/journal.pone.0037917. 22662249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Albano E., Clot P., Morimoto M., Tomasi A., Ingelman-Sundberg M., French S.W. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology (Baltimore, Md.) 1996;23(1):155–163. doi: 10.1002/hep.510230121. 8550035 [DOI] [PubMed] [Google Scholar]
- 204.Clot P., Albano E., Eliasson E., Tabone M., Aricò S., Israel Y., Moncada C., Ingelman-Sundberg M. Cytochrome P4502E1 hydroxyethyl radical adducts as the major antigen in autoantibody formation among alcoholics. Gastroenterology. 1996;111(1):206–216. doi: 10.1053/gast.1996.v111.pm8698201. 8698201 [DOI] [PubMed] [Google Scholar]
- 205.Cheshchevik V.T., Lapshina E.A., Dremza I.K., Zabrodskaya S.V., Reiter R.J., Prokopchik N.I., Zavodnik I.B. Rat liver mitochondrial damage under acute or chronic carbon tetrachloride-induced intoxication: protection by melatonin and cranberry flavonoids. Toxicology and Applied Pharmacology. 2012;261(3):271–279. doi: 10.1016/j.taap.2012.04.007. 22521486 [DOI] [PubMed] [Google Scholar]
- 206.Kim H.S., Quon M.J., Kim J.A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biology. 2014;2:187–195. doi: 10.1016/j.redox.2013.12.022. 24494192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zou X., Yan C., Shi Y., Cao K., Xu J., Wang X., Chen C., Luo C., Li Y., Gao J., Pang W., Zhao J., Zhao F., Li H., Zheng A., Sun W., Long J., Szeto I.M., Zhao Y., Dong Z., Zhang P., Wang J., Lu W., Zhang Y., Liu J., Feng Z. Mitochondrial dysfunction in obesity-associated nonalcoholic fatty liver disease: the protective effects of pomegranate with its active component punicalagin. Antioxidants and Redox Signaling. 2014 doi: 10.1089/ars.2013.5538. 24393106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pallauf K., Giller K., Huebbe P., Rimbach G. Nutrition and healthy ageing: calorie restriction or polyphenol-rich “MediterrAsian” diet? Oxidative Medicine and Cellular Longevity. 2013;2013:707421. doi: 10.1155/2013/707421. 24069505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Davinelli S., Sapere N., Zella D., Bracale R., Intrieri M., Scapagnini G. Pleiotropic protective effects of phytochemicals in Alzheimer's disease. Oxidative Medicine and Cellular Longevity. 2012;2012:386527. doi: 10.1155/2012/386527. 22690271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Basli A., Soulet S., Chaher N., Mérillon J.M., Chibane M., Monti J.P., Richard T. Wine polyphenols: potential agents in neuroprotection, oxid. Medicine. 2012;2012:805762. doi: 10.1155/2012/805762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rodrigues C.F., Ascenção K., Silva F.A., Sarmento B., Oliveira M.B., Andrade J.C. Drug-delivery systems of green tea catechins for improved stability and bioavailability. Current Medicinal Chemistry. 2013;20(37):4744–4757. doi: 10.2174/09298673113209990158. 23834175 [DOI] [PubMed] [Google Scholar]
- 212.Bailey S.M., Robinson G., Pinner A., Chamlee L., Ulasova E., Pompilius M., Page G.P., Chhieng D., Jhala N., Landar A., Kharbanda K.K., Ballinger S., Darley-Usmar V. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2006;291(5):G857–G867. doi: 10.1152/ajpgi.00044.2006. 16825707 [DOI] [PubMed] [Google Scholar]
- 213.Andringa K.K., King A.L., Eccleston H.B., Mantena S.K., Landar A., Jhala N.C., Dickinson D.A., Squadrito G.L., Bailey S.M. Analysis of the liver mitochondrial proteome in response to ethanol and S-adenosylmethionine treatments: novel molecular targets of disease and hepatoprotection. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2010;298(5):G732–G745. doi: 10.1152/ajpgi.00332.2009. 20150243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kharbanda K.K., Todero S.L., King A.L., Osna N.A., McVicker B.L., Tuma D.J., Wisecarver J.L., Bailey S.M. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. International Journal of Hepatology. 2012;2012:962183. doi: 10.1155/2012/962183. 22187660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Santamaria E., Avila M.A., Latasa M.U., Rubio A., Martin-Duce A., Lu S.C., Mato J.M., Corrales F.J. Functional proteomics of nonalcoholic steatohepatitis: mitochondrial proteins as targets of S-adenosylmethionine. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(6):3065–3070. doi: 10.1073/pnas.0536625100. 12631701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Caballero F., Fernández A., Matías N., Martínez L., Fucho R., Elena M., Caballeria J., Morales A., Fernández-Checa J.C., García-Ruiz C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-l-methionine and glutathione. Journal of Biological Chemistry. 2010;285(24):18528–18536. doi: 10.1074/jbc.M109.099333. 20395294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Basaranoglu M., Basaranoglu G., Sentürk H. From fatty liver to fibrosis: a tale of “second hit”. World Journal of Gastroenterology: WJG. 2013;19(8):1158–1165. doi: 10.3748/wjg.v19.i8.1158. 23483818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhao K., Zhao G.M., Wu D., Soong Y., Birk A.V., Schiller P.W., Szeto H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. Journal of Biological Chemistry. 2004;279(33):34682–34690. doi: 10.1074/jbc.M402999200. 15178689 [DOI] [PubMed] [Google Scholar]
- 219.Sheu S.S., Nauduri D., Anders M.W. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochimica et Biophysica Acta. 2006;1762(2):256–265. doi: 10.1016/j.bbadis.2005.10.007. 16352423 [DOI] [PubMed] [Google Scholar]
- 220.Szeto H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS Journal. 2006;8(2):E277–E283. doi: 10.1007/BF02854898. 16796378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Melov S., Doctrow S.R., Schneider J.A., Haberson J., Patel M., Coskun P.E., Huffman K., Wallace D.C., Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. Journal of Neuroscience. 2001;21(21):8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. 11606622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Reily C., Mitchell T., Chacko B.K., Benavides G., Murphy M.P., Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biology. 2013;1(1):86–93. doi: 10.1016/j.redox.2012.11.009. 23667828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Murphy M.P. Antioxidants as therapies: can we improve on nature? Free Radical Biology and Medicine. 2014;66:20–23. doi: 10.1016/j.freeradbiomed.2013.04.010. 23603661 [DOI] [PubMed] [Google Scholar]
- 224.Jauslin M.L., Meier T., Smith R.A., Murphy M.P. Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2003;17(13):1972–1974. doi: 10.1096/fj.03-0240fje. 12923074 [DOI] [PubMed] [Google Scholar]
- 225.Chacko B.K., Reily C., Srivastava A., Johnson M.S., Ye Y., Ulasova E., Agarwal A., Zinn K.R., Murphy M.P., Kalyanaraman B., Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(+/)-(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochemical Journal. 2010;432(1):9–19. doi: 10.1042/BJ20100308. 20825366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Mukhopadhyay P., Horváth B., Zsengellér Z., Zielonka J., Tanchian G., Holovac E., Kechrid M., Patel V., Stillman I.E., Parikh S.M., Joseph J., Kalyanaraman B., Pacher P. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radical Biology and Medicine. 2012;52(2):497–506. doi: 10.1016/j.freeradbiomed.2011.11.001. 22120494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Vergeade A., Mulder P., Vendeville-Dehaudt C., Estour F., Fortin D., Ventura-Clapier R., Thuillez C., Monteil C. Mitochondrial impairment contributes to cocaine-induced cardiac dysfunction: prevention by the targeted antioxidant MitoQ. Free Radical Biology and Medicine. 2010;49(5):748–756. doi: 10.1016/j.freeradbiomed.2010.05.024. 20566328 [DOI] [PubMed] [Google Scholar]
- 228.Mukhopadhyay P., Horváth B., Zsengellėr Z., Bátkai S., Cao Z., Kechrid M., Holovac E., Erdėlyi K., Tanchian G., Liaudet L., Stillman I.E., Joseph J., Kalyanaraman B., Pacher P. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radical Biology and Medicine. 2012;53(5):1123–1138. doi: 10.1016/j.freeradbiomed.2012.05.036. 22683818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Chacko B.K., Srivastava A., Johnson M.S., Benavides G.A., Chang M.J., Ye Y., Jhala N., Murphy M.P., Kalyanaraman B., Darley-Usmar V.M. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology (Baltimore, Md.) 2011;54(1):153–163. doi: 10.1002/hep.24377. 21520201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gane E.J., Weilert F., Orr D.W., Keogh G.F., Gibson M., Lockhart M.M., Frampton C.M., Taylor K.M., Smith R.A., Murphy M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver International: Official Journal of the International Association for the Study of the Liver. 2010;30(7):1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. 20492507 [DOI] [PubMed] [Google Scholar]
- 231.Smith R.A., Murphy M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Annals of the New York Academy of Sciences. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. 20649545 [DOI] [PubMed] [Google Scholar]
- 232.Zhong W., McClain C.J., Cave M., Kang Y.J., Zhou Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2010;298(5):G625–G633. doi: 10.1152/ajpgi.00350.2009. 20167873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kirpich I.A., Feng W., Wang Y., Liu Y., Barker D.F., Barve S.S., McClain C.J. The type of dietary fat modulates intestinal tight junction integrity, gut permeability, and hepatic Toll-like receptor expression in a mouse model of alcoholic liver disease. Alcoholism, Clinical and Experimental Research. 2012;36(5):835–846. doi: 10.1111/j.1530-0277.2011.01673.x. 22150547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Dunagan M., Chaudhry K., Samak G., Rao R.K. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2012;303(12):G1356–G1364. doi: 10.1152/ajpgi.00526.2011. 23064762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kirpich I.A., Feng W., Wang Y., Liu Y., Beier J.I., Arteel G.E., Falkner K.C., Barve S.S., McClain C.J. Ethanol and dietary unsaturated fat (corn oil/linoleic acid enriched) cause intestinal inflammation and impaired intestinal barrier defense in mice chronically fed alcohol. Alcohol (Fayetteville, N.Y.) 2013;47(3):257–264. doi: 10.1016/j.alcohol.2013.01.005. 23453163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ge X., Lu Y., Leung T.M., Sørensen E.S., Nieto N. Milk osteopontin, a nutritional approach to prevent alcohol-induced liver injury. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2013;304(10):G929–G939. doi: 10.1152/ajpgi.00014.2013. 23518682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Forsyth C.B., Voigt R.M., Shaikh M., Tang Y., Cederbaum A.I., Turek F.W., Keshavarzian A. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2013;305(2):G185–G195. doi: 10.1152/ajpgi.00354.2012. 23660503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Abdelmegeed M.A., Banerjee A., Jang S., Yoo S.H., Yun J.W., Gonzalez F.J., Keshavarzian A., Song B.J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radical Biology and Medicine. 2013;65:1238–1245. doi: 10.1016/j.freeradbiomed.2013.09.009. 24064383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cresci G.A., Bush K., Nagy L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcoholism, Clinical and Experimental Research. 2014;38(6):1489–1501. doi: 10.1111/acer.12428. 24890666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Spruss A., Bergheim I. Dietary fructose and intestinal barrier: potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. Journal of Nutritional Biochemistry. 2009;20(9):657–662. doi: 10.1016/j.jnutbio.2009.05.006. 19679262 [DOI] [PubMed] [Google Scholar]
- 241.Spruss A., Kanuri G., Stahl C., Bischoff S.C., Bergheim I. Metformin protects against the development of fructose-induced steatosis in mice: role of the intestinal barrier function. Laboratory Investigation: a Journal of Technical Methods and Pathology. 2012;92(7):1020–1032. doi: 10.1038/labinvest.2012.75. 22525431 [DOI] [PubMed] [Google Scholar]
- 242.Ritze Y., Bárdos G., Claus A., Ehrmann V., Bergheim I., Schwiertz A., Bischoff S.C. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PloS One. 2014;9(1):e80169. doi: 10.1371/journal.pone.0080169. 24475018 [DOI] [PMC free article] [PubMed] [Google Scholar]