

Abstract
Prostaglandins, particularly PGE2, are important to adult bone and joint health, but how prostaglandins act on growth plate cartilage to affect bone growth is unclear. We show that growth plate cartilage is distinct from articular cartilage with respect to cyclooxygenase (COX)-2 mRNA expression; although articular chondrocytes express very little COX-2, COX-2 expression is high in growth plate chondrocytes and is increased by IGF-I. In bovine primary growth plate chondrocytes, ATDC5 cells, and human metatarsal explants, inhibition of COX activity with nonsteroidal antiinflammatory drugs (NSAIDs) inhibits chondrocyte proliferation and ERK activation by IGF-I. This inhibition is reversed by prostaglandin E2 and prostacyclin (PGI2) but not by prostaglandin D2 or thromboxane B2. Inhibition of COX activity in young mice by ip injections of NSAIDs causes dwarfism. In growth plate chondrocytes, inhibition of proliferation and ERK activation by NSAIDs is reversed by forskolin, 8-bromoadenosine, 3′,5′-cAMP and a prostacyclin analog, iloprost. The inhibition of proliferation and ERK activation by celecoxib is also reversed by 8CPT-2Me-cAMP, an activator of Epac, implicating the small G protein Rap1 in the pathway activated by iloprost. These results imply that prostacyclin is required for proper growth plate development and bone growth.
Although a large number of autocrine and paracrine factors regulate the pace of development at the mammalian growth plate, IGF-I is the major systemic factor driving the growth of long bones (1–5). Within the growth plate, chondrocytes resting within the reserve zone are recruited to the proliferative zone, where IGF-I stimulates cell division, which occurs along the long axis of the bone. Chondrocytes then transition from the proliferative to the prehypertrophic zone, where they cease proliferation and begin differentiating. Terminally differentiated cells are found in the hypertrophic zone, where glycogen accumulation leads to dramatic cell hypertrophy. Optimal growth requires a fine balance between proliferation and hypertrophy within the growth plate throughout the growth phase of an animal, but exactly how IGF-I controls this balance is incompletely understood. We have previously demonstrated that IGF-I changes the ratio of activities of the MAPKs, ERK1/2, and p38; relatively high ERK1/2 activity drives proliferation, whereas relatively high p38 activity favors differentiation (6). The kinase cascades in which ERK1/2 and p38 participate presumably have indirect transcriptional effects, but little is known about the downstream transcriptional targets of IGF-I in these cells.
We have noted from microarray analyses of chondrocytes treated with IGF-I that the growth factor significantly increases the expression of cyclooxygenase (COX)-2 in primary bovine growth plate chondrocytes (our unpublished data). COX-1 and COX-2 catalyze the same rate-limiting step in the synthesis of prostaglandins, the conversion of arachidonic acid to prostaglandin (PG) H2 (7, 8). PGH2 is further metabolized by specific isomerases to produce a variety of prostanoids, such as prostaglandins, prostacyclins, and thromboxanes. COX-1 is constitutively expressed in almost all tissues. COX-2 is normally undetectable in most tissues, but its expression is rapidly induced by injury and proinflammatory factors, and in some tissues, mitogens increase COX-2 expression (9–11).
Prostaglandins play an important role in bone metabolism; principle among these is thought to be PGE2, which stimulates bone formation (1, 12, 13). Chondrocytes synthesize and release PGE2, which is thought to act in a paracrine manner in cartilage (14–17). In articular chondrocytes, PGE2 enhances chondrocyte differentiation (18), stimulates collagen synthesis (16), and increases the expression of RANK ligand (19). The effects of nonsteroidal antiinflammatory drugs (NSAIDs) have been studied in normal and osteoarthritic articular cartilage. The earliest studies showed that NSAIDs suppress both collagen and noncollagen protein biosynthesis in cartilage (20, 21), and later studies showed that NSAIDs also suppress DNA synthesis and cell proliferation of articular cartilage (22–24). Some authors have suggested that certain NSAIDs may actually protect articular cartilage against stress (25–27). Growth plate chondrocytes release PGE2, which enhances their proliferation (28), but very little is known about the role of prostaglandins in growth plate chondrocyte development.
We demonstrate that, although COX-2 expression is low in articular chondrocytes, the growth plate expresses high levels of COX-2 mRNA, and IGF-I increases COX-2 expression and activity in these cells; the ability of IGF-I to stimulate chondrocyte proliferation and ERK activation is dependent on the presence of PGE2 or PGI2; PGI2 is more potent than PGE2 in supporting proliferation and ERK activation; and PGI2 signals via cAMP activation of the exchange protein directly activated by cAMP (Epac)/Rap1 pathway. We demonstrate with the use of in vitro, ex vivo, and in vivo methods that NSAIDs currently in wide clinical use impair growth in young mice and cause abnormal growth plate development in both murine and human tissue.
Materials and Methods
Growth factors, hormones, and enzyme inhibitors
Recombinant human IGF-I and insulin were from Sigma-Aldrich. Calf serum, transferrin, and selenium were from GIBCO, Inc. Ibuprofen was from Sigma and celecoxib from LKT Laboratories, Inc. Prostaglandins, iloprost, butaprost, Cay10598, and PGE2, and PGI2 enzyme immunoassay (EIA) kits were from Cayman Chemical. Forskolin (FSK), 3-isobutyl-1-methylxanthine (IBMX), 2′,5′-dideoxyadenosine (ddAd), and 8-bromoadenosine 3′,5′-cAMP (8-Br) were from Sigma; 8CPT-2Me-cAMP (8CPT) was from Tocris/R&D Systems.
Harvesting and separation of chondrocytes
Growth plate chondrocytes were obtained from metacarpals of male dairy cattle aged 6–10 months and fractionated by continuous density gradient centrifugation as described (29).
Cells were removed from the gradient in 4 fractions of equal volume, with fraction 1 containing the lowest-density hypertrophic zone cells, and fraction 4 containing the highest-density proliferative zone cells. Articular cartilage was harvested from the same bones and processed as above but without density gradient centrifugation.
Cell proliferation assays
Primary chondrocytes and ATDC5 cells (obtained from RIKEN) were plated in serum-free DMEM/F12 medium on 60-mm plastic plates at a density of 1 × 105 cells per well and maintained in a humidified incubator at 37°C and 5% CO2. All cell samples were plated and assayed in triplicate. Cells were incubated with various concentrations of NSAID, 100-ng/mL IGF-I, prostaglandins, prostaglandin analogs, or other small molecule modifiers of signaling pathways as indicated for 48 hours, at which time the cells were trypsinized and counted on a hemocytometer.
RNA isolation, cDNA synthesis, and real-time RT-PCR
All RNA extractions were performed with RNA STAT-60 (TelTest, Inc) per the manufacturer's instructions. Genomic DNA was removed using DNA-Free (Ambion), and 5 μg of RNA reverse transcribed with the High-Capacity cDNA Archive kit (Applied Biosystems). Real-time RT-PCR was performed using the Roche LightCycler 480 following the protocol supplied by the manufacturer. 18S RNA was detected using TaqMan with the LightCycler 480 Probes Master Mix (Roche) and 100nM each primer and probe. All other targets, bovine prostaglandin-endoperoxide synthase (PTGS) (COX1), PTGS2 (COX2), and mouse Cox1 and COX2, were analyzed using double-stranded DNA dye SYBR Green with the LightCycler 480 SYBR Green Master Mix (Roche) and 200nM each primer. All primers spanned an intron/exon boundary, and all RT-PCR were confirmed to produce only a single PCR product by comparison of the melt curves at the completion of each PCR. Primer pairs are listed in Supplemental Table 1. Relative gene expression for each mRNA was calculated by the ΔΔCT method using the untreated sample as calibrator.
For absolute quantitation of COX1/2 message, a 136-bp fragment of bovine COX-1 and a 157-bp fragment of bovine COX2 were generated by PCR from cDNA made from bovine reserve zone cells and inserted into PCRII-TOPO (Invitrogen). A 187-bp fragment of 18s cDNA was inserted into PCR2.1-TOPO. Standard curves were generated for all 3 constructs for use in real-time PCR reactions.
Western blot analysis for ERK activation
Isolated bovine proliferative zone growth plate chondrocytes were plated at 1 × 106 cells per 60-mm plate in serum-free DMEM:F12 plus 1-mg/mL BSA, with or without additions as indicated and allowed to rest overnight. Cells were then treated as indicated, followed by receiving either 100-ng/mL IGF-I or no further treatment for 15 minutes. Whole-cell lysates were prepared in lysis buffer plus protease inhibitors and phosphatase inhibitors (1mM NaF and 1mM Na3VO4). Equal amounts of total protein were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to Hybond-P membranes (Amersham). Membranes were blocked and all antibody incubations were performed in StartingBlock Blocking buffer (Thermo Scientific) diluted 1:4 in Tris-buffered saline plus 0.05% Tween 20. Primary antibodies used were: antiphospho-ERK1/2 (1:5000; Invitrogen) and antitotal-ERK1/2 (1:20,000; Sigma). After washing with Tris-buffered saline plus 0.05% Tween 20, membranes were incubated with a horseradish peroxidase-labeled secondary antibody at 1:5000, and bands were visualized by chemiluminescence and Hyperfilm (Amersham).
Measurement of prostaglandin production
Where indicated, conditioned media from cell proliferation experiments were collected and assayed for PGE2 and PGI2 content by EIA according to the manufacturer's instructions. Prostaglandin content was normalized to total cell count for each sample.
Human metatarsal explant culture
Metatarsals from a 6-month-old boy were obtained after amputation due to fibular hemimelia with clubfoot; use of the tissue was approved by the University of Texas Southwestern Institutional Review Board. Thin sections of bone including growth plate were maintained in culture for 24 hours in BGJb medium with or without 100-ng/mL IGF-I and increasing concentrations of ibuprofen or celecoxib. Bromodeoxyuridine (BrdU) at 100μM was added for 4 hours before the explants were fixed in 4% paraformaldehyde/0.01M PBS (pH 7.4) for 24 hours at 4°C, followed by decalcification for 2 weeks in 0.5M EDTA (pH 8.0) before imbedding in paraffin. Cell proliferation was assessed with the BrdU Labeling and Detection kit II from Roche Diagnostics.
Mice treated with NSAIDs
All animal studies were approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center under approval number 2010–0097. All animal euthanasia was performed via CO2 inhalation. C57/B6 mice were purchased from The Jackson Laboratory. All mice were kept in a pathogen-free environment on a 12-hour light, 12-hour dark cycle. Litters were culled to a maximum of 7 pups per litter; once weaned, mice were housed at a maximum of 4 per cage. Water and standard chow were provided ad libitum. Starting either at postnatal day (P)7 or P14, mice were treated once daily for a total of 7 days with the indicated amount of NSAID by ip injection. Because COX inhibitors are not soluble in aqueous solution, minimal volumes of dimethyl sulfoxide (DMSO) were used for injection; control animals received DMSO only. The maximum amount of DMSO received was 10 μL per 5 g body weight per day. Starting at the time of injections, weight and nose-to-rump length was measured daily until 4 weeks of age; individuals performing measurements were blinded to treatment group.
For determination of serum levels of COX inhibitors, adult male mice were injected at the same weight-adjusted dose as the experimental mice once daily for 7 days. On day 8, the mice were killed by CO2 inhalation, and blood was obtained by cardiac puncture. Serum from 5 mice in each treatment group was pooled and sent to NMS Labs for assay of ibuprofen and celecoxib concentrations. Because 3 mL were required for each determination, older and thus larger mice were used for this analysis.
Histology
Tissues from mice were fixed in 4% paraformaldehyde/0.01M PBS (pH 7.4) for 24 hours at 4°C and were decalcified for 2 weeks in 0.5M EDTA (pH 8.0) before imbedding in paraffin. Seven-millimeter sections were cut and stained with hematoxylin and eosin (H&E). Immunohistochemical staining used rabbit antirat collagen X at a 1:50 dilution (Abcam). Immunostaining was performed with the Cell and Tissue Staining kit from R&D Systems according to the manufacturer's instructions. Assessment of growth plate morphology was performed by a blinded reviewer using ImageJ. Staining for apoptosis was done with the TACS 2 dTd-DAB In Situ Apoptosis Detection kit (Trevigen, Inc).
BrdU uptake
Mice were treated as indicated then injected ip with BrdU (100 μg/g body weight) 4 hours before killing. Tibiae were fixed in 4% paraformaldehyde/0.01M PBS (pH 7.4) for 24 hours at 4°C, decalcified in EDTA for 2 weeks, and embedded in paraffin. BrdU uptake in cells was determined by BrdU labeling with the BrdU Labeling and Detection kit II from Roche Diagnostics. Percentage of cells staining positively for BrdU was assessed by a blinded reviewer using ImageJ.
Statistical analyses
For COX expression and cell proliferation studies, ANOVA was performed, followed by Student-Newman-Keuls post hoc test for multiple comparisons with correct for type 1 error, and differences were considered significant at P < .05. To compare mouse growth curves, repeated-measures ANOVA was performed; and to determine at which day the growth curves became significantly different, t tests for individual days were used. All analyses were performed with SigmaPlot 12.5 (Systat Software, Inc).
Results
PTGS2 (COX-2) expression is high in growth plate chondrocytes and is increased by IGF-I
Although far more is known about the effects of the prostaglandin PGE2 in adult bone, PGE2 is known to be released from growth plate chondrocytes in vitro (17, 30–32). To confirm and extend this observation, we used real-time RT-PCR to examine IGF-I-regulated expression of PTGS1 and PTGS2, encoding COX-1 and COX-2, respectively. Both isozymes are widely expressed, but although COX-2 mRNA and protein expression are regulated by a variety of factors, COX-1 expression is constitutive (33, 34). Articular chondrocytes have been reported to have negligible levels of COX-2 mRNA that increase with exposure to IL-1 (35). We examined the expression of COX-1 and COX-2 mRNA in primary bovine articular and growth plate chondrocytes incubated with IGF-I or vehicle. In articular chondrocytes, the expression of COX-2 mRNA was indeed negligible, but COX-1 was measurable at about 2 × 105 copies/mcg 18s RNA (Figure 1A). In contrast, in primary bovine growth plate cells, COX-2 mRNA expression was approximately 800-fold that of COX-1 at baseline and increased to 4000-fold that of COX-1 with IGF-I incubation. Incubation of both primary growth plate chondrocytes and ATDC5 cells with IGF-I for 24 hours increased PGE2 synthesis by more than 2-fold (Figure 1B).
Figure 1.
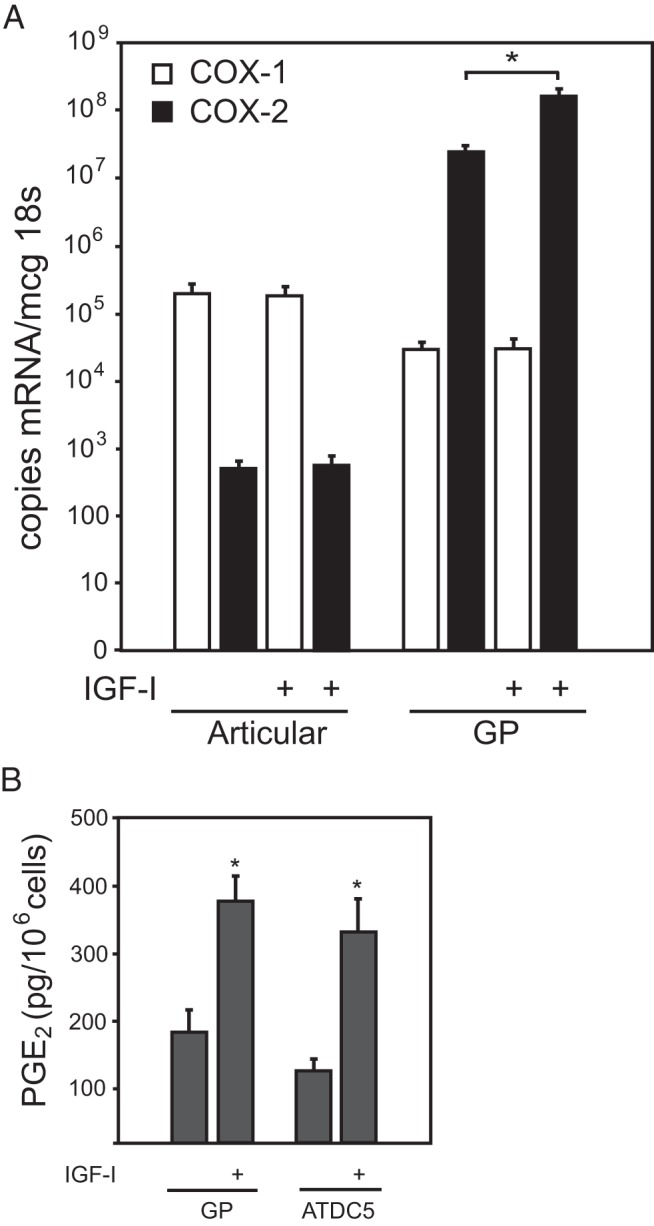
COX1/2 expression and regulation in chondrocytes. A, Primary bovine articular chondrocytes or primary bovine growth plate chondrocytes from the reserve zone in serum-free medium received 24 hours of IGF-I (100 ng/mL) or vehicle only. Quantitative real-time PCR was used to determine the copy number of COX-1 and COX-2 mRNA per μg of 18s, n = 4 per sample, P < .001 by Student's t test (compared with vehicle). B, Primary growth plate or ATDC5 cells were incubated with IGF-I or vehicle only as above, and PGE2 in the medium of cells was quantitated by EIA and normalized to the total number of cells. *, P < .001 by t test (compared with vehicle).
In vitro, COX inhibitors suppress IGF-I-stimulated chondrocyte proliferation
We speculated that the stimulatory effect of IGF-I on chondrocyte proliferation might be mediated in part by increased COX-2 expression. Therefore, ATDC5cells were treated with IGF-I in combination with 2 COX enzyme inhibitors used commonly in clinical practice, ibuprofen and celecoxib; results seen in ATDC5 cells were then confirmed in isolated bovine proliferative zone cells. Ibuprofen has IC50 for COX-1 and COX-2 of 5μM and 75μM, respectively, indicating that it more effectively inhibits COX-1 at lower concentrations but will inhibit both enzymes if the concentration is above the IC50 for COX-2 (36, 37). The reverse is true for celecoxib, which has an IC50 for COX-1 of 2.5μM and a lower IC50 for COX-2 of 0.4μM (38–40). Thus, ibuprofen is a relatively COX-1-specific inhibitor, whereas celecoxib is relatively specific for COX-2. Both ibuprofen and celecoxib blocked IGF-I-stimulated proliferation in both primary chondrocytes (Figure 2A) and in ATDC5 cells (Figure 2B) at concentrations near or above the IC50 for COX-2 (eg, 50μM ibuprofen or 2μM celecoxib for primary chondrocytes). The bovine cells appear to be more sensitive to the NSAIDs than are the murine cells; this may represent a species difference or a difference between primary cells (bovine) and a cell line (murine ATDC5 cells).
Figure 2.
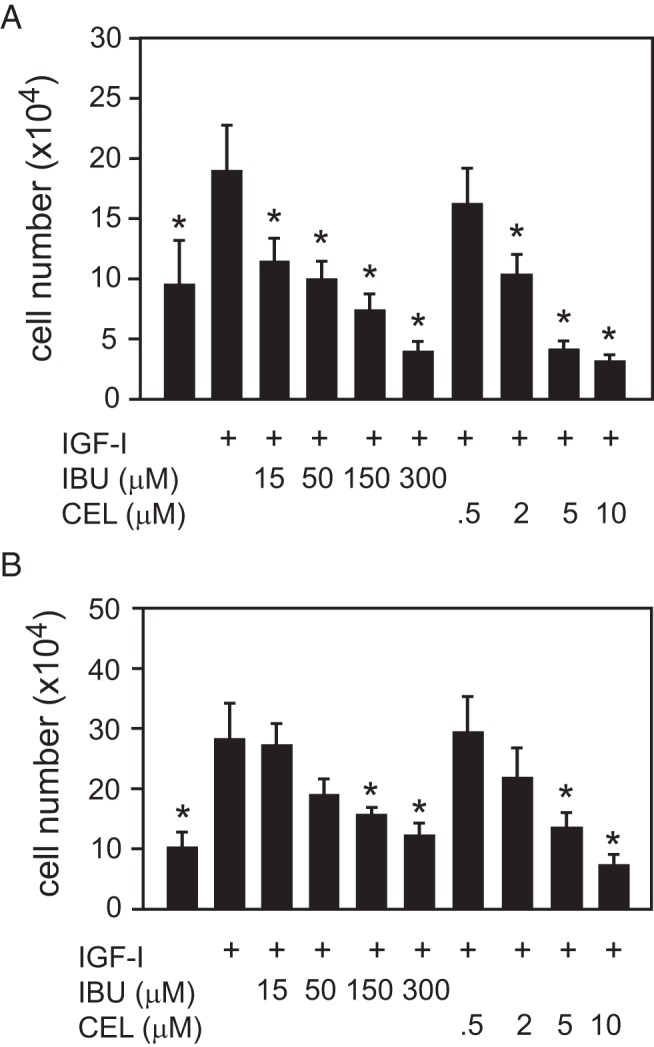
Dose-dependent inhibition of growth plate chondrocyte proliferation by NSAIDs. A, Primary bovine growth plate chondrocytes from the RZ in serum-free medium were treated with 100-ng/mL IGF-I and increasing concentrations of either ibuprofen (IBU) or celecoxib (CEL) for 48 hours, and cell proliferation was assessed by cell counts. B, Identical conditions were used to assess proliferation in ATDC5 cells. Data points represent the mean ± SD from 6 samples. *, P < .05 by ANOVA with Newman-Keuls post hoc correction (compared with IGF-I only sample).
In vivo, mice treated with NSAIDs are dwarfed, and COX inhibitors block effects of IGF-I on human metatarsal explants
In a study of COX inhibition on eye development, young rats were reported to grow poorly on high-dose ibuprofen (41). To determine whether COX inhibition could affect the growth of young mice, male and female C57/B6 mice were treated with either ibuprofen (15 μg/g), celecoxib (10 μg/g), or vehicle once daily by ip injection starting at P7. These doses were chosen because they are similar to the oral doses used in children, which are approximately 10 mg/kg body weight. The serum level of ibuprofen after 7 days of injections in adult male C57/B6 mice was 8.9 μg/mL (45μM); the serum level of celecoxib was 1.2 μg/mL (3μM). These levels are similar to those seen in humans after single oral doses of these medications (42–44).
Both the ibuprofen- and celecoxib-treated mice were significantly smaller than the mice that received vehicle only (Figure 3, A and B). The difference between vehicle and ibuprofen-treated mice was significant by day 5–6 (by post hoc t tests for each day); the difference between vehicle and celecoxib-treated mice was significant by day 3 of treatment. The NSAID-treated mice remained significantly smaller into adulthood. To determine whether growth suppression also occurred in somewhat older mice, the same treatments were given starting at P14. The ibuprofen-treated mice were significantly smaller than the vehicle-only mice by day 6 of injections, and the celecoxib mice by day 3; none of the NSAID-treated mice showed any catch-up growth by P28 (Figure 3C).
Figure 3.

NSAIDs interfere with somatic growth and growth plate development in vivo in mice, and ex vivo in human tissue. Male (A) and female (D) C57/B6 mice were treated once daily from P7 to P13 (underlined days) by ip injection with 15-μg/g·d ibuprofen (IBU) in DMSO, or DMSO only. Maximum volume received was 10 μL per 5 g body weight. Nose-to-rump length was measured daily. Male (B) and female (E) mice were treated with 10-μg/g·d celecoxib (CEL) or vehicle for the same period of time. Male (C) and female (F) mice were treated with IBU, CEL, or vehicle for 7 days starting at P14. Repeated measures ANOVA was used to determine significance between curves, and t tests within each day used to determine the day at which the difference became significant. G–L, Male mice treated with IBU and CEL at the same doses from P14 to P20 were killed on P21 4 hours after receiving a single ip dose of BrdU, and 1 tibia from each mouse was excised, fixed in paraformaldehyde, and decalcified in EDTA. Panels G (vehicle treated), H (IBU treated), and I (CEL treated) are tibial sections stained for ColX and counterstained with H&E. Brackets are included to indicate the size of the hypertrophic zone. Panels J (vehicle treated), K (IBU treated), and L (CEL treated) were stained for BrdU for assessment of cell proliferation. M, Human metatarsal sections cultured in BJGb for 24 hours; sections received either no treatment, or IGF-I alone or with increasing concentrations of either IBU or CEL. BrdU was added for 4 hours before fixation and cell proliferation visualized by staining with anti-BrdU antisera.
Male mice at P7 were treated as above with either ibuprofen or celecoxib for 7 days, and on P14, they received BrdU 4 hours before killing. Cell proliferation in excised tibias was assessed by BrdU staining and general morphology by H&E staining (Figure 3, D–I). The percentage of the growth plate made up of hypertrophic zone cells was wider in the NSAID-treated mice (30% in control, n = 5; 38% in NSAID-treated, n = 7; P = .002) and in the celecoxib-treated mice, the cells of all zones appeared abnormally large. BrdU staining was reduced in sections from NSAID-treated mice as compared with the vehicle only control, suggesting that chondrocyte proliferation is suppressed by NSAIDs (Figure 3, E, G, and I). The percentage of cells within the hypertrophic zone staining positively for DNA fragmentation as a marker of apoptosis was not significantly different between the NSAID-treated and control mice (data not shown).
Additionally, human metatarsal specimens from a 6-month-old male were maintained for 24 hours in serum-free BJGb media. The explants received either 100-ng/mL IGF-I, IGF-I plus NSAIDs (either ibuprofen or celecoxib) at increasing concentrations, or vehicle alone. BrdU was added 4 hours before the explants were fixed, and staining for BrdU was performed to assess for cell proliferation. Figure 3J shows what appears to be a dose-responsive decrease in BrdU uptake with increasing concentrations of either ibuprofen or celecoxib.
Rescue of inhibition of proliferation by prostacyclin in vitro
COX-1 and COX-2 catalyze the same key rate-limiting step in the synthesis of prostaglandins, the conversion of arachidonic acid to PGH2. PGH2 is further metabolized by specific isomerases to produce a variety of prostaglandins, prostacyclins, and thromboxanes.
To demonstrate that the inhibition of chondrocyte proliferation caused by NSAIDs was indeed due to depletion of specific endogenous prostaglandins, primary bovine growth plate chondrocytes were incubated with 100-ng/mL IGF-I plus or minus various prostaglandins. The prostaglandins PGE2 and PGI2 at 1μM were able to reverse the inhibitory effect of celecoxib on IGF-I-stimulated chondrocyte proliferation, whereas PGD2 and TXB2 had no effect (Figure 4A). PGI2 was consistently more effective than PGE2 at the same concentration. Nearly identical results were seen in ATDC5 cells (data not shown).
Figure 4.

Iloprost reversal of CEL-induced IGF-I resistance requires increased intracellular cAMP and Epac activation. A, Primary bovine chondrocytes were incubated in serum-free medium and indicated additions for 48 hours; after which cell proliferation was assessed by cell counting after trypsinization. IGF-I (100 ng/mL), celecoxib (CEL) (5μM), 1μM PGs. Individual t tests were performed for the relevant pairwise comparisons. B, Primary chondrocytes in serum-free medium, with or without CEL (5μM), were allowed to rest overnight. Cells received 1μM PGE2, PGI2, PGD2, TXB2, or vehicle for 10 minutes, then either 100-ng/mL IGF-I or no further treatment for 15 minutes. Cell lysates were analyzed by Western blotting for total and phospho-ERK1/2 as a measure of ERK activity. The blot shown is representative of 3 separate experiments. C, Cells were assessed for proliferation as in A; IGF-I (100 ng/mL), CEL (5 μM), butaprost (1μM), and Cay10598 (1μM) are PGE2 analogs that activate EP2 and EP4, respectively; iloprost (1μM) is an analog of PGI2 (prostacyclin). D, Cell proliferation was assessed as above; IGF-I (100 ng/mL), CEL (5μM), FSK (5μM), IBMX (50μM), 8-Br (50μM), or ddAd (1μM) were added simultaneously. E, Cell proliferation was assessed as above; IGF-I (100 ng/mL), CEL (5μM), iloprost 1μM), the protein kinase A inhibitor H89 (10μM), ddAd (1μM), or 8CPT (25μM), an activator of Epac, were added simultaneously. F, Increasing concentrations of H89 were used with IGF-I (100 ng/mL), and proliferation was assessed as above. For a and c-f, n = 18. *, P < .05 by ANOVA with Newman-Keuls post hoc correction (as compared with vehicle sample).
Because activation of the MAPKs ERK1/2 is absolutely required for chondrocyte proliferation (6, 45–47), we questioned whether NSAIDs might affect ERK activity in these cells. As seen in Figure 4B, 5μM celecoxib blunts the activation of ERK1/2 by IGF-I (lane 4). Although neither PGD2 nor TXB2 had an effect on ERK activity, both PGE2 and PGI2, when added for 10 minutes at 1μM, effectively reversed the ERK inhibition caused by an overnight incubation with 5μM celecoxib (lanes 5 and 6). Neither PGE2 nor PGI2 activated ERK in the absence of IGF-I (lanes 9 and 10).
PGE2 acts through 4 receptors, EP1–EP4, and all 4 have been reported to be expressed in growth plate cartilage (28, 30). PGI2 acts via a single receptor, IP. IP expression has been reported in adult osteoblasts (48), but its expression at the growth plate has not been explored. To better delineate which prostaglandin receptors are involved in the actions of IGF-I in chondrocytes, cells exposed to celecoxib and IGF-I were also treated with specific prostaglandin analogs. Butaprost is a PGE2 analog specific for EP2, Cay10598 is a PGE2 analog specific for EP4, and iloprost is a PGI2 analog. As seen in Figure 4C, butaprost partly relieved the inhibition of proliferation caused by celecoxib, but the prostacyclin analog iloprost was consistently more effective at the same concentration.
The prostacyclin analog iloprost uses Epac/Rap1
Prostaglandin receptors are G protein-coupled receptors; PGE2 receptors EP2/4 and the prostacyclin receptor IP are linked to Gαs, and receptor activation leads to activation of adenylate cyclase and increased intracellular cAMP levels, whereas activation of EP3, which is linked to Gαi, leads to reduced cAMP levels. EP1 is linked to Gαq, and its activation causes increased intracellular calcium levels. To determine whether in fact the rescue of celecoxib inhibition of proliferation by iloprost is due to increased cAMP, various small molecules were used to manipulate intracellular cAMP levels. FSK, an activator of adenylate cyclase, fully reversed the inhibitory effect of celecoxib on proliferation in growth plate chondrocytes, as did the phosphodiesterase inhibitor IBMX and the stable cAMP analog 8-Br (Figure 4D). Moreover, an inhibitor of adenylate cyclase, ddAd completely blocked the ability of iloprost to restore proliferative capacity to celecoxib-treated cells (Figure 4E).
The ras-raf-MEK-ERK pathway may be affected by intracellular cAMP levels via activation of protein kinase A, but depending on the cell type, protein kinase A may be either stimulatory or inhibitory to the ERK pathway (49–52). Another source of cross talk between Gαs-linked G protein-coupled receptor and ERK is via activation by cAMP of Epac, a guanine nucleotide exchange factor for Rap1, which can directly increase B-raf activity towards MEK (53–55). Figure 4E shows that although inhibition of protein kinase A with 10μM H89 blocks iloprost action in chondrocytes, a specific activator of Epac (8CPT) simulates the ability of iloprost to reverse the inhibition of proliferation by celecoxib. We found that the effects of the protein kinase A inhibitor H89 in chondrocytes is dose-dependent; low doses tended to increase IGF-I-stimulated proliferation slightly, whereas higher doses (such as 10μM) blocked chondrocyte proliferation (Figure 4F).
Celecoxib inhibition of proliferation is via suppression of ERK activation
Because, as seen in Figure 4B, COX inhibition prevented the activation of ERK by IGF-I, and this blockade was reversed by prostacyclin, we questioned whether ERK activation by IGF-I is cAMP dependent in chondrocytes. Indeed, preincubation of chondrocytes with the adenylate cyclase inhibitor ddAd completely blocked the ability of IGF-I to increase ERK activity (Figure 5A). Note that low-dose H89 increased IGF-I-stimulated ERK activity. As shown in Figure 5B, high doses of H89 result in increased activation of ERK by IGF-I, suggesting that the inhibitory effect of H89 on proliferation seen in these cells (see Figure 4F) is likely to be the result of excessive, prolonged ERK activation, rather than ERK inhibition.
Figure 5.

Iloprost acts via cAMP/Epac/Rap1 to reverse celecoxib-induced IGF-I resistance. A–C, Primary bovine chondrocytes were incubated in serum-free medium and indicated additions for 1 hour before the addition of IGF-I (100 ng/mL) or vehicle for 15 minutes. Cell lysates were analyzed by Western blotting for total and phospho-ERK1/2 as a measure of ERK activity. D, Cells in serum-free medium were incubated with increasing concentrations of CEL for 12 hours, then IGF-I (100 ng/mL) was added for 15 minutes, and cell lysates were analyzed by Western blotting for total and phospho-ERK1/2. All blots shown are representative of at least 3 separate experiments. The media from cells treated as above were assessed for PGE2 and PGI2 content by EIA; n = 6, * P < .05 by ANOVA with Newman-Keuls post hoc correction (as compared with IGF-I only sample).
To demonstrate that iloprost antagonizes celecoxib by restoring the ability of IGF-I to activate ERK, cells were incubated with 5μM celecoxib overnight before being treated with either FSK, iloprost, or the Epac activator 8CPT. Figure 5C shows that FSK, iloprost, and the Epac activator each completely restored the ability of IGF-I to activate ERK in the face of COX inhibition by celecoxib.
Growth plate chondrocytes secrete PGI2 in response to IGF-I stimulation
Figure 5D shows that chondrocytes exposed to increasing concentrations of celecoxib overnight displayed blunting of ERK activation by IGF-I, as well as decreased PGE2 and PGI2 synthesis. In both baseline and IGF-I-stimulated states, the growth plate cells consistently secreted more PGI2 than PGE2 by a factor of about 2-fold.
Discussion
Children with chronic inflammatory conditions such as juvenile idiopathic arthritis are often treated with NSAIDs, which inhibit the COX enzymes, as well as corticosteroids that suppress COX-2 expression. The poor growth often seen in these children is generally thought to be due to the direct effects of their inflammatory disease in combination with the deleterious effects of corticosteroid therapy, although exactly how corticosteroids restrict growth is incompletely understood. Nonselective COX inhibitors, such as ibuprofen and naproxen, and selective COX-2 inhibitors, such as celecoxib, are an accepted part of the treatment of chronic inflammatory diseases in children (7, 8, 56, 57) and adults (58, 59). NSAIDs are not thought to affect growth, but although the effects of prostaglandins and NSAIDs on articular chondrocytes has been studied, very little is known about the role of prostaglandins in growth plate chondrocyte development and long bone growth.
Using in vitro, ex vivo, and in vivo techniques, we demonstrate that the presence of specific prostaglandins (PGI2 and PGE2) is required for IGF-I action in growth plate chondrocytes and for proper growth plate development and long bone growth. COX-2 inhibition negatively affects growth plate development in mice by blocking the ability of IGF-I to activate ERK1/2 and stimulate chondrocyte proliferation. The fact that PGI2 (and to a lesser extent PGE2) and their analogs are able to reverse this blockade suggests that the ability of NSAIDs to suppress chondrocyte proliferation is due to direct inhibition of COX activity. At least in vitro, we found that the ERK-inhibiting effects of celecoxib on chondrocytes are rapidly reversed by a PGI2 analog, suggesting that the dwarfism in mice treated with NSAIDs is due to direct inhibition of COX activity.
In bovine growth plate chondrocytes and the murine ATDC5 cell line, the ability of NSAIDs to inhibit proliferation is at least partly due to the inhibition of the MAPKs ERK1/2. In other cell types, such as human cholangiocarcinoma, nonsmall cell lung cancer, colon carcinoma, and gastric epithelial cells, PGE2 is required or permissive for mitogen-stimulated ERK activity and cell proliferation (60–63). In cholangiocarcinoma and nonsmall cell lung cancer cells, PGE2 activation of ERK is dependent on activation of protein kinase C by increased intracellular calcium (60, 61), which is consistent with PGE2 acting through EP1. Activation of protein kinase C by PGE2 has also been reported in growth plate chondrocytes (17, 30). In contrast, we found that in growth plate chondrocytes the proliferation-supporting activity of PGI2 (and PGE2) required increased intracellular cAMP and that cAMP is acting through the Epac/Rap1 pathway and not via protein kinase A to activate ERK (see Figure 6B). These results add to recent advances in our understanding of cAMP signaling in chondrocytes. Reduced chondrocyte proliferation and resistance to parathyroid hormone related peptide is seen in mice lacking genes for both Gsα and Gq/11α, whereas deletion of only one or the other small G protein does not reduce chondrocyte proliferation (64). The rare skeletal dysplasia acrodysostosis is caused by mutations in phosphodiesterase 4D or a regulatory subunit of protein kinase A, cAMP-dependent protein kinase regulatory subunit alpha (PRKAR1A) (65). Both mutations are thought to cause resistance to parathyroid hormone-related peptide by reducing protein kinase A activity in chondrocytes. However, the inclusion of Epac/Rap1 in the regulation of chondrocyte proliferation suggests that not all cAMP signaling in chondrocytes is dependent on protein kinase A.
Figure 6.
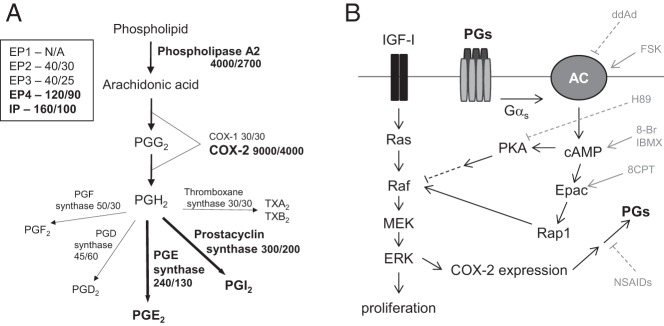
A, Diagram of the prostaglandin synthesis pathway including relative expression values (arbitrary units) for synthetic enzymes and receptors from our previously reported microarray analysis of bovine growth plate chondrocytes (29). First number shown represents expression in the proliferative zone, the second number represents expression in the hypertrophic zone. Note that prostaglandin receptor EP1 was not represented on the array. B, Diagram of the interaction in growth plate chondrocytes between pathways downstream of IGF-I and the prostaglandin receptors, which are G protein coupled. Targets of inhibitors and activators used in the experiments are shown. Adapted from Gerits et al (76).
Unlike what is seen in articular chondrocytes, we showed that growth plate chondrocytes express high levels of COX-2 mRNA in the unstimulated state. Although others have reported the production of PGE2 by growth plate chondrocytes (28), the present report reveals that these cells actually produce more PGI2 than PGE2 and that PGI2 is more effective at reversing the proliferation-suppressing effects of NSAIDs. The 4 PGE2 receptors, EP1–EP4, have been reported in growth plate chondrocytes (28, 30). The fact that the EP2-specific PGE2 analog butaprost, but not an EP4-specific analog (CAY10598), partially rescued NSAID suppression of cell growth suggests that EP2 but not EP4 is involved in the regulation of growth plate chondrocyte proliferation by PGE2. To our knowledge, the PGI2 receptor IP has not been previously described at the growth plate, although IP has been reported to be expressed in osteoblasts (48). Iloprost is known to bind EP1 (66, 67), and thus, it is possible that the effects seen with iloprost could be due to activation of this PGE2 receptor. Iloprost is also known to directly stimulate peroxisome proliferator-activated receptor-γ (68–70). However, EP1 signals via increased intracellular Ca2+, and the effects of iloprost in growth plate chondrocytes are mediated by increased cAMP, consistent with the prostacyclin analog acting through the IP receptor. In Figure 6A is shown relative expression values for prostaglandin receptors and synthetic enzymes taken from our previous microarray analysis of bovine reserve and hypertrophic zone cells (29). Although probes for EP1 were not present on the array, the results suggest that EP4 and IP are the most highly expressed of the prostaglandin receptors.
PGE2 has been reported to stimulate chick or rat chondrocyte proliferation directly (28, 71), and these results stand in contrast to the present study, where PGE2 or PGI2 alone did not stimulate ERK activation or chondrocyte proliferation but were instead required for IGF-I to stimulate ERK activity and cell proliferation. This difference is likely a result of the fact that we used completely serum-free conditions for all cell proliferation and ERK activation studies, whereas the other studies employed FBS concentrations as high as 1%.
Mice with global deletions for either COX1 or COX2 exist and are reported to be normally grown (72–74); deletion of both genes is early embryonic lethal. Presumably mice lacking COX-2 maintain local levels of PGI2 at the growth plate because COX-1 is present, but the growth plates of these mice have not been examined. Mice with a global deletion of the PGI2 receptor were also reported to be normally grown (75). This may be due to the fact that local PGE2 can compensate for the lack of response to PGI2, but the growth plates of these mice have not been examined either.
To the best of our knowledge, this mechanism-based toxicity of NSAIDs on somatic growth has not been previously reported. Although our studies were mainly conducted in bovine chondrocytes and in mice, our limited ex vivo data demonstrating marked antiproliferative effects of both ibuprofen and celecoxib in human metatarsal explants suggest that our other findings may pertain to humans as well, at least as regards the relevant mechanisms. Because marked growth suppression with use of NSAIDs is not seen in humans, such an effect must be relatively mild in vivo, perhaps reflecting differing pharmacodynamics in humans and mice.
Acknowledgments
We thank Harry Kim, MD, at the Texas Scottish Rite Hospital for Children for providing the human metatarsal samples.
This work was supported by NIH Grants K08DK073447 and R03DK089151 from the National Institute of Diabetes and Digestive and Kidney Diseases (to M.R.H.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- 8-Br
- 8-bromoadenosine 3′,5′-cAMP
- BrdU
- bromodeoxyuridine
- COX
- cyclooxygenase
- 8CPT
- 8CPT-2Me-cAMP
- ddAd
- 2′,5′-dideoxyadenosine
- DMSO
- dimethyl sulfoxide
- EIA
- enzyme immunoassay
- Epac
- exchange protein directly activated by cAMP
- FSK
- forskolin
- H&E
- hematoxylin and eosin
- IBMX
- 3-isobutyl-1-methylxanthine
- NSAID
- nonsteroidal antiinflammatory drug
- P
- postnatal day
- PG
- prostaglandin
- PTGS
- prostaglandin-endoperoxide synthase.
References
- 1. Flanagan AM, Chambers TJ. Stimulation of bone nodule formation in vitro by prostaglandins E1 and E2. Endocrinology. 1992;130:443–448. [DOI] [PubMed] [Google Scholar]
- 2. Olney RC, Mougey EB. Expression of the components of the insulin-like growth factor axis across the growth-plate. Mol Cell Endocrinol. 1999;156:63–71. [DOI] [PubMed] [Google Scholar]
- 3. Sims NA, Clément-Lacroix P, Da Ponte F, et al. Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest. 2000;106:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olney RC, Wang J, Sylvester JE, Mougey EB. Growth factor regulation of human growth plate chondrocyte proliferation in vitro. Biochem Biophys Res Commun. 2004;317:1171–1182. [DOI] [PubMed] [Google Scholar]
- 5. Hutchison MR, Bassett MH, White PC. Insulin-like growth factor-I and fibroblast growth factor, but not growth hormone, affect growth plate chondrocyte proliferation. Endocrinology. 2007;148:3122–3130. [DOI] [PubMed] [Google Scholar]
- 6. Hutchison MR. BDNF alters ERK/p38 MAPK activity ratios to promote differentiation in growth plate chondrocytes. Mol Endocrinol. 2012;26:1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dueckers G, Guellac N, Arbogast M, et al. Evidence and consensus based GKJR guidelines for the treatment of juvenile idiopathic arthritis. Clin Immunol. 2012;142:176–193. [DOI] [PubMed] [Google Scholar]
- 8. DeWitt EM, Kimura Y, Beukelman T, et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2012;64:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DuBois RN, Awad J, Morrow J, Roberts LJ, 2nd, Bishop PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-α and phorbol ester. J Clin Invest. 1994;93:493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fletcher BS, Kujubu DA, Perrin DM, Herschman HR. Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J Biol Chem. 1992;267:4338–4344. [PubMed] [Google Scholar]
- 11. Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA. 1992;89:7384–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weinreb M, Suponitzky I, Keila S. Systemic administration of an anabolic dose of PGE2 in young rats increases the osteogenic capacity of bone marrow. Bone. 1997;20:521–526. [DOI] [PubMed] [Google Scholar]
- 13. Igarashi K, Hirafuji M, Adachi H, Shinoda H, Mitani H. Role of endogenous PGE2 in osteoblastic functions of a clonal osteoblast-like cell, MC3T3-E1. Prostaglandins Leukot Essent Fatty Acids. 1994;50:169–172. [DOI] [PubMed] [Google Scholar]
- 14. Wang X, Li F, Fan C, Wang C, Ruan H. Analysis of isoform specific ERK signaling on the effects of interleukin-1β on COX-2 expression and PGE2 production in human chondrocytes. Biochem Biophys Res Commun. 2010;402:23–29. [DOI] [PubMed] [Google Scholar]
- 15. Heinecke LF, Grzanna MW, Au AY, Mochal CA, Rashmir-Raven A, Frondoza CG. Inhibition of cyclooxygenase-2 expression and prostaglandin E2 production in chondrocytes by avocado soybean unsaponifiables and epigallocatechin gallate. Osteoarthritis Cartilage. 2010;18:220–227. [DOI] [PubMed] [Google Scholar]
- 16. Di Battista JA, Doré S, Martel-Pelletier J, Pelletier JP. Prostaglandin E2 stimulates incorporation of proline into collagenase digestible proteins in human articular chondrocytes: identification of an effector autocrine loop involving insulin-like growth factor I. Mol Cell Endocrinol. 1996;123:27–35. [DOI] [PubMed] [Google Scholar]
- 17. Schwartz Z, Swain LD, Kelly DW, Brooks B, Boyan BD. Regulation of prostaglandin E2 production by vitamin D metabolites in growth zone and resting zone chondrocyte cultures is dependent on cell maturation. Bone. 1992;13:395–401. [DOI] [PubMed] [Google Scholar]
- 18. Lowe GN, Fu YH, McDougall S, et al. Effects of prostaglandins on deoxyribonucleic acid and aggrecan synthesis in the RCJ 3.1C5.18 chondrocyte cell line: role of second messengers. Endocrinology. 1996;137:2208–2216. [DOI] [PubMed] [Google Scholar]
- 19. Moreno-Rubio J, Herrero-Beaumont G, Tardio L, Alvarez-Soria MA, Largo R. Nonsteroidal antiinflammatory drugs and prostaglandin E(2) modulate the synthesis of osteoprotegerin and RANKL in the cartilage of patients with severe knee osteoarthritis. Arthritis Rheum. 2010;62:478–488. [DOI] [PubMed] [Google Scholar]
- 20. Marcelon G, Cros J, Guiraud R. Activity of anti-inflammatory drugs on an experimental model of osteoarthritis. Agents Actions. 1976;6:191–194. [DOI] [PubMed] [Google Scholar]
- 21. Fontagne J, Loizeau M, Adolphe M, Lechat P. Effect of indomethacin on collagen biosynthesis by rabbit articular chondrocytes in monolayer cultures. Int J Tissue React. 1984;6:233–241. [PubMed] [Google Scholar]
- 22. Bassleer C, Magotteaux J, Geenen V, Malaise M. Effects of meloxicam compared to acetylsalicylic acid in human articular chondrocytes. Pharmacology. 1997;54:49–56. [DOI] [PubMed] [Google Scholar]
- 23. Dingle JT. The effects of NSAID on the matrix of human articular cartilages. Z Rheumatol. 1999;58:125–129. [DOI] [PubMed] [Google Scholar]
- 24. Chang JK, Wu SC, Wang GJ, Cho MH, Ho ML. Effects of non-steroidal anti-inflammatory drugs on cell proliferation and death in cultured epiphyseal-articular chondrocytes of fetal rats. Toxicology. 2006;228:111–123. [DOI] [PubMed] [Google Scholar]
- 25. Iimoto S, Watanabe S, Takahashi T, Shimizu A, Yamamoto H. The influence of Celecoxib on matrix synthesis by chondrocytes under mechanical stress in vitro. Int J Mol Med. 2005;16:1083–1088. [PubMed] [Google Scholar]
- 26. Jeffrey JE, Aspden RM. Cyclooxygenase inhibition lowers prostaglandin E2 release from articular cartilage and reduces apoptosis but not proteoglycan degradation following an impact load in vitro. Arthritis Res Ther. 2007;9:R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alvarez-Soria MA, Herrero-Beaumont G, Moreno-Rubio J, Calvo E, Santillana J, Egido J, Largo R. Long-term NSAID treatment directly decreases COX-2 and mPGES-1 production in the articular cartilage of patients with osteoarthritis. Osteoarthritis Cartilage. 2008;16:1484–1493. [DOI] [PubMed] [Google Scholar]
- 28. Brochhausen C, Neuland P, Kirkpatrick CJ, Nusing RM, Klaus G. Cyclooxygenases and prostaglandin E2 receptors in growth plate chondrocytes in vitro and in situ–prostaglandin E2 dependent proliferation of growth plate chondrocytes. Arthritis Res Ther. 2006;8:R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hutchison MR, Bassett MH, White PC. SCF, BDNF, and Gas6 are regulators of growth plate chondrocyte proliferation and differentiation. Mol Endocrinol. 2010;24:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sylvia VL, Del Toro F, Jr, Hardin RR, Dean DD, Boyan BD, Schwartz Z. Characterization of PGE(2) receptors (EP) and their role as mediators of 1α,25-(OH)(2)D(3) effects on growth zone chondrocytes. J Steroid Biochem Mol Biol. 2001;78:261–274. [DOI] [PubMed] [Google Scholar]
- 31. Li TF, Zuscik MJ, Ionescu AM, et al. PGE2 inhibits chondrocyte differentiation through PKA and PKC signaling. Exp Cell Res. 2004;300:159–169. [DOI] [PubMed] [Google Scholar]
- 32. Welting TJ, Caron MM, Emans PJ, et al. Inhibition of cyclooxygenase-2 impacts chondrocyte hypertrophic differentiation during endochondral ossification. Eur Cell Mater. 2011;22:420–436; discussion 436–427. [DOI] [PubMed] [Google Scholar]
- 33. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. [DOI] [PubMed] [Google Scholar]
- 34. Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. [DOI] [PubMed] [Google Scholar]
- 35. Blanco FJ, Guitian R, Moreno J, de Toro FJ, Galdo F. Effect of antiinflammatory drugs on COX-1 and COX-2 activity in human articular chondrocytes. J Rheumatol. 1999;26:1366–1373. [PubMed] [Google Scholar]
- 36. Adams SS, Bough RG, Cliffe EE, et al. Some aspects of the pharmacology, metabolism, and toxicology of ibuprofen. I. Pharmacology and metabolism. Rheumatol Phys Med. 1970;10(suppl 10):19–26. [DOI] [PubMed] [Google Scholar]
- 37. Spangler RS. Cyclooxygenase 1 and 2 in rheumatic disease: implications for nonsteroidal anti-inflammatory drug therapy. Semin Arthritis Rheum. 1996;26:435–446. [DOI] [PubMed] [Google Scholar]
- 38. Penning TD, Talley JJ, Bertenshaw SR, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib). J Med Chem. 1997;40:1347–1365. [DOI] [PubMed] [Google Scholar]
- 39. van Ryn J, Pairet M. Selective cyclooxygenase-2 inhibitors: pharmacology, clinical effects and therapeutic potential. Expert Opin Investig Drugs. 1997;6:609–614. [DOI] [PubMed] [Google Scholar]
- 40. McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA. 1999;96:272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beharry KD, Modanlou HD, Hasan J, et al. Comparative effects of early postnatal ibuprofen and indomethacin on VEGF, IGF-I, and GH during rat ocular development. Invest Ophthalmol Vis Sci. 2006;47:3036–3043. [DOI] [PubMed] [Google Scholar]
- 42. Sörgel F, Fuhr U, Minic M, et al. Pharmacokinetics of ibuprofen sodium dihydrate and gastrointestinal tolerability of short-term treatment with a novel, rapidly absorbed formulation. Int J Clin Pharmacol Ther. 2005;43:140–149. [DOI] [PubMed] [Google Scholar]
- 43. Brown RD, Wilson JT, Kearns GL, Eichler VF, Johnson VA, Bertrand KM. Single-dose pharmacokinetics of ibuprofen and acetaminophen in febrile children. J Clin Pharmacol. 1992;32:231–241. [DOI] [PubMed] [Google Scholar]
- 44. Paulson SK, Vaughn MB, Jessen SM, et al. Pharmacokinetics of celecoxib after oral administration in dogs and humans: effect of food and site of absorption. J Pharmacol Exp Ther. 2001;297:638–645. [PubMed] [Google Scholar]
- 45. Macrae VE, Ahmed SF, Mushtaq T, Farquharson C. IGF-I signalling in bone growth: inhibitory actions of dexamethasone and IL-1β. Growth Horm IGF Res. 2007;17:435–439. [DOI] [PubMed] [Google Scholar]
- 46. Yonekura A, Osaki M, Hirota Y, et al. Transforming growth factor-β stimulates articular chondrocyte cell growth through p44/42 MAP kinase (ERK) activation. Endocr J. 1999;46:545–553. [DOI] [PubMed] [Google Scholar]
- 47. Ciarmatori S, Kiepe D, Haarmann A, Huegel U, Tönshoff B. Signaling mechanisms leading to regulation of proliferation and differentiation of the mesenchymal chondrogenic cell line RCJ3.1C5.18 in response to IGF-I. J Mol Endocrinol. 2007;38:493–508. [DOI] [PubMed] [Google Scholar]
- 48. Fortier I, Patry C, Lora M, Samadfan R, de Brum-Fernandes AJ. Immunohistochemical localization of the prostacyclin receptor (IP) human bone. Prostaglandins Leukot Essent Fatty Acids. 2001;65:79–83. [DOI] [PubMed] [Google Scholar]
- 49. Burgering BM, Pronk GJ, van Weeren PC, Chardin P, Bos JL. cAMP antagonizes p21ras-directed activation of extracellular signal-regulated kinase 2 and phosphorylation of mSos nucleotide exchange factor. EMBO J. 1993;12:4211–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262:1069–1072. [DOI] [PubMed] [Google Scholar]
- 51. Graves LM, Bornfeldt KE, Raines EW, et al. Protein kinase A antagonizes platelet-derived growth factor-induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells. Proc Natl Acad Sci USA. 1993;90:10300–10304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Frödin M, Peraldi P, Van Obberghen E. Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J Biol Chem. 1994;269:6207–6214. [PubMed] [Google Scholar]
- 53. Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. [DOI] [PubMed] [Google Scholar]
- 54. Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. [DOI] [PubMed] [Google Scholar]
- 55. Yao H, York RD, Misra-Press A, Carr DW, Stork PJ. The cyclic adenosine monophosphate-dependent protein kinase (PKA) is required for the sustained activation of mitogen-activated kinases and gene expression by nerve growth factor. J Biol Chem. 1998;273:8240–8247. [DOI] [PubMed] [Google Scholar]
- 56. Espinosa M, Gottlieb BS. Juvenile idiopathic arthritis. Pediatr Rev. 2012;33:303–313. [DOI] [PubMed] [Google Scholar]
- 57. Ji P, Chowdhury BA, Yim S, Sahajwalla CG. Dosing regimen determination for juvenile idiopathic arthritis: a review of studies during drug development. J Pharm Sci. 2012;101:2621–2634. [DOI] [PubMed] [Google Scholar]
- 58. Kiely PD, Brown AK, Edwards CJ, et al. Contemporary treatment principles for early rheumatoid arthritis: a consensus statement. Rheumatology (Oxford). 2009;48:765–772. [DOI] [PubMed] [Google Scholar]
- 59. Adebajo A non-steroidal anti-inflammatory drugs for the treatment of pain and immobility-associated osteoarthritis: consensus guidance for primary care. BMC Fam Pract. 2012;13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang L, Jiang L, Sun Q, et al. Prostaglandin E2 enhances mitogen-activated protein kinase/Erk pathway in human cholangiocarcinoma cells: involvement of EP1 receptor, calcium and EGF receptors signaling. Mol Cell Biochem. 2007;305:19–26. [DOI] [PubMed] [Google Scholar]
- 61. Krysan K, Reckamp KL, Dalwadi H, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005;65:6275–6281. [DOI] [PubMed] [Google Scholar]
- 62. Pozzi A, Yan X, Macias-Perez I, et al. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem. 2004;279:29797–29804. [DOI] [PubMed] [Google Scholar]
- 63. Sasaki E, Tominaga K, Watanabe T, et al. COX-2 is essential for EGF induction of cell proliferation in gastric RGM1 cells. Dig Dis Sci. 2003;48:2257–2262. [DOI] [PubMed] [Google Scholar]
- 64. Chagin AS, Vuppalapati KK, Kobayashi T, et al. G-protein stimulatory subunit α and Gq/11α G-proteins are both required to maintain quiescent stem-like chondrocytes. Nat Commun. 2014;5:3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Linglart A, Fryssira H, Hiort O, et al. PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab. 2012;97:E2328–E2338. [DOI] [PubMed] [Google Scholar]
- 66. Abramovitz M, Adam M, Boie Y, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. [DOI] [PubMed] [Google Scholar]
- 67. Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bishop-Bailey D, Hla T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Δ12, 14-prostaglandin J2. J Biol Chem. 1999;274:17042–17048. [DOI] [PubMed] [Google Scholar]
- 69. Reginato MJ, Krakow SL, Bailey ST, Lazar MA. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J Biol Chem. 1998;273:1855–1858. [DOI] [PubMed] [Google Scholar]
- 70. Lim H, Dey SK. A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology. 2002;143:3207–3210. [DOI] [PubMed] [Google Scholar]
- 71. Schwartz Z, Gilley RM, Sylvia VL, Dean DD, Boyan BD. The effect of prostaglandin E2 on costochondral chondrocyte differentiation is mediated by cyclic adenosine 3′,5′-monophosphate and protein kinase C. Endocrinology. 1998;139:1825–1834. [DOI] [PubMed] [Google Scholar]
- 72. Langenbach R, Morham SG, Tiano HF, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. [DOI] [PubMed] [Google Scholar]
- 73. Morham SG, Langenbach R, Loftin CD, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. [DOI] [PubMed] [Google Scholar]
- 74. Dinchuk JE, Car BD, Focht RJ, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. [DOI] [PubMed] [Google Scholar]
- 75. Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. [DOI] [PubMed] [Google Scholar]
- 76. Gerits N, Kostenko S, Shiryaev A, Johannessen M, Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cell Signal. 2008;20:1592–1607. [DOI] [PubMed] [Google Scholar]