

Abstract
One would assume that the anti-inflammatory activity of α1-anti-trypsin (AAT) is the result of inhibiting neutrophil enzymes. However, AAT exhibits tolerogenic activities that are difficult to explain by serine-protease inhibition or by reduced inflammatory parameters. Targets outside the serine-protease family have been identified, supporting the notion that elastase inhibition, the only functional factory release criteria for clinical-grade AAT, is over-emphasized. Non-obvious developments in the understanding of AAT biology disqualify it from being a straightforward anti-inflammatory agent: AAT does not block dendritic cell activities, nor does it promote viral and tumour susceptibilities, stunt B lymphocyte responses or render treated patients susceptible to infections; accordingly, outcomes of elevated AAT do not overlap those attained by immunosuppression. Aside from the acute-phase response, AAT rises during the third trimester of pregnancy and also in advanced age. At the molecular level, AAT docks onto cholesterol-rich lipid-rafts and circulating lipid particles, directly binds interleukin (IL)-8, ADAM metallopeptidase domain 17 (ADAM17) and danger-associated molecular pattern (DAMP) molecules, and its activity is lost to smoke, high glucose levels and bacterial proteases, introducing a novel entity – ‘relative AAT deficiency’. Unlike immunosuppression, AAT appears to help the immune system to distinguish between desired responses against authentic threats, and unwanted responses fuelled by a positive feedback loop perpetuated by, and at the expense of, inflamed injured innocent bystander cells. With a remarkable clinical safety record, AAT treatment is currently tested in clinical trials for its potential benefit in a variety of categorically distinct pathologies that share at least one common driving force: cell injury.
Keywords: acute-phase proteins, diabetes, transplantation
Introduction
From its name, one would assume that the activity of α1-anti-trypsin (AAT) is primarily biochemical and that its anti-inflammatory actions are chiefly the result of inhibiting inflammatory neutrophil enzymes: elastase, cathepsin G and proteinase-3 (PR-3). Indeed, individuals with genetic AAT-deficiency lack control over inflammatory mediators such as interleukin (IL)-1β, IL-6, tumour necrosis factor (TNF)-α and IL-8 1 and exhibit elevated rates of various types of vasculitis 2,3; accordingly, direct introduction of AAT to human peripheral blood mononuclear cells (PBMCs) reduces inflammatory responses 4. Thus, patients with AAT-deficiency are treated with lifelong slow-drip weekly infusions of plasma-derived affinity-purified human AAT.
During the past decade, studies have reported tolerogenic activities that are difficult to explain by serine-protease inhibition or by reduced inflammatory parameters. In addition, targets outside the serine-protease family have been identified, and evidence for protease-independent activities have accumulated, further supporting the notion that the contribution of elastase inhibition, the only functional factory release criteria for clinical-grade human AAT, is over-emphasized. In the present review, the aspect of functions independent of elastase inhibition will be discussed at length.
With these non-obvious developments in the understanding of AAT biology, it has become imperative to establish a comprehensive appreciation of the effect of AAT on the immune system, molecule-by-molecule, cell-by-cell, pathway-by-pathway.
The current search for AAT's immune system ‘footprint’ leans heavily towards deciphering the mechanism of its immune tolerance activity. Although a naturally occurring anti-inflammatory molecule, the blockade of inflammation is not sufficient to explain its effects on specific immune cells and immune pathways (Table 1). AAT does not block dendritic cell (DC) activities but, rather, diverts DCs towards a tolerogenic profile; it does not promote viral and tumour susceptibilities, suggesting that essential NK cell responses remain intact; AAT does not stunt B lymphocyte responses but rather limits antibody isotype switching; it does not render treated patients susceptible to bacterial infections and has bacterial burden-reducing properties, without killing bacteria in culture dishes. AAT not only inhibits inflammatory cytokine release, but also promotes greater levels of inflammation-driven anti-inflammatory agents. Thus, outcomes of elevated AAT levels do not overlap with those attained by anti-inflammatory agents, immunosuppression or even by serine-protease inhibition.
Table 1.
Major immune responses under AAT therapy
| Disease model | Treatment route | Outcome | Immune cells | Cell-specific outcomes | Ref. |
|---|---|---|---|---|---|
| Experimental autoimmune encephalomyelitis (EAE) | Transgenic hAAT | Increased survival, improved disease markers | T cells | Reduced CNS infiltration | 5 |
| Tregs | Expanded in spleen, blood and lymph nodes | ||||
| Islet transplantation | Exogenous, transgenic hAAT or in-vivo transfection with hAAT plasmid | Improved islet survival, development of tolerance towards islet allografts | Tregs | Expanded in blood and at graft site | 6–9 |
| Collagen-induced arthritis (CIA) | Exogenous hAAT or in-vivo transfection with hAAT plasmid | Delayed onset and ameliorated disease development | B cells | Reduced serum levels of autoimmune antibodies | 10 |
| Skin transplantation | Exogenous hAAT | Not followed | DC | Turn semi-mature with low CD40 expression, intact inducible CCR7 and intact migration to lymph nodes | 8 |
| GVHD | Exogenous AAT | Increased survival | Tregs, T cells | Increased Treg proportion and reduced T effector cells | 11 |
| Autoimmune diabetes (NOD mice) | Exogenous hAAT | Increased survival, reduced disease markers | β cells, Tcells | Reduced β cell apoptosis, reduced T cell infiltration to the pancreatic islets | 12,13 |
| Crohn's disease (SAMP-1 mice) | Exogenous hAAT | Improved disease markers | T cells, B cells | Reduced inflammation associated colon damage. Reduced lymphocyte infiltration | 14 |
| Cancer (B16 melanoma model) | Exogenous hAAT | Intact anti-cancer cell responses | NK cells | Intact NK cell degranulation and cancer cell killing | 15 |
| Antigen vaccination | Exogenous or transgenic hAAT | Not followed | B cells | Reduced B cell proliferation and antigen-specific IgG, elevated antigen-specific IgM | 16 |
AAT = α1-anti-trypsin; hAAT = human AAT; NK = natural killer; Ig = immunoglobulin; SAMP-1 = senescence accelerated mouse prone 1; NOD = non-obese diabetic; GVHD = graft-versus-host disease; CNS = central nervous system; Tregs = regulatory T cells.
Instead, AAT appears to allow responses to pathogenic threats while restraining bystander cell injury and its damaging potential to evoke self-recognizing adaptive immune responses. In this way, AAT holds its own category as a tolerogenic immune modulator that may promote the distinction between authentic and non-authentic threats.
AAT and the acute-phase response: downplaying bystander cell injury
To best grasp the intriguing profile of an AAT-augmented immune system, one could begin with dissecting the hallmark environment of the principal condition, the acute-phase response, that incorporates physiological excess in circulating AAT.
AAT is released into the circulation by liver hepatocytes in a steady-state manner, reaching circulating levels that are second only to albumin. AAT is also released in an inducible manner upon activating its inflammation-, cortisol- and hypoxia-responsive promoter. Inducible plasma levels of AAT are four- to sixfold higher than its steady-state levels, and persist for more than a week in accordance with the dynamic of each underlying trigger, be it infection, tissue injury or, interestingly, the third trimester of pregnancy and advanced age 17.
However, even during the acute-phase response, while rising AAT levels allow effective immune responses towards authentic threats, bystander cells appear to be protected. For example, in three distinct models of sterile tissue injury, one can observe protection of stress-inflicted non-functioning cells that reside in the so-called penumbra area, i.e. around the point of immediate pathological impact (as illustrated in Fig. 1). Thus, AAT allows cardiomyocyte function outside heart tissue that died due to a blocked coronary artery 18, as well as nerves outside a site that died due to cerebral artery blockade 19,20 and insulin-producing β cells directly adjacent to β cells that expired due to an immune attack or to toxin-related damage 6,7,12,21,22.
Figure 1.
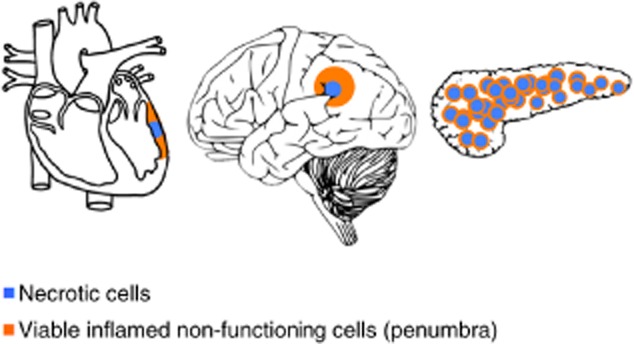
The penumbra effect illustrated. The outcome of an injured section of tissue separates into the immediate centre of cell death and a surrounding perimeter of viable cells that are stunted due to the spread of inflammation, as depicted in a blocked coronary artery (left), stroke (middle) and pancreatic islet injury (right). Blue = necrotic area; orange = viable stunted cells.
One of the functions of AAT is straightforward protease inhibition. Protease/anti-protease imbalance has, in general, deleterious effects, as recently established in autoimmune diabetes 23. In one scenario, serine-proteases activate IL-1 precursor molecules that leak out of necrotic cells 24. In another, protease-activated receptors (PARs) induce inflammation even prior to measurable cytokine release; agonists of PAR2, for example, induce neutrophil infiltration and compromise the epithelial barrier 25.
Protease inhibition holds another aspect in the biology of AAT: it is the most efficient method of terminating the activity of AAT. In contrast, oxidation of AAT is reversible under reducing conditions, such as serum. This might be part of the reason behind the observation that an AAT recombinant protein that lacks elastase inhibition capacity is several-fold more potent than native AAT in blocking inflammation 26. That said, gene therapy techniques have established that minute circulating native AAT levels that are too low to shift serum anti-elastase homeostasis can protect from inflammatory spread, suggesting that non-inhibitory AAT may still contain protective activities 7,27,28.
Some activities of AAT appear to extend beyond serine-protease inhibition. AAT inhibits caspase-3 and matrix metallopeptidase 9 (MMP-9), both belonging to separate protease families, resulting in anti-apoptotic outcomes under various conditions and in various cell types 29–31. AAT also inhibits neutrophil calpain I, with direct implications on neutrophil behaviour 32 and, to a lesser extent, caspase-1 18. The entry of AAT into cells is described later in the present review.
How can a serine-protease inhibitor inhibit other proteases? It appears that AAT binds to, rather than is cleaved by, several proteases, most typically those that are membrane-associated. For example, the inhibition of membrane-associated ADAM metallopeptidase domain 17 (ADAM17) by AAT is independent of elastase inhibition by AAT 26,33. The conservation of membrane-associated TNF-α may be considered superior to other anti-TNF-α approaches, in which the TNF-α pathway is broadly blocked. As ADAM17 also inhibits CD154 release, AAT may interfere with some aspects of the CD40 pathway, which are described below with respect to B lymphocyte responses 34.
Functions that are unrelated to protease inhibition mainly involve direct binding partners, which do not necessitate the protease-binding site of AAT. AAT disrupts neutrophil migration by binding to IL-8 33 and, accordingly, AAT-deficient individuals present with excessive lung neutrophils 35. In contrast, patients with cystic fibrosis (CF) that are treated with inhaled AAT exhibit reduced numbers of lung neutrophils and a lower degree of inflammatory markers 36. AAT also binds to retinoic acid 37, potentially interfering with inflammatory disorders 38. AAT was shown recently to bind to both TNF-α receptors, albeit at a low affinity, suggesting that it may become more relevant upon up-regulation of AAT levels, such that occur during an acute-phase response 39. The binding of AAT to gp96 and HSP70 has been described recently 40,41, and suggests a role in blunting immune responses to adjuvant-like danger-associated molecular pattern (DAMP) molecules, as illustrated in Fig. 2. The notion that human AAT (hAAT) might interact with circulating damage molecules emerged from the fact that we harbour somewhat high concentrations of circulating hAAT under normal conditions, and more so during acute-phase responses. That our bodies constantly contain injured and dying cells is a well-established clinical occurrence; we routinely measure liver enzymes, which arrive from injured hepatocytes in the absence of an apparent disease; we measure circulating lactate dehydrogenase (LDH) and free DNA that originate from a vast array of injured cells; and we measure elevated heart muscle enzymes during an acute ischaemic cardiac event in a time-dependent manner. Conversely, protective circulating molecules and cell populations are not measured routinely and we assume that homeostasis is being kept by immune-regulatory cells, anti-inflammatory mediators, inhibitory ligands, receptors and anti-proteases, such as serine protease inhibitors (SERPINs).
Figure 2.
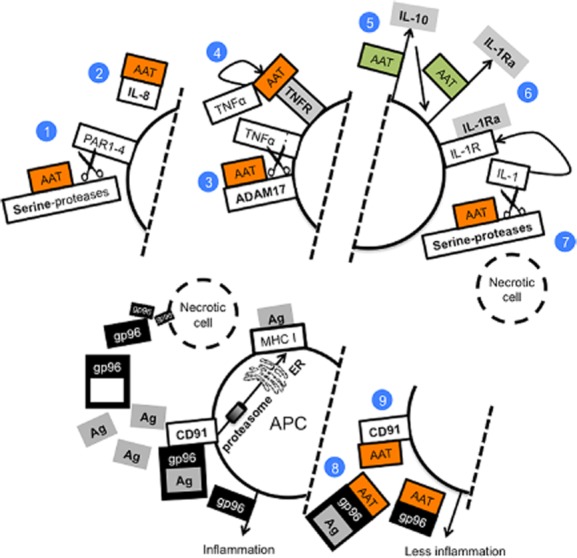
Mechanisms illustrated are selected experimentally supported concepts. (1) Inhibition of serine-proteases interferes with activation of immune-related protease-activated receptors (PARs); (2) direct binding to interleukin (IL)-8 interferes with neutrophil recruitment; (3) inhibition of ADAM metallopeptidase domain 17 (ADAM17) interferes with release of tumour necrosis factor (TNF)-α; (4) binding to TNF receptors (TNFR) at high concentrations interferes with TNF-α-induced pathways; (5) up-regulation of IL-10 release by multiple cell types exerts an anti-inflammatory environment, and joins α1-anti-trypsin (AAT) in (6) up-regulation of IL-1Ra, which will interfere with the IL-1 pathway. (7) Release of pro-IL-1β from necrotic cells results in active IL-1β upon engagement with serine-proteases, the targets of AAT. (8) Necrotic cells contribute potent adjuvants to the immune system; gp96 chaperones antigens and then binds to CD91, which promotes their processing and loading on major histocompatibility complex (MHC) class I 42. AAT was shown to both (8) bind to gp96, thus neutralizing its inflammatory activity 40, and to (9) CD91 43.
Aside from inhibiting inflammatory cytokine release, AAT increases IL-1Ra and IL-10 expression, although only when an underlying inflammatory trigger is present 44. A negative feedback loop between IL-1-induced AAT and AAT-driven IL-1Ra is thus formed. Interestingly, the IL-1Ra promoter requires the binding of the nuclear factor kappa B (NF-κB) family member p65 45. Thus, one might find reports of AAT blocking NF-κB nuclear translocation to conflict with its ability to induce IL-1Ra 46. However, Abecassis et al. have recently reported a unique p65 cellular distribution pattern and possible covalent modifications in the presence of AAT, such that allow inflammation-driven p65-mediated IL-1Ra induction without consequent NF-κB-related inflammatory gene activation 47. IL-10 induction by AAT shares similarities with IL-1Ra induction by AAT in that, typically, a resting cell would be unresponsive to added AAT. IL-10 induction is consistent with a rise in cyclic adenosine monophosphate (cAMP) levels by AAT 48,49. IL-10 will then also induce IL-1Ra production 50.
The systemic rise in AAT levels during the acute-phase response highlights its significance as an angiogenesis and wound-healing modulator, and agrees with reports of anti-viral and bacterial burden-reducing activities that are very much sought-after during a pathogen-related systemic inflammatory flare. Thus, it is not unexpected that sterile immune responses are highly responsive to AAT therapy, including lung 51 and renal 52 ischaemia–reperfusion injury-related responses. Interestingly, both lung and renal epithelial cells produce AAT during inflammatory conditions 53.
The systemic rise in AAT levels during the third trimester of pregnancy highlights its significance as a tolerogenic agent. Pregnancy complications correlate with either non-rising or non-functional AAT levels, including premature rupture of membranes (suggestive of compromised tissue integrity) and spontaneous abortions (associated with immune intolerance) 54–57. Thus, while serum AAT may present as normal in these patients, it might not be the desired protective levels expected during the third trimester. Such ‘relative’ deficiencies appear in two more important conditions: diabetes (AAT is neutralized by glycation) and bacterial sepsis (AAT is cleaved by bacterial proteases) 58.
AAT and neutrophils: selective functional blockade favours bacterial clearance and lessens tissue injury
Neutrophils are crucial in clearing bacterial and fungal infections both in the circulation and upon rapid extravasation. As stimulated neutrophils release serine-proteases, venular basement membranes that contain matrix protein low-expression regions act as serine-protease-sensitive exit points for neutrophils 59. Indeed, AAT reduces neutrophil chemotaxis across multiple stimuli 60,61. Unexpectedly, a recombinant AAT that lacks the ability to inhibit elastase is also able to reduce neutrophil infiltration 60. With these attributes it might be predicted that AAT paralyzes neutrophils, yet this appears not to be the case.
AAT becomes inactive upon direct engagement with neutrophil-derived serine-proteases, as well as neutrophil metalloproteases 62,63 and collagenases 64. AAT is neutralized further by oxygen radicals 65. Thus, interstitial AAT is inactive at the very centre of a site of neutrophil activity, but inhibitory at its perimeter. AAT may thus preserve responses directed at pathogens and promote an environment of tissue-friendly neutrophils.
None the less, this does not explain bacterial load reduction in AAT studies, a particularly perplexing observation considering the phenotype of neutrophils in its presence. It appears that AAT prevents host protein cleavage targets from bacterial protease degradation: membrane complement receptor 1 (on neutrophils), C3b (on opsonized bacteria) 66,67 and CXCR1, an IL-8 receptor 68. In addition, AAT carries anti-bacterial amounts of nitric oxide once its single-surface cysteine residue is nitrosylated (illustrated in Fig. 3), as occurs when AAT passes through a nitric oxide-rich environment 69. Lastly, an unexpected observation suggests that AAT exerts an initial narrow inflammatory surge 70, a phenomenon suggested to be CD14-dependent; indeed, anti-CD14 antibody reduces lipopolysaccharide (LPS)-stimulated AAT-related early cytokine release 71.
Figure 3.
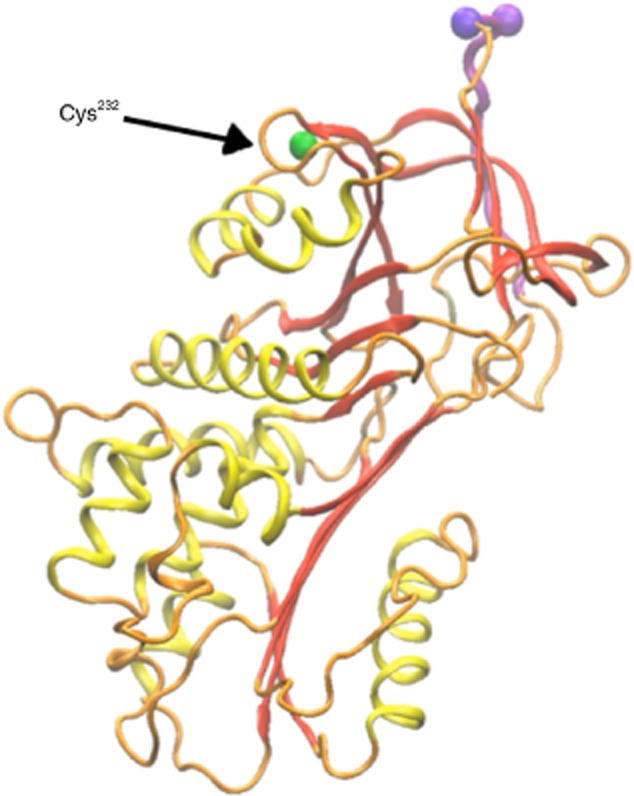
In-silico depiction of the single surface cysteine residue within the sequence of α1-anti-trypsin (AAT). Orange = wire-diagram of the protein-sequence with secondary structures highlighted in yellow and red, and the protease-binding domain in purple. Non-exposed amino acids that are positioned under the surface of the molecule are represented by white beads. Green = cysteine at position 232.
AAT modifies macrophage and DC phenotypes towards a tolerogenic profile
AAT docks onto monocytic cholesterol-rich lipid-rafts 72. In fact, it is interesting to note that circulating AAT is detected bound to LDL and high-density lipoprotein (HDL) particles 73,74. This phenomenon may provide an initial unifying paradigm for the observed effects of AAT on immune cells, according to which lipid-raft-related activities would be inhibited by AAT while lipid-raft-independent pathways are left intact 7,8. For example, macrophage and DC lipid-rafts are home to Toll-like receptor (TLR)-2 and TLR-4, both down-regulated by AAT 71. Accordingly, AAT reduces LPS-induced cytokine and nitric oxide release, as well as LPS-induced lethality in vivo 46,75.
Interestingly, macrophages and neutrophils express AAT 76,77. Macrophages employ a macrophage-specific promoter, located approximately 2 Kb upstream of the hepatocyte-specific promoter. Activation of the two promoters is mutually exclusive; the macrophage promoter is silent in hepatocytes and the hepatocyte promoter is silent in macrophages. Conversely, neutrophils release stored AAT. The implications of macrophage- and neutrophil-derived AAT are under investigation.
AAT has a consistent effect on DCs, as it has on macrophages, with a single interesting exception: inflammation-driven cell migration. DC migration is not inhibited by AAT. Ozeri et al. reported that stimulated DCs express lower levels of CD40, CD86 and major histocompatibility complex (MHC) class II in the presence of AAT, produce more IL-10 and promote the expansion of CD4+ forkhead box protein 3 (FoxP3+) T cells 8. However, as DCs must reach the draining lymph node in order to skew an alloantigen-responsive T cell towards a regulatory T cell (Treg), Ozeri et al. were concerned that AAT might interfere with inflammation-driven DC migration. However, their study shows that AAT-treated stimulated DCs maintain migratory capacity, rapidly reaching lymph nodes while containing elevated levels of IL-10 and apparently communicating an IL-10-expressing phenotype to local DCs. This phenomenon was corroborated by the finding of uninterrupted inducible CCR7 levels on the surface of DCs, such that allow their migration towards CCL19/21-rich draining lymph nodes. Together with vascular endothelial growth factor (VEGF), CCR7 represents an inflammation-driven molecule that is inducible, despite the predominating anti-inflammatory conditions exerted by AAT 78.
Another aspect of monocyte immunomodulation arose from one of the recent autoimmune diabetes clinical trials that tested the outcomes of AAT therapy in recent-onset autoimmune diabetes patients 79. AAT was infused once weekly for 8 consecutive weeks and glycaemic parameters were followed for 18 months. According to the study, almost half the participants displayed greater insulin production compared to their individual entry levels, although a placebo-controlled study is required in order to support these outcomes. None the less, the beneficial effect of AAT in specific patients correlated with less inflammatory circulating myeloid cells. This is a highly intriguing aspect of AAT therapy, as the changes appeared only 9 months after the AAT infusions, separating its vastly reported immediate islet-protective attributes 80 from unrelated immune responses. Specifically, this particular time-point agrees with the concept that bone marrow myeloid cells employ PR-3 to complete one of several differentiation stages 81. PR-3 is a membrane-associated elastin degrading protease that is also known to be the antigen recognized by cytoplasmic anti-neutrophil cytoplasmic autoantibodies (c-ANCA); it is readily inhibited by AAT 82. Circulating c-ANCA levels correlate with neutrophil activation in Wegener's disease (WG); interestingly, AAT-deficient WG patients exhibit a significantly more aggressive form of vasculitis compared to other WG patients 83. It has been suggested that a blockade in PR-3 may also affect the differentiation of other myeloid cells, with implications concerning the aggressiveness of subsequent circulating myeloid cells 81. The influence of AAT on the bone marrow cell profile is discussed further in a specific section in the present review.
AAT and B lymphocytes: imposing a divergence of activation pathways
Information on AAT and B lymphocytes is scarce. Hadzic et al. show that tonsillar B lymphocytes stimulated with Moraxella catarrhalis exhibit reduced proliferation rates and diminished IL-6 release 84. AAT might thus be considered to be an inhibitor of B lymphocyte responses; however, this is not the case.
Mizrahi et al. have recently dissected a spectrum of B lymphocyte responses in the presence of AAT 16. According to their findings, AAT selectively reduces B lymphocyte activation-related properties, such as proliferation and activation marker expression following stimulation with both T cell-dependent and -independent antigens in vitro, as well as in an allogeneic skin transplantation model in vivo. Unexpectedly, antigen-specific antibody production remained intact in the presence of elevated AAT levels, yet the profile achieved displayed an abrupt reciprocal change in antibody isotypes, resulting in an IgM-high/IgG-low profile. Indeed, isotype switching is tightly regulated by CD40, a pathway readily inhibited by AAT in these and other studies.
The authors also establish that AAT expands IL-10-producing B lymphocytes, a regulatory cell population considered to play a role in alloantigen tolerance 85. In addition, their study shows that these cells are required for AAT to expand Tregs 16. Although further studies are required, B lymphocytes appear to be important targets of AAT, especially in light of their involvement in the pathogenesis of autoimmune diseases and allograft rejection.
AAT and NK cell functions: one of the few stones left unturned
While natural killer (NK) cells partake in the immune response to viral infections and to tumour cells, they also appear to play a role in the pathogenesis of autoimmune diabetes and in the response to allogeneic transplants 53,86,87. Together with the positive outcomes of AAT treatment in both these unwanted immune processes, NK cells represent a potential cellular target of AAT. Guttman et al. have recently established an effect of AAT on DCs that may influence NK cell activation indirectly 15. Importantly, while direct NK cell activation remained intact in the presence of AAT, DC-derived IL-15-related NK cell responses were altered; indeed, the group described that DCs presented less IL-15 in the presence of AAT.
As NK cells are of an innate nature, one would suspect that AAT would interfere with NK cell activities, yet the rates of tumour incidences are actually lower in AAT-treated individuals 88–91. Evidence for anti-viral and anti-tumour effects of AAT are consistent across research models 92–94 and AAT-treated deficient individuals exemplify this upon comparison with AAT-untreated-deficient patients 95,96. Thus, it is hypothesized that the effect of AAT on NK cells is suppressive when the targets are healthy cells and the immune signal is primarily antigen-presenting cell (APC)-mediated, and is permissive upon encounter with viral-infected or transformed cells.
AAT and viral infections: merging anti-inflammatory and anti-viral properties
In an in-vitro study performed on primary Rhesus monkey kidney cells, AAT inhibited H1N1 influenza virus cell infection; in mice, upon infection with the virus, AAT provided lower mortality rates, as well as a significant decrease in baseline levels of inflammatory cytokines 97. Some aspects of the anti-viral profile exerted by AAT are related most probably to protease inhibition, inclusive both of viral and host proteases. For example, AAT prevents viral haemagglutinin activation by host serine-proteases, as well as subsequent viral infection.
The anti-viral activity of AAT may contain aspects outside protease inhibition. The fact that HIV replication in whole blood is obtained only after dilution with culture medium has raised the possibility of the presence of circulating anti-retroviral substances 98,99. Indeed, Shapiro et al. describe inhibitory outcomes when introducing AAT to HIV assays 100, and Münch et al. describe anti-viral properties in fractionated human serum as contained in a fraction subsequently sequenced and identified as AAT 101. Reports since then have described AAT as a rate-limiting factor in HIV replication 102,103, and that AAT therapy elevates CD4+ T cell numbers in HIV-infected patients 104. Apparently, an amino acid sequence within human AAT, buried under the surface of the molecule until AAT is cleaved, contains anti-viral properties 105 (illustrated in Fig. 4).
Figure 4.
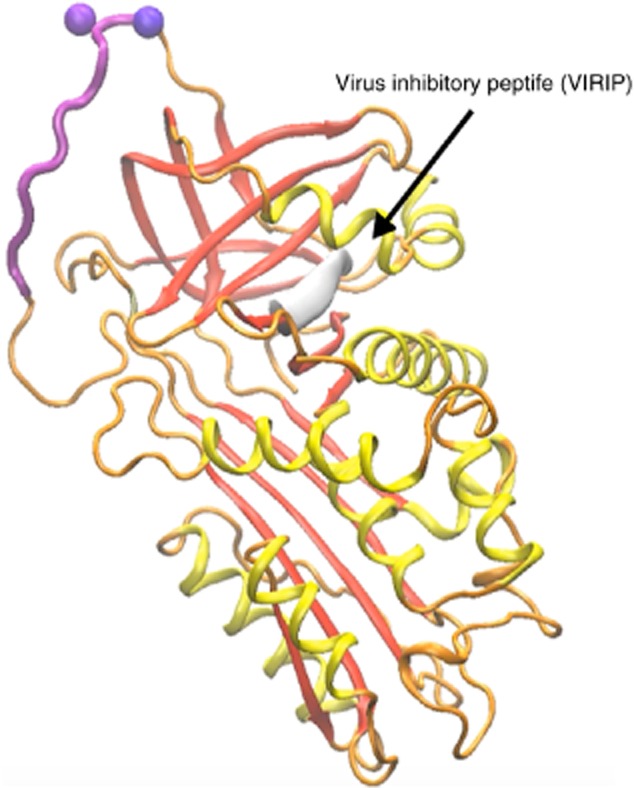
In-silico depiction of the HIV inhibiting peptide within the sequence of α1-anti-trypsin (AAT). Orange = wire-diagram of the protein sequence with secondary structures highlighted in yellow and red, and the protease-binding domain in purple. Non-exposed amino acids that are positioned under the surface of the molecule are represented by white beads. Grey sleeve = virus inhibitory peptide (VIRIP).
AAT protects epithelial and endothelial barriers
Human lung epithelial cells secrete AAT 106. While the absence of this protection in genetic AAT deficiency may result in elastase-related emphysema, it is also possible that intracelluar aggregates of mutated AAT trigger inflammation. None the less, AAT therapy arrests the development of emphysema 107, supporting the notion that the activities of AAT may indeed extend to overall protection from cell injury.
Intestinal and corneal epithelia also secrete AAT 108,109. Intestinal epithelial cells engage in a dynamic cross-talk with the intestinal immune system, helping in the discrimination between invasive pathogens and harmless antigens. Because IL-1β causes an increase in intestinal epithelial tight junction permeability, local AAT has the potential to counteract loss of intestinal barrier function. In support of this, AAT therapy provides dramatic outcomes in an animal model of Crohn's disease, and intestinal biopsies from patients with Crohn's disease demonstrate lack of local AAT expression 14. The blood–brain barrier is another example of barrier protection by AAT, as demonstrated in an in-vivo stroke model 20.
AAT emerges as a pivotal modulator during the wound-healing process, facilitating rapid wound size reduction in primary human fibroblasts 78. Wound healing requires inflammation, but untamed inflammation can cause cell injury. Neutrophil infiltration into wound sites exemplifies this duality; neutrophils are abundant in the early phase of a wound, in accordance with their decontamination capacity, although their proteases degrade components of the extracellular matrix, compromising the scaffold required for subsequent infiltrating reconstructive cells. Based on the ability of AAT to allow bacterial killing, AAT may thus be considered to be diverting damaging activity at a wound site towards greater decontamination power, with minimal destructive outcomes.
Macrophages facilitate the removal of neutrophils, promoting angiogenesis, fibroblast proliferation and extracellular matrix synthesis. The phenotype of such macrophages includes expression of anti-inflammatory mediators and the production of growth factors, as reinforced by AAT 110. Other cells that partake in wound-healing also appear to relate to AAT in a beneficial manner. AAT promotes DC production of TGF-β and IL-10 8, and its anti-apoptotic activities may facilitate the survival of young labile migrating fibroblasts, myocytes and endothelial cells 111–113. The ability of AAT to neutralize DAMPs allows the wound site to continue its recovery process while downplaying adjuvant-like excitation of the adaptive immune response.
Unlike epithelial cells, endothelial cells uptake AAT by pinocytosis 114. As the promoter of AAT is hypoxia-responsive, AAT is produced in a context-specific manner by local cells and subsequently advances early VEGF expression; AAT also inhibits expression of anti-angiogenic factors and prevents degradation of VEGF 78. Interestingly, emphysema can be replicated in animal models by blocking VEGF 29, suggesting that the pathogenesis of emphysema in AAT deficiency may also be attributed to capillary bed collapse.
The unexpected response of the adaptive immune system to AAT
AAT skews the T cell population towards Tregs in various animal models, but never in a single-cell T lymphocyte culture 5–7,10–12,27,28. Indeed, it is only when AAT-treated DCs are introduced to naive T cells that the expansion of Tregs is found 8. Unlike typical immunosuppression, without a direct inhibitory effect on IL-2 AAT appears to at least allow for inducible Treg expansion 6,12. In addition, Treg expansion requires a particular cytokine milieu afforded by AAT, i.e. low IL-1β, low IL-6 and high TGF-β levels. The increase in IL-10 production by DCs and B lymphocytes may also contribute to Treg expansion in multi-cellular immune compartments 8,11,16.
These attributes may play a role in protection of islets from alloimmune and autoimmune responses, but they are less fitting for the setting-up of the xenoimmune response. Accordingly, Ashkenazi et al. show that AAT alone is incapable of withholding the xenoimmune response 115. However, when assessing the combination of AAT and temporary T cell depletion, a treatment that exerts duration-limited results as monotherapy, xenografts exhibited significantly extended survival rates. The mechanism behind such a synergy may relate to the prominent role of AAT-responsive macrophages in the CD4+ T cell-orientated xenoimmune response. It is also possible that post-depletion T cell repopulation, suggested to preferentially favour regulatory cells, was further skewed towards Treg expansion in the presence of AAT.
Serving as an important example of an unwanted alloimmune response, graft-versus-host disease (GVHD) presents as a pathology with a surprisingly positive response to AAT 11,116. The major targets of AAT in the context of GVHD would most probably be the severely injured gastrointestinal and/or skin epithelial cells that perpetuate GVHD. Given that AAT does not block T cell responses directly, AAT preserves graft-versus-tumour and graft-versus-leukaemia activities 11,116, providing the premise for two clinical trials that currently examine AAT therapy for individuals with treatment-resistant GVHD.
Immunosuppression and AAT: predicted outcomes of combination therapies
Perhaps the most striking difference between AAT and immunosuppression is that the latter holds the risk of infectious and cancerous complications, and is usually a lifelong treatment. In contrast, lifelong administration of AAT at doses that reach high plasma levels, even in AAT-deficient patients, has rendered no treated individual immunocompromised. Also, the emergence of immune tolerance has rarely been experienced in humans using immunosuppression, while the outcomes of an 8-week-long AAT treatment protocol in patients with recent onset autoimmune diabetes appears to have provided a hint of persistent immunomodulation, although one should assess these outcomes together with the currently ongoing placebo-controlled studies that examine AAT and autoimmune diabetes 79,80.
The possibility of combination therapy has been gaining attention in recent years. Based on the little we know of the mechanisms employed by AAT, it would most probably be counterproductive to combine AAT with an IL-2 blocker, as IL-2 is required for promoting Treg expansion. Similarly, it would probably be futile to combine AAT with TNF-α blockers, as these approaches are predicted to be additive, at best. Similarly, co-stimulation blockade is expected to be additive, considering that AAT also inhibits CD40 pathway and reduces inducible CD80/CD86 levels; this combination was tested recently by Ashkenazi et al. 115, establishing lack of synergy. Conversely, the combination of AAT with temporary T cell depletion (or in the clinical set-up, anti-thymocyte globulin) has provided a clear synergy, most probably as two independent non-overlapping areas of the immune system are targeted, and the elective immunocyte repopulation activity serves as a window of opportunity for re-educating the immune response.
Discussion
The recent exponential growth in studies that examine the relevance of AAT to immune disorders in non-AAT-deficient individuals represents an authentic curiosity as to the capabilities of this evolutionarily finely sculpted molecule. When one learns about the works of AAT one gains insight into pivotal immune processes through a fresh lens, that of exploiting a natural resource, a product of biology, a regimen that is not foreign to our cells and molecules that have co-evolved alongside our complex defence systems – from age-old chemokines and inflammatory agents, to innate immune cells and, finally, to the late-appearing adaptive immune system. Indeed, the lack of a direct effect on T cells agrees with the concept that AAT preceded T cells in evolution, dominating eras of biology in which the interests were those such as pathogen invasion, inflammation, cell injury and wound healing.
Harnessing AAT to modulate the immune system was met initially with scepticism. How can a temporary rise in the levels of a constitutively expressed protein provide such benefits? How can an anti-protease relate to such complexities as T lymphocyte responses in the context of autoimmune diseases and allograft rejection? Sporadic reports were apparently present all along. In 1978, Nature reported that AAT acts on innate immunocytes but not on T lymphocytes; in 1983 there were reports of improved prognosis in patients who were admitted with acute myocardial infarction (MI), provided that they were in the midst of a measurable acute-phase response 117; in 1987, there were reports of AAT being inactivated by excessive glycation in patients with autoimmune diabetes.
The biology of AAT, representing a one-gene disease, and its secretion pathway being exemplary in its category, as well as the attempts to correct intracellular AAT aggregates in liver cells of patients with genetic AAT deficiency and the efforts to supply AAT-deficient individuals with recombinant forms of AAT – from forms of AAT produced by transgenic tobacco leaves 118 and tomatoes 119 to transgenic rice 120 and sheep 121 – have dominated scientific literature in the past half-century. It is only quite recently that publications relating to a protective role for AAT in autoimmune diabetes, multiple sclerosis, rheumatoid arthritis, allo- and xenograft transplantation, Crohn's diseases, GVHD, ischaemia–reperfusion injury, stroke, acute MI and bacterial and viral infections have surfaced. Relative AAT deficiency has only recently received attention, raising the proposition that novel clinical indications for AAT therapy might be recognized in the near future 122.
Thirty years after the first infusion of AAT was administered to patients with genetic AAT deficiency, it is being evaluated in non-deficient individuals 97,122–124; it is being tested for controlling CF inflammation in an inhaler form; in recent-onset autoimmune diabetes under six clinical trials; and its benefit in treatment-resistant GVHD in two clinical trials and during acute MI in one clinical trial in which patients receive an AAT drip at the limited but critical period of door-to-balloon time.
It will require elaborate structure–function studies, cell- and pathway-specific analyses and extensive functional arrays in order to identify its mechanisms of action, particularly in light of the comprehensive efforts to generate non-plasma-derived formulations of AAT in order to supply the anticipated pharmaceutical rise in demand 26,60,125. In the meantime, elevated levels of AAT during acute-phase responses are perhaps the best indicator for its relevance as a naturally occurring protective agent during excessive inflammatory conditions, one that is skilled at helping the immune system to distinguish between the desired response against an authentic threat and the unwanted response fuelled by the positive feedback loop of inflamed injured cells, further perpetuated by, and at the expense of, injured innocent bystander cells.
Acknowledgments
The authors thank Dr. Stas Engel for providing computerized three-dimensional prediction images.
Disclosure
The authors declare no competing interests.
References
- Malerba M, Ricciardolo F, Radaeli A. Neutrophilic inflammation and IL-8 levels in induced sputum of alpha-1-antitrypsin PiMZ subjects. Thorax. 2006;61:129–133. doi: 10.1136/thx.2005.043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad A, Segelmark M. Primary systemic vasculitis with severe alpha1-antitrypsin deficiency revisited. Scand J Rheumatol. 2014;43:242–245. doi: 10.3109/03009742.2013.846405. [DOI] [PubMed] [Google Scholar]
- Stone H, Pye A, Stockley RA. Disease associations in alpha-1-antitrypsin deficiency. Respir Med. 2014;108:338–343. doi: 10.1016/j.rmed.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Pott GB, Chan ED, Dinarello CA, Shapiro L. Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol. 2009;85:886–895. doi: 10.1189/jlb.0208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Shahaf G, Ozeri E. Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis. 2011;26:107–113. doi: 10.1007/s11011-011-9239-9. [DOI] [PubMed] [Google Scholar]
- Lewis EC, Mizrahi M, Toledano M. Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA. 2008;105:16236–16241. doi: 10.1073/pnas.0807627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahaf G, Moser H, Ozeri E, Mizrahi M, Abecassis A, Lewis EC. Alpha-1-antitrypsin gene delivery reduces inflammation, increases T-regulatory cell population size and prevents islet allograft rejection. Mol Med. 2011;17:1000–1011. doi: 10.2119/molmed.2011.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozeri E, Mizrahi M, Shahaf G, Lewis EC. Alpha-1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol. 2012;189:146–153. doi: 10.4049/jimmunol.1101340. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yan HJ, Zhou SY. The immunoregulation effect of alpha 1-antitrypsin prolong beta-cell survival after transplantation. PLOS ONE. 2014;9:e94548. doi: 10.1371/journal.pone.0094548. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Grimstein C, Choi YK, Wasserfall CH. Alpha-1 antitrypsin protein and gene therapies decrease autoimmunity and delay arthritis development in mouse model. J Transl Med. 2011;9:21. doi: 10.1186/1479-5876-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawara I, Sun Y, Lewis EC. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci USA. 2012;109:564–569. doi: 10.1073/pnas.1117665109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulmanda M, Bhasin M, Hoffman L. Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci USA. 2008;105:16242–16247. doi: 10.1073/pnas.0808031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Lu Y, Campbell-Thompson M. Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes. 2007;56:1316–1323. doi: 10.2337/db06-1273. [DOI] [PubMed] [Google Scholar]
- Collins CB, Aherne CM, Ehrentraut SF. Alpha-1-antitrypsin therapy ameliorates acute colitis and chronic murine ileitis. Inflamm Bowel Dis. 2013;19:1964–1973. doi: 10.1097/MIB.0b013e31829292aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman O, Yossef R, Freixo-Lima G, Rider P, Porgador A, Lewis EC. Alpha1-antitrypsin modifies general NK cell interactions with dendritic cells and specific interactions with islet beta-cells in favor of protection from autoimmune diabetes. Immunology. 2014 doi: 10.1111/imm.12403. . doi: 10.1111/imm.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi M, Cal P, Rosenthal M. Human alpha1-antitrypsin modifies B-lymphocyte responses during allograft transplantation. Immunology. 2013;140:362–373. doi: 10.1111/imm.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczek L, Michalska W, Bartlomiejczyk I. Trypsin, elastase, plasmin and MMP-9 activity in the serum during the human ageing process. Age Ageing. 2008;37:318–323. doi: 10.1093/ageing/afn039. [DOI] [PubMed] [Google Scholar]
- Toldo S, Seropian IM, Mezzaroma E. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia–reperfusion injury. J Mol Cell Cardiol. 2011;51:244–251. doi: 10.1016/j.yjmcc.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Moldthan HL, Hirko AC, Thinschmidt JS. Alpha 1-antitrypsin therapy mitigated ischemic stroke damage in rats. J Stroke Cerebrovasc Dis. 2014;23:e355–363. doi: 10.1016/j.jstrokecerebrovasdis.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Dang Q, Lapergue B, Tran-Dinh A. High-density lipoproteins limit neutrophil-induced damage to the blood–brain barrier in vitro. J Cereb Blood Flow Metab. 2013;33:575–582. doi: 10.1038/jcbfm.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulmanda M, Bhasin M, Fan Z. Alpha 1-antitrypsin reduces inflammation and enhances mouse pancreatic islet transplant survival. Proc Natl Acad Sci USA. 2012;109:15443–15448. doi: 10.1073/pnas.1018366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci USA. 2005;102:12153–12158. doi: 10.1073/pnas.0505579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xiao Y, Zhong L. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes. 2014;63:4239–4248. doi: 10.2337/db14-0480. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Oppenheim JJ, Matsushima K. Human pre-interleukin 1 alpha and beta: structural features revealed by limited proteolysis. Chem Pharm Bull (Tokyo) 1991;39:1513–1517. doi: 10.1248/cpb.39.1513. [DOI] [PubMed] [Google Scholar]
- Bunnett NW. Protease-activated receptors: how proteases signal to cells to cause inflammation and pain. Semin Thromb Hemost. 2006;32(Suppl. 1):39–48. doi: 10.1055/s-2006-939553. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee Y, Hong K. Effect of recombinant alpha1-antitrypsin Fc-fused (AAT-Fc) protein on the inhibition of inflammatory cytokine production and streptozotocin-induced diabetes. Mol Med. 2013;19:65–71. doi: 10.2119/molmed.2012.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Tang M, Wasserfall C. Alpha1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum Gene Ther. 2006;17:625–634. doi: 10.1089/hum.2006.17.625. [DOI] [PubMed] [Google Scholar]
- Song S, Goudy K, Campbell-Thompson M. Recombinant adeno-associated virus-mediated alpha-1 antitrypsin gene therapy prevents type I diabetes in NOD mice. Gene Ther. 2004;11:181–186. doi: 10.1038/sj.gt.3302156. [DOI] [PubMed] [Google Scholar]
- Petrache I, Fijalkowska I, Zhen L. A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med. 2006;173:1222–1228. doi: 10.1164/rccm.200512-1842OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache I, Fijalkowska I, Medler TR. Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol. 2006;169:1155–1166. doi: 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YP, Yan C, Garner WL. Proteolytic activation of matrix metalloproteinase-9 in skin wound healing is inhibited by alpha-1-antichymotrypsin. J Invest Dermatol. 2008;128:2334–2342. doi: 10.1038/jid.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Omari M, Korenbaum E, Ballmaier M. Acute-phase protein alpha1-antitrypsin inhibits neutrophil calpain I and induces random migration. Mol Med. 2011;17:865–874. doi: 10.2119/molmed.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergin DA, Reeves EP, Meleady P. Alpha-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120:4236–4250. doi: 10.1172/JCI41196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoub D, Benslimane N, Al-Zoobi L, Hassan G, Nadiri A, Mourad W. CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J Biol Chem. 2013;288:36083–36093. doi: 10.1074/jbc.M113.506220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhani F, Paone G, Smith NK, Krein P, Barnes P, Brantly ML. Lung neutrophil burden correlates with increased pro- inflammatory cytokines and decreased lung function in individuals with alpha(1)-antitrypsin deficiency. Chest. 2000;117:250S–251. doi: 10.1378/chest.117.5_suppl_1.250s. [DOI] [PubMed] [Google Scholar]
- Griese M, Latzin P, Kappler M. Alpha1-antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;29:240–250. doi: 10.1183/09031936.00047306. [DOI] [PubMed] [Google Scholar]
- Karnaukhova E. Interactions of alpha1-proteinase inhibitor with small ligands of therapeutic potential: binding with retinoic acid. Amino Acids. 2010;38:1011–1020. doi: 10.1007/s00726-009-0309-9. [DOI] [PubMed] [Google Scholar]
- Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35:13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergin DA, Reeves EP, Hurley K. The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med. 2014;6:217ra1. doi: 10.1126/scitranslmed.3007116. [DOI] [PubMed] [Google Scholar]
- Ochayon DE, Mizrahi M, Shahaf G, Baranovski BM, Lewis EC. Human alpha1-antitrypsin binds to heat-shock protein gp96 and protects from endogenous gp96-mediated injury in vivo. Front Immunol. 2013;4:320. doi: 10.3389/fimmu.2013.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotti P, Pagetta A. A heat shock protein70 fusion protein with alpha1-antitrypsin in plasma of type 1 diabetic subjects. Biochem Biophys Res Commun. 2004;315:297–305. doi: 10.1016/j.bbrc.2004.01.058. [DOI] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- Congote LF. Serpin A1 and CD91 as host instruments against HIV-1 infection: are extracellular antiviral peptides acting as intracellular messengers? Virus Res. 2007;125:119–134. doi: 10.1016/j.virusres.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Tilg H, Vannier E, Vachino G, Dinarello CA, Mier JW. Antiinflammatory properties of hepatic acute phase proteins: preferential induction of interleukin 1 (IL-1) receptor antagonist over IL-1 beta synthesis by human peripheral blood mononuclear cells. J Exp Med. 1993;178:1629–1636. doi: 10.1084/jem.178.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MF, Jr, Eidlen D, Brewer MT, Eisenberg SP, Arend WP, Gutierrez-Hartmann A. Human IL-1 receptor antagonist promoter. Cell type-specific activity and identification of regulatory regions. J Immunol. 1992;149:2000–2007. [PubMed] [Google Scholar]
- Chan ED, Pott GB, Silkoff PE, Ralston AH, Bryan CL, Shapiro L. Alpha-1-antitrypsin inhibits nitric oxide production. J Leukoc Biol. 2012;92:1251–1260. doi: 10.1189/jlb.0212071. [DOI] [PubMed] [Google Scholar]
- Abecassis A, Schuster R, Shahaf G. Alpha1-antitrypsin increases interleukin-1 receptor antagonist production during pancreatic islet graft transplantation. Cell Mol Immunol. 2014;11:377–386. doi: 10.1038/cmi.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janciauskiene SM, Nita IM, Stevens T. Alpha1-antitrypsin, old dog, new tricks. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282:8573–8582. doi: 10.1074/jbc.M607976200. [DOI] [PubMed] [Google Scholar]
- Kalis M, Kumar R, Janciauskiene S, Salehi A, Cilio CM. Alpha 1-antitrypsin enhances insulin secretion and prevents cytokine-mediated apoptosis in pancreatic beta-cells. Islets. 2010;2:185–189. doi: 10.4161/isl.2.3.11654. [DOI] [PubMed] [Google Scholar]
- Carl VS, Gautam JK, Comeau LD, Smith MF., Jr Role of endogenous IL-10 in LPS-induced STAT3 activation and IL-1 receptor antagonist gene expression. J Leukoc Biol. 2004;76:735–742. doi: 10.1189/jlb.1003526. [DOI] [PubMed] [Google Scholar]
- Gao W, Zhao J, Kim H. Alpha1-antitrypsin inhibits ischemia reperfusion-induced lung injury by reducing inflammatory response and cell death. J Heart Lung Transplant. 2014;33:309–315. doi: 10.1016/j.healun.2013.10.031. [DOI] [PubMed] [Google Scholar]
- Daemen MA, Heemskerk VH, van't Veer C. Functional protection by acute phase proteins alpha(1)-acid glycoprotein and alpha(1)-antitrypsin against ischemia/reperfusion injury by preventing apoptosis and inflammation. Circulation. 2000;102:1420–1426. doi: 10.1161/01.cir.102.12.1420. [DOI] [PubMed] [Google Scholar]
- Zager RA, Johnson AC, Frostad KB. Rapid renal alpha-1 antitrypsin gene induction in experimental and clinical acute kidney injury. PLOS ONE. 2014;9:e98380. doi: 10.1371/journal.pone.0098380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar T, Shahaf G, Sheiner E. Low levels of circulating alpha-1 antitrypsin are associated with spontaneous abortions. J Matern Fetal Neonatal Med. 2013;26:1782–1787. doi: 10.3109/14767058.2013.801955. [DOI] [PubMed] [Google Scholar]
- Twina G, Sheiner E, Shahaf G. Lower circulation levels and activity of alpha-1 antitrypsin in pregnant women with severe preeclampsia. J Matern Fetal Neonatal Med. 2012;25:2667–2670. doi: 10.3109/14767058.2012.705397. [DOI] [PubMed] [Google Scholar]
- Baron J, Sheiner E, Abecassis A. Alpha1-antitrypsin insufficiency is a possible contributor to preterm premature rupture of membranes. J Matern Fetal Neonatal Med. 2012;25:934–937. doi: 10.3109/14767058.2011.600369. [DOI] [PubMed] [Google Scholar]
- Salem SY, Shahaf G, Sheiner E. Diminished activity of circulating alpha1-antitrypsin is associated with pre-gestational isolated obesity. J Matern Fetal Neonatal Med. 2014 doi: 10.3109/14767058.2014.925442. ; doi: 10.3109/14767058.2014.925442. [DOI] [PubMed] [Google Scholar]
- Braun J, Dalhoff K, Schaaf B, Wood WG, Wiessmann KJ. Characterization of protein-antiproteinase imbalance in bronchoalveolar lavage from patients with pneumonia. Eur Respir J. 1994;7:127–133. doi: 10.1183/09031936.94.07010127. [DOI] [PubMed] [Google Scholar]
- Wang S, Voisin MB, Larbi KY. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203:1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonigk D, Al-Omari M, Maegel L. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci USA. 2013;110:15007–15012. doi: 10.1073/pnas.1309648110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley RA, Shaw J, Afford SC, Morrison HM, Burnett D. Effect of alpha-1-proteinase inhibitor on neutrophil chemotaxis. Am J Respir Cell Mol Biol. 1990;2:163–170. doi: 10.1165/ajrcmb/2.2.163. [DOI] [PubMed] [Google Scholar]
- Ottonello L, Dapino P, Scirocco M, Dallegri F, Sacchetti C. Proteolytic inactivation of alpha-1-antitrypsin by human neutrophils: involvement of multiple and interlinked cell responses to phagocytosable targets. Eur J Clin Invest. 1994;24:42–49. doi: 10.1111/j.1365-2362.1994.tb02058.x. [DOI] [PubMed] [Google Scholar]
- Ottonello L, Dapino P, Dallegri F. Inactivation of alpha-1-proteinase inhibitor by neutrophil metalloproteinases. Crucial role of the myeloperoxidase system and effects of the anti-inflammatory drug nimesulide. Respiration. 1993;60:32–37. doi: 10.1159/000196170. [DOI] [PubMed] [Google Scholar]
- Knauper V, Reinke H, Tschesche H. Inactivation of human plasma alpha 1-proteinase inhibitor by human PMN leucocyte collagenase. FEBS Lett. 1990;263:355–357. doi: 10.1016/0014-5793(90)81412-h. [DOI] [PubMed] [Google Scholar]
- Taggart C, Cervantes-Laurean D, Kim G. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–27265. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- Tosi MF, Zakem H, Berger M. Neutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J Clin Invest. 1990;86:300–308. doi: 10.1172/JCI114699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadallah S, Hess C, Miot S, Spertini O, Lutz H, Schifferli JA. Elastase and metalloproteinase activities regulate soluble complement receptor 1 release. Eur J Immunol. 1999;29:3754–3761. doi: 10.1002/(SICI)1521-4141(199911)29:11<3754::AID-IMMU3754>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hartl D, Latzin P, Hordijk P. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13:1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Akaike T, Maeda H. S-nitrosylated human alpha(1)-protease inhibitor. Biochim Biophys Acta. 2000;1477:90–97. doi: 10.1016/s0167-4838(99)00264-2. [DOI] [PubMed] [Google Scholar]
- Subramaniyam D, Steele C, Kohnlein T. Effects of alpha 1-antitrypsin on endotoxin-induced lung inflammation in vivo. Inflamm Res. 2010;59:571–578. doi: 10.1007/s00011-010-0164-x. [DOI] [PubMed] [Google Scholar]
- Nita IM, Serapinas D, Janciauskiene SM. Alpha1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int J Biochem Cell Biol. 2007;39:1165–1176. doi: 10.1016/j.biocel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Subramaniyam D, Zhou H, Liang M, Welte T, Mahadeva R, Janciauskiene S. Cholesterol rich lipid raft microdomains are gateway for acute phase protein, SERPINA1. Int J Biochem Cell Biol. 2010;42:1562–1570. doi: 10.1016/j.biocel.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Diffenderfer MR, Schaefer EJ. The composition and metabolism of large and small LDL. Curr Opin Lipidol. 2014;25:221–226. doi: 10.1097/MOL.0000000000000067. [DOI] [PubMed] [Google Scholar]
- Mashiba S, Wada Y, Takeya M. In vivo complex formation of oxidized alpha(1)-antitrypsin and LDL. Arterioscler Thromb Vasc Biol. 2001;21:1801–1808. doi: 10.1161/hq1101.098232. [DOI] [PubMed] [Google Scholar]
- Libert C, Van Molle W, Brouckaert P, Fiers W. Alpha1-antitrypsin inhibits the lethal response to TNF in mice. J Immunol. 1996;157:5126–5129. [PubMed] [Google Scholar]
- Perlino E, Cortese R, Ciliberto G. The human alpha 1-antitrypsin gene is transcribed from two different promoters in macrophages and hepatocytes. EMBO J. 1987;6:2767–2771. doi: 10.1002/j.1460-2075.1987.tb02571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakko P, Kirby M, du Bois RM, Gillissen A, Ferrans VJ, Crystal RG. Activated neutrophils secrete stored alpha 1-antitrypsin. Am J Respir Crit Care Med. 1996;154:1829–1833. doi: 10.1164/ajrccm.154.6.8970377. [DOI] [PubMed] [Google Scholar]
- Bellacen K, Kalay N, Ozeri E, Shahaf G, Lewis EC. Revascularization of pancreatic islet allografts is enhanced by alpha-1-antitrypsin under anti-inflammatory conditions. Cell Transplant. 2013;22:2119–2133. doi: 10.3727/096368912X657701. [DOI] [PubMed] [Google Scholar]
- Gottlieb PA, Alkanani AK, Michels AW. Alpha-antitrypsin therapy downregulates toll like receptor-induced IL-1beta responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J Clin Endocrinol Metab. 2014;99:E1418–1426. doi: 10.1210/jc.2013-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freixo-Lima G, Ventura H, Medini M, Bar L, Strauss P, Lewis EC. Mechanistic evidence in support of alpha1-antitrypsin as a therapeutic approach for type 1 diabetes. J Diabetes Sci Technol. 2014;8:1193–1203. doi: 10.1177/1932296814547096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitomi A, Ishikawa T, Tajima K, Konaka Y, Yagita M. Bone marrow transplantation with a reduced-intensity conditioning regimen in a patient with Wegener granulomatosis and therapy-related leukemia. Int J Hematol. 2006;83:262–265. doi: 10.1532/IJH97.05148. [DOI] [PubMed] [Google Scholar]
- Duranton J, Bieth JG. Inhibition of proteinase 3 by [alpha]1-antitrypsin in vitro predicts very fast inhibition in vivo. Am J Respir Cell Mol Biol. 2003;29:57–61. doi: 10.1165/rcmb.2002-0258OC. [DOI] [PubMed] [Google Scholar]
- Crystal RG, Brantly ML, Hubbard RC, Curiel DT, States DJ, Holmes MD. The alpha 1-antitrypsin gene and its mutations. Clinical consequences and strategies for therapy. Chest. 1989;95:196–208. doi: 10.1378/chest.95.1.196. [DOI] [PubMed] [Google Scholar]
- Hadzic R, Nita I, Tassidis H, Riesbeck K, Wingren AG, Janciauskiene S. Alpha1-antitrypsin inhibits Moraxella catarrhalis MID protein-induced tonsillar B cell proliferation and IL-6 release. Immunol Lett. 2006;102:141–147. doi: 10.1016/j.imlet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Redfield RR, III, Rodriguez E, Parsons R. Essential role for B cells in transplantation tolerance. Curr Opin Immunol. 2011;23:685–691. doi: 10.1016/j.coi.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur C, Enk J, Kassem SA. Recognition and killing of human and murine pancreatic beta cells by the NK receptor NKp46. J Immunol. 2011;187:3096–3103. doi: 10.4049/jimmunol.1101269. [DOI] [PubMed] [Google Scholar]
- Villard J. The role of natural killer cells in human solid organ and tissue transplantation. J Innate Immun. 2011;3:395–402. doi: 10.1159/000324400. [DOI] [PubMed] [Google Scholar]
- Yang P, Bamlet WR, Sun Z. Alpha1-antitrypsin and neutrophil elastase imbalance and lung cancer risk. Chest. 2005;128:445–452. doi: 10.1378/chest.128.1.445. [DOI] [PubMed] [Google Scholar]
- Yang P, Sun Z, Krowka MJ. Alpha1-antitrypsin deficiency carriers, tobacco smoke, chronic obstructive pulmonary disease, and lung cancer risk. Arch Intern Med. 2008;168:1097–1103. doi: 10.1001/archinte.168.10.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Yang P. Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression. Lancet Oncol. 2004;5:182–190. doi: 10.1016/S1470-2045(04)01414-7. [DOI] [PubMed] [Google Scholar]
- Yang P, Wentzlaff KA, Katzmann JA. Alpha1-antitrypsin deficiency allele carriers among lung cancer patients. Cancer Epidemiol Biomarkers Prev. 1999;8:461–465. [PubMed] [Google Scholar]
- Xu Y, Zhang J, Han J. Curcumin inhibits tumor proliferation induced by neutrophil elastase through the upregulation of alpha1-antitrypsin in lung cancer. Mol Oncol. 2012;6:405–417. doi: 10.1016/j.molonc.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Akawi ZJ, Nusier MK, Zoughool FE. Relationship between alpha-1 antitrypsin deficient genotypes S and Z and lung cancer in Jordanian lung cancer patients. Saudi Med J. 2006;27:181–184. [PubMed] [Google Scholar]
- Huang H, Campbell SC, Nelius T. Alpha1-antitrypsin inhibits angiogenesis and tumor growth. Int J Cancer. 2004;112:1042–1048. doi: 10.1002/ijc.20494. [DOI] [PubMed] [Google Scholar]
- Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118:1480–1485. doi: 10.1378/chest.118.5.1480. [DOI] [PubMed] [Google Scholar]
- Kohnlein T, Welte T. Alpha-1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. Am J Med. 2008;121:3–9. doi: 10.1016/j.amjmed.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Wanner A, Arce AD, Pardee E. Novel therapeutic uses of alpha-1 antitrypsin: a window to the future. COPD. 2012;9:583–588. doi: 10.3109/15412555.2012.717125. [DOI] [PubMed] [Google Scholar]
- Pantaleo G, Graziosi C, Demarest JF. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- Embretson J, Zupancic M, Ribas JL. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Pott GB, Ralston AH. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001;15:115–122. doi: 10.1096/fj.00-0311com. [DOI] [PubMed] [Google Scholar]
- Munch J, Standker L, Adermann K. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell. 2007;129:263–275. doi: 10.1016/j.cell.2007.02.042. [DOI] [PubMed] [Google Scholar]
- Ferreira TC, Sampaio EP, Arganaraz GA, Gondim MV, Shapiro L, Arganaraz ER. Increased prevalence of the alpha-1-antitrypsin (A1AT) deficiency-related S gene in patients infected with human immunodeficiency virus type 1. J Med Virol. 2014;86:23–29. doi: 10.1002/jmv.23759. [DOI] [PubMed] [Google Scholar]
- Bristow CL, Babayeva MA, LaBrunda M, Mullen MP, Winston R. Alpha1 proteinase inhibitor regulates CD4+ lymphocyte levels and is rate limiting in HIV-1 disease. PLOS ONE. 2012;7:e31383. doi: 10.1371/journal.pone.0031383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow CL, Modarresi R, Babayeva MA. A feedback regulatory pathway between LDL and alpha-1 proteinase inhibitor in chronic inflammation and infection. Discov Med. 2013;16:201–218. [PubMed] [Google Scholar]
- Jia Q, Jiang X, Yu F. Short cyclic peptides derived from the C-terminal sequence of alpha1-antitrypsin exhibit significant anti-HIV-1 activity. Bioorg Med Chem Lett. 2012;22:2393–2395. doi: 10.1016/j.bmcl.2012.02.037. [DOI] [PubMed] [Google Scholar]
- Lockett AD, Brown MB, Santos-Falcon N. Active trafficking of alpha 1 antitrypsin across the lung endothelium. PLOS ONE. 2014;9:e93979. doi: 10.1371/journal.pone.0093979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res. 2010;11:136. doi: 10.1186/1465-9921-11-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskovic G, Twining SS. Local control of alpha1-proteinase inhibitor levels: regulation of alpha1-proteinase inhibitor in the human cornea by growth factors and cytokines. Biochim Biophys Acta. 1998;1403:37–46. doi: 10.1016/s0167-4889(98)00018-4. [DOI] [PubMed] [Google Scholar]
- Geboes K, Ray MB, Rutgeerts P, Callea F, Desmet VJ, Vantrappen G. Morphological identification of alpha-I-antitrypsin in the human small intestine. Histopathology. 1982;6:55–60. doi: 10.1111/j.1365-2559.1982.tb02701.x. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikari Y, Mulvihill E, Schwartz SM. Alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, and alpha 2-macroglobulin are the antiapoptotic factors of vascular smooth muscle cells. J Biol Chem. 2001;276:11798–11803. doi: 10.1074/jbc.M008503200. [DOI] [PubMed] [Google Scholar]
- Aldonyte R, Hutchinson TE, Jin B. Endothelial alpha-1-antitrypsin attenuates cigarette smoke induced apoptosis in vitro. COPD. 2008;5:153–162. doi: 10.1080/15412550802092936. [DOI] [PubMed] [Google Scholar]
- Ikari Y, Fujikawa K, Yee KO, Schwartz SM. Alpha(1)-proteinase inhibitor, alpha(1)-antichymotrypsin, or alpha(2)-macroglobulin is required for vascular smooth muscle cell spreading in three-dimensional fibrin gel. J Biol Chem. 2000;275:12799–12805. doi: 10.1074/jbc.275.17.12799. [DOI] [PubMed] [Google Scholar]
- Sohrab S, Petrusca DN, Lockett AD. Mechanism of alpha-1 antitrypsin endocytosis by lung endothelium. FASEB J. 2009;23:3149–3158. doi: 10.1096/fj.09-129304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi E, Baranovski BM, Shahaf G, Lewis EC. Pancreatic islet xenograft survival in mice is extended by a combination of alpha-1-antitrypsin and single-dose anti-CD4/CD8 therapy. PLOS ONE. 2013;8:e63625. doi: 10.1371/journal.pone.0063625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcondes AM, Li X, Tabellini L. Inhibition of IL-32 activation by alpha-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood. 2011;118:5031–5039. doi: 10.1182/blood-2011-07-365247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilutz H, Siegel Y, Paran E, Cristal N, Quastel MR. Alpha 1-antitrypsin in acute myocardial infarction. Br Heart J. 1983;49:26–29. doi: 10.1136/hrt.49.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadai M, Bally J, Vitel M. High-level expression of active human alpha1-antitrypsin in transgenic tobacco chloroplasts. Transgenic Res. 2009;18:173–183. doi: 10.1007/s11248-008-9209-0. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Singh R, Sanyal I, Amla DV. Expression of modified gene encoding functional human alpha-1-antitrypsin protein in transgenic tomato plants. Transgenic Res. 2008;17:881–896. doi: 10.1007/s11248-008-9173-8. [DOI] [PubMed] [Google Scholar]
- McDonald KA, Hong LM, Trombly DM, Xie Q, Jackman AP. Production of human alpha-1-antitrypsin from transgenic rice cell culture in a membrane bioreactor. Biotechnol Prog. 2005;21:728–734. doi: 10.1021/bp0496676. [DOI] [PubMed] [Google Scholar]
- Wright G, Carver A, Cottom D. High level expression of active human alpha-1-antitrypsin in the milk of transgenic sheep. Biotechnology. 1991;9:830–834. doi: 10.1038/nbt0991-830. [DOI] [PubMed] [Google Scholar]
- Lewis EC. Expanding the clinical indications for alpha(1)-antitrypsin therapy. Mol Med. 2012;18:957–970. doi: 10.2119/molmed.2011.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco I, Lara B, de Serres F. Efficacy of alpha1-antitrypsin augmentation therapy in conditions other than pulmonary emphysema. Orphanet J Rare Dis. 2011;6:14. doi: 10.1186/1750-1172-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JW, Campber R. Sharing a precious resource: an example of the promise of therapeutics for rare diseases to treat common conditions. COPD. 2012;9:581–582. doi: 10.3109/15412555.2012.741936. [DOI] [PubMed] [Google Scholar]
- Turner AM. Alpha-1 antitrypsin deficiency: new developments in augmentation and other therapies. BioDrugs. 2013;27:547–558. doi: 10.1007/s40259-013-0042-5. [DOI] [PubMed] [Google Scholar]