

Abstract
Today, the comparative analysis of DNA molecules mainly uses information inferred from nucleotide substitutions. Insertion/deletion (INDEL) mutations, in contrast, are largely considered uninformative and discarded, due to our lacking knowledge on their evolution. However, including rather than discarding INDELs would be relevant to any research area in ecology and evolution that uses molecular data. As a practical approach to better understanding INDEL evolution in general, we propose the study of recent INDEL (reINDEL) mutations – mutations where both ancestral and derived state are seen in the sample. The precondition for reINDEL identification is knowledge about the pedigree of the individuals sampled. Sound reINDEL knowledge will allow the improved modeling needed for including INDELs in the downstream analysis of molecular data. Both microsatellites, currently still the predominant marker system in the analysis of populations, and sequences generated by next-generation sequencing, a promising and rapidly developing range of technologies, offer the opportunity for reINDEL identification. However, a 2013 sample of animal microsatellite studies contained unexpectedly few reINDELs identified. As most likely explanation, we hypothesize that reINDELs are underreported rather than absent and that this underreporting stems from common reINDEL unawareness. If our hypothesis applies, increased reINDEL awareness should allow gathering data rapidly. We recommend the routine reporting of either the absence or presence of reINDELs together with standardized key information on the nature of mutations when they are detected and the use of the keyword “reINDEL” to increase visibility in both instances of successful and unsuccessful search.
Keywords: INDEL, microsatellite, models of evolution, molecular marker, next-generation sequencing, pedigree
Introducing reINDELs: Recent Insertion/Deletion Mutations
The comparative analysis of DNA molecules looks back on a history of over 40 years. Increasingly complex models of nucleotide substitution patterns at point mutations have been developed (Sullivan and Joyce 2005) and are routinely applied on DNA sequence markers. Another type of mutation, the gain or loss of nucleotides within a defined locus, termed insertion/deletion (INDEL) mutation, has received much less attention (Lunter et al. 2006). This lack of attention is not due to a lack of relevance – on the contrary, INDELs constitute a considerable fraction of mutations in coding and noncoding parts of the genome, are responsible for copy number variants, and are the signature of transposable elements (Korbel et al. 2007; Huang et al. 2010). Rather, the lack of attention is due to a lack of profound knowledge on the evolution of INDELs (Ogden and Rosenberg 2007 and references therein, Sunday and Hart 2013), for which reason they are usually considered uninformative and removed during data preparation, except in the context of gene finding (Kellis et al. 2004) and microsatellite analyses (Ellegren 2004).
The special case of recent INDEL, henceforth reINDEL mutations, that is, mutations where both ancestral and derived state are seen in the sample, allows us witnessing INDEL evolution in real time. Knowledge of reINDELs based on broad sampling should therefore facilitate a better understanding of INDEL evolution generally. The preconditions for reINDEL identification are that we know the pedigree in our sample and that our sample is large (Ellegren 2000; Schlötterer 2000); examples of such situations include studies on extrapair paternity, intraspecific brood parasitism, and eusocial societies, ideally with a single female reproductive (see Fig.1 for an example study system). Whenever we find previously unknown allelic size variation in genotype data for such systems (see Box 1 for microsatellites as an example), reINDELs can be identified easily. For example, a validated allele detected in a singly mated female's offspring that is unknown from the parents can be deduced to represent a recent mutation. The reINDEL can then be described in base-pair length and understood as either insertion or deletion.
Figure 1.

Eusocial insects are among study systems that facilitate generating large population-genetic or population-genomic data sets for individuals with known pedigree and that are thus ideal for studying reINDELs. Here, one of the 1,000–10,000 workers (Steiner et al. 2004) of a colony of the ant Lasius austriacus carries a worker sister at the pupal stage, all these workers stemming from the same mother and father (Steiner et al. 2007). Photograph copyright B.C. Schlick-Steiner & F.M. Steiner.
Box 1. Microsatellites as a fast road to understanding reINDELs
Microsatellites are noncoding, codominantly inherited DNA loci consisting of simple sequence repeats (SSRs), sometimes also termed short tandem repeats (STRs) or simple sequence length polymorphisms (SSLPs). Due to frequent INDEL mutations via loss and gain of repeat units, they commonly exhibit high variation. The design of studies with known pedigree allows precise expectations on the number and size of alleles in the data set based on the knowledge of the parental alleles.
The vast majority of recent mutations in microsatellites will cause variation in allele size (Estoup et al. 2002), and any deviation from the original expectations thus is a reINDEL candidate. For correct data interpretation, validation of allele calling to exclude a scoring error and re-genotyping of the respective individual to exclude a PCR error are necessary.
Hitherto studies on microsatellite evolution have shed some light on, for example, the factors increasing slippage events and multistep changes (Primmer et al. 1996; Chakraborty et al. 1997; Schlötterer 2000; Eckert et al. 2002; Beal et al. 2012) and the existence of differences between male and female germline (Anmarkrud et al. 2011). However, many questions remain such as about the influence of base composition of repeat motifs, about mating and/or sex-determination systems, about cross-taxa and cross-loci variation, and about differences between experimental and natural populations (Ellegren 2000, 2004; Schlötterer 2000; Leclercq et al. 2010; Anmarkrud et al. 2011).
Thinking about the increase of accuracy the improvement of nucleotide models of evolution brought about in phylogenetic applications, we can expect comparable advances in microsatellite-based analyses. Currently existing microsatellite models such as the stepwise mutation, the generalized stepwise, or the K-allele model (reviewed in Estoup et al. 2002) are either rather simplistic or make rather unrealistic assumptions, both of which can lead to poor performance in empirical tests (see, e.g., Balloux and Lugon-Moulin 2002 on stepwise-mutation-model-based Rst values or, Peery et al. 2012 on the reliability of microsatellite-based bottleneck tests for detecting recent population declines). However, with profound knowledge about microsatellite evolution, future models might incorporate the full range of relevant aspects for which maximum likelihood or Bayesian approaches can make good estimates (cf. Caliebe et al. 2010; Wu and Drummond 2011; Nikolic and Chevalet 2014). Potential implementations include different mutability rates according to motif sequence, motif and allele size, allele-frequency dependence, different probabilities for expansion or contraction events, and different rates for male and female germline. More realistic models will aid multiple research areas in ecology and evolution: kinship, parentage, and behavior analyses will be more accurate facilitating a better understanding of mating systems, dispersal patterns, and social organization; better estimations of effective population sizes and detection of bottlenecks will aid nature conservation research; hybridization and backcrossing patterns will be more correctly mirrored, which in turn will increase our understanding of these evolutionary forces; aberrant modes of reproduction might be elucidated, phylogeography exploring the recent past improved, and signals of selection better recognized.
The Relevance of (recent) INDEL Mutations to the Study of Ecology and Evolution
A better understanding of INDEL evolution, achievable via the study of reINDELs, will allow the inclusion of INDELs as genetic variation in our analyses rather than their discard. The gain will be manifold. Our phylogenetic trees and genetic networks will be based on more and more solid information, and our population-genetic and population-genomic inferences will be more accurate (see Box 1 for microsatellite examples). This will have far-reaching effects on any research area using molecular data to make ecological and evolutionary inferences, such as speciation research, sociobiology, the study of species interactions, conservation genetics, invasion biology, and climate change biology.
From the obviously vast range of potential implications of including INDEL information in the data analyses, we illustrate one aspect in more detail: sex differences in germline mutability. There is broad consensus on the existence of a male mutation bias in vertebrates and some plants caused by the higher number of cell divisions during spermatogenesis than during oogenesis (Kirkpatrick and Hall 2004). Anyway, the underlying factors shaping the extent of male mutation bias are still poorly understood (Bartosch-Harlid et al. 2003; Goetting-Minesky and Makova 2006), information from invertebrates is scarce, and recent observations indicate that the mutation rate can be highly different among closely related species (Venn et al. 2014). Thus, more information on patterns at individual loci and in different organisms is needed (Ellegren 2007). Such information will then open up additional research avenues; for example, such bias may even influence the long-term persistence of populations with skewed sex allocation (Cotton and Wedekind 2010).
Studying reINDELs in the NGS Era
The advent of next-generation sequencing (NGS), the massive parallel sequencing of DNA, has opened up a previously unthinkable array of opportunities (Andrew et al. 2013). For ecology and evolution including applied fields, possibly the most important consequence is that NGS makes available genomics for the study of nonmodel organisms (Hudson 2008; Tautz et al. 2010; Williams et al. 2014). Compared with population genetics, the promises of population genomics include improved identification of adaptive molecular variation as well as improved inferences about population demography and evolutionary history (Luikart et al. 2003; Stapley et al. 2010; Williams et al. 2014).
Mutation rates in microsatellites tend to vary considerably within taxa, from 10−6 to 10−2 per site and generation, relatively homogenously across the tree of life (Bhargava and Fuentes 2010). In contrast, estimations of genome-wide rates of nonmicrosatellite INDEL mutations per site and generation are in the range of 2.1 × 10−10 (Sacharomyces cerevisiae; Lang and Murray 2008) to 1.2 × 10−8 (Caenorhabditis elegans; Denver et al. 2004), that is, two to eight orders of magnitude lower than the estimated rates of mutations in microsatellites. Anyway, the sheer amount of data in NGS projects compensates for the lower mutation rate. For example, RADseq (Baird et al. 2008), one of the more established NGS methods for population genomics looking at just fractions of the genome in the range of typically 1%, targets thousands or tens of thousands of loci in a single analysis, compared with some dozens or, rarely, hundreds in the most comprehensive microsatellite studies. Some NGS data sets have been found to contain considerable amounts of INDELs indeed (e.g., Baldwin et al. 2012). Currently, it is common to filter out INDELs in an early stage of the bioinformatics pipeline (e.g., Toonen et al. 2013), but there is growing awareness that INDELs in NGS data are not just a nuisance in the alignment process but also a valuable source of information (e.g., Pacurar et al. 2012; Smolina et al. 2014).
The unparalleled promises of NGS have caused many researchers in ecology and evolution to switch from traditional, locus-based to whole-genome-based approaches or to plan doing so. However, to complete the transition, multiple challenges coming with the new technologies need be overcome (Sboner et al. 2011; DeWoody et al. 2013; Poisot et al. 2013; Andrews and Luikart 2014; Mesak et al 2014). The transition is slower in the study of nonmodel organisms than in that of model organisms (McCormack et al. 2013), given that for nonmodel organisms, resources tend to be more limited both in terms of relevant genomic background information (Nevado et al. 2014) and money.
In any case, not the technology per se but the relevance of the question addressed and the stringency in testing the hypotheses raised make the quality of research. For various research questions, using NGS technology might be like using a sledgehammer to crack a nut (cf. Brewer et al. 2014) – viewed matter-of-factly, there are both advantages and disadvantages to microsatellites compared with NGS techniques, and microsatellites are still being used massively (Box 2). In short, some believe in the fast replacement of microsatellites by NGS approaches (Andrew et al. 2013), others in the persistence of microsatellites in the study of populations also in the future (e.g., Zalapa et al. 2012; Dawson et al. 2013; Butler et al. 2014).
Box 2. Microsatellites versus next-generation sequencing in the study of populations
Today, researchers addressing population-level questions can decide among a range of classical population genetic and next-generation sequencing (NGS) approaches. No approach is “inherently better” (Karl et al. 2012) than any other one. The decision is not easy, and the criteria to be considered range from scientific to resource related. In compiling a list of characteristics (Table B1), we considered just microsatellites among classical approaches due to their common use but two different approaches among NGS techniques, representing the two extremes in effort and information. These are RADseq (Baird et al. 2008), the most frequently used among approaches analyzing just a fraction of the genome, and whole-genome resequencing (Huang et al. 2009), the approach using the maximum of information that can be used. We used recent protocols and manuals and our own experience; we aimed at covering a wide range of characteristics and at objectivity but take responsibility for any failure in doing so.
Table B1.
Characteristics (as of October 2014) of microsatellites versus two selected next-generation sequencing (NGS) approaches in studying populations, RADseq (Baird et al. 2008) and whole-genome resequencing (Huang et al. 2009). All sequencing information is based on the assumption that Illumina (http://www.illumina.com/) technology is used, except the sequencing for developing microsatellite loci, for which the use of Roche 454 (http://www.454.com/) technology is assumed. Where feasible, we classified using a five-step scale of very low, low, intermediate, high, and very high, for each characteristic calibrated relatively across the three techniques. Secondary bioinformatics: sequence alignment and variant calling; tertiary bioinformatics: further downstream steps of sequence annotation and interpretation (Wright et al. 2011; primary bioinformatics, that is, base calling, usually is performed by the software of the sequencing machine)
| NGS-based population genomics |
|||
|---|---|---|---|
| Characteristics | Microsatellite-based population genetics | RADseq | Whole-genome resequencing |
| Establishment of resources | |||
| Type of establishment required | Development of loci | n/a | De-novo whole-genome sequencing |
| DNA quality / amount needed | High / ca. hundred ng | Very high / several μg | |
| Problems due to DNA contaminants | Intermediate | Very high | |
| Sequencing effort | 0.01–0.05× coverage | 30–100× coverage | |
| Need of secondary / tertiary bioinformatics expertise | Intermediate / low | Very high / very high | |
| Total time / expenses | Few weeks / intermediate | Few months to year / high to very high | |
| Application | |||
| DNA quality / amount needed | Very low / several ng per locus | High / 250 ng | High / several hundred ng |
| Problems due to DNA contaminants | Very low | Very high | Very high |
| Need of secondary / tertiary bioinformatics expertise | Low / depends on research question | High / depends on research question | High / depends on research question |
| Total time / expenses per individual | Few days per locus / very low | Weeks / intermediate | Weeks / very high |
| Numbers of individuals that can be analyzed | Very high | Intermediate to high | Low |
| Information accessed | |||
| Genome coverage | Very low | Low | Very high |
| Evenness of distribution of loci over genome | Unpredictable | High | Very high |
| Number of research questions that can be tackled | Intermediate | High | Very high |
| Appropriateness for basic / complex research questions | Very high / low | Intermediate / intermediate | Very low / very high |
We also performed a Web-of-Science-based analysis of the numbers of annually published original articles on microsatellites and population genomics, from 1995, when the first population-genomic paper came out, to 2013 (Fig.B1; Appendix 1) and made two inferences. First, microsatellites still represent the major research approach with about 4500 contributions in 2013 and thus the 47-fold of population-genomic papers. Second, population genomics should indeed have a bright future with a growth rate better explained by an exponential than a linear function. In contrast, microsatellites' growth rate is, over the observed period, better explained by a linear than an exponential function, and, in fact, the number of contributions on microsatellites seems to level off in recent years.
Figure B1.
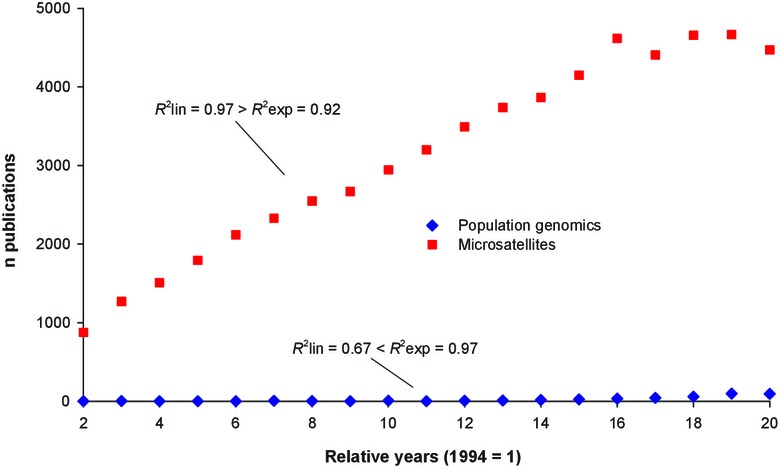
The annual numbers of publications on microsatellites and population genomics published 1995–2013 included in Web of Science. The results of regression analyses comparing the R² of linear (lin) and exponential (exp) functions are added. See Appendix 1 for the database query protocols used and for the regression analysis results. Years are given as relative years as used in the regression analyses: 2 = 1995, 20 = 2013.
Currently Few reINDEL Reports – we Hypothesize Little reINDEL Awareness
Can we simply use the published literature to study reINDELs? We did a standardized analysis of microsatellite literature (see Appendix 2 for details) and found unexpectedly few reINDEL reports. In detail, in 16 animal microsatellite studies from 2013 in which sufficient information on pedigree was available, between 264 and 93,140 microsatellite alleles were seen per study. One study reported the absence of reINDELs explicitly (Liu et al. 2013). No study reported proven reINDELs. One study (32,788 alleles seen) reported 17 putative reINDELs, but did not report the validation of allele calling to exclude a scoring error and re-genotyping of the respective individual to exclude a PCR error (Mayer and Pasinelli 2013). In that study, the position of the loci on autosomes was indicated as was the sex that introduced the mutations, but no information was provided on the identity of the locus or loci, mutation type(s), and allele size(s).
We also calculated the binomial proportion confidence interval to identify mutation rates in line with the number of microsatellite reINDELs reported in the 16 papers surveyed using a confidence level of 0.95, as implemented in a custom Fortran program: 17 reINDELs of 32,788 alleles are in line with mutation rates of 3 × 10−4 to 8 × 10−4 which is well in the middle range of the reported per-locus-per-generation mutation rates of microsatellites of 10−6 to 10−2 (Bhargava and Fuentes 2010). On the other hand, zero reINDELs of the combined 184,660 alleles seen in the 15 studies without reINDEL reports (eight animal classes) are compatible with, at most, a mutation rate of 2 × 10−5. There are two explanations thinkable for a mutation rate at the lower end of the rate range known: (1) The mutation rate could be very low indeed, or (2) researchers may be insufficiently reINDEL aware. We are not able to decide definitively in favor of one of these explanations but for two reasons hypothesize that (2) applies. Firstly, microsatellite loci used in the 15 studies lacking reINDEL detection were all chosen for maximum polymorphism by the authors, rendering (1) rather implausible. Secondly, 14 of the 15 studies lacking successful reINDEL detection lack any statement about the absence of or scanning for reINDELs.
Recommendation
Apparently, an increase of reINDEL awareness is needed, irrespective of the future prime methods in ecology and evolution. To prevent further loss of reINDEL information, we appeal to researchers to scan their data for reINDELs and report them together with a few easy-to-convey and crucial details on the nature of locus and mutation. We recommend the standard reINDEL report to contain the following information: report of absence, or, alternatively, (1) identity of locus affected including GenBank accession number, (2) (putative) ancestral allele and derived allele, (3) sex of (putative) ancestral-allele donor, as well as (4) pipeline of reINDEL validation. Importantly, we suggest that in both instances, absence and presence of reINDELs, the result of the reINDEL search should be reported: Only when reINDEL absence is exported explicitly, will it be possible to use absence data in mutability calculations, in contrast to our inability of doing so with the results of our literature analysis.
By managing to efficiently tap all sources for reINDEL knowledge from now on, we will rapidly create a comprehensive set of information including on insertion/deletion lengths, flanking regions, and chromosomal locations, suitable for the development of INDEL mutation models. No matter whether the study of reINDELs proposed here will be based on microsatellite or NGS data, it seems that the time for advanced INDEL modeling is near.
Acknowledgments
We thank two anonymous referees, Nolan Kane, and especially the Associate Editor at Ecology and Evolution for input that helped improve the paper.
Appendix 1: Database queries using Web of Science to quantify the number of annual publications on microatel-slites and population genomics 1995–2013, as of 14 October 2014
(a) Protocol to quantify the number of publications on microsatellites
All Research Areas (SU) from Life Sciences & Biomedicine were used.
TS=(microsatellite OR microsatellites OR SSR OR SSRs OR “simple sequence repeat” OR “simple sequence repeats” OR SSLP OR SSLPs OR “simple sequence length polymorphism” OR “simple sequence length polymorphisms” OR STR OR STRs OR “short tandem repeat” OR “short tandem repeats”) AND SU=(Agriculture OR Allergy OR Anatomy & Morphology OR Anesthesiology OR Anthropology OR Behavioral Sciences OR Biochemistry & Molecular Biology OR Biodiversity & Conservation OR Biophysics OR Biotechnology & Applied Microbiology OR Cardiovascular System & Cardiology OR Cell Biology OR Critical Care Medicine OR Dentistry, Oral Surgery & Medicine OR Dermatology OR Developmental Biology OR Emergency Medicine OR Endocrinology & Metabolism OR Entomology OR Environmental Sciences & Ecology OR Evolutionary Biology OR Fisheries OR Food Science & Technology OR Forestry OR Gastroenterology & Hepatology OR General & Internal Medicine OR Genetics & Heredity OR Geriatrics & Gerontology OR Health Care Sciences & Services OR Hematology OR Immunology OR Infectious Diseases OR Integrative & Complementary Medicine OR Legal Medicine OR Life Sciences Biomedicine Other Topics OR Marine & Freshwater Biology OR Mathematical & Computational Biology OR Medical Ethics OR Medical Informatics OR Medical Laboratory Technology OR Microbiology OR Mycology OR Neurosciences & Neurology OR Nursing OR Nutrition & Dietetics OR Obstetrics & Gynecology OR Oncology OR Ophthalmology OR Orthopedics OR Otorhinolaryngology OR Paleontology OR Parasitology OR Pathology OR Pediatrics OR Pharmacology & Pharmacy OR Physiology OR Plant Sciences OR Psychiatry OR Public, Environmental & Occupational Health OR Radiology, Nuclear Medicine & Medical Imaging OR Rehabilitation OR Reproductive Biology OR Research & Experimental Medicine OR Respiratory System OR Rheumatology OR Sport Sciences OR Substance Abuse OR Surgery OR Toxicology OR Transplantation OR Tropical Medicine OR Urology & Nephrology OR Veterinary Sciences OR Virology OR Zoology) AND LANGUAGE: (English) AND DOCUMENT TYPES: (Article).
Indexes=SCI-EXPANDED Timespan = 1994–2013.
(b) Protocol to quantify the number of publications on population genomics
All Research Areas (SU) from Life Sciences & Biomedicine were used.
Advanced Search: TS=(“population genomic*”) AND SU=(Agriculture OR Allergy OR Anatomy & Morphology OR Anesthesiology OR Anthropology OR Behavioral Sciences OR Biochemistry & Molecular Biology OR Biodiversity & Conservation OR Biophysics OR Biotechnology & Applied Microbiology OR Cardiovascular System & Cardiology OR Cell Biology OR Critical Care Medicine OR Dentistry, Oral Surgery & Medicine OR Dermatology OR Developmental Biology OR Emergency Medicine OR Endocrinology & Metabolism OR Entomology OR Environmental Sciences & Ecology OR Evolutionary Biology OR Fisheries OR Food Science & Technology OR Forestry OR Gastroenterology & Hepatology OR General & Internal Medicine OR Genetics & Heredity OR Geriatrics & Gerontology OR Health Care Sciences & Services OR Hematology OR Immunology OR Infectious Diseases OR Integrative & Complementary Medicine OR Legal Medicine OR Life Sciences Biomedicine Other Topics OR Marine & Freshwater Biology OR Mathematical & Computational Biology OR Medical Ethics OR Medical Informatics OR Medical Laboratory Technology OR Microbiology OR Mycology OR Neurosciences & Neurology OR Nursing OR Nutrition & Dietetics OR Obstetrics & Gynecology OR Oncology OR Ophthalmology OR Orthopedics OR Otorhinolaryngology OR Paleontology OR Parasitology OR Pathology OR Pediatrics OR Pharmacology & Pharmacy OR Physiology OR Plant Sciences OR Psychiatry OR Public, Environmental & Occupational Health OR Radiology, Nuclear Medicine & Medical Imaging OR Rehabilitation OR Reproductive Biology OR Research & Experimental Medicine OR Respiratory System OR Rheumatology OR Sport Sciences OR Substance Abuse OR Surgery OR Toxicology OR Transplantation OR Tropical Medicine OR Urology & Nephrology OR Veterinary Sciences OR Virology OR Zoology) AND LANGUAGE: (English) AND DOCUMENT TYPES: (Article).
Indexes=SCI-EXPANDED Timespan=1994–2013.
(c) Regression analyses of the annual number of publications on microsatellites and population genomics included in Web of Science 1995–2013, using linear regression functions (of the form y = a + b × x) and exponential functions (of the form y = d + a × eb·x)
See Appendix 1a,b for the database-query protocols used.
| Linear regressions |
Exponential regressions |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Research approach | Intercept | Slope | R² | P | d | a | b | R² | P |
| Microsatellites | 736.2 | 216.9 | 0.97 | <.0001 | −30593.9 | 31294.8 | 0.006 | 0.92 | <.0001 |
| Population genomics | −27.9 | 4.6 | 0.67 | <.0001 | −1.0 | 0.4 | 0.279 | 0.97 | <.0001 |
Appendix 2: Standardized literature analysis of the frequency of reINDEL reports in publications with animal microsatellite data published 2013
(a) Selection of 15 journals
On 20 November 2013, we searched Web of Science (WoS) using Advanced Search. In detail, we used “microsat* OR SSR*” under Topic (TS) to identify microsatellite studies. We added the set of 16 search terms “(extrapair … monandr*)” below under TS to identify those studies that potentially included the relevant information on pedigree to facilitate the recognition of reINDELs. The set of ten Research Areas (SU) below was used to identify animal studies. Just Article was searched under Document Types to retrieve just primary research articles.
The complete search query read:
TS=((microsat* OR SSR*) AND (extrapair OR eusocial OR paternity OR maternity OR pedigree* OR patriline* OR matriline* OR colon* OR parthenogen* OR clonal* OR nest* OR sibling* OR “mated once” OR “single-mated” OR monogyn* OR monandr*)) AND SU=(Behavioral Sciences OR Biodiversity & Conservation OR Entomology OR Environmental Sciences & Ecology OR Evolutionary Biology OR Genetics & Heredity OR Marine & Freshwater Biology OR Parasitology OR Reproductive Biology OR Zoology) AND Document Types=(Article).
Databases=SCI-EXPANDED Timespan=2013.
The results were sorted in descending order by record count for Source Titles under Results Analysis, two inappropriate journals were excluded (Tree Genetics Genomes, American Journal of Physical Anthropology) and the top 15 were selected among the remaining journals.
(b) Retrieval of primary research articles from the 15 journals selected
Based on the results from (a), we searched WoS using Advanced Search on 20 November 2013:
TS=((microsat* OR SSR*) AND (extrapair OR eusocial OR paternity OR maternity OR pedigree* OR patriline* OR matriline* OR colon* parthenogen* OR clonal* OR nest* OR sibling* OR “mated once” OR “single-mated” OR monogyn* OR monandr*)) AND SU=(Behavioral Sciences OR Biodiversity & Conservation OR Entomology OR Environmental Sciences & Ecology OR Evolutionary Biology OR Genetics & Heredity OR Marine & Freshwater Biology OR Parasitology OR Reproductive Biology OR Zoology) AND SO=(Molecular Ecology OR Conservation Genetics OR Journal of Heredity OR Ecology and Evolution OR Behavioral Ecology OR Conservation Genetics Resources OR Heredity OR Behavioral Ecology and Sociobiology OR Evolution OR Aquaculture OR Hydrobiologia OR Insectes Sociaux OR Journal of Evolutionary Biology OR Biological Invasions OR Evolutionary Applications) AND Document Types=(Article).
Databases=SCI-EXPANDED Timespan=2013.
(c) Selection of definitive set of papers
From the 97 papers retrieved under (b), we selected as definitive set of papers those that fulfilled the following criteria:
Primary research article, that is, not meta-analysis or review article; despite our search for just Article under Document Type under (b), not all results were primary research articles indeed.
Empirical, that is, not simulated data.
Animals analyzed.
Number of alleles seen discernible, that is, number of individuals successfully analyzed using microsatellites, number of microsatellite loci successfully genotyped, and ploidy level discernible.
Sufficient information on pedigree available, either via inference from independent data or from microsatellite data under monogyny/monandry or clonality.
(d) Analysis of definitive set of papers
We retrieved information from the 16 papers selected under (c) on the following:
Taxonomic affiliation of the animals analyzed at the level of Class.
Means of allele scoring (automatic, manual, combined).
Number of alleles seen.
Number of reINDELs reported, as putative or proven.
In case of reINDEL report, the presence/absence of statements on identity of locus affected, position of locus on auto-/allosome, mutation type, direction of mutation in case of size-shift mutation, allele size, sex.
In case of no reINDEL report, the presence/absence of statement that no reINDEL was detected.
(e) Results from (c) and (d)
Ref = Reference; Incl = inclusion in definitive set of papers (1 = yes, 0 = no); Scor = means of scoring (no = no information, aut = automatic, man = manual); Class = taxonomic affiliation at the level of Class; n all = n alleles seen; n put = number of putative reINDELs (successful validation of allele calling to exclude scoring errors); n prov = number of proven reINDELs (successful re-genotyping of individuals to exclude PCR errors); Loc? = information given on identity of locus affected? (1 = yes, 0 = no, n/a = not applicable); Pos = information given on position of locus on auto/allosome? (1 = yes, 0 = no, n/a = not applicable); Typ? = information given on mutation type (expansion, contraction, flanking-region SNP)? (1 = yes, 0 = no, n/a = not applicable); Dir? = information given on direction of mutation in case of size-shift mutation? (1 = yes, 0 = no, n/a = not applicable); Siz? = information given on allele size? (1 = yes, 0 = no, n/a = not applicable); Sex? = information given on sex? (1 = yes, 0 = no, n/a = not applicable); No? = statement made that no reINDEL was detected? (1 = yes, 0 = no, n/a = not applicable).
| Ref | Incl | Scor | Class | n all | n put | n prov | Loc? | Pos? | Typ? | Dir? | Siz? | Sex? | No? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aquaculture 388:14–23 | 0 | ||||||||||||
| Aquaculture 400:77–84 | 0 | ||||||||||||
| Aquaculture 404:139–149 | 0 | ||||||||||||
| Aquaculture 404:95–104 | 0 | ||||||||||||
| Behav Ecol. 24:1022–1029 | 0 | ||||||||||||
| Behav Ecol. 24:1128–1137 | 0 | ||||||||||||
| Behav Ecol. 24:1306–1311 | 0 | ||||||||||||
| Behav Ecol. 24:1356–1362 | 1 | no | Aves | 2320 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Behav Ecol. 24:29–38 | 0 | ||||||||||||
| Behav Ecol. 24:540–546 | 1 | aut | Actinopterygii | 572 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Behav Ecol. 24:949–954 | 0 | ||||||||||||
| Behav Ecol Sociobiol. 67:113–122 | 0 | ||||||||||||
| Behav Ecol Sociobiol. 67:243–255 | 0 | ||||||||||||
| Behav Ecol Sociobiol. 67:399–408 | 0 | ||||||||||||
| Behav Ecol Sociobiol. 67:621–627 | 1 | aut + man | Insecta | 9116 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Behav Ecol Sociobiol. 67:727–735 | 0 | ||||||||||||
| Biol Invasions 15:1331–1342 | 0 | ||||||||||||
| Biol Invasions 15:199–212 | 0 | ||||||||||||
| Biol Invasions 15:2281–2297 | 0 | ||||||||||||
| Conserv Genet. 14:1019–1028 | 0 | ||||||||||||
| Conserv Genet. 14:1029–1042 | 1 | man | Reptilia | 6110 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Conserv Genet. 14:1099–1110 | 0 | ||||||||||||
| Conserv Genet. 14:171–183 | 0 | ||||||||||||
| Conserv Genet. 14:21–30 | 0 | ||||||||||||
| Conserv Genet. 14:559–571 | 0 | ||||||||||||
| Conserv Genet. 14:601–613 | 0 | ||||||||||||
| Conserv Genet. 14:625–636 | 0 | ||||||||||||
| Conserv Genet. 14:65–77 | 0 | ||||||||||||
| Conserv Genet. 14:875–883 | 0 | ||||||||||||
| Conserv Genet. 14:953–962 | 0 | ||||||||||||
| Conserv Genet Res. 5:181–183 | 0 | ||||||||||||
| Conserv Genet Res. 5:199–201 | 0 | ||||||||||||
| Conserv Genet Res. 5:507–510 | 0 | ||||||||||||
| Conserv Genet Res. 5:555–560 | 0 | ||||||||||||
| Conserv Genet Res. 5:667–669 | 0 | ||||||||||||
| Conserv Genet Res. 5:749–753 | 0 | ||||||||||||
| Conserv Genet Res. 5:863–866 | 0 | ||||||||||||
| Ecol Evol. 3:1569–1579 | 0 | ||||||||||||
| Ecol Evol. 3:1765–1779 | 0 | ||||||||||||
| Ecol Evol. 3:2933–2946 | 1 | aut | Amphibia | 93,140 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Ecol Evol. 3:3152–3165 | 0 | ||||||||||||
| Ecol Evol. 3:3379–3387 | 1 | no | Anthozoa | 1140 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Ecol Evol. 3:474–481 | 0 | ||||||||||||
| Ecol Evol. 3:694–705 | 1 | aut | Aves | 32,788 | 17 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | n/a |
| Ecol Evol. 3:80–88 | 0 | ||||||||||||
| Evolution 67:1169–1180 | 0 | ||||||||||||
| Evolution 67:2299–2308 | 0 | ||||||||||||
| Evolution 67:2561–2576 | 0 | ||||||||||||
| Evolution 67:2701–2713 | 0 | ||||||||||||
| Evolution 67:646–660 | 0 | ||||||||||||
| Evol Appl. 6:165–179 | 0 | ||||||||||||
| Evol Appl. 6:34–45 | 0 | ||||||||||||
| Evol Appl. 6:524–534 | 0 | ||||||||||||
| Heredity 110:111–122 | 0 | ||||||||||||
| Heredity 110:355–362 | 0 | ||||||||||||
| Heredity 110:439–448 | 0 | ||||||||||||
| Heredity 110:560–569 | 0 | ||||||||||||
| Heredity 111:321–329 | 0 | ||||||||||||
| Heredity 111:338–344 | 0 | ||||||||||||
| Hydrobiologia 700:33–45 | 0 | ||||||||||||
| Hydrobiologia 714:61–70 | 0 | ||||||||||||
| Hydrobiologia 715:113–123 | 0 | ||||||||||||
| Hydrobiologia 715:37–50 | 0 | ||||||||||||
| Insect Soc. 60:135–145 | 0 | ||||||||||||
| Insect Soc. 60:203–211 | 1 | no | Insecta | 1260 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Insect Soc. 60:231–241 | 1 | aut | Insecta | 720 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Insect Soc. 60:337–344 | 0 | ||||||||||||
| J Evol Biol. 26:108–117 | 0 | ||||||||||||
| J Evol Biol. 26:1330–1340 | 0 | ||||||||||||
| J Evol Biol. 26:1727–1737 | 0 | ||||||||||||
| J Evol Biol. 26:889–899 | 0 | ||||||||||||
| J Hered. 104:127–133 | 0 | ||||||||||||
| J Hered. 104:182–191 | 1 | no | Mammalia | 7776 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| J Hered. 104:217–222 | 1 | aut | Actinopterygii | 264 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 1 |
| J Hered. 104:301–311 | 0 | ||||||||||||
| J Hered. 104:371–379 | 1 | no | Chondrichthyes | 2400 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| J Hered. 104:386–398 | 0 | ||||||||||||
| J Hered. 104:465–475 | 0 | ||||||||||||
| J Hered. 104:532–546 | 0 | ||||||||||||
| J Hered. 104:692–703 | 0 | ||||||||||||
| J Hered. 104:92–104 | 0 | ||||||||||||
| Mol Ecol. 22:1158–1170 | 0 | ||||||||||||
| Mol Ecol. 22:1282–1294 | 0 | ||||||||||||
| Mol Ecol. 22:1447–1462 | 1 | no | Insecta | 46,560 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Mol Ecol. 22:1546–1557 | 0 | ||||||||||||
| Mol Ecol. 22:1640–1649 | 0 | ||||||||||||
| Mol Ecol. 22:1998–2010 | 1 | aut + man | Insecta | 1040 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Mol Ecol. 22:2787–2796 | 1 | aut + man | Mammalia | 816 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Mol Ecol. 22:3391–3402 | 0 | ||||||||||||
| Mol Ecol. 22:3721–3736 | 0 | ||||||||||||
| Mol Ecol. 22:3916–3932 | 0 | ||||||||||||
| Mol Ecol. 22:4499–4515 | 0 | ||||||||||||
| Mol Ecol. 22:4549–4561 | 0 | ||||||||||||
| Mol Ecol. 22:5001–5015 | 0 | ||||||||||||
| Mol Ecol. 22:5027–5039 | 1 | aut | Aves | 11,426 | 0 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | 0 |
| Mol Ecol. 22:5430–5440 | 0 | ||||||||||||
| Mol Ecol. 22:74–86 | 0 | ||||||||||||
| Total | 16 | 217,448 | 17 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
Conflict of Interest
None declared.
References
- Andrew RL, Bernatchez L, Bonin A, Buerkle CA, Carstens BC, Emerson BC, et al. A road map for molecular ecology. Mol. Ecol. 2013;22:2605–2626. doi: 10.1111/mec.12319. [DOI] [PubMed] [Google Scholar]
- Andrews KR. Luikart G. Recent novel approaches for population genomics data analysis. Mol. Ecol. 2014;23:1661–1667. doi: 10.1111/mec.12686. [DOI] [PubMed] [Google Scholar]
- Anmarkrud JA, Kleven O, Augustin J, Bentz KH, Blomqvist D, Fernie KJ, et al. Factors affecting germline mutations in a hypervariable microsatellite: a comparative analysis of six species of swallows (Aves: Hirundinidae) Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011;708:37–43. doi: 10.1016/j.mrfmmm.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin S, Revanna R, Thomson S, Pither-Joyce M, Wright K, Crowhurst R, et al. A toolkit for bulk PCR-based marker design from next-generation sequence data: application for development of a framework linkage map in bulb onion (Allium cepa L.) BMC Genom. 2012;13:637. doi: 10.1186/1471-2164-13-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balloux F. Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol. Ecol. 2002;11:155–165. doi: 10.1046/j.0962-1083.2001.01436.x. [DOI] [PubMed] [Google Scholar]
- Bartosch-Harlid A, Berlin S, Smith NGC, Moller AP. Ellegren H. Life history and the male mutation bias. Evolution. 2003;57:2398–2406. doi: 10.1554/03-036. [DOI] [PubMed] [Google Scholar]
- Beal MA, Glenn TC, Lance SL. Somers CM. Characterization of unstable microsatellites in mice: no evidence for germline mutation induction following gamma-radiation exposure. Environ. Mol. Mutagen. 2012;53:599–607. doi: 10.1002/em.21726. [DOI] [PubMed] [Google Scholar]
- Bhargava A. Fuentes FF. Mutational dynamics of microsatellites. Mol. Biotechnol. 2010;44:250–266. doi: 10.1007/s12033-009-9230-4. [DOI] [PubMed] [Google Scholar]
- Brewer MS, Cotoras DD, Croucher PJP. Gillespie RG. New sequencing technologies, the development of genomics tools, and their applications in evolutionary arachnology. J. Arachnol. 2014;42:1–15. [Google Scholar]
- Butler IA, Siletti K, Oxley PR. Kronauer DJC. Conserved microsatellites in ants enable population genetic and colony pedigree studies across a wide range of species. PLoS ONE. 2014;9:e107334. doi: 10.1371/journal.pone.0107334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliebe A, Jochens A, Krawczak M. Rösler U. A Markov chain description of the stepwise mutation model: local and global behaviour of the allele process. J. Theor. Biol. 2010;266:336–342. doi: 10.1016/j.jtbi.2010.06.033. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Kimmel M, Stivers DN, Davison LJ. Deka R. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc. Natl Acad. Sci. USA. 1997;94:1041–1046. doi: 10.1073/pnas.94.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton S. Wedekind C. Male mutation bias and possible long-term effects of human activities. Conserv. Biol. 2010;24:1190–1197. doi: 10.1111/j.1523-1739.2010.01524.x. [DOI] [PubMed] [Google Scholar]
- Dawson D, Ball A, Spurgin L, Martin-Galvez D, Stewart IRK, Horsburgh G, et al. High-utility conserved avian microsatellite markers enable parentage and population studies across a wide range of species. BMC Genom. 2013;14:176. doi: 10.1186/1471-2164-14-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M. Thomas WK. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature. 2004;430:679–682. doi: 10.1038/nature02697. [DOI] [PubMed] [Google Scholar]
- DeWoody JA, Abts KC, Fahey AL, Ji YZ, Kimble SJA, Marra NJ, et al. Of contigs and quagmires: next-generation sequencing pitfalls associated with transcriptomic studies. Mol. Ecol. Resour. 2013;13:551–558. doi: 10.1111/1755-0998.12107. [DOI] [PubMed] [Google Scholar]
- Eckert KA, Yan G. Hile SE. Mutation rate and specificity analysis of tetranucleotide microsatellite DNA alleles in somatic human cells. Mol. Carcinog. 2002;34:140–150. doi: 10.1002/mc.10058. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Microsatellite mutations in the germline: implications for evolutionary inference. Trends Genet. 2000;16:551–558. doi: 10.1016/s0168-9525(00)02139-9. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc. Biol. Sci. 2007;274:1–10. doi: 10.1098/rspb.2006.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoup A, Jarne P. Cornuet J-M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2002;11:1591–1604. doi: 10.1046/j.1365-294x.2002.01576.x. [DOI] [PubMed] [Google Scholar]
- Goetting-Minesky MP. Makova KD. Mammalian male mutation bias: Impacts of generation time and regional variation in substitution rates. J. Mol. Evol. 2006;63:537–544. doi: 10.1007/s00239-005-0308-8. [DOI] [PubMed] [Google Scholar]
- Huang XH, Feng Q, Qian Q, Zhao Q, Wang L, Wang AH, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CRL, Schneider AM, Lu YQ, Niranjan T, Shen PL, Robinson MA, et al. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;141:1171–1182. doi: 10.1016/j.cell.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson ME. Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol. Ecol. Resour. 2008;8:3–17. doi: 10.1111/j.1471-8286.2007.02019.x. [DOI] [PubMed] [Google Scholar]
- Karl SA, Toonen RJ, Grant WS. Bowen BW. Common misconceptions in molecular ecology: echoes of the modern synthesis. Mol. Ecol. 2012;21:4171–4189. doi: 10.1111/j.1365-294X.2012.05576.x. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Birren B, Berger B. Lander ES. Methods in comparative genomics: Genome correspondence, gene identification and regulatory motif discovery. J. Comput. Biol. 2004;11:319–355. doi: 10.1089/1066527041410319. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M. Hall DW. Male-biased mutation, sex linkage, and the rate of adaptive evolution. Evolution. 2004;58:437–440. [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI. Murray AW. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics. 2008;178:67–82. doi: 10.1534/genetics.107.071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq S, Rivals E. Jarne P. DNA slippage occurs at microsatellite loci without minimal threshold length in humans: a comparative genomic approach. Genome Biol. Evol. 2010;2:325–335. doi: 10.1093/gbe/evq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Tatarenkov A, O'rear TA, Moyle PB. Avise JC. Molecular evidence for multiple paternity in a population of the viviparous tule perch Hysterocarpus traski. J. Hered. 2013;104:217–222. doi: 10.1093/jhered/ess105. [DOI] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S. Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Lunter G, Ponting CP. Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput. Biol. 2006;2:2–12. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C. Pasinelli G. New support for an old hypothesis: density affects extra-pair paternity. Ecol. Evol. 2013;3:694–705. doi: 10.1002/ece3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Hird SM, Zellmer AJ, Carstens BC. Brumfield RT. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenet. Evol. 2013;66:526–538. doi: 10.1016/j.ympev.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Mesak F, Tatarenkov A, Earley RL. Avise JC. Hundreds of SNPs versus dozens of SSRs: Which dataset better characterizes natural clonal lineages in a self-fertilizing fish. Front. Eco. Evol. 2014;2:74. [Google Scholar]
- Nevado B, Ramos-Onsins SE. Perez-Enciso M. Resequencing studies of nonmodel organisms using closely related reference genomes: optimal experimental designs and bioinformatics approaches for population genomics. Mol. Ecol. 2014;23:1764–1779. doi: 10.1111/mec.12693. [DOI] [PubMed] [Google Scholar]
- Nikolic N. Chevalet C. Detecting past changes of effective population size. Evol. Appl. 2014;7:663–681. doi: 10.1111/eva.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden TH. Rosenberg MS. How should gaps be treated in parsimony? A comparison of approaches using simulation. Mol. Phylogenet. Evol. 2007;42:817–826. doi: 10.1016/j.ympev.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Pacurar DI, Pacurar ML, Street N, Bussell JD, Pop TI, Gutierrez L, et al. A collection of INDEL markers for map-based cloning in seven Arabidopsis accessions. J. Exp. Bot. 2012;63:2491–2501. doi: 10.1093/jxb/err422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Beer E, Robinson S, et al. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012;21:3403–3418. doi: 10.1111/j.1365-294X.2012.05635.x. [DOI] [PubMed] [Google Scholar]
- Poisot T, Pequin B. Gravel D. High-throughput sequencing: a roadmap toward community ecology. Ecol. Evol. 2013;3:1125–1139. doi: 10.1002/ece3.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primmer CR, Ellegren H, Saino N. Moller AP. Directional evolution in germline microsatellite mutations. Nat. Genet. 1996;13:391–393. doi: 10.1038/ng0896-391. [DOI] [PubMed] [Google Scholar]
- Sboner A, Mu XJ, Greenbaum D, Auerbach RK. Gerstein MB. The real cost of sequencing: higher than you think! Genome Biol. 2011;12:10. doi: 10.1186/gb-2011-12-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C. Evolutionary dynamics of microsatellite DNA. Chromosoma. 2000;109:365–371. doi: 10.1007/s004120000089. [DOI] [PubMed] [Google Scholar]
- Smolina I, Kollias S, Poortvliet M, Nielsen TG, Lindeque P, Castellani C, et al. Genome- and transcriptome-assisted development of nuclear insertion/deletion markers for Calanus species (Copepoda: Calanoida) identification. Mol. Ecol. Resour. 2014;14:1072–1079. doi: 10.1111/1755-0998.12241. [DOI] [PubMed] [Google Scholar]
- Stapley J, Reger J, Feulner PGD, Smadja C, Galindo J, Ekblom R, et al. Adaptation genomics: the next generation. Trends Ecol. Evol. 2010;25:705–712. doi: 10.1016/j.tree.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Steiner FM, Schlick-Steiner BC, Schödl S, Espadaler X, Seifert B, Christian E, et al. Phylogeny and bionomics of Lasius austriacus (Hymenoptera, Formicidae) Insectes Soc. 2004;51:24–29. [Google Scholar]
- Steiner FM, Schlick-Steiner BC, Moder K, Stauffer C, Arthofer W, Buschinger A, et al. Abandoning aggression but maintaining self-nonself discrimination as a first stage in ant supercolony formation. Curr. Biol. 2007;17:1903–1907. doi: 10.1016/j.cub.2007.09.061. [DOI] [PubMed] [Google Scholar]
- Sullivan J. Joyce P. Model selection in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2005;36:445–466. [Google Scholar]
- Sunday JM. Hart MW. Sea star populations diverge by positive selection at a sperm-egg compatibility locus. Ecol. Evol. 2013;3:640–654. doi: 10.1002/ece3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Ellegren H. Weigel D. Next generation molecular ecology. Mol. Ecol. 2010;19:1–3. doi: 10.1111/j.1365-294X.2009.04489.x. [DOI] [PubMed] [Google Scholar]
- Toonen RJ, Puritz JB, Forsman ZH, Whitney JL, Fernandez-Silva I, Andrews KR, et al. ezRAD: a simplified method for genomic genotyping in non-model organisms. PeerJ. 2013;1:e203. doi: 10.7717/peerj.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venn O, Turner I, Mathieson I, De Groot N, Bontrop R. Mcvean G. Strong male bias drives germline mutation in chimpanzees. Science. 2014;344:1272–1275. doi: 10.1126/science.344.6189.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AV, Nevill PG. Krauss SL. Next generation restoration genetics: applications and opportunities. Trends Plant Sci. 2014;19:529–537. doi: 10.1016/j.tplants.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Wright C, Burton H, Hall A, Moorthie S, Pokorska-Bocci A, Sagoo G, et al. Next steps in the sequence. The implications of whole genome sequencing for health in the UK. Cambridge, U.K: PHG Foundation; 2011. [Google Scholar]
- Wu CH. Drummond AJ. Joint inference of microsatellite mutation models, population history and genealogies using transdimensional Markov Chain Monte Carlo. Genetics. 2011;188:151–164. doi: 10.1534/genetics.110.125260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalapa JE, Cuevas H, Zhu HY, Steffan S, Senalik D, Zeldin E, et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012;99:193–208. doi: 10.3732/ajb.1100394. [DOI] [PubMed] [Google Scholar]