

Abstract
Purpose
The Ataxia-telangiectasia mutated (ATM) gene encodes a multifunctional kinase, which is linked to important cellular functions. Women heterozygous for ATM mutations have an estimated relative risk of developing breast cancer of 3.8. However, the pattern of ATM mutations and their role in breast cancer etiology has been controversial and remains unclear. In the present study, we investigated the frequency and spectrum of ATM mutations in a series of sporadic breast cancers and controls from the Brazilian population.
Methods
Using PCR-Single Strand Conformation Polymorphism (SSCP) analysis and direct DNA sequencing, we screened a panel of 100 consecutive, unselected sporadic breast tumors and 100 matched controls for all 62 coding exons and flanking introns of the ATM gene.
Results
Several polymorphisms were detected in 12 of the 62 coding exons of the ATM gene. These polymorphisms were observed in both breast cancer patients and the control population. In addition, evidence of potential ATM mutations was observed in 7 of the 100 breast cancer cases analyzed. These potential mutations included six missense variants found in exon 13 (p.L546V), exon 14 (p.P604S), exon 20 (p.T935R), exon 42 (p.G2023R), exon 49 (p.L2307F), and exon 50 (p.L2332P) and one nonsense mutation in exon 39 (p.R1882X), which was predicted to generate a truncated protein.
Conclusions
Our results corroborate the hypothesis that sporadic breast tumors may occur in carriers of low penetrance ATM mutant alleles and these mutations confer different levels of breast cancer risk.
Electronic supplementary material
The online version of this article (doi:10.1186/s40064-015-0787-z) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, ATM gene, Mutations, Polymorphisms
Background
The Ataxia-telangiectasia mutated (ATM) gene encodes a multifunctional kinase, which is linked to important cellular functions (Derheimer and Kastan 2010; Shiloh and Ziv 2013). Individuals carrying two ATM alleles with loss-of-function mutations are affected with Ataxia-Telangiectasia (A-T), which is an autosomal recessive syndrome characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, immunodeficiency, hypersensitivity to ionizing radiation, and cancer susceptibility (Gatti et al. 1991; Savitsky et al. 1995a; Ambrose and Gatti 2013). The estimated incidence of A-T is 1:40,000 to 1:100,000 live births (Easton, 1994). Patients with A-T have an increased risk of developing cancer compared with non-A-T individuals (Taylor and Byrd 2005; Mavrou et al. 2008). The frequency of individuals heterozygous for a mutation in ATM is estimated to be 0.5 - 1% in the general population (Swift et al. 1986; Easton 1994). Individuals heterozygous for A-T mutations, who are phenotypically normal, may exhibit increased radio- and chemo-sensitivity and an increased relative risk of carcinogenesis (Swift et al. 1991; West et al. 1995). An increased relative risk of breast cancer was identified for female A-T heterozygotes; while, the proportion of sporadic breast cancers in the general population that is due to ATM mutations remains controversial (Swift et al. 1987; Ahmed and Rahman 2006; Mavrou et al. 2008). Somatically occurring ATM mutations are also prevalent in a number of sporadic human cancers, most notably leukemias and carcinomas of the breast and lung (Cremona and Behrens, 2014).
The ATM gene is located on chromosome 11q22-q23, spanning approximately 150 kb of genomic DNA (Savitsky et al. 1995a; Savitsky et al. 1995b). ATM is structurally complex, containing 66 exons, and encodes a protein of 3056 amino acids (~350 kDa) that is related to the phosphatidylinositol 3-kinase (PI3-kinase) superfamily (Savitsky et al. 1995a, Lavin et al. 1995). The COOH-terminal region of the ATM protein shows similarity to several checkpoint/damage response proteins from other organisms, including TOR1 and TOR2, TEL1p, ATR, RAD3p, MEC1pC, MEI-41, and DNA-PK (Mavrou et al. 2008; Bensimon et al. 2011). ATM-knockout mice show A-T-like phenotypes, including neurological abnormalities, immunodeficiency, chromosomal instability, radiosensitivity, and the development of lymphomas (Xu et al. 1996; Barlow et al. 1996; Liao and Van Dyke. 1999; Deng and Brodie 2001; Shiloh and Ziv 2012). ATM belongs to the class of caretaker tumor suppressor genes that defend genome integrity (Levitt and Hickson 2002). Irradiated A-T cells had defects in the activation of the G1/S, S phase, and G2 checkpoints (Beamish and Lavin 1994; Derheimer and Kastan 2010). ATM is believed to be involved in multiple cellular processes that occur in response to DNA damage, including cell cycle check point control, DNA repair, and apoptosis (Derheimer and Kastan 2010; Shiloh and Ziv 2013). ATM is one of the main sensors for DNA damage evoked by genotoxic agents, such as ionizing radiation (Rotman and Shiloh 1999; Thompson and Schild 2002; Liang et al. 2009). Members of the PI3-kinase superfamily of serine/threonine protein kinases, such as ATM and DNA-dependent protein kinase (DNA-PK) have been implicated in cell cycle checkpoint control in response to DNA damage (Derheimer and Kastan 2010; Sperka et al. 2012). ATM is considered an important transducer of DNA damage signaling via phosphorylation of downstream effectors, which results in a kinase cascade that works to promote genetic stability (Canman and Lim 1998; Khanna et al. 1998; Khosravi et al. 1999; Cortez et al. 1999; Li et al. 2000; Maya et al. 2001).
The size and complexity of the ATM gene make the search for mutations and identification of genotype-phenotype associations difficult, especially for the missense mutations primarily identified in heterozygous subjects. The nature and distribution of the ATM mutations found in A-T patients are variable with no specific hot spot (Yuille and Coignet 1998; Sandoval et al. 1999; Mavrou et al. 2008). More than 80% of A-T patients have ATM-truncating mutations; however, exon-skipping and missense mutations are also observed (Renwick et al. 2006; Mavrou et al. 2008). Detected mutations are believed to cause structural or functional effects on the final protein, abolishing ATM function. On the other hand, the majority of ATM variants described so far in sporadic leukemia and breast cancers are missense base substitutions (Prokopcova et al. 2007; Broeks et al. 2008), making it difficult to distinguish missense mutations that affect protein function from sequence polymorphisms with no effect on normal ATM protein activity.
While the spectrum of ATM mutations in classical A-T patients is well established, the pattern of ATM mutations and their role in breast cancer etiology remains unclear (Broeks et al. 2008; Milne 2009). The aim of the present study was to investigate the frequency and the spectrum of ATM mutations in a series of sporadic breast cancer and control samples from the Brazilian population.
Results
DNA samples from a series of 100 breast cancer patients (tumor and normal tissues) and 100 control individuals were screened for ATM gene mutations. Sixty-four sets of primers were used to amplify all coding exons and flanking intron sequences of the ATM gene (Vorechovsky et al. 1996). The amplified DNA fragments were screened by PCR-Single Strand Conformation Polymorphism (SSCP) analysis and the samples showing band mobility shifts were further analyzed by direct DNA sequencing. This analysis led to the identification of 24 distinct sequence variants (Tables 1 and 2). The polymorphism p.P1054R (Table 1), which has been implicated in breast cancer and linked to F858L (Larson et al. 1997–1998; Fletcher et al., 2010), could not be detected by SSCP screening. The presence of this polymorphism in our population could be confirmed only by re-sequencing some of the breast cancer cases and controls showing the polymorphism p.F858L (Additional file 1); therefore, the SSCP technique had low sensitivity for this mutation. The majority of variants observed were base substitutions within the ATM coding region. One base substitution, one deletion, and two insertions were observed in the introns of the ATM gene.
Table 1.
ATM gene variants detected in controls and breast cancer patients
| Position Exon/Intron | Nucleotide change | Residue | Variation type | Functional impact* |
|---|---|---|---|---|
| 7 | c.378 T > A | p.D126E | Non-Synonymous | Neutral |
| 7 | c.1287-18delT | N/A | Non-coding | - |
| 9 | c.735C > T | p.V245V | Synonymous | Neutral |
| 13 | c.1744 T > C | p.F582L | Non-Synonymous | Neutral |
| 15 | c.2119 T > C | p.S707P | Non-Synonymous | Neutral |
| 16 | c.2193C > T | p.Y731Y | Synonymous | Neutral |
| 18 | c.2442C > A | p.D814E | Non-Synonymous | Neutral |
| 19 | c.2572 T > C | p.F858L | Non-Synonymous | Damaging |
| 20 | c.2685A > G | p.L895L | Synonymous | Neutral |
| 20 | c.2805G > C | p.T935T | Synonymous | Neutral |
| 23 | c.3118A > G | p.M1040V | Non-Synonymous | Neutral |
| 24 | c.3161C > G | p.P1054R | Non-Synonymous | Damaging |
| 32 | c.4578C > T | p.P1526P | Synonymous | Neutral |
| 39 | c.5557G > A | p.D1853N | Non-Synonymous | Neutral |
| 55 | c.8536 + 13insT | N/A | Non-coding | - |
| 56 | c.8653 + 30insT | N/A | Non-coding | - |
| 62 | c.9372 + 8A > C | N/A | Non-coding | - |
http://genetics.bwh.harvard.edu/pph2/; http://provean.jcvi.org/genome_submit_2.php;
http://chromium.liacs.nl/LOVD2/home.php?select_db=ATM (Last update January 2012); http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=472; http://www.ensembl.org/Homo_sapiens/Gene/Variation_Gene/Table?db=core;g=ENSG00000149311;r=11:108093211–108239829; * http://www.1000genomes.org/1000-genomes-browsers; http://evs.gs.washington.edu/EVS/.
Table 2.
Potential mutations in the ATM gene identified in Brazilian breast cancer patients
| Exon position | Nucleotide change | Residue | Functional impact* | LOH | Sample | Age*** (years) | Clinical stage |
|---|---|---|---|---|---|---|---|
| 13 | c.1636C > G | p.L546V | Damaging | No | M227 | 46 | I |
| 14 | c.1810C > T | p.P604S | Damaging | Yes** | M231 | 50 | IV |
| 20 | c.2804C > G | p.T935R | Neutral | Yes** | M214 | 51 | III |
| 39 | c.5647C > T | p.R1882X | Protein truncation | Yes** | M115 | 75 | III |
| 42 | c.6067G > A | p.G2023R | Damaging | No | M294 | 45 | II |
| 49 | c.6919C > T | p.L2307F | Damaging | No | 72TL | 67 | II |
| Yes | M214 | 51 | |||||
| 50 | c.6995 T > C | p.L2332P | Neutral | Yes | 112TL | 25 | II |
| No | M227 | 46 |
http://chromium.liacs.nl/LOVD2/home.php?select_db=ATM (Last update January 2012); http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=472;
http://www.ensembl.org/Homo_sapiens/Gene/Variation_Gene/Table?db=core;g=ENSG00000149311;r=11:108093211–108239829; http://www.1000genomes.org/1000-genomes-browsers.
*http://genetics.bwh.harvard.edu/pph2/; http://provean.jcvi.org/genome_submit_2.php.
**LOH, indicates loss of the wild type allele.
***age at diagnosis.
Seventeen of the ATM variants were detected in DNA samples from both breast cancer patients and the control population and were, thus, classified as polymorphisms. The polymorphisms observed in patients and controls are summarized in Table 1. Among these, five of the base substitutions were synonymous changes that did not affect amino acid sequences and 8 were non-synonymous changes (Table 1).
Seven unique DNA sequence variants were observed exclusively in samples from breast cancer patients and were classified as potential ATM mutations (Table 2). The alterations observed were distributed throughout the ATM coding sequence with no specific clustering, which would have suggested no mutational hot spot. The spectrum of the ATM variants observed only in breast tumors consisted of six missense mutations affecting exons 13, 14, 20, 42, 49, and 50 and one nonsense mutation affecting exon 39. Three of the base substitutions resulted in conservative amino acid changes (p.L546V, case M227; p.P604S, case M231; and p.L2307F, cases 72TL and M214) and three led to non-conservative changes (p.T935R, case M214; p.G2023R, case M294; and p.L2332P, cases 112TL and M214). Analyses of DNA from Case M115 identified a nonsense mutation in exon 39 (p.R1882X), which was not previously reported in sporadic breast cancer patients and was predicted to lead to protein truncation (Table 2). Two breast tumors demonstrated two distinct ATM variants that were classified as potential causal missense mutations, but several breast tumors and controls displayed multiple ATM variants classified as polymorphisms. The SIFT and PolyPhen algorithms were used to evaluate the potential functional effect of the missense variants, which could be classified as damaging (potential disease-causing mutation) or neutral variants (Tables 1 and 2). Two of the 13 (15.4%) variants identified in both control and breast cancer patients and 4 of the 6 (67%) potential missense mutations identified only in breast cancer patients were classified as damaging.
DNA samples from the primary breast cancer patients were also examined for the occurrence of loss of heterozygosity (LOH) for markers D11S897, D11S1818, and D11S2000 that span approximately 10 cM of the chromosomal region 11q22-23 where the ATM gene is located. Allelic loss was detected in 36/90 (40%) tumors that were informative (heterozygous) for at least one of the markers examined. The frequency of allelic loss at each of the markers analyzed is listed in Table 3. Case M115 had the nonsense mutation in exon 39 (p.R1882X) as well as LOH for marker D11S1818 (Figure 1), suggesting that ATM function was abolished in this tumor. Three of the tumors with potential ATM mutations also showed LOH for the microsatellite markers on chromosomal region 11q23 (Table 2). However, the microsatellite markers used were not located within the ATM gene. We sequenced all DNA samples from normal and tumor tissues that had potential ATM mutations (Table 2). Sequencing results showed evidence of wild type allele loss in cases M214 and M231 harboring potential mutations (Figure 2) and in case M115 with a nonsense mutation. In addition, two patients showed two missense variants, classified as potential ATM mutations. Each patient harbor one variant classified as deleterious based on the prediction of functional impact (M214, p.L2307F; M227, p.L546V) and one variant predicted to be neutral (M214, p.T935R; M227, p.L2332P). We also identified three tumors harboring ATM variants classified as polymorphisms, such as p.S707P, p.F858L, and p.M1040V, with evidence of wild type allele loss.
Table 3.
Summary of Loss of Heterozygosity (LOH) on chromosome 11q in primary breast tumors
| Locus | No. of cases analyzed | Allelic loss/informative cases (%)* |
|---|---|---|
| D11S897 | 100 | 17/71 (24) |
| D11S1818 | 100 | 26/69 (38) |
| D11S2000 | 100 | 22/84 (26) |
*Informative cases: individuals heterozygous for the microsatellite marker examined.
Figure 1.
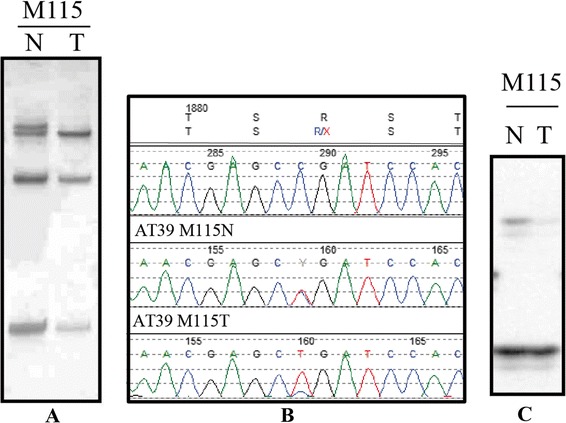
ATM nonsense mutation in a sporadic breast cancer patient. A, SSCP analysis of exon 39 (case M115); B, sequence profile for exon 39 showing a nonsense mutation R1882X; C, loss of heterozygosity at marker D11S1818; N, normal DNA; T, tumor DNA.
Figure 2.
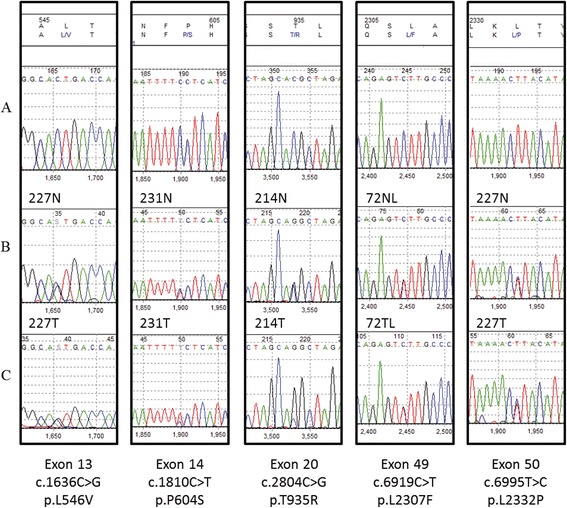
Representative examples of the sequencing analysis of potential ATM mutations in sporadic breast cancer patients. A, upper panel: reference sequence; B, middle panel: sequencing electropherograms from normal tissue samples from breast cancer patients (227 N, 231 N, 214 N, and 72NL); C, lower panel: sequencing electropherograms from tumor tissue samples from breast cancer patients (227 T, 231 T, 214 T, and 72TL). Case 227 showed two potential ATM missense mutations without evidence of LOH; Cases 214 and 231 showed potential ATM mutations with evidence of loss of the wild type allele.
Discussion
Epidemiological studies have suggested that A-T heterozygous individuals are at increased relative risk for cancer, especially breast cancer (Thompson et al. 2005; Ahmed and Rahman 2006). Hence, several studies have investigated the frequency, relative risk, and clinical significance of ATM mutations in breast cancer patients (Ahmed and Rahman 2006; Tavtigian et al. 2009; Graña et al. 2011). In the present study, we examined DNA from a series of 100 breast cancer patients without a family history of breast cancer and 100 healthy individuals for ATM gene mutations. To the best of our knowledge, this is the first study to investigate the occurrence of ATM mutations in a Brazilian breast cancer population. We were able to identify variants occurring only in the breast cancer population. Overall, 7% of the patient population had ATM gene alterations classified as potential mutations, with the majority being missense variants. Low penetrance, potentially deleterious ATM missense variants, which were associated with an increased risk for breast cancer, have been found in other breast cancer populations (Scott et al. 2002; Thorstenson et al. 2003; Graña et al. 2011). Using SIFT and PolyPhen tools to predict the possible impact of the amino acid changes on the ATM function, we found 4 of the 6 variants identified only in breast cancer patients could be classified as potentially deleterious mutations that might impair ATM function, suggesting a causality to the disease. Further experimental and clinical studies are needed to better understand the association of these variants with breast cancer risk. In addition, we also identified a sporadic breast cancer patient carrying a nonsense mutation, with loss of the wild-type allele, which could be a biologically significant ATM mutation. Although this variant, p.R1882X, has been described in Brazilian A-T patients (Mitui et al., 2003; Coutinho et al. 2004); to date, it has not been described in sporadic breast cancer patients.
To our knowledge the exact prevalence of A-T heterozygotes in the Brazilian population is unknown. However, there was a high prevalence for nonsense, splicing site, or frameshift ATM mutations in 27 Brazilian families with classical A-T, and these led to truncated proteins (Coutinho et al. 2004). We identified one individual carrying a nonsense mutation that was predicted to generate a truncated protein, a phenomenon frequently observed in classical A-T (Sandoval et al. 1999; Demuth et al. 2011). This finding is consistent with the frequency of 0.2 – 1% of A-T carriers in the general population, as predicted from epidemiological studies (Easton, 1994). The breast cancer patient carrying a nonsense mutation was constitutively heterozygous for the mutation, but a DNA sample from her tumor showed LOH for the wild-type allele, which is consistent with the tumor suppressor role predicted for the ATM gene in the pathogenesis of breast cancer. We identified two patients with two missense variants classified as potential ATM mutations. Each patient harbored one variant classified as deleterious (M214, pL2307F; M227, pL546V) and another variant classified as neutral (M214, p.T935R; M227, pL2332P). In addition, we identified some tumors with ATM variants classified as polymorphisms with evidence of LOH of the wild type allele, suggesting there was selection of the polymorphic allele during the tumorigenic process. Loss of the wild-type allele of the ATM gene has also been reported in tumors of individuals with familial breast cancer (Bay et al. 1999) and early-onset breast cancer (Izatt et al. 1999). Reduced levels of ATM mRNA and protein have also been reported in sporadic breast tumors, implicating ATM gene inactivation in the development and progression of breast cancer (Ding et al. 2004; Bhattacharya et al. 2013; Bueno et al. 2014). There has been a high frequency of DNA methylation observed in advanced breast tumors and attributed to reduced ATM expression and function (Vo et al. 2004). Furthermore, recent studies demonstrated that over-expression of microRNAs, such as miR181 and miR421, may be involved in ATM down-regulation in breast cancer (Fang et al. 2010; Bueno et al. 2014).
More than 70% of the ATM mutations described in classical A-T families are frameshift or nonsense mutations leading to protein truncation (Cavaciutti et al. 2005; Goldgar et al. 2011). To date, most of the ATM mutations observed in breast cancer patients, including the data reported here, are missense mutations distributed throughout the ATM gene, but mainly outside of the PI-3 K domain or other putative functional domains defined by sequence similarity (Savitsky et al. 1995a; Bosotti et al. 2000; Thorstenson et al. 2001; Tavtigian et al. 2009). Thus, it is difficult to define clear genotype-phenotype associations, and the clinical implications of these mutations remain unknown.
In vitro studies have shown that lymphocytes from ATM mutation-carriers are more radiosensitive than lymphocytes from normal donors (West et al. 1995), suggesting that even low radiation dosages might be deleterious for such individuals, increasing their risk for breast cancer or inducing severe complications. Many clinical studies have tested the hypothesis that ATM heterozygote breast cancer patients exhibit enhanced radiosensitivity; yet, most of those studies failed to show a clear association between ATM mutation status and increased radiosensitivity (Ramsay et al. 1996; Appleby et al. 1997; Ramsay et al. 1998; Weissberg et al. 1998; Shayeghi et al. 1998). Iannuzzi et al. (2002) reported that breast cancer patients harboring ATM missense mutations are more susceptible to develop subcutaneous late responses after radiation therapy. Using an ATM kinase assay, Scott et al. (2002) demonstrated that only 1 of 5 missense mutations detected in breast cancer patients abrogated ATM kinase activity when expressed in AT1ABR cells. The ATM protein is a large serine-threonine protein kinase that plays a key role in the cellular response induced by ionizing radiation by directly interacting and phosphorylating key components of different DNA damage response pathways such as TP53, MDM2, and BRCA1. Therefore, we cannot rule out the potential effect of these missense mutations on interindividual ionizing radiation sensitivity and cancer risk (Banin et al. 1998; Kastan and Lim 2000; Wang et al. 2000). Recently, Byrd et al. (2012) reported on a breast cancer patient with a severe reaction to radiotherapy who was a carrier of biallelic ATM mutations, including a missense mutation c.8672C > A (p.Gly2891Asp), and displayed residual ATM kinase activity. The expression of a missense mutated ATM protein, especially resulting from a non-conservative missense mutation, may alter the ATM protein stability and function (Gatti et al. 1999). In addition, low levels of ATM protein expression have been associated with severe adverse normal tissue reaction in breast cancer patients, suggesting that ATM protein levels may represent an independent predictive biomarker for clinical radiosensitivity (Fang et al. 2010).
The limitations of the present study are the small number of samples and low sensitivity of the methodology; although, we identified several ATM polymorphisms in both breast cancer patients and the control population. In an effort to better understand the etiology and to identify factors associated with the predisposition to breast cancer, several groups have recently used next-generation sequencing (NGS) to determine the pattern of mutations associated with the disease. Despite the large number of studies using NGS, no investigation has specifically focused on the ATM gene. Studies using exome sequencing technologies in breast cancer showed a high prevalence of mutations and variants in genes such as BRCA1, BRCA2, TP53, and PIK3CA, but few reported findings for ATM (Network CGA 2012; Banerji et al. 2012). A study evaluating 691 sporadic breast tumors, identified mutations affecting genes such as PIK3CA, FGFR2, TP53, and ERBB2, but no alterations in the ATM gene (Wilkerson et al. 2014). On the other hand, studies evaluating hereditary breast cancer have identified mutations and variants of ATM in a subset of cases (Snape et al. 2012; Kurian, et al. 2014). Thus, at present, data related to the ATM gene in sporadic breast tumors are limited; thus, it is difficult to make an adequate assessment of its role in sporadic breast cancer. Our group recently analyzed exome sequencing data from breast cancer samples, including one of the cases (M294) included here, and our preliminary (unpublished) results identified a larger number of ATM alterations compared with the data obtained from SSCP screening and direct DNA sequencing. Using SSCP analysis, we identified two missense variants in case M294 (p.L895L and p.G2023R); however, using exome sequencing, we found five missense variants (p.Y731Y, pP872S, pL895L, p.N1983S, and p.G2023R) and many INDELs and base substitutions in the introns of the ATM gene in this same case (Additional file 2). Thus, the NGS technique is a promising approach to better estimate the number of ATM variants related to breast cancer.
Conclusions
Our results support the hypothesis that ATM heterozygotes carrying low penetrance ATM mutant alleles are at risk for breast cancer. There is a potential increased sensitivity of these heterozygotes to ionizing radiation; therefore, identifying these carriers may have an impact on the treatment offered to these individuals. We have identified several ATM variants in this study that may potentially act as disease-causing missense mutations. However, further functional studies to evaluate the biological effects of the potential ATM missense mutations observed in breast cancer patients and large case–control studies using NGS (amplicon and/or exome sequencing) are needed. These investigations should contribute to the understanding of the role played by the ATM gene in the etiology and pathobiology of breast cancer.
Materials and methods
Samples
Paired normal and tumor DNA samples were obtained from 100 sporadic breast cancer patients with no family history of A-T at Hospital do Cancer A.C. Camargo, São Paulo, Brazil (from 1993 to 1998). The age of the patients at the time of operation ranged from 25 to 78 years (median 48 years). Tumor samples were dissected to remove residual normal tissue before freezing and stored in liquid nitrogen. The largest diameter of the tumors was recorded. The number of lymph node metastases was determined by microscopic examination of an average of 24 lymph nodes per patient. Tumor metastasis to the lymph nodes was detected in 58 patients. All cases were submitted for histopathological review of tumor sections to confirm the diagnosis. All tumors were classified as invasive ductal carcinomas according to the World Health Organization Histological Typing of Breast Tumors classification, and the clinical disease stage of each patient was determined according to the 5th Edition of the UICC TNM classification of malignant tumors. In addition, DNA was extracted from one hundred control blood samples from healthy and unrelated women at Hemocentro-Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo. The Institutional Ethics Committee approved this study and all subjects studied (patients and matched controls) signed an informed consent for participation.
DNA extraction
Normal and tumoral tissue was ground to a powder using a frozen tissue pulverizer (Termovac). The powder was resuspended in 1 mL of lysis buffer (10 mM Tris–HCl, pH 7.6, 1 mM EDTA, and 0.6% SDS) with 100 μg/mL proteinase K, and incubated at 37°C overnight. High molecular weight DNA was extracted with phenol-chloroform and precipitated with ethanol. Blood samples were collected in EDTA (5 mL) and DNA was extracted as described by Lahiri & Nurnberger (1991) and adapted by Salazar et al. (1998).
Loss of heterozygosity (LOH) analysis
The LOH analysis for the chromosomal region 11q23 was performed using three polymorphic microsatellite markers (D11S897, D11S1818, and D11S2000). Primer sequences for these markers were obtained from the Genome Data Base (http://www.ncbi.nlm.nih.gov/probe/). Allelic loss assays were performed as previously described (Miracca et al. 1999). Allelic loss, determined by densitometric scan (UltroScan XL; Pharmacia), was considered to be complete or partial if the intensity of one allele was reduced by at least 40% in the tumor compared with normal DNA samples from the same patient. LOH was scored for informative (heterozygous) patients only.
Mutation analysis
Mutation analyses were performed by Single Strand Conformation Polymorphism (PCR-SSCP) and direct DNA sequencing analysis, as previously described (Miracca et al. 1999). Briefly, sixty-four sets of primers were used to amplify exons 2 to 63 of the ATM gene, as previously described by Vorechovsky et al. (1996). Radiolabeled PCR reactions were performed in 25 μL volumes using 50–100 ng of genomic DNA template, containing 0.1 mCi of [α32P-dCTP] (Amersham, specific activity, 3000 Ci mmol−1). Amplification products were diluted ten-fold in a buffer containing 95% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol, heated at 83°C for 5 min, and applied (3 μL per lane) on two 6% polyacrylamide non-denaturing gels, one containing 5% glycerol and the other 10% glycerol. Electrophoresis was performed at 6 W for 14–16 h at room temperature with two cooling fans. Band shift mobility was detected by autoradiography of dried gels using Kodak X-Omat XAR film with an intensifying screen for 12–48 h at −70°C. DNA samples with evidence of sequence variants in the SSCP analysis were characterized by direct sequencing using the Big Dye Terminator kit (Life Technologies™) according to the manufacturer’s specifications. Sequencing reaction products were purified by ethanol/EDTA precipitation and resuspended in Hi-Di™ formamide (Life Technologies™). Samples were separated by electrophoresis on the ABI Prism 3730 DNA Analyzer (Life Technologies™) and the results were evaluated in Mutation SurveyorR DNA Variant Analysis Software version 3.30 (Softgenetics LLC). The variants identified were evaluated using the following public ATM databases to determine if changes were novel or already known: http://chromium.liacs.nl/LOVD2/home.php?select_db=ATM; NCBI dbSNP - http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=472; Ensembl - http://www.ensembl.org/Homo_sapiens/Info/Index; and EVS - http://evs.gs.washington.edu/EVS/. In addition, the algorithms PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://provean.jcvi.org/genome_submit_2.php) were used to predict the possible impact of amino acid changes on the function of the ATM protein.
Acknowledgments
We thank Sibeli Salaorni for technical support and Ursula Urias dos Santos for reading the manuscript. This work was supported by FAPESP – Fundação de Amparo a Pesquisa do Estado de São Paulo (96/4027-3) and by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, 305408/2009-7 and 577587/2008-0).
Additional files
Representative example of the sequencing analysis of a breast cancer patient harboring both variants p.F858L and p.P1054R.
Summary of the ATM variants observed by exome sequencing analysis in Tumor and Normal tissue of one breast cancer patient (M294) Chromosomal coordinates of the ATM variants observed by exome sequencing analysis of Tumor and Normal tissues from one breast cancer patient (M294). The results were analyzed using the Golden Helix GenomeBrowser (www.goldenhelix.com/GenomeBrowse/).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Nagai MA conceived the study’s aims and design and contributed to the data analysis, manuscript preparation, manuscript editing, and review. Feiloter HE and Mulligan LM contributed to the data analysis, manuscript preparation, manuscript editing, and review. Miracca EC carried out sequencing experiments, data acquisition, the literature review, and manuscript preparation. Mangone FR carried out the literature review, data acquisition, data analysis, and manuscript preparation. All authors read and approved the manuscript.
Contributor Information
Flavia Rotea Mangone, Email: flavia@lim24.fm.usp.br.
Elisabete C Miracca, Email: ecmiracca@yahoo.com.br.
Harriet E Feilotter, Email: hf4@queensu.ca.
Lois M Mulligan, Email: mulligal@queensu.ca.
Maria Aparecida Nagai, Email: nagai@usp.br.
References
- Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25(43):5906–5911. doi: 10.1038/sj.onc.1209873. [DOI] [PubMed] [Google Scholar]
- Ambrose M, Gatti RA. Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood. 2013;121(20):4036–4045. doi: 10.1182/blood-2012-09-456897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby JM, Barber JB, Levine E, Varley JM, Taylor AM, Stankovic T, Heighway J, Warren C, Scott D. Absence of mutations in the ATM gene in breast cancer patients with severe responses to radiotherapy. Br J Cancer. 1997;6(12):1546–1549. doi: 10.1038/bjc.1997.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Piña V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86(1):159–171. doi: 10.1016/S0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Bay JO, Uhrhammer N, Pernin D, Presneau N, Tchirkov A, Vuillaume M, Laplace V, Grancho M, Verrelle P, Hall J, Bignon YJ. High incidence of cancer in a family segregating a mutation of the ATM gene: possible role of ATM heterozygosity in cancer. Hum Mutat. 1999;14(6):485–492. doi: 10.1002/(SICI)1098-1004(199912)14:6<485::AID-HUMU7>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Beamish H, Lavin MF. Radiosensitivity in ataxia-telangiectasia: anomalies in radiation-induced cell cycle delay. Int J Radiat Biol. 1994;65(2):175–184. doi: 10.1080/09553009414550211. [DOI] [PubMed] [Google Scholar]
- Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Bhattacharya N, Mukherjee N, Singh RK, Sinha S, Alam N, Roy A, Roychoudhury S, Panda CK. Frequent alterations of MCPH1 and ATM are associated with primary breast carcinoma: clinical and prognostic implications. Ann Surg Oncol. 2013;20(Suppl 3):S424–S432. doi: 10.1245/s10434-012-2715-6. [DOI] [PubMed] [Google Scholar]
- Bosotti R, Isacchi A, Sonnhammer EL. FAT: a novel domain in PIK-related kinases. Trends Biochem Sci. 2000;25(5):225–227. doi: 10.1016/S0968-0004(00)01563-2. [DOI] [PubMed] [Google Scholar]
- Broeks A, Braaf LM, Huseinovic A, Schmidt MK, Russell NS, Van Leeuwen FE, Hogervorst FB, Veer LJ VT. The spectrum of ATM missense variants and their contribution to contralateral breast cancer. Breast Cancer Res Treat. 2008;107(2):243–248. doi: 10.1007/s10549-007-9543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno RC, Canevari RA, Villacis RA, Domingues MA, Caldeira JR, Rocha RM, Drigo SA, Rogatto SR. ATM down-regulation is associated with poor prognosis in sporadic breast carcinomas. Ann Oncol. 2014;25(1):69–75. doi: 10.1093/annonc/mdt421. [DOI] [PubMed] [Google Scholar]
- Byrd PJ, Srinivasan V, Last JI, Smith A, Biggs P, Carney EF, Exley A, Abson C, Stewart GS, Izatt L, Taylor AM. Severe reaction to radiotherapy for breast cancer as the presenting feature of ataxia telangiectasia. Br J Cancer. 2012;106(2):262–268. doi: 10.1038/bjc.2011.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGA Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS. The role of ATM in DNA damage responses and cancer. Oncogene. 1998;17(25):3301–3308. doi: 10.1038/sj.onc.1202577. [DOI] [PubMed] [Google Scholar]
- Cavaciutti E, Laugé A, Janin N, Ossian K, Hall J, Stoppa-Lyonnet D, Andrieu N. Cancer risk according to type and location of ATM mutation in ataxia-telangiectasia families. Genes Chromosom Cancer. 2005;42:1–9. doi: 10.1002/gcc.20101. [DOI] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Coutinho G, Mitui M, Campbell C, Costa Carvalho BT, Nahas S, Sun X, Huo Y, Lai CH, Thorstenson Y, Tanouye R, Raskin S, Kim CA, Llerena J, Jr, Gatti RA. Five haplotypes account for fifty-five percent of ATM mutations in Brazilian patients with ataxia telangiectasia: seven new mutations. Am J Med Genet A. 2004;126A(1):33–40. doi: 10.1002/ajmg.a.20570. [DOI] [PubMed] [Google Scholar]
- Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2014;33:3351–3360. doi: 10.1038/onc.2013.275. [DOI] [PubMed] [Google Scholar]
- Demuth I, Dutrannoy V, Marques W, Jr, Neitzel H, Schindler D, Dimova PS, Chrzanowska KH, Bojinova V, Gregorek H, Graul-Neumann LM, von Moers A, Schulze I, Nicke M, Bora E, Cankaya T, Oláh E, Kiss C, Bessenyei B, Szakszon K, Gruber-Sedlmayer U, Kroisel PM, Sodia S, Goecke TO, Dork T, Digweed M, Sperling K, Sa J, Lourenco CM, Varon R. New mutations in the ATM gene and clinical data of 25 AT patients. Neurogenetics. 2011;12:273–282. doi: 10.1007/s10048-011-0299-0. [DOI] [PubMed] [Google Scholar]
- Deng CX, Brodie SG. Knockout mouse models and mammary tumorigenesis. Semin Cancer Biol. 2001;11(5):387–394. doi: 10.1006/scbi.2001.0394. [DOI] [PubMed] [Google Scholar]
- Derheimer FA, Kastan MB. Multiple roles in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584:3675–3681. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SL, Sheu LF, Yu JC, Yang TL, Chen BF, Leu FJ, Shen CY. Abnormality of the DNA double-strand-break checkpoint/repair genes ATM, BRCA1 and TP53 in breast cancer is related to tumour grade. Br J Cancer. 2004;90:1995–2001. doi: 10.1038/sj.bjc.6601804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF. Cancer risks in A-T heterozygotes. Int J Radiat Biol. 1994;66(6 Suppl):S177–S182. doi: 10.1080/09553009414552011. [DOI] [PubMed] [Google Scholar]
- Fang Z, Kozlov S, McKay MJ, Woods R, Birrell G, Sprung CN, Murrell DF, Wangoo K, Teng L, Kearsley JH, Lavin MF, Graham PH, Clarke RA. Low levels of ATM in breast cancer patients with clinical radiosensitivity. Genome Integr. 2010;1(1):9. doi: 10.1186/2041-9414-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher O, Johnson N, Dos Santos Silva I, Orr N, Ashworth A, Nevanlinna H, Heikkinen T, Aittomäki K, Blomqvist C, Burwinkel B, Bartram CR, Meindl A, Schmutzler RK, Cox A, Brock I, Elliott G, Reed MW, Southey MC, Smith L, Spurdle AB, Hopper JL, Couch FJ, Olson JE, Wang X, Fredericksen Z, Schürmann P, Waltes R, Bremer M, Dörk T, Devilee P, kConFab Investigators; AOCS Group Breast cancer association consortium. Missense variants in ATM in 26,101 breast cancer cases and 29,842 controls. Cancer Epidemiol Biomarkers Prev. 2010;19(9):2143–2151. doi: 10.1158/1055-9965.EPI-10-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine. 1991;70(2):99–117. doi: 10.1097/00005792-199103000-00003. [DOI] [PubMed] [Google Scholar]
- Gatti RA, Tward A, Concannon P. Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab. 1999;68(4):419–423. doi: 10.1006/mgme.1999.2942. [DOI] [PubMed] [Google Scholar]
- Goldgar DE, Healey S, Dowty JG, Silva L, Cheng X, Spurdle AB, Terry MB, Daly MJ, Buys SM, Southey MC, Andrulis I, John EM, Khanna KK, Hopper JL, Oefner PJ, Lakhani S, Chenevix-Trench G. Rare variants in the ATM gene and risk of breast cancer. Breast Can Res. 2011;13(R73):1–9. doi: 10.1186/bcr2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graña B, Fachal L, Darder E, Balmaña J, Ramón Y, Cajal T, Blanco I, Torres A, Lázaro C, Diez O, Alonso C, Santamariña M, Velasco A, Teulé A, Lasa A, Blanco A, Izquierdo A, Borràs J, Gutiérrez-Enríquez S, Vega A, Brunet J. Germline ATM mutational analysis in BRCA1/BRCA2 negative hereditary breast cancer families by MALDI-TOF mass spectrometry. Breast Cancer Res Treat. 2011;128(2):573–579. doi: 10.1007/s10549-011-1462-x. [DOI] [PubMed] [Google Scholar]
- Iannuzzi CM, Atencio DP, Green S, Stock RG, Rosenstein BS. ATM mutations in female breast cancer patients predict for an increase in radiation-induced late effects. Int J Radiat Oncol Biol Phys. 2002;52(3):606–613. doi: 10.1016/S0360-3016(01)02684-0. [DOI] [PubMed] [Google Scholar]
- Izatt L, Greenman J, Hodgson S, Ellis D, Watts S, Scott G, Jacobs C, Liebmann R, Zvelebil MJ, Mathew C, Solomon E. Identification of germline missense mutations and rare allelic variants in the ATM gene in early-onset breast cancer. Genes Chromosom Cancer. 1999;26(4):286–294. doi: 10.1002/(SICI)1098-2264(199912)26:4<286::AID-GCC2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1(3):179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, Lavin MF. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. 1998;20(4):398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci U S A. 1999;96(26):14973–14977. doi: 10.1073/pnas.96.26.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32(19):2001–2009. doi: 10.1200/JCO.2013.53.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri D, Nurnberger J. A rapid non-enzimatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acid Res. 1991;19:5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson GP, Zhang G, Ding S, Foldenauer K, Udar N, Gatti RA, Neuberg D, Lunetta KL, Ruckdeschel JC, Longmate J, Flanagan S, Krontiris TG. An allelic variant at the ATM locus is implicated in breast cancer susceptibility. Genet Test. 1997;1(3):165–170. doi: 10.1089/gte.1997.1.165. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Khanna KK, Beamish H, Spring K, Watters D, Shiloh Y. Relationship of the ataxia-telangiectasia protein ATM to phosphoinositide 3-kinase. Trends Biochem Sci. 1995;20(10):382–383. doi: 10.1016/S0968-0004(00)89083-0. [DOI] [PubMed] [Google Scholar]
- Levitt NC, Hickson ID. Caretaker tumour suppressor genes that defend genome integrity. Trends Mol Med. 2002;8(4):179–186. doi: 10.1016/S1471-4914(02)02298-0. [DOI] [PubMed] [Google Scholar]
- Li S, Ting NS, Zheng L, Chen PL, Ziv Y, Shiloh Y, Lee EY, Lee WH. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature. 2000;406(6792):210–215. doi: 10.1038/35018134. [DOI] [PubMed] [Google Scholar]
- Liang Y, Lin SY, Brunicardi C, Goss J, Li K. DNA damage response pathways in tumor suppression and cancer treatment. World J Surg. 2009;33:661–666. doi: 10.1007/s00268-008-9840-1. [DOI] [PubMed] [Google Scholar]
- Liao MJ, Van Dyke T. Critical role for Atm in suppressing V(D)J recombination-driven thymic lymphoma. Genes Dev. 1999;13(10):1246–1250. doi: 10.1101/gad.13.10.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrou A, Tsangaris GT, Roma E, Kolialexi A. The ATM gene and ataxia telangiectasia. Anticancer Res. 2008;28:401–406. [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, Kastan MB, Katzir E, Oren M. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15(9):1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RL. Variants in the ATM gene and breast cancer susceptibility. Genome Med. 2009;1(1):12. doi: 10.1186/gm12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miracca EC, Kowalski LP, Nagai MA. High prevalence of p16 genetic alterations in head and neck tumours. Br J Cancer. 1999;81(4):677–683. doi: 10.1038/sj.bjc.6690747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitui M, Campbell C, Coutinho G, Sun X, Lai CH, Thorstenson Y, Castellvi-Bel S, Fernandez L, Monros E, Carvalho BT, Porras O, Fontan G, Gatti RA. Independent mutational events are rare in the ATM gene: haplotype prescreening enhances mutation detection rate. Hum Mutat. 2003;22(1):43–50. doi: 10.1002/humu.10232. [DOI] [PubMed] [Google Scholar]
- Prokopcova J, Kleibl Z, Banwell CM, Pohlreich P. The role of ATM in breast cancer development. Breast Cancer Res Treat. 2007;104:121–128. doi: 10.1007/s10549-006-9406-6. [DOI] [PubMed] [Google Scholar]
- Ramsay J, Birrell G, Lavin M. Breast cancer and radiotherapy in ataxia-telangiectasia heterozygote. Lancet. 1996;347(9015):1627. doi: 10.1016/S0140-6736(96)91114-9. [DOI] [PubMed] [Google Scholar]
- Ramsay J, Birrell G, Lavin M. Testing for mutations of the ataxia telangiectasia gene in radiosensitive breast cancer patients. Radiother Oncol. 1998;47(2):125–128. doi: 10.1016/S0167-8140(98)00014-0. [DOI] [PubMed] [Google Scholar]
- Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D. The breast cancer susceptibility collaboration (UK), Easton DF, Stratton MR, rahman N. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Gen. 2006;38(8):873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- Rotman G, Shiloh Y. ATM: a mediator of multiple responses to genotoxic stress. Oncogene. 1999;18(45):6135–6144. doi: 10.1038/sj.onc.1203124. [DOI] [PubMed] [Google Scholar]
- Salazar LA, Hirata MH, Cavalli SS, Machado MO, Hirata RD. Optimized procedure for DNA isolation from fresh and cryopreserved clotted human blood usefulin clinical molecular testing. Clin Chem. 1998;44(8):1748–1750. [PubMed] [Google Scholar]
- Sandoval N, Platzer M, Rosenthal A, Dork T, Bendix R, Skawran B, Stuhrmann M, Wegner RD, Sperling K, Banin S, Shiloh Y, Baumer A, Bernthaler U, Sennefelder H, Brohm M, Weber BH, Schindler D. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum Mol Genet. 1999;8(1):69–79. doi: 10.1093/hmg/8.1.69. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS, Shiloh Y, Rotman G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet. 1995;4(11):2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- Scott SP, Bendix R, Chen P, Clark R, Dork T, Lavin MF. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc Natl Acad Sci U S A. 2002;99(2):925–930. doi: 10.1073/pnas.012329699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayeghi M, Seal S, Regan J, Collins N, Barfoot R, Rahman N, Ashton A, Moohan M, Wooster R, Owen R, Bliss JM, Stratton MR, Yarnold J. Heterozygosity for mutations in the ataxia telangiectasia gene is not a major cause of radiotherapy complications in breast cancer patients. Br J Cancer. 1998;78(7):922–927. doi: 10.1038/bjc.1998.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein: the importance of being active. J Cell Biol. 2012;198(3):273–275. doi: 10.1083/jcb.201207063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotixic stress, and more. Mol. Cell Biol. 2013;14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- Snape K, Ruark E, Tarpey P, Renwick A, Turnbull C, Seal S, Murray A, Hanks S, Douglas J, Stratton MR, Rahman N. Predisposition gene identification in common cancers by exome sequencing: insights from familial breast cancer. Breast Cancer Res Treat. 2012;134(1):429–433. doi: 10.1007/s10549-012-2057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperka T, Wang J, Rudolph L. DNA damage checkpoints in stem cells, ageing and cancer. Mol Cell Biol. 2012;13:579–590. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. 1986;39(5):573–583. [PMC free article] [PubMed] [Google Scholar]
- Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med. 1991;325(26):1831–1836. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med. 1987;316(21):1289–1294. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Oefner PJ, Babikyan D, Hartmann A, Healey S, Le Calvez-Kelm F, Lesueur F, Byrnes GB, Chuang SC, Forey N, Feuchtinger C, Gioia L, Hall J, Hashibe M, Herte B, McKay Chopin S, Thomas A, Vallée MP, Voegele C, Webb PM, Whiteman DC. Australian cancer study; breast cancer family registries (BCFR); Kathleen cuningham foundation consortium for research into familial aspects of breast cancer (kConFab), sangrajrang S, hopper JL, Southey MC, andrulis IL, john EM, chenevix-trench G (2009) rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am J Hum Genet. 2009;85(4):427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58(10):1009–1015. doi: 10.1136/jcp.2005.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97(11):813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res. 2002;509(1–2):49–78. doi: 10.1016/S0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- Thorstenson YR, Roxas A, Kroiss R, Jenkins MA, Yu KM, Bachrich T, Muhr D, Wayne TL, Chu G, Davis RW, Wagner TM, Oefner PJ. Contributions of ATM mutations to familial breast and ovarian cancer. Cancer Res. 2003;63(12):3325–3333. [PubMed] [Google Scholar]
- Thorstenson YR, Shen P, Tusher VG, Wayne TL, Davis RW, Chu G, Oefner PJ. Global analysis of ATM polymorphism reveals significant functional constraint. Am J Hum Genet. 2001;69(2):396–412. doi: 10.1086/321296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo QN, Kim WJ, Cvitanovic L, Boudreau DA, Ginzinger DG, Brown KD. The ATM gene is a target for epigenetic silencing in locally advanced breast cancer. Oncogene. 2004;23(58):9432–9437. doi: 10.1038/sj.onc.1208092. [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Rasio D, Luo L, Monaco C, Hammarström L, Webster AD, Zaloudik J, Barbanti Brodani G, James M, Russo G, Croce CM, Negrini M. The ATM gene and susceptibility to breast cancer: analysis of 38 breast tumors reveals no evidence for mutation. Cancer Res. 1996;56(12):2726–2732. [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14(8):927–939. [PMC free article] [PubMed] [Google Scholar]
- Weissberg JB, Huang DD, Swift M. Radiosensitivity of normal tissues in ataxia-telangiectasia heterozygotes. Int J Radiat Oncol Biol Phys. 1998;42(5):1133–1136. doi: 10.1016/S0360-3016(98)00295-8. [DOI] [PubMed] [Google Scholar]
- West CM, Elyan SA, Berry P, Cowan R, Scott D. A comparison of the radiosensitivity of lymphocytes from normal donors, cancer patients, individuals with ataxia-telangiectasia (A-T) and A-T heterozygotes. Int J Radiat Biol. 1995;68(2):197–203. doi: 10.1080/09553009514551101. [DOI] [PubMed] [Google Scholar]
- Wilkerson MD, Cabanski CR, Sun W, Hoadley KA, Walter V, Mose LE, Troester MA, Hammerman PS, Parker JS, Perou CM, Hayes DN. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res. 2014;42(13):e107. doi: 10.1093/nar/gku489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10(19):2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Yuille MA, Coignet LJ. The ataxia telangiectasia gene in familial and sporadic cancer. Recent Results Cancer Res. 1998;154:156–173. doi: 10.1007/978-3-642-46870-4_9. [DOI] [PubMed] [Google Scholar]